Dernière mise à jour le 01/06/2026
IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : AGONISTES SELECTIFS DES RECEPTEURS 5-HT1 - N02CC01
Ce médicament est indiqué dans le traitement de la crise de migraine avec ou sans aura (sensation subjective passagère qui précède la crise de migraine, très variable d’un sujet à l’autre et qui touche l’audition, la vue ).
La forme solution pour pulvérisation nasale est particulièrement adaptée à la crise de migraine s’accompagnant de nausées et de vomissements.
Présentations
> 2 ampoule(s) en verre de 0,1 ml avec embout nasal polypropylène
Code CIP : 343 383-4 ou 34009 343 383 4 2
Déclaration de commercialisation : 01/09/1999
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 6,92 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 7,94 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 17/04/2013 | Renouvellement d'inscription (CT) | Le service médical rendu par IMIGRANE reste important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 30/10/2025
IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour une ampoule
Une ampoule correspond à une pulvérisation unique de 10 mg de sumatriptan.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution pour pulvérisation nasale.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Adultes (à partir de 18 ans)
La dose recommandée est d’une pulvérisation de 20 mg administrée dans une seule narine.
Cependant, en raison de la variabilité inter et intra individuelle des patients, aussi bien des crises de migraine que de l’absorption de sumatriptan, une dose de 10 mg peut s’avérer efficace chez certains patients.
Pour les patients non soulagés par la dose de 10 mg, la dose de 20 mg peut s’avérer nécessaire lors de la crise suivante.
En l’absence de soulagement après la première dose, il n’est pas recommandé d’administrer une deuxième dose au cours de la même crise. Toutefois, cette crise peut être traitée avec du paracétamol, de l’acide acétylsalicylique, ou des anti-inflammatoires non-stéroïdiens. Le sumatriptan pourra être utilisé pour les crises suivantes.
Si un patient a été soulagé après la première prise mais que les symptômes réapparaissent, une seconde dose peut être utilisée dans les 24 heures suivantes à condition de respecter un intervalle d’au moins 2 heures entre les 2 administrations.
Ne pas utiliser plus de 2 pulvérisations de 20 mg de sumatriptan solution pour pulvérisation nasale par 24 heures.
Adolescents (12-17 ans)
L’utilisation de sumatriptan chez les adolescents doit se faire selon les recommandations d’un médecin spécialiste de la migraine, en tenant compte des recommandations nationales.
La dose recommandée est d’une pulvérisation de 10 mg de sumatriptan solution pour pulvérisation nasale administrée dans une seule narine.
En l’absence de soulagement après la première dose, il n’est pas recommandé d’administrer une deuxième dose au cours de la même crise. Toutefois, cette crise peut être traitée avec du paracétamol, de l’acide acétylsalicylique, ou des anti-inflammatoires non-stéroïdiens. Le sumatriptan pourra être utilisé pour les crises suivantes.
Si un patient a été bien soulagé à la première prise mais que les symptômes réapparaissent, une seconde dose peut être utilisée dans les 24 heures suivantes à condition de respecter un intervalle d’au moins 2 heures entre les 2 administrations.
Ne pas utiliser plus de deux pulvérisations de 10 mg de sumatriptan solution pour pulvérisation nasale par 24 heures.
Enfant (de moins de 12 ans)
L’utilisation du sumatriptan par voie nasale n’est pas recommandée chez les enfants de moins de 12 ans en raison du manque de données sur la sécurité et l’efficacité du sumatriptan chez l’enfant.
Patient âgé (de plus de 65 ans)
Il n’existe pas de données concernant l’utilisation du sumatriptan par voie nasale chez les patients de plus de 65 ans. La cinétique chez les patients âgés n’a pas été suffisamment étudiée. C’est pourquoi, l’utilisation du sumatriptan n’est pas recommandée tant que des données complémentaires ne seront pas disponibles.
Mode d’administration
Le sumatriptan ne doit pas être utilisé en prophylaxie.
Ne pas dépasser la dose recommandée de sumatriptan.
Il est recommandé d’utiliser le sumatriptan en monothérapie dans le traitement de la crise de migraine et de ne pas le donner de façon concomitante avec de l’ergotamine ou des dérivés (y compris le méthysergide) (voir rubrique 4.3).
Il est conseillé de prendre le sumatriptan aussi précocement que possible dès la survenue de la crise migraineuse. Son efficacité est identique quel que soit le stade de la crise durant lequel il est administré.
Le sumatriptan ne doit pas être donné aux patients ayant des antécédents d’infarctus du myocarde ou une pathologie cardiaque ischémique, un vasospasme coronarien (angor de Printzmetal), une pathologie vasculaire périphérique ou aux patients présentant des symptômes de pathologie cardiaque ischémique ou des signes compatibles avec une pathologie cardiaque ischémique.
Le sumatriptan ne doit pas être administré aux patients présentant des antécédents d’accident vasculaire cérébral (AVC) ou d’accident ischémique transitoire (AIT).
Le sumatriptan ne doit pas être administré aux patients ayant une insuffisance hépatique sévère.
L'utilisation du sumatriptan est contre-indiquée chez les patients ayant une hypertension modérée ou sévère.
L'utilisation du sumatriptan est contre-indiquée chez les patients ayant une hypertension légère traitée non contrôlée.
L’association du sumatriptan avec l’ergotamine ou les dérivés de l’ergotamine ou de tout autre triptan/agoniste 5-HydroxyTryptamine1 (5-HT1) (y compris le méthysergide) est contre-indiquée (voir rubrique 4.5).
L’association du sumatriptan avec les inhibiteurs de la monoamine oxydase (IMAO) est contre-indiquée.
Le sumatriptan ne doit pas être utilisé dans les 2 semaines suivant l'arrêt d'un traitement par les inhibiteurs de la monoamine oxydase.
4.4. Mises en garde spéciales et précautions d'emploi
Le sumatriptan ne doit être utilisé qu'après avoir établi un diagnostic certain de migraine.
Le sumatriptan n'est pas indiqué dans le traitement des migraines hémiplégiques, basilaires ou ophtalmoplégiques.
Avant de traiter avec du sumatriptan, il est nécessaire d'exclure les pathologies neurologiques potentiellement graves (par exemple : AVC, AIT) si le patient présente des symptômes atypiques ou s’il n’a pas reçu le diagnostic approprié pour l’utilisation de sumatriptan.
Après administration, la prise de sumatriptan peut être associée à des symptômes transitoires comprenant des douleurs thoraciques ou une sensation d'oppression pouvant être intense et pouvant s'étendre au niveau de la gorge (voir rubrique 4.8). Si la symptomatologie évoque une ischémie cardiaque, il ne faut pas prendre de doses supplémentaires de sumatriptan et des explorations appropriées devront être réalisées.
Le sumatriptan ne doit pas être administré aux patients ayant des facteurs de risque de maladie cardiaque ischémique, y compris les gros fumeurs ou les patients utilisant des thérapies de substitution à base de nicotine, sans un bilan cardiovasculaire préalable (voir rubrique 4.3). Une attention particulière doit être portée aux femmes ménopausées et aux hommes de plus de 40 ans présentant ces facteurs de risque. Cependant, ce bilan peut ne pas identifier tous les patients qui ont une maladie cardiovasculaire et, dans de très rares cas, des évènements cardiaques graves sont survenus chez des patients sans maladie cardiovasculaire sous-jacente et chez les adolescents (voir rubrique 4.8).
Le sumatriptan devra être donné avec prudence chez les patients présentant une hypertension légère contrôlée. En effet, une augmentation transitoire de la pression artérielle ainsi que des résistances vasculaires périphériques ont été observées chez une petite proportion de patients (voir section 4.3).
Après commercialisation, de rares cas de syndrome sérotoninergique (incluant une modification de l'état mental, des manifestations neurovégétatives et des troubles neuromusculaires) ont été décrits après utilisation concomitante d'un inhibiteur sélectif de la recapture de la sérotonine (ISRS) et du sumatriptan. Des cas de syndrome sérotoninergique ont également été rapportés après administration concomitante de triptans et d’inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSN). Si l'association du sumatriptan et d'un ISRS/IRSN est cliniquement justifiée, il est conseillé d'assurer une surveillance appropriée du patient (voir rubrique 4.5).
Le sumatriptan doit être utilisé avec précaution chez les patients ayant des antécédents de convulsions ou présentant d’autres facteurs de risque susceptibles d’abaisser le seuil épileptogène, car des cas de convulsions ont été rapportés en association avec le sumatriptan (voir rubrique 4.8).
Le sumatriptan doit être administré avec précaution chez les patients présentant des facteurs pouvant modifier l'absorption, le métabolisme ou l'élimination du médicament comme par exemple chez l'insuffisant hépatique (Score de Child-Pugh grade A ou B ; voir rubrique 5.2 – Populations spéciales de patients) ou l’insuffisant rénal (voir rubrique 5.2).
Chez les patients ayant une hypersensibilité connue aux sulfamides, des réactions allergiques peuvent être observées après administration de sumatriptan. Ces réactions vont de l'allergie cutanée aux réactions anaphylactiques. La démonstration d'une allergie croisée est limitée, cependant la prudence est recommandée avant d'utiliser le sumatriptan chez ces patients.
La fréquence des effets indésirables peut être augmentée par l’association des triptans à des préparations contenant du millepertuis (Hypericum perforatum).
L'utilisation prolongée d'un traitement antalgique pour traiter les céphalées peut entraîner une aggravation de celles-ci. Dans ce cas, qu'il soit avéré ou suspecté, un avis médical est nécessaire et le traitement doit être interrompu. Le diagnostic de céphalée par abus médicamenteux (CAM) doit être suspecté chez les patients présentant des céphalées fréquentes ou quotidiennes malgré (ou à cause de) l'utilisation régulière d'un traitement antimigraineux.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Il n'a pas été mis en évidence d'interaction avec le propranolol, la flunarizine, le pizotifène ou l'alcool. Les données concernant l'interaction du sumatriptan avec les médicaments contenant de l'ergotamine ou un autre triptan/agoniste du récepteur 5-HT1 sont limitées. Le risque accru de vasospasme coronarien est théoriquement possible. L'administration concomitante de ces produits est donc contre-indiquée (voir rubrique 4.3). Le délai devant s’écouler entre l’utilisation du sumatriptan et des médicaments contenant de l’ergotamine ou un autre triptan/agoniste du récepteur 5-HT1 n’est pas connu. Il dépendra aussi des doses et du type de produits à base d’ergotamine ou de tout autre triptan/agoniste du récepteur 5-HT1 utilisé. Les effets peuvent être additifs. Il est conseillé d’attendre au moins 24 heures après l’utilisation de médicaments contenant de l’ergotamine ou un autre triptan/agoniste du récepteur 5-HT1, avant l’administration du sumatriptan. Inversement, il est conseillé d’attendre au moins 6 heures après l’utilisation du sumatriptan, avant l’administration d’un médicament contenant de l’ergotamine et au moins 24 heures avant l’administration d’un autre triptan/agoniste du récepteur 5-HT1.Une interaction peut se produire entre le sumatriptan et les IMAO. L'administration concomitante de ces deux produits est donc contre-indiquée (voir rubrique 4.3).
Après commercialisation, de rares cas de syndrome sérotoninergique (incluant une modification de l'état mental, des manifestations neurovégétatives et des troubles neuromusculaires) ont été décrits après utilisation concomitante d'un ISRS et du sumatriptan. Des cas de syndrome sérotoninergique ont également été rapportés après administration concomitante de triptans et d’IRSN (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
Des données d’après commercialisation, provenant de l’utilisation du sumatriptan au cours du premier trimestre de la grossesse chez plus de 1000 femmes, sont disponibles.
Bien que ces données soient insuffisantes pour tirer des conclusions définitives, elles ne montrent pas une augmentation du risque tératogène. L’expérience de l’utilisation du sumatriptan au cours du deuxième et troisième trimestre de la grossesse est limitée.
Les études chez l’animal n’indiquent pas d’effets tératogènes directs ou des effets nuisibles sur le développement péri et post-natal. Toutefois, la viabilité embryofoetale peut être altérée chez le lapin (voir rubrique 5.3).
En conséquence, l’administration de sumatriptan ne doit être envisagée que si le bénéfice attendu pour la mère est supérieur aux risques possibles pour le fœtus.
Allaitement
Le sumatriptan est excrété dans le lait maternel, avec des doses relatives moyennes pour le nourrisson de < 4 % après administration d'une dose unique de sumatriptan. L’exposition du nourrisson peut être minimisée en évitant l’alimentation au lait maternel dans les 12 heures suivant le traitement. Le lait collecté pendant cette période doit être éliminé.
Des cas de douleur mammaire et/ou du mamelon ont été rapportés après l’utilisation du sumatriptan chez des femmes qui allaitent (voir rubrique 4.8). La douleur était généralement transitoire et disparaissait en 3 à 12 heures.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Liés à la forme pharmaceutique
Des effets indésirables rapportés chez l’adulte ont également été observés chez l’adolescent. Ces effets incluent de très rares cas de vasospasme coronaire et d’infarctus du myocarde (voir rubrique 4.4).
Affections du système nerveux
Très fréquent : dysgueusie/goût désagréable
Affections respiratoires, thoraciques et médiastinales
Fréquent : après administration de sumatriptan en pulvérisation nasale, des cas d’irritation légère et transitoire ou de sensation de brûlure au niveau du nez ou de la gorge et d’épistaxis ont été rapportés.
Généraux
Certains des symptômes rapportés comme un effet indésirable peuvent être associés aux symptômes de la migraine.
Affections du système immunitaire
Fréquence indéterminée : réactions d'hypersensibilité allant d’une allergie cutanée (comme l’urticaire) à une réaction anaphylactique.
Affections du système nerveux
Fréquents : vertiges, somnolence, troubles de la sensibilité dont paresthésie et hypoesthésie.
Fréquence indéterminée :
· crises convulsives, bien que certaines de ces convulsions soient survenues chez des patients présentant soit des antécédents de convulsions soit des conditions prédisposant aux convulsions. Quelques cas ont été rapportés chez des patients qui n’avaient pas de conditions apparentes prédisposant aux convulsions.
· tremblements, dystonie, nystagmus, scotome.
Affections oculaires
Fréquence indéterminée : troubles visuels tels que papillotements, diplopie, baisse de la vision. Perte de la vision, dont certains cas peuvent être permanents. Toutefois, des troubles visuels peuvent également survenir au cours de la crise de migraine elle-même.
Affections cardiaques
Fréquence indéterminée : bradycardie, tachycardie, palpitations, arythmies cardiaques, signes ischémiques transitoires à l'ECG, vasospasme des artères coronaires, angor, infarctus du myocarde (voir rubriques 4.3 et 4.4).
Affections vasculaires
Fréquents : augmentations transitoires de la pression artérielle survenant juste après le traitement, flush.
Fréquence indéterminée : hypotension et syndrome de Raynaud.
Affections respiratoires, thoraciques et médiastinales
Fréquent : dyspnée.
Affections gastro-intestinales
Fréquents : des nausées et vomissements sont survenus chez certains patients, sans que l'on puisse déterminer si ces symptômes sont liés au sumatriptan ou à la pathologie sous-jacente.
Fréquence indéterminée : colites ischémiques, diarrhées, dysphagie.
Affections musculo-squelettiques et systémiques
Fréquents :
· sensation de lourdeur (habituellement transitoire, mais pouvant être intense et intéresser n'importe quelle partie du corps, y compris la poitrine et la gorge).
· myalgie.
Fréquence indéterminée : raideur de la nuque, arthralgie.
Troubles généraux et anomalies au site d'administration
Fréquents : douleur, sensation de chaleur ou de froid, de pression ou d'oppression (ces effets sont habituellement transitoires, mais ils peuvent être intenses et intéresser n'importe quelle partie du corps, y compris la poitrine et la gorge) ; sensations de faiblesse, fatigue (ces deux effets sont le plus souvent d'intensité faible à modérée et transitoires).
Fréquence indéterminée : douleur traumatique provoquée, douleur inflammatoire provoquée.
Investigations
Très rares : des perturbations mineures des tests hépatiques ont été occasionnellement observées.
Affections psychiatriques
Fréquence indéterminée : anxiété.
Affections de la peau et du tissu sous-cutané
Fréquence indéterminée : hyperhidrose.
Affections des organes de reproduction et du sein
Fréquence rare : Douleur mammaire
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de Santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Des doses uniques allant jusqu'à 40 mg par voie nasale et dépassant 16 mg par voie sous-cutanée et 400 mg par voie orale n'ont pas entraîné d'effets indésirables autres que ceux mentionnés.
Au cours des études cliniques, des volontaires ont reçu 20 mg de sumatriptan par voie nasale 3 fois par jour pendant 4 jours sans effets indésirables significatifs.
Traitement
En cas de surdosage, le patient doit être mis sous surveillance pendant au moins 10 heures et un traitement symptomatique standard doit être administré, si nécessaire. L'effet de l'hémodialyse ou de la dialyse péritonéale sur les concentrations plasmatiques de sumatriptan n'est pas connu.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : AGONISTES SELECTIFS DES RECEPTEURS 5 HT1, Code ATC : N02CC01.
Le sumatriptan est un agoniste sélectif des récepteurs vasculaires à la 5-hydroxytryptamine-1 (5HT1d) sans effet sur les autres sous-types de récepteurs 5HT (5HT2 à 5HT7). Les récepteurs vasculaires 5HT1d sont localisés principalement au niveau des vaisseaux sanguins crâniens et induisent une vasoconstriction.
Chez l'animal, le sumatriptan est responsable d'une vasoconstriction sélective de la circulation artérielle carotidienne, mais ne modifie pas le flux sanguin cérébral. La circulation artérielle carotidienne vascularise les tissus extracrâniens et intracrâniens tels que les méninges et on pense que la dilatation et/ou la formation d’œdèmes au niveau de ces vaisseaux pourraient correspondre au mécanisme de la migraine chez l'homme. De plus, les résultats des études chez l'animal indiquent que le sumatriptan inhibe l'activité du nerf trijumeau. Ces deux actions (vasoconstriction crânienne et inhibition de l'activité du nerf trijumeau) pourraient contribuer à l'action anti-migraineuse du sumatriptan chez l'homme.
Les résultats de cinq études cliniques comparant les doses de 10 mg/0,1 ml et de 20 mg/0,1 ml et portant sur plus de 3 000 patients montrent :
· Un début de soulagement de la céphalée à partir de la 30ème minute avec la dose de 10 mg/0,1 ml et à partir de la 15ème minute avec la dose de 20 mg/0,1 ml.
· Un soulagement de la céphalée (disparition complète ou céphalée légère) 2 heures après administration chez 43 à 54 % des patients avec la dose de 10 mg/0,1 ml et 55 à 64 % des patients avec la dose de 20 mg/0,1 ml.
5.2. Propriétés pharmacocinétiques
La pharmacocinétique du sumatriptan par voie nasale ne semble pas être significativement modifiée par la crise de migraine.
Absorption
Après administration nasale, Ie sumatriptan est rapidement absorbé, la concentration plasmatique maximale étant atteinte en 1 - 1,5 heures. Après une dose de 20 mg, la concentration plasmatique maximale moyenne est de 13 ng/ml.
La biodisponibilité moyenne après administration par voie nasale est d'environ 16 % de celle après administration sous-cutanée, à cause en partie du métabolisme pré-systémique.
La liaison aux protéines plasmatiques est faible (14-21 %), le volume de distribution total moyen est de 170 litres.
Métabolisme
Le principal métabolite, l'analogue acide indolacétique du sumatriptan, est principalement excrété dans les urines, où il est retrouvé sous forme d'acide libre ou de glucuroconjugué. II n'a pas d'activité 5HT1 ou 5HT2 connue. Les métabolites mineurs n'ont pas été identifiés.
Elimination
La demi-vie d'élimination est d'environ 2 heures. La clairance plasmatique moyenne est d'environ 1160 ml/min et la clairance plasmatique rénale moyenne est d'environ 260 ml/min.
La clairance non rénale représente environ 80 % de la clairance totale. Le sumatriptan est éliminé tout d'abord par métabolisme oxydatif dû à la monoamine-oxydase A.
Populations spéciales de patients
Insuffisance hépatique
Après administration par voie orale, la clairance pré-systémique est réduite chez les patients insuffisants hépatiques, provoquant une augmentation des taux plasmatiques de sumatriptan. Une augmentation similaire pourrait survenir après administration nasale (voir rubrique 4.4).
Adolescents (12- 17 ans)
Une étude pharmacocinétique chez des sujets adolescents (12-17 ans) indique que la concentration plasmatique maximale moyenne est de 13,9 ng/ml et que la demi-vie moyenne d'élimination est de 2 heures après administration de 20 mg par voie nasale. Un modèle de cinétique de population a permis de montrer que la clairance et le volume de distribution augmentent avec la taille de l'adolescent, entraînant une exposition plus importante chez les adolescents de poids corporel plus faible.
Patients âgés
La cinétique chez le sujet âgé n'a pas été suffisamment étudiée pour justifier la mention de possibles différences entre les cinétiques chez des volontaires âgés et chez des volontaires jeunes.
5.3. Données de sécurité préclinique
Lors d’une étude de fertilité chez le rat, une réduction du succès de l’insémination a été observée à des taux d’exposition bien supérieurs à l’exposition maximale chez l’homme. Chez le lapin, une embryolétalité sans anomalie tératogène marquée a été observée. La pertinence de ces résultats chez l’homme n’est pas connue.
Les études réalisées chez l’animal et sur des modèles in vitro ont montré que le sumatriptan était dépourvu d’activité génotoxique et carcinogène.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30°C.
Conserver le conditionnement primaire dans l’emballage extérieur, à l’abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
0,1 ml (1 pulvérisation) en ampoule (verre) avec bouchon (chlorobutyl) + embout nasal (polypropylène) : boîte de 2
6.6. Précautions particulières d’élimination et de manipulation
Description de l'étui
· L'étui contient deux pulvérisateurs pour administration nasale, emballés séparément sous blister. Chaque pulvérisateur contient une dose de sumatriptan.
· N'ouvrir le blister qu’au moment de l’utilisation du pulvérisateur ; chaque pulvérisateur est commercialisé dans un blister pour vous aider à le maintenir propre et intact. Si le pulvérisateur est enlevé du blister ou si, il est maintenu dans le blister ouvert, il est possible que son fonctionnement en soit altéré.
· Garder dans l'étui les pulvérisateurs maintenus dans leur blister. Cet étui permet de les protéger de la lumière et de tout risque d'endommagement.
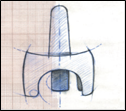
Description des différentes parties du pulvérisateur nasal
L’embout arrondi : partie à introduire dans la narine. La solution pour pulvérisation provient de l’interstice situé sur le dessus de l’embout.
Le dispositif de maintien : partie à maintenir durant l’utilisation du pulvérisateur nasal.
Le poussoir bleu : après actionnement de celui-ci, la totalité de la dose de solution pour pulvérisation est introduite en une fois dans la narine. Le poussoir ne fonctionne qu’une fois, aussi ne pas appuyer avant d’avoir introduit l’embout dans la narine car l’on risque de perdre la dose.
 1
1
Voir rubrique description ci-dessous.
Instruction concernant la manipulation :
Le pulvérisateur nasal ne doit être retiré du blister qu’immédiatement avant utilisation.
 2
2
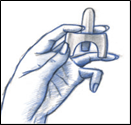
· Dans un premier temps, adopter une position confortable, la position assise étant tout à fait possible ;
· Se moucher en cas de rhume ou de nez bouché ;
· Retirer le pulvérisateur nasal de son blister.
 3
3
· Maintenir le pulvérisateur nasal avec précaution ;
· A ce stade, ne pas encore appuyer sur le poussoir bleu.
 4
4
Etape précédant l’introduction du pulvérisateur dans le nez.
· Presser fermement sur une narine pour la boucher ;
· Expirer lentement par la bouche.
 5
5
Etape d’introduction du pulvérisateur dans le nez, prêt à l’emploi.
· Introduire l’embout dans l’autre narine d’environ 1 cm ;
· Incliner légèrement la tête vers l’arrière comme indiqué sur le dessin et fermer la bouche ;
· Inspirer lentement par le nez et en même temps presser fermement le poussoir bleu avec le pouce ;
· Le poussoir peut opposer une légère résistance et l’on peut entendre un déclic lors de son utilisation ;
· Retirer le pulvérisateur du nez ainsi que l’index qui maintient l’autre narine bouchée ;
· Inspirer doucement par le nez pour permettre au produit de bien rester dans la narine ;
· Expirer par la bouche ;
· Après utilisation du pulvérisateur, le nez peut sembler pris et l’on peut également ressentir un léger goût dans la bouche. Ces sensations sont normales et passeront rapidement ;
· Après une seule utilisation, le pulvérisateur est vide. Le jeter dans un endroit sûr.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
23 RUE FRANCOIS JACOB
92500 RUEIL-MALMAISON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 30/10/2025
IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale
Sumatriptan
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
2. Quelles sont les informations à connaître avant d’utiliser IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale ?
3. Comment utiliser IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : AGONISTES SELECTIFS DES RECEPTEURS 5-HT1 - N02CC01
Ce médicament est indiqué dans le traitement de la crise de migraine avec ou sans aura (sensation subjective passagère qui précède la crise de migraine, très variable d’un sujet à l’autre et qui touche l’audition, la vue ).
La forme solution pour pulvérisation nasale est particulièrement adaptée à la crise de migraine s’accompagnant de nausées et de vomissements.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale ?
N’utilisez jamais IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale
· si vous êtes allergique à la substance active ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous avez des antécédents d'infarctus du myocarde.
· si vous souffrez de certaines maladies cardiovasculaires ou d’antécédents de maladies cardiovasculaires tels que l'angine de poitrine (caractérisée par des douleurs violentes localisées au niveau de la poitrine et pouvant s'étendre dans le bras gauche), des troubles de la circulation périphérique (notamment au niveau des doigts et des orteils).
· si vous avez des antécédents d'accident vasculaire cérébral (AVC) ou de mini AVC appelé accident ischémique transitoire (AIT).
· si vous souffrez d’hypertension artérielle modérée ou sévère et d’hypertension artérielle légère non contrôlée par un traitement.
· si vous avez une maladie grave du foie.
· si vous l’utilisez en association à certains autres médicaments utilisés également dans le traitement de la migraine (ergotamine et dérivés de l'ergotamine, y compris le méthysergide ou les médicaments de la famille des triptans/agonistes 5-HT1).
· si vous l’utilisez en association aux IMAO (iproniazide, nialamide, toloxatone, moclobémide (médicaments utilisés dans le traitement de la dépression), sélégiline (médicament utilisé dans le traitement de la maladie de Parkinson)). Le sumatriptan ne doit pas être utilisé dans les deux semaines suivant l'arrêt d'un traitement par IMAO.
EN CAS DE DOUTE, IL EST INDISPENSABLE DE DEMANDER L'AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN.
Avertissements et précautions
Mises en garde spéciales
· Ce médicament n'a pas été suffisamment étudié chez les sujets de plus de 65 ans et de moins de 12 ans, c’est pourquoi il n’est pas recommandé d’utiliser ce médicament chez ces personnes.
· Le diagnostic de migraine doit avoir été clairement établi par votre médecin.
· Ce médicament n’est pas indiqué au cours des migraines accompagnées de paralysie, de paralysie oculaire ou de migraine basilaire.
· Une sensation d'oppression ou des douleurs au niveau de la poitrine, parfois intenses, pouvant s'étendre au niveau de la gorge, peuvent survenir dans les minutes suivant l'administration du médicament ; contactez alors votre médecin et ne prenez pas de dose supplémentaire de ce médicament.
· La fréquence d’effets indésirables peut être augmentée par l’association avec des préparations contenant du millepertuis.
· Comme avec les autres traitements de la crise de migraine, si vous prenez trop d’IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale vos maux de tête peuvent s’aggraver et/ou survenir plus souvent. Si cela se produit, votre médecin pourra vous demander d’arrêter d’utiliser IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale.
· Ce médicament ne doit pas être administré en cas d'allergie connue aux sulfamides (risque d'allergie croisée).
· Ce médicament doit être utilisé avec prudence en cas d’hypertension légère contrôlée par un traitement.
Précautions d'emploi
Prévenez le médecin, en cas de :
· Présence de facteurs de risque cardiovasculaire (femme après la ménopause, âge (homme de plus de 40 ans), tabagisme ou prise de médicaments destinés à arrêter de fumer, ...) ;
· Antécédents de crises convulsives. Ce médicament est susceptible d'augmenter le risque de survenue de convulsions ;
· Traitement concomitant par certains antidépresseurs notamment la fluvoxamine, la fluoxétine, la paroxétine, le citalopram, la sertraline, l’escitalopram, le milnacipran ou la venlafaxine. Si vous présentez plusieurs des symptômes suivants : diarrhée, accélération du rythme cardiaque, fièvre, sueurs, tremblements ou contractions musculaires involontaires, agitation, confusion mentale, hallucinations, prévenez immédiatement votre médecin.
Ceux-ci peuvent être les signes d’une réaction appelée syndrome sérotoninergique (voir rubrique « Prise ou utilisation d'autres médicaments »).
· Maladie du rein ou du foie.
Adressez-vous à votre médecin ou pharmacien avant d’utiliser IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale.
Enfants
Sans objet.
Autres médicaments et IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale
Vous ne devez pas prendre ce médicament en même temps que certains autres médicaments utilisés dans le traitement de la migraine (notamment l'ergotamine et les dérivés de l'ergotamine, y compris le méthysergide ou les médicaments de la famille des triptans/agonistes 5-HT1) ; un délai de 24 heures doit être respecté entre l'arrêt de ces médicaments et l'administration de sumatriptan.
De même, l'ergotamine et les dérivés de l'ergotamine, y compris le méthysergide ne doivent pas être administrés dans les 6 heures qui suivent une administration de sumatriptan. Quant aux médicaments de la famille des triptans/agonistes 5-HT1 il faut attendre 24 heures après la prise de sumatriptan avant de les administrer.
En revanche, vous pouvez prendre du paracétamol, de l'acide acétylsalicylique ou des anti-inflammatoires non stéroïdiens.
Vous ne devez pas prendre ce médicament en même temps que certains médicaments utilisés dans le traitement de la dépression (IMAO (iproniazide, nialamide, toloxatone, moclobémide)) ou le traitement de la maladie de Parkinson (sélégiline) ; respectez un délai de 2 semaines entre l'arrêt de l'IMAO et l'instauration du traitement par le sumatriptan.
Si vous prenez ce médicament alors que vous êtes traité par un antidépresseur tel que la fluoxétine, la paroxétine, le citalopram, la sertraline, l'escitalopram, le milnacipran ou la venlafaxine, prévenez votre médecin immédiatement. Cette association peut provoquer rarement une réaction liée à un excès de sérotonine (appelé syndrome sérotoninergique) produisant les symptômes suivants : diarrhée, accélération du rythme cardiaque, fièvre, sueurs, tremblements ou contractions musculaires involontaires, agitation, confusion mentale, hallucinations.
Vous devez informer votre médecin de tout médicament que vous prenez déjà ou que vous avez récemment pris, même s'il s'agit d'un médicament obtenu sans ordonnance en particulier pour traiter la migraine, la dépression, ou l'aide à l'arrêt du tabac.
IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale avec des aliments et boissons
Sans objet.
Grossesse et allaitement
L'utilisation de ce médicament est déconseillée pendant la grossesse ou en cas d'allaitement, sauf avis contraire de votre médecin.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Si vous découvrez que vous êtes enceinte pendant le traitement, consultez votre médecin : lui seul pourra adapter le traitement à votre état.
Vous devez éviter d’allaiter votre enfant pendant 12 heures suite à la prise d’IMIGRANE. Le lait collecté pendant cette période doit être éliminé.
Certaines femmes qui allaitent signalent de la douleur mammaire et/ou au mamelon après l’utilisation du sumatriptan. La douleur est généralement temporaire et disparaît en 3 à 12 heures.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Ce médicament ainsi que la migraine peuvent entraîner une somnolence. Dans ce cas, conduire un véhicule ou utiliser une machine peut être dangereux.
IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale contient
Sans objet.
3. COMMENT UTILISER IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale ?
Posologie
Ce médicament peut être administré chez l'adolescent (à partir de 12 ans) et chez l'adulte (de 18 à 65 ans).
· Adultes de plus de 18 ans et de moins de 65 ans :
Ne pas administrer plus de 2 pulvérisations par 24 heures et bien respecter un délai de 2 heures entre les 2 administrations.
· Adolescents entre 12 ans et 17 ans :
Le dosage de 20 mg/0,1 ml n’est pas adapté pour l’adolescent.
Ne pas administrer plus de deux pulvérisations de 10 mg de sumatriptan solution pour pulvérisation nasale par 24 heures.
· Traitement du mal de tête au cours de la crise de migraine : une seule pulvérisation administrée dans une seule narine.
· En l'absence de soulagement, au cours d'une crise, l'administration d'une seconde dose n'apporte pas de bénéfice supplémentaire.
· En cas de soulagement après la première prise puis de réapparition de la douleur dans les 24 heures, une seconde dose pourra être administrée, à condition de respecter un intervalle d'au moins 2 heures entre les 2 administrations.
Mode d’administration
Pulvérisation nasale.
Description de l'étui
· L'étui contient deux pulvérisateurs pour administration nasale, emballés séparément sous blister. Chaque pulvérisateur contient une dose de sumatriptan.
· N'ouvrez le blister que si vous êtes sur le point d'utiliser un pulvérisateur ; chaque pulvérisateur est commercialisé dans un blister pour vous aider à le maintenir propre et intact. Si vous enlevez un pulvérisateur du blister ou si vous le maintenez dans le blister ouvert, il est possible que son fonctionnement en soit altéré.
· Gardez dans l'étui les pulvérisateurs maintenus dans leur blister. Cet étui permet de les protéger de la lumière et de tout risque d'endommagement.
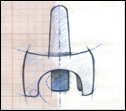
Description des différentes parties du pulvérisateur nasal
· L’embout arrondi : partie à introduire dans la narine. La solution pour pulvérisation provient de l’interstice situé sur le dessus de l’embout.
· Le dispositif de maintien : partie à maintenir durant l’utilisation du pulvérisateur nasal.
· Le poussoir bleu : après actionnement de celui-ci, la totalité de la dose de solution pour pulvérisation est introduite en une fois dans la narine. Le poussoir ne fonctionne qu’une fois, aussi ne pas appuyer avant d’avoir introduit l’embout dans la narine car l’on risque de perdre la dose.
 1
1
Voir rubrique description ci-dessous.
Instruction concernant la manipulation :
Le pulvérisateur nasal ne doit être retiré du blister qu’immédiatement avant utilisation.
 2
2
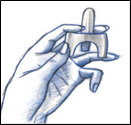
· Dans un premier temps, adopter une position confortable, la position assise étant tout à fait possible ;
· Se moucher en cas de rhume ou de nez bouché ;
· Retirer le pulvérisateur nasal de son blister.
 3
3
· Maintenir le pulvérisateur nasal avec précaution ;
· A ce stade, ne pas encore appuyer sur le poussoir bleu.
 4
4
Etape précédant l’introduction du pulvérisateur dans le nez.
· Presser fermement sur une narine pour la boucher ;
· Expirer lentement par la bouche.
 5
5
Etape d’introduction du pulvérisateur dans le nez, prêt à l’emploi.
· Introduire l’embout dans l’autre narine d’environ 1 cm ;
· Incliner légèrement la tête vers l’arrière comme indiqué sur le dessin et fermer la bouche ;
· Inspirer lentement par le nez et en même temps presser fermement le poussoir bleu avec le pouce ;
· Le poussoir peut opposer une légère résistance et l’on peut entendre un déclic lors de son utilisation ;
· Retirer le pulvérisateur du nez ainsi que l’index qui maintient l’autre narine bouchée ;
· Inspirer doucement par le nez pour permettre au produit de bien rester dans la narine ;
· Expirer par la bouche ;
· Après utilisation du pulvérisateur, le nez peut sembler pris et l’on peut également ressentir un léger goût dans la bouche. Ces sensations sont normales et passeront rapidement ;
· Après une seule utilisation, le pulvérisateur est vide. Le jeter dans un endroit sûr.
· Il est recommandé d’administrer le médicament le plus rapidement possible dès l’apparition des maux de tête ;
· Ne pas utiliser ce médicament à titre préventif.
Fréquence d'administration
· Il est recommandé de prendre le sumatriptan dès l'apparition des maux de tête ou des symptômes pouvant être associés à une crise de migraine comme les nausées, les vomissements ou une sensibilité gênante à la lumière (photophobie).
Durée du traitement
· Ne pas utiliser ce médicament à titre préventif.
Avertir immédiatement un médecin qui prendra les dispositions nécessaires.
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez d’utiliser IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale
Sans objet.
Si vous arrêtez d’utiliser IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Ce médicament peut entraîner chez certaines personnes des effets plus ou moins gênants.
Réactions allergiques : demander de l’aide immédiatement
Les effets indésirables suivants peuvent survenir mais leur fréquence est inconnue :
Manifestations allergiques plus ou moins sévères incluant une éruption cutanée avec ou sans démangeaison, une respiration sifflante, un gonflement des paupières, du visage ou des lèvres, ou un choc.
Si vous ressentez un de ces symptômes juste après avoir pris IMIGRANE,
è N’en prenez plus et contactez immédiatement votre médecin
Effets indésirables liés à la forme pharmaceutique
Très fréquent : goût désagréable
Fréquent : irritation légère et transitoire ou sensation de brûlure au niveau du nez ou de la gorge et épistaxis (saignements du nez).
Effets indésirables généraux
Certains des symptômes indiqués ci-dessous peuvent être associés aux symptômes de la migraine.
Effets indésirables fréquents :
(affectent jusqu’à 1 personne sur 10)
· Douleur, sensations de lourdeur, de pression ou d’oppression dans n’importe quelle partie du corps, notamment la poitrine ou la gorge ou sensations inhabituelles telles que engourdissements, fourmillements, diminution de la sensibilité, sensations de chaleur ou de froid. Ces effets peuvent être intenses mais sont le plus souvent transitoires.
Si ces effets persistent ou s’aggravent (en particulier la douleur à la poitrine),
è Contactez immédiatement votre médecin ou l’hôpital. Chez un petit nombre de patients, ces symptômes peuvent être liés à une crise cardiaque.
Autres effets indésirables fréquents :
· Nausées, vomissements qui peuvent aussi être dus à la crise de migraine.
· Fatigue ou somnolence.
· Vertiges, sensation de faiblesse, bouffées de chaleur.
· Augmentation transitoire de la tension artérielle après le traitement (hypertension).
· Difficulté respiratoire.
· Douleurs musculaires.
Effets indésirables rares :
(affectent jusqu’à 1 personne sur 1 000)
· Douleur mammaire.
Effets indésirables très rares :
(affectent jusqu’à 1 personne sur 10 000)
· Perturbations mineures des enzymes du foie. Si vous faites un test sanguin pour vérifier vos fonctions hépatiques, parlez à votre médecin de la prise d’IMIGRANE.
Certains patients peuvent être affectés par d’autres effets indésirables mais leur fréquence exacte n’est pas connue :
· Crises convulsives, tremblements, dystonie (contraction douloureuse des muscles), raideur de la nuque.
· Troubles de la vision (qui peuvent aussi être dus à la crise de migraine elle-même) : papillotement, baisse de la vision, vision double, mouvements oscillatoires et quelquefois rotatoires du globe oculaire, lacune fixe dans le champ visuel, pertes de la vision qui peuvent être permanentes dans certains cas.
· Problèmes cardiaques : ralentissement ou accélération du rythme des battements du cœur (bradycardie ou tachycardie), palpitations, troubles du rythme cardiaque, angine de poitrine (sensation de douleur au niveau de la poitrine), infarctus du myocarde.
· Diminution de la tension artérielle (hypotension).
· Troubles de la circulation des doigts et des orteils, souvent déclenchés par le froid (Syndrome de Raynaud).
· Lésions intestinales d’origine vasculaire (colites ischémiques).
· Diarrhée.
· Si vous avez eu une blessure récente ou si vous avez une inflammation (rhumatisme ou inflammation du côlon, par exemple), vous pouvez ressentir une douleur ou une aggravation de la douleur au niveau de la blessure ou de l’inflammation.
· Douleurs articulaires.
· Anxiété.
· Difficultés à avaler.
· Sudation excessive.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de Santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance – Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale ?
Tenir ce médicament hors de la vue et de la portée des enfants.
A conserver à une température ne dépassant pas 30°C.
Conserver le conditionnement primaire dans l’emballage extérieur, à l’abri de la lumière.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l'environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient IMIGRANE 10 mg/0,1 ml, solution pour pulvérisation nasale
· La substance active est :
Sumatriptan ................ .10,00 mg
Pour une ampoule.
· Les autres composants sont :
Dihydrogénophosphate de potassium, hydrogénophosphate de sodium anhydre, acide sulfurique, hydroxyde de sodium, eau purifiée.
Ce médicament se présente sous la forme d’une solution pour pulvérisation nasale, boîte de 2 ampoules.
Titulaire de l’autorisation de mise sur le marché
23 RUE FRANCOIS JACOB
92500 RUEIL-MALMAISON
Exploitant de l’autorisation de mise sur le marché
23 RUE FRANCOIS JACOB
92500 RUEIL-MALMAISON
GLAXOSMITHKLINE MANUFACTURING S.P.A
STRADA PROVINCIALE ASOLANA, 90
43056 SAN POLO DI TORRILE
PARME
ITALIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).