Dernière mise à jour le 01/06/2026
MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose
Présentations
> 60 récipient(s) unidose(s) polyéthylène basse densité (PEBD) de 0,4 ml
Code CIP : 222 146-1 ou 34009 222 146 1 0
Déclaration de commercialisation : 18/02/2013
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 6,10 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 7,12 €
- Taux de remboursement :30%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Modéré | Avis du 07/06/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par MONOKETO reste modéré dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 18/07/2012 | Inscription (CT) | Absence d'amélioration du service médical rendu (ASMR V) |
ANSM - Mis à jour le : 07/12/2023
MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Kétotifène........................................................................................................................ 0,100 mg
Sous forme de fumarate de kétotifène............................................................................... 0,138 mg
Pour 0,4 ml.
Chaque goutte contient 9,5 microgrammes de fumarate de kétotifène.
Pour la liste complète des excipients, voir rubrique 6.1
Collyre en solution en récipient unidose.
Solution claire, incolore à légèrement colorée en jaune.
4.1. Indications thérapeutiques
Traitement symptomatique de la conjonctivite allergique saisonnière.
4.2. Posologie et mode d'administration
Le contenu du récipient unidose est suffisant pour le traitement des deux yeux.
Le collyre est stérile jusqu'à la première ouverture du récipient unidose. Afin d'éviter tout risque de contamination, il convient de ne pas toucher quelque surface que ce soit avec l'embout de l'unidose.
La sécurité et l'efficacité de MONOKETO n'ont pas été établies chez les enfants âgés de moins de 3 ans.
Hypersensibilité au kétotifène ou à l'un des composants du collyre.
4.4. Mises en garde spéciales et précautions d'emploi
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L'utilisation du kétotifène par voie orale peut potentialiser les effets des dépresseurs du système nerveux central, des antihistaminiques et de l'alcool. Bien que cela n'ait pas été observé avec MONOKETO, la possibilité de telles interactions ne peut être exclue.
4.6. Fertilité, grossesse et allaitement
Grossesse
On ne dispose pas de données cliniques pertinentes concernant l'exposition des femmes enceintes au kétotifène en collyre. Des études effectuées chez l'animal utilisant des doses materna-toxiques par voie orale, ont montré une augmentation de la mortalité pré et post-natale, mais pas d'effet tératogène. Les taux systémiques observés après instillation oculaire sont très largement inférieurs à ceux observés après administration orale. Néanmoins, la prudence sera de rigueur lorsqu'on prescrira ce médicament aux femmes enceintes.
Allaitement
Bien que les études chez l'animal après administration orale montrent un passage dans le lait maternel, il est peu probable que l'administration topique chez l'être humain produise des quantités détectables dans le lait maternel. MONOKETO peut être utilisé pendant l'allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables sont classés par ordre de fréquence selon la convention suivante : très fréquent (≥1/10) ; fréquent (≥1/100, <1/10) ; peu fréquent (≥1/ 1000, <1/100) ; rare (≥1/10 000, <1/1 000) ; très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). A la posologie recommandée, les effets indésirables suivants sont rapportés :
Affections oculaires
Fréquent : irritation oculaire, douleur oculaire, kératite ponctuée, érosion ponctuée de l'épithélium cornéen.
Peu fréquent : vision trouble (durant l'instillation), sécheresse oculaire, irritation des paupières, conjonctivite, photophobie, hémorragie conjonctivale.
Affections du système nerveux
Peu fréquent : céphalées.
Troubles généraux et anomalies au site d'administration
Peu fréquent : somnolence.
Affections de la peau et du tissu sous-cutané
Peu fréquent : éruption cutanée, eczéma, urticaire.
Affections gastro-intestinales
Peu fréquent : sécheresse buccale.
Affections du système immunitaire
Peu fréquent : réaction d'hypersensibilité.
Les effets indésirables suivants ont été observés après la mise sur le marché : réactions d'hypersensibilité incluant des réactions allergiques locales (principalement dermatite de contact, gonflement des yeux, irritation et œdème de la paupière), réactions allergiques systémiques incluant gonflement/œdème du visage (parfois associées à une dermatite de contact) et exacerbation d'états allergiques préexistants tels que l'asthme et l'eczéma.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Aucun cas de surdosage n'a été rapporté.
L'absorption orale du contenu d'une unidose équivaut à 0, 1 mg de kétotifène, ce qui correspond à 5 % de la posologie orale quotidienne recommandée pour un enfant de 3 ans. Les résultats cliniques n'ont pas mis en évidence de signe ou symptôme grave après absorption de doses allant jusqu'à 20 mg de kétotifène par voie orale.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Médicaments ophtalmologiques, autres anti-allergiques, code ATC: S01GX08.
Le kétotifène est un antagoniste des récepteurs H1 à l'histamine. Les études réalisées in vivo chez l'animal et les études réalisées in vitro suggèrent des mécanismes d'action additionnels telles la stabilisation de la membrane mastocytaire et l'inhibition de l'infiltration, de l'activation et de la dégranulation des éosinophiles.
5.2. Propriétés pharmacocinétiques
Au cours d'une étude pharmacocinétique menée auprès de 18 volontaires sains traités avec MONOKETO, les taux plasmatiques de kétotifène après instillation oculaire de doses répétées pendant 14 jours, étaient, dans la plupart des cas, au-dessous de la limite quantifiable (20 pg/ml). Après administration orale, le kétotifène est éliminé de manière biphasique, avec une demi-vie initiale de distribution de 3 à 5 heures et une demi-vie d'élimination de 21 heures. Environ 1 % de la substance est éliminée sous forme inchangée dans l'urine en 48 heures, et 60 à 70 % sous forme de métabolites. Le métabolite principal est le glucuronide-N-kétotifène, pratiquement inactif.
5.3. Données de sécurité préclinique
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité, cancérogenèse et des fonctions de reproduction et de développement, n'ont pas révélé de risque particulier pour l'homme lié à l'utilisation du collyre MONOKETO.
Glycérol, hydroxyde de sodium, eau pour préparations injectables.
Avant ouverture du sachet : 2 ans.
Après ouverture du sachet mais dans le carton d'origine : 3 mois.
Après ouverture du récipient unidose, le contenu doit être utilisé immédiatement.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
6.5. Nature et contenu de l'emballage extérieur
0,4 ml en récipient unidose en PEBD transparent. Deux barrettes de 5 récipients unidoses conditionnées dans un sachet.
Boîte de 20, 60 ou 100 récipients unidoses.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les récipients unidoses doivent être jetés après utilisation.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
22 Allée Camille Muffat
INEDI 5
06200 NICE
HORUS PHARMA
148 AVENUE GEORGES GUYNEMER
CAP VAR
06700 SAINT-LAURENT DU VAR
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 222 145 5 9 : 20 récipients(s) unidose(s) (PEBD) de 0,4 ml.
· 34009 222 146 1 0 : 60 récipients(s) unidose(s) (PEBD) de 0,4 ml.
· 34009 222 147 8 8 : 100 récipients(s) unidose(s) (PEBD) de 0,4 ml.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II
ANSM - Mis à jour le : 07/12/2023
MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose
Kétotifène
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose ?
3. Comment utiliser MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose ?
6. Contenu de l'emballage et autres informations.
1. QU’EST-CE QUE MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose ET DANS QUELS CAS EST-IL UTILISE ?
MONOKETO contient un principe actif anti-allergique, le kétotifène.
Ce collyre est préconisé dans le traitement symptomatique de la conjonctivite allergique saisonnière.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose?
N'utilisez jamais MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose :
· si vous êtes allergique à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d'utiliser MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose.
Enfants et adolescents
Sans objet.
Autres médicaments et MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose
Si vous devez utiliser un autre collyre en même temps que MONOKETO, attendez au moins 5 minutes entre les deux instillations.
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Ceci est particulièrement important si vous prenez des médicaments utilisés pour le traitement de la dépression ou des allergies (antihistaminique).
MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose avec des aliments, boissons et de l’alcool
MONOKETO peut augmenter l'effet de l'alcool.
MONOKETO peut être utilisé pendant l'allaitement.
Conduite de véhicules et utilisation de machines
Si vous présentez une vision trouble ou des signes de somnolence après l'utilisation de MONOKETO, vous devez attendre la disparition de ces symptômes avant de conduire des véhicules ou d'utiliser des machines.
MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose contient :
Sans objet.
3. COMMENT UTILISER MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose?
La posologie habituelle chez l'adulte, la personne âgée et l'enfant à partir de 3 ans est de 1 goutte dans l'œil ou les yeux malade(s) deux fois par jour (le matin et le soir).
Un récipient unidose contient suffisamment de solution pour une instillation dans chaque œil.
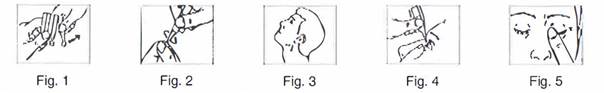
Mode d'emploi
1. Lavez-vous les mains.
2. Ouvrez le sachet et retirez la bande de récipients unidoses.
3. Détachez un des récipients unidoses de la bande (Figure 1).
4. Remettez les autres récipients unidoses dans l'emballage et refermez le sachet en repliant le bord. Replacez le sachet dans la boîte.
5. Ouvrez le récipient unidose en tournant l'embout. Ne touchez pas l'embout après avoir ouvert le récipient (Figure 2).
6. Penchez la tête en arrière (Figure 3).
7. Tirez votre paupière inférieure vers le bas avec le doigt en tenant le récipient dans l'autre main. Pressez sur le récipient pour faire tomber une goutte dans l'œil (Figure 4).
8. Fermez les yeux et appuyez le bout du doigt sur le coin intérieur de l'œil pendant 1 à 2 minutes environ. Cela empêchera la goutte de passer dans la gorge par le canal lacrymal et pratiquement la totalité de la goutte restera ainsi dans l'œil (Figure 5). Si nécessaire, répétez les étapes 6 à 8 pour traiter l'autre œil.
9. Jetez le récipient unidose après utilisation
Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez plus d'informations à votre médecin ou à votre pharmacien.
Utilisation chez les enfants et les adolescents
Sans objet.
Si vous avez utilisé plus de MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose que vous n'auriez dû :
Il n'y a pas de risques si vous avez avalé accidentellement MONOKETO ou si vous avez instillé plus d'une goutte dans l'œil. En cas de doute, demandez conseil à votre médecin.
Si vous oubliez d'utiliser MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose :
Si vous avez oublié d'utiliser MONOKETO, vous devez instiller une goutte de collyre dès que vous vous en rendez compte. Reprenez ensuite le schéma d'administration habituel. N'instillez pas de dose double pour compenser la dose que vous avez oubliée de prendre.
Si vous arrêtez d'utiliser MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose :
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables suivants ont été décrits :
Fréquent (affectant moins de 1 patient sur 10)
· irritation ou douleur oculaire
· inflammation de l'œil
· douleur oculaire, vision trouble, sensibilité anormale à la lumière
Peu fréquent (affectant moins de 1 patient sur 100)
· vision trouble lors de l'instillation du collyre dans l'œil
· sécheresse oculaire
· irritation des paupières
· conjonctivite
· augmentation de la sensibilité des yeux à la lumière
· saignement visible dans le blanc de l'œil
· maux de tête
· somnolence
· éruption cutanée (avec éventuellement des démangeaisons)
· eczéma (éruption avec rougeur, démangeaisons et sensation de brûlure)
· sécheresse buccale
· réaction allergique (incluant gonflement du visage et des paupières) et aggravation d'états allergiques existants tels que l'asthme et l'eczéma
Effets indésirables supplémentaires chez les enfants et les adolescents
Sans objet.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose?
Tenir ce médicament hors de la vue et de la portée des enfants.
N'utilisez pas ce médicament après la date de péremption indiquée sur l'emballage et le récipient unidose après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25°C.
Les récipients unidoses sont d'autant mieux conservés s'ils restent dans le sachet non ouvert. Après ouverture du sachet, le récipient unidose peut être conservé 3 mois à condition qu'il soit conservé dans le carton d'origine.
Vous devez utiliser le contenu du récipient unidose dès l'ouverture de celui-ci et jeter la solution restante après utilisation ; ceci afin d'éviter tout risque de contamination microbienne.
Ne jetez aucun médicament au tout-à-l'égout ou avec les ordures ménagères. Demandez à votre pharmacien d'éliminer les médicaments que vous n'utilisez plus. Ces mesures contribueront à protéger l'environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient MONOKETO 0,25 mg/ml, collyre en solution en récipient unidose
· La substance active est :
Kétotifène................................................................................................................... 0,100 mg
Sous forme de fumarate de kétotifène.............................................................................. 0,138 mg*
Pour 0,4 ml.
Chaque goutte contient 9,5 microgrammes de fumarate de kétotifène.
· Les autres composants sont:
Glycérol, hydroxyde de sodium, eau pour préparations injectables.
Titulaire de l’autorisation de mise sur le marché
HORUS PHARMA
22 Allée Camille Muffat
INEDI 5
06200 NICE
HORUS PHARMA
148 AVENUE GEORGES GUYNEMER
CAP VAR
06700 SAINT-LAURENT DU VAR
Exploitant de l’autorisation de mise sur le marché
HORUS PHARMA
22 Allée Camille Muffat
INEDI 5
06200 Nice
HORUS PHARMA
148 AVENUE GEORGES GUYNEMER
CAP VAR
06700 SAINT-LAURENT DU VAR
WERKSSTRASSE 3
92551 STULLN
ALLEMAGNE
ou
LABORATOIRE UNITHER
1 RUE DE L’ARQUERIE
50200 coutances
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
Janvier 2022
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).