Dernière mise à jour le 01/06/2026
FIBROGAMMIN 62,5 Ul/ml, poudre et solvant pour solution injectable/perfusion
Indications thérapeutiques
Classe pharmacothérapeutique : antihémorragiques - code ATC : B02B D07
Ce médicament est un substitut de la coagulation.
Fibrogammin est indiqué chez l’adulte et l’enfant :
· pour le traitement prophylactique de le déficit congénital en facteur XIII et
· pour le traitement périopératoire des hémorragies chirurgicales liées à un déficit congénital en facteur XIII.
Présentations
> 1 flacon(s) en verre de poudre - 1 flacon(s) en verre de solvant de 4 ml avec dispositif(s) de transfert avec dispositif(s) d'administration
Code CIP : 575 416-9 ou 34009 575 416 9 9
Déclaration de commercialisation : 09/03/2010
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre de poudre - 1 flacon(s) en verre de solvant de 20 ml avec dispositif(s) de transfert avec dispositif(s) d'administration
Code CIP : 575 417-5 ou 34009 575 417 5 0
Déclaration de commercialisation : 09/03/2010
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 10/03/2010 | Inscription (CT) | Le service médical rendu de FIBROGAMMIN est important dans l'indication de l'AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| I (Majeur) | Avis du 10/03/2010 | Inscription (CT) | En l'absence d'alternative thérapeutique validée par une AMM et sur la base de l'expérience clinique rapportée sur l'intérêt de ce médicament, la Commission considère que FIBROGAMMIN apporte une amélioration du service médical rendu majeure (de niveau I) dans la prise en charge des patients atteints de déficit congénital en facteur XIII. |
Autres informations
- Titulaire de l'autorisation : CSL BEHRING GmbH
- Conditions de prescription et de délivrance :
- délivrance réservée aux ETS pour les malades qui y sont traités
- délivrance réservée aux pharmacies à usage intérieur
- liste I
- prescription initiale hospitalière semestrielle
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 688 523 5
ANSM - Mis à jour le : 26/09/2025
FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient nominalement 250 ou 1250 UI de facteur XIII de coagulation humain plasmatique.
Fibrogammin contient approximativement 62,5 UI/ml (250 UI/4 ml et 1250 UI/20 ml) de facteur XIII de coagulation humain plasmatique après reconstitution, respectivement, avec 4 et 20 ml d’eau pour préparation injectable.
L’activité spécifique de Fibrogammin est approximativement de 3,1 à 13,3 UI/mg de protéine.
Excipient(s) à effet notoire :
Sodium
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable/ perfusion.
Poudre blanche et claire, solvant incolore.
4.1. Indications thérapeutiques
Fibrogammin est indiqué chez l’adulte et l’enfant :
· pour le traitement prophylactique du déficit congénital en facteur XIII et
· pour le traitement périopératoire des hémorragies chirurgicales liées à un déficit congénital en facteur XIII.
4.2. Posologie et mode d'administration
1 ml correspond à 62,5 UI et 100 UI est équivalent à 1,6 ml, respectivement.
Important :
La quantité à administrer et la fréquence d’administration doivent toujours être déterminées en fonction de l’efficacité clinique dans chaque cas individuel.
Dosage
La posologie doit être individualisée en fonction du poids du patient, des valeurs du laboratoire et de l’état clinique du patient.
Schéma posologique pour la prophylaxie de routine dans le traitement de la carence en facteur XIII congénitale
Dose initiale
· 40 unités internationales (UI) par kilogramme de poids corporel.
· La vitesse d'injection ne doit pas dépasser 4 ml par minute.
Doses ultérieures :
· La posologie doit être guidée par le plus bas niveau récent de l'activité en facteur XIII, avec une administration tous les 28 jours (4 semaines) pour maintenir un niveau bas d'activité en FXIII d'environ 5 à 20 %.
· Les ajustements posologiques recommandés de ± 5 UI par kg doivent être fondés sur les niveaux d'activité en FXIII comme indiqué dans le tableau 1 et l'état clinique du patient.
· Ces ajustements doivent être fondés sur la base d'une analyse sensible spécifique de détermination du taux de facteur XIII. Un exemple d’ajustements de la dose à l'aide du test standard d'activité Berichrom FXIII est décrit dans le tableau 1 ci-dessous.
Tableau 1 : Ajustement de la dose par l’utilisation du test standard Berichrom FXIII
|
Niveau d’activité du facteur XIII (%) |
Ajustement de posologie |
|
Niveau <5 % |
Augmentation de 5 unités par kg |
|
Niveau entre 5 % et 20 % |
Pas d’ajustement |
|
2 mesures >20 % |
Diminution de 5 unités par kg |
|
1 mesure >25 % |
Diminution de 5 unités par kg |
L’activité exprimée en unités est déterminée en utilisant le test d'activité Berichrom FXIII, par rapport à la norme internationale en vigueur pour le facteur XIII de coagulation plasmatique. Par conséquent, une unité est égale à une unité internationale.
Prophylaxie avant intervention chirurgicale
Après la dernière dose prophylactique de routine du patient, si une intervention chirurgicale est prévue :
· Entre 21 et 28 jours plus tard : administrer la dose prophylactique complète au patient immédiatement avant la chirurgie et la dose prophylactique suivante 28 jours plus tard.
· Entre 8 et 21 jours plus tard : une dose supplémentaire (totale ou partielle) peut être administrée avant la chirurgie. La dose doit être fondée sur le niveau d'activité en FXIII du patient, de son état clinique et doit être ajusté en fonction de la demi-vie de Fibrogammin.
· Dans les 7 jours suivant la dernière dose : une administration supplémentaire peut ne pas être nécessaire.
Les ajustements de posologie peuvent être différents de ces recommandations et doivent être individualisées en fonction des niveaux d'activité du FXIII et de l'état clinique du patient. Tous les patients doivent être étroitement surveillés pendant et après la chirurgie.
Ainsi, il est recommandé de contrôler l'augmentation de FXIII-activité avec un dosage du FXIII. Dans le cas d'une intervention chirurgicale majeure et d’hémorragies graves, le but est d'obtenir près des valeurs normales (70 % à 140 %).
Population pédiatrique
La posologie et le mode d'administration chez les enfants et les adolescents est basée sur le poids corporel et est donc généralement basées sur les mêmes lignes directrices que pour les adultes. La dose et/ ou la fréquence d'administration pour chaque individu doivent toujours être fondées sur l’efficacité clinique et sur l’activité en FXIII (voir également rubriques 5.1 et 5.2.)
Population âgée
La posologie et mode d'administration chez les personnes âgées (> 65 ans) n'a pas été documenté dans des études cliniques.
Mode d’administration
Après reconstitution la solution doit être claire ou légèrement opalescente. Avant administration, la préparation doit être amenée à la température ambiante ou corporelle. Injecter ou perfuser le produit avec le set d’administration fourni, par voie intraveineuse, lentement, dans une ligne d’injection/de perfusion dédiée (fournie avec le produit) à un débit confortable pour le patient. Le débit d’injection ou de perfusion ne doit pas dépasser approximativement 4 ml par minute.
Surveiller le patient à la recherche d’une éventuelle réaction immédiate. En cas de survenue d’une réaction paraissant liée à l’administration de Fibrogammin, le débit de perfusion devra être ralenti ou la perfusion interrompue, selon l’état clinique du patient.
Pour des instructions sur la reconstitution du médicament avant administration, voir la rubrique 6.6.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Des réactions allergiques de type hypersensibilité sont possibles avec Fibrogammin. Si des symptômes d’hypersensibilité (tels que urticaires, urticaires généralisées, oppression thoracique, une respiration sifflante, une hypotension et l’anaphylaxie) apparaissent, l’administration de Fibrogammin doit être interrompue immédiatement. En cas de choc, le traitement médical standard relatif à l’état de choc doit être mise en œuvre.
En cas de thromboses récentes, des précautions doivent être prises en raison de l’effet stabilisant du facteur XIII de la fibrine.
Immunogénicité
Le développement d'anticorps inhibiteurs contre le facteur XIII a été détecté chez les patients recevant Fibrogammin. Par conséquent, les patients doivent être surveillés pour le développement possible d'anticorps inhibiteurs. La présence d'anticorps inhibiteurs peut se manifester par une réponse inadéquate au traitement. Si le taux de facteur XIII plasmatique attendu n’est pas atteint ou si l’hémorragie n’est pas contrôlée par la dose adéquate, un dosage doit être réalisé afin de rechercher la présence d’un inhibiteur du facteur XIII.
Remarque à l’attention des patients sous régime hyposodé
Ce médicament contient 124,4 à 195,4 mg de sodium par dose, ce qui équivaut à 8 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
Sécurité virale
Les mesures habituelles de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection clinique des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de plasma et l’inclusion dans le procédé de fabrication d’étapes efficaces pour l’inactivation/élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents ou à d’autres types d’agents infectieux.
Les mesures prises sont considérées comme efficaces contre les virus enveloppés tels que le virus de l'immunodéficience humaine (VIH), le virus de l’hépatite C (VHC) et le virus de l’hépatite B (VHB) et contre les virus non enveloppés tels que le virus de l’hépatite A et parvovirus B19.
Il est fortement recommandé, qu’à chaque administration de Fibrogammin à un patient, le nom et le numéro de lot du produit soient enregistrés afin de maintenir un lien entre le patient et le numéro du lot du médicament.
Une vaccination appropriée (hépatites A et B) des patients recevant des facteurs de coagulation (dont Fibrogammin) est recommandée.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction du facteur XIII de coagulation humain avec d’autres médicaments n’est connue.
4.6. Fertilité, grossesse et allaitement
Aucune étude de reprotoxicité n’a été réalisée avec Fibrogammin chez l’animal.
Grossesse
Des données limitées sur l'utilisation clinique de Fibrogammin pendant la grossesse n'ont pas montré d'effets négatifs sur le déroulement de la grossesse et le développement péri ou post-natale. L'utilisation de Fibrogammin peut être envisagée au cours de la grossesse, le cas échéant.
Allaitement
Il n'existe pas de données sur l'excrétion de Fibrogammin dans le lait maternel. Toutefois, en fonction de sa taille moléculaire l’excrétion dans le lait est peu probable, et en raison de son caractère protéique, l'absorption de molécules intactes par le nourrisson est également peu probable. Par conséquent, Fibrogammin peut être utilisé pendant l'allaitement.
Fertilité
Il n'existe pas de données concernant les effets de Fibrogammin sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les réactions indésirables suivantes sont basées sur l'expérience post-commercialisation.
Tableau récapitulatif des effets indésirables :
Le tableau présenté ci-dessous est établi conformément à la classification des systèmes d’organes MedDRA (classes de systèmes d’organes et termes préconisés). Les fréquences ont été estimées d’après la convention suivante : très fréquent (≥ 1/10), fréquent (≥1/100 à <1/10), peu fréquent (≥1/1000 à <1/100), rare (≥1/10 000 à <1/1000), très rare (<1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classification MedDRA Classe de système d’organes |
Effets indésirables |
Fréquence |
|
Affections du système immunitaire |
Réactions allergiques anaphylactiques (telles que urticaire, urticaire généralisée, rash, hypotension, dyspnée) |
Rare |
|
Développement d’inhibiteurs du FXIII |
Très rare |
|
|
Troubles généraux et administration au site administration |
Augmentation de la température corporelle |
Rare |
Si une réaction allergique-anaphylactique survient, l’administration de Fibrogammin doit être immédiatement interrompue et un traitement approprié initié. Les standards habituels de traitement de l’état de choc doivent être suivis.
Population pédiatrique
Le profil de sécurité pour les patients pédiatriques n'est pas différent de celle des adultes dans les études cliniques.
Pour les informations relatives à la sécurité virale, voir rubrique 4.4.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
Les conséquences d’un surdosage sont inconnues.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antihémorragiques, code ATC : B02B D07.
Le facteur XIII relie le groupe aminé de la lysine à la glutamine par le biais de sa fonction enzymatique (activité transamidase) conduisant ainsi à la création d’un réseau de molécules de fibrine. Le réseau de fibrine et sa stabilisation favorisent la pénétration de fibroblastes et confortent la cicatrisation.
Population pédiatrique
Dans les études cliniques qui ont inclus les sujets de plus de 18 ans ayant un déficit congénital FXIII, l'administration prophylactique par Fibrogammin tous les 28 jours a réussi à maintenir un niveau de 5 à 20 % de l'activité de FXIII.
5.2. Propriétés pharmacocinétiques
Le produit est administré par voie intraveineuse et est de ce fait immédiatement biodisponible conduisant à une concentration plasmatique correspondante à la dose administrée.
Elimination
Chez les patients atteints de déficit congénital en facteur XIII, la demi-vie biologique de Fibrogammin a été évaluée à 6,6±2,29 jours (moyenne ± SD). Fibrogammin est métabolisé de la même façon que le facteur XIII de coagulation endogène.
Un aperçu des paramètres pharmacocinétiques (population adulte / 18 ans et plus) est donné dans le tableau suivant :
|
Paramètres |
Médiane (min. – max.) |
|
ASC ss, 0 – inf. (unitésxh/ml) |
182,9 (133,5 – 300,2) |
|
Css, max (unités/ml)* |
0,9 (0,6 – 1,2) |
|
Css, min (unités/ml)* |
0,07 (0,0 – 0,16) |
|
Tmax (h) |
1,2 (0,7 – 4,2) |
|
Demi-vie (jours) |
7,8 (3,1 – 11,02) |
|
Cl [ml/h/kg] |
0,22 (0,13 – 0,30) |
|
Vss [ml/kg] |
49,4 (31,65 – 62,91) |
|
TMR (jours) |
11,7 (5,7 – 17,02) |
ASC ss, (0-inf.): aire sous la courbe à l’état d’équilibre, courbe de concentration du T0 à l’infini
* 100% d’activité corresponde à 1 unité/ ml
Css, max = concentration maximale à l’état d’équilibre
Css, min = concentration minimale à l’état d’équilibre
Tmax : temps pour atteindre le pic plasmatique
Cl : clairance
Vss = volume de distribution à l’état d’équilibre
TMR = temps moyen résiduel
Population pédiatrique
Sur les 188 sujets de l’étude clinique relative au concentré de facteur XIII (humain), 117 étaient des sujets âgés de 18 ans ou moins au moment de l'intégration (1 mois à <2 ans, n = 17; 2 à <12 ans, n = 62 ; 12 à <16 ans, n = 30; 17 à 18 ans, n = 8). Dans l’étude PK de pharmacocinétique de 2002, 5 des 14 sujets étaient âgés de 2 à 18 ans (2-11 ans, n = 3; 12-16 ans, n = 2; 17-18 ans, n = 0). Les sujets de moins de 16 ans ont une demi-vie plus courte et une clairance plus rapide (demi-vie: 5,7 ± 1,00 jours; clairance: 0,291 ± 0,12 ml/h/kg) que les adultes (demi-vie: 7,1 ± 2,74 jours, la clairance: 0,22 ± 0,07 ml/h/kg).
Le produit a une demi-vie plus courte et une clairance plus rapide chez les enfants que chez les adultes. Cependant, comme dans tous les groupes d'âge de dosage est déterminée individuellement par poids du sujet et ajustés par l'activité de FXIII, et pas nécessairement liée à l'âge.
5.3. Données de sécurité préclinique
L’étude de la toxicité à dose unique et répétée chez l’animal n’a pas révélé de potentiel toxique de Fibrogammin.
Aucune étude sur la reproduction ou le développement fœtoembryonnaire n’a été menée.
Albumine humaine
Glucose monohydraté
Chlorure de sodium
Hydroxyde de sodium (pour l’ajustement du pH).
Solvant :
Eau pour préparation injectable
Ne pas utiliser après la date d’expiration mentionnée sur l’emballage et le flacon.
La stabilité chimique et physique a été démontrée pendant 24 h à une température ne dépassant pas 25°C. D’un point de vue microbiologique, le produit reconstitué doit être utilisé immédiatement. S’il n’est pas administré immédiatement, la durée de conservation ne doit pas dépasser 4 heures à une température ne dépassant 25°C. Ne pas réfrigérer ou congeler le médicament reconstitué.
6.4. Précautions particulières de conservation
A conserver au réfrigérateur (entre + 2 °C et + 8 °C).
Ne pas congeler.
Conserver le flacon dans l’emballage extérieur à l’abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
250 UI
Poudre : flacon injectable en verre tubulaire incolore de type 1, muni d’un bouchon (bouchon en bromobutyle), un cerclage (aluminium) et un opercule en plastique
Solvant (eau pour préparation injectable) : flacon injectable en verre tubulaire incolore de type 1, muni d’un bouchon (chlorobutyle ou bromobutyle) et d’une capsule de sertissage (aluminium/polypropylène)
1250 UI
Poudre : flacon injectable en verre moulé incolore de type 2, muni d’un bouchon (bouchon en bromobutyle), un cerclage (aluminium) et un opercule en plastique
Solvant (eau pour préparation injectable) : flacon injectable en verre tubulaire incolore de type 1, muni d’un bouchon (chlorobutyle ou bromobutyle) et d’une capsule de sertissage (aluminium/polypropylène)
Présentations
Etui de 250 UI
· 1 flacon de poudre
· 1 flacon avec 4 ml d’eau pour préparation injectable
· 1 dispositif de transfert avec filtre 20/20 (Mix2Vial)
· Set d'administration (boîte intérieure):
· 1 seringue jetable de 5 ml
· 1 nécessaire de ponction veineuse
· 2 tampons alcoolisés
· 1 pansement non stérile
Etui de 1250 UI
· 1 flacon de poudre
· 1 flacon avec 20 ml d’eau pour préparation injectable
· 1 dispositif de transfert avec filtre 20/20 (Mix2Vial)
· Set d'administration (boîte intérieure):
· 1 seringue jetable de 20 ml
· 1 nécessaire de ponction veineuse
· 2 tampons alcoolisés
· 1 pansement non stérile
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Instructions générales
· La solution doit être claire ou légèrement opalescente. Après filtration/prélèvement (voir ci-dessous) le produit reconstitué doit être inspecté visuellement afin de mettre en évidence la présence de particules ou un changement de coloration avant administration.
· La reconstitution et le prélèvement doivent être faits sous conditions aseptiques.
· N’utilisez pas de solutions troubles ou contenant des résidus (dépôts/ particules).
Reconstitution
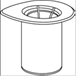
Amener le solvant à température ambiante. Retirer les capuchons protecteurs des flacons de produit et de solvant et nettoyer les bouchons avec une solution antiseptique puis laisser sécher avant l’ouverture de l’emballage du Mix2Vial.
|
|
1. Ouvrir l’emballage du dispositif Mix2Vial en retirant l’opercule. N'enlevez pas le Mix2Vial de l’emballage. |
|
|
2. Placer le flacon de solvant sur une surface plane et propre et le maintenir fermement. En tenant le dispositif Mix2Vial à travers son emballage, pousser l’extrémité bleue tout droit à travers le bouchon du flacon de solvant. |
|
|
3. Retirer avec précaution l’emballage du dispositif Mix2Vial en tenant les bords et en tirant verticalement vers le haut. Bien s’assurer que vous avez seulement retiré l’emballage et que le dispositif Mix2Vial est bien resté en place. |
|
|
4. Poser le flacon de produit sur une surface plane et rigide. Retourner l’ensemble flacon de diluant-dispositif Mix2Vial et pousser la partie transparente de l’adaptateur tout droit à travers le bouchon du flacon de produit. Le solvant coule automatiquement dans le flacon de poudre. |
|
|
5. En maintenant la partie produit reconstitué d’une main et la partie solvant de l’autre, séparer les flacons en dévissant dans le sens opposé des aiguilles d’une montre le dispositif Mix2Vial. Jeter le flacon de solvant avec la partie bleue attachée de l’adaptateur Mix2Vial. |
|
|
6. Agiter délicatement le flacon de produit avec la partie transparente de l’adaptateur attaché dessus jusqu’à ce que la substance soit totalement dissoute. Ne pas secouer. |
|
|
7. Remplir d’air une seringue stérile vide. Tout en maintenant verticalement le flacon de produit reconstitué, connecter la seringue en vissant dans le sens des aiguilles d’une montre au Luer Lock du dispositif Mix2Vial. Injecter l’air dans le flacon de produit. |
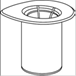
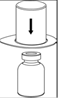
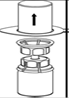
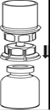
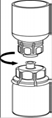
Prélèvement et administration
|
|
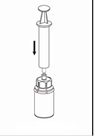
8. Tout en maintenant le piston de la seringue appuyé, retourner l’ensemble et prélever la solution dans la seringue en tirant lentement sur le piston. |
|
|
9. Une fois la solution transférée dans la seringue, tenir le corps de la seringue fermement (en maintenant le piston de la seringue dirigé vers le bas) et déconnecter la partie transparente de l’adaptateur Mix2Vial de la seringue en dévissant dans le sens opposé des aiguilles d’une montre. |
Veiller à ce que du sang ne pénètre pas dans la seringue remplie de produit en raison d'un risque de coagulation dans la seringue et d'administration de caillots de fibrine au patient.
Dans le cas où des volumes importants de Fibrogammin sont nécessaires, il est possible d’utiliser plusieurs flacons de Fibrogammin pour une seule perfusion via un dispositif de perfusion disponible dans le commerce.
La solution de Fibrogammin ne doit pas être diluée.
La solution reconstituée doit être administrée par la ligne d’injection/de perfusion fournie par injection intraveineuse lente, sans dépasser la vitesse de perfusion recommandée de 4 ml par minute.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
EMIL VON BEHRING STRASSE 76
35041 MARBURG
ALLEMAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 575 417-5 ou 34009 575 417 5 0 : poudre en flacon (verre de type I) muni d’un bouchon (caoutchouc), un cerclage (aluminium) et un opercule (plastique) + 20 ml de solvant en flacon (verre de type II) avec un dispositif de transfert et un set d’administration – boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à une prescription initiale hospitalière de six mois (les établissements de transfusion sanguine autorisés à dispenser des médicaments dérivés du sang aux malades qui y sont traités, inclus).
La délivrance est réservée aux pharmacies à usage intérieur des établissements de santé ou aux établissements de transfusion sanguine pour les malades qui y sont traités.
ANSM - Mis à jour le : 26/09/2025
FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion
Facteur XIII de coagulation humain
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
2. Quelles sont les informations à connaître avant d'utiliser FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion ?
3. Comment utiliser FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : antihémorragiques - code ATC : B02B D07
Ce médicament est un substitut de la coagulation.
Fibrogammin est indiqué chez l’adulte et l’enfant :
· pour le traitement prophylactique de le déficit congénital en facteur XIII et
· pour le traitement périopératoire des hémorragies chirurgicales liées à un déficit congénital en facteur XIII.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion?
N’utilisez jamais FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion :
· si vous êtes allergique à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser FIBROGAMMIN.
· Chez les patients présentant des antécédents d’allergies au produit (associés à des symptômes tels que : urticaire généralisée, rash, chute de la pression sanguine, dyspnée), des antihistamines et des corticoïdes peuvent être administrés en prophylaxie.
Si des réactions allergiques locales ou généralisées apparaissent, la perfusion doit être arrêtée immédiatement. En cas de choc, le traitement symptomatique relatif à l’état de choc devra être instauré.
Des réactions allergiques de type hypersensibilité sont possibles avec Fibrogammin. Si des symptômes d’hypersensibilité (tels que urticaires, urticaires généralisées, oppression thoracique, une respiration sifflante, une hypotension et l’anaphylaxie) apparaissent, l’administration de Fibrogammin doit être interrompue immédiatement. En cas de choc, le traitement médical standard relatif à l’état de choc doit être mise en œuvre.
En cas de thrombose récente (apparition d’un caillot obstruant un vaisseau), une surveillance médicale particulière doit être exercée en raison de l’effet stabilisant de la fibrine (du caillot) par le facteur XIII.
Immunogénicité
Le développement d'anticorps inhibiteurs contre le facteur XIII a été détecté chez les patients recevant Fibrogammin. Par conséquent, les patients doivent être surveillés pour le développement possible d'anticorps inhibiteurs. La présence d'anticorps inhibiteurs peut se manifester par une réponse inadéquate au traitement. Si le taux de facteur XIII plasmatique attendu n’est pas atteint ou si l’hémorragie n’est pas contrôlée par la dose adéquate, un dosage doit être réalisé afin de rechercher la présence d’un inhibiteur du facteur XIII
Remarque à l’attention des patients sous régime hyposodé :
Ce médicament contient 124,4 à 195,4 mg de sodium (composant principal du sel de cuisine/table) par dose. Cela équivaut à 8 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
Autres médicaments et FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion
Aucune interaction du facteur XIII de coagulation humain avec d’autres médicaments n’est connue.
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
FIBROGAMMIN ne doit pas être mélangé avec d’autres médicaments ou solvants, à l’exception de ceux mentionnés dans la rubrique 3, et doit être administré par une ligne de perfusion séparée.
Informations importantes concernant certains composants de FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/ perfusion :
Remarque à l’attention des patients sous régime hyposodé : Fibrogammin contient du chlorure de sodium.
Ceci est à prendre en compte chez les patients suivant un régime hyposodé strict.
Grossesse, allaitement et fertilité
Grossesse
Des données limitées sur l'utilisation clinique de Fibrogammin pendant la grossesse n'ont pas montré d'effets négatifs sur le déroulement de la grossesse et le développement péri ou post-natale. L'utilisation de Fibrogammin peut être envisagée au cours de la grossesse, le cas échéant.
Allaitement
Il n'existe pas de données sur l'excrétion de Fibrogammin dans le lait maternel. Toutefois, en fonction de sa taille moléculaire l’excrétion dans le lait est peu probable, et en raison de son caractère protéique, l'absorption de molécules intactes par le nourrisson est également peu probable. Par conséquent, Fibrogammin peut être utilisé pendant l'allaitement.
Fertilité
Il n'existe pas de données concernant les effets de Fibrogammin sur la fertilité.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Sécurité virale
Lorsque des médicaments sont préparés à partir de sang ou de plasma humain, des mesures de prévention de la transmission d’agents infectieux sont mises en place. Celles-ci comprennent :
· une sélection soigneuse des donneurs de sang et de plasma de façon à exclure les donneurs risquant d’être porteurs d’infections
· le contrôle de chaque don et des mélanges de plasma pour la présence de virus/d’infection
· la mise en œuvre dans le procédé de fabrication des étapes capables d’éliminer ou d’inactiver les virus.
Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également à tous les virus inconnus ou émergents ou autres types d’agents infectieux.
Les mesures prises sont considérées comme efficaces pour lutter contre le risque d’infection par les virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B, le virus de l’hépatite C et contre les virus non enveloppés tels que virus de l’hépatite A et le parvovirus B19.
Il est fortement recommandé, qu’à chaque administration de Fibrogammin à un patient, le nom et le numéro de lot du produit soient enregistrés afin de maintenir un lien entre le patient et le numéro du lot du médicament.
Une vaccination appropriée (hépatites A et B) des patients recevant des facteurs de coagulation (dont FIBROGAMMIN) est recommandée.
3. COMMENT UTILISER FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion ?
Posologie
1 ml correspond à 62,5 UI et 100 UI est équivalent à 1,6 ml, respectivement.
Important
La quantité à administrer et la fréquence d’administration doivent toujours être déterminées en fonction de l’efficacité clinique dans chaque cas individuel.
Dosage
La posologie doit être individualisée en fonction du poids du patient, des valeurs du laboratoire et de l’état clinique du patient.
Schéma posologique pour la prophylaxie de routine dans le traitement de la carence en facteur XIII congénitale
Dose initiale
· 40 unités internationales (UI) par kilogramme de poids corporel.
· La vitesse d'injection ne doit pas dépasser 4 ml par minute.
Doses ultérieures
· La posologie doit être guidée par le plus bas niveau récent de l'activité en facteur XIII, avec une administration tous les 28 jours (4 semaines) pour maintenir un niveau bas d'activité en FXIII d'environ 5 à 20%.
· Les ajustements posologiques recommandés de ± 5 UI par kg doivent être fondés sur les niveaux d'activité en FXIII comme indiqué dans le tableau 1 et l'état clinique du patient.
· Ces ajustements doivent être fondés sur la base d'une analyse sensible spécifique de détermination du taux de facteur XIII. Un exemple d’ajustements de la dose à l'aide du test standard d'activité Berichrom FXIII est décrit dans le tableau 1 ci-dessous.
Tableau 1 : Ajustement de la dose par l’utilisation du test standard Berichrom FXIII
|
Niveau d’activité du facteur XIII (%) |
Ajustement de posologie |
|
Niveau <5% |
Augmentation de 5 unités par kg |
|
Niveau entre 5% et 20% |
Pas d’ajustement |
|
2 mesures >20% |
Diminution de 5 unités par kg |
|
1 mesure >25% |
Diminution de 5 unités par kg |
L’activité exprimée en unités est déterminée en utilisant le test d'activité Berichrom FXIII, par rapport à la norme internationale en vigueur pour le facteur XIII de coagulation plasmatique. Par conséquent, une unité est égale à une unité internationale.
Prophylaxie avant intervention chirurgicale :
Après la dernière dose prophylactique de routine du patient, si une intervention chirurgicale est prévue :
· Entre 21 et 28 jours plus tard : administrer la dose prophylactique complète au patient immédiatement avant la chirurgie et la dose prophylactique suivante 28 jours plus tard.
· Entre 8 et 21 jours plus tard : une dose supplémentaire (totale ou partielle) peut être administrée avant la chirurgie. La dose doit être fondée sur le niveau d'activité en FXIII du patient, de son état clinique et doit être ajusté en fonction de la demi-vie de Fibrogammin.
· Dans les 7 jours suivant la dernière dose : une administration supplémentaire peut ne pas être nécessaire.
Les ajustements de posologie peuvent être différents de ces recommandations et doivent être individualisées en fonction des niveaux d'activité du FXIII et de l'état clinique du patient. Tous les patients doivent être étroitement surveillés pendant et après la chirurgie.
Ainsi, il est recommandé de contrôler l'augmentation de FXIII-activité avec un dosage du FXIII Dans le cas d'une intervention chirurgicale majeure et d’hémorragies graves, le but est d'obtenir près des valeurs normales (70% à 140%).
Utilisation chez les enfants et les adolescents
La posologie et le mode d'administration chez les enfants et les adolescents est basée sur le poids corporel et est donc généralement basées sur les mêmes lignes directrices que pour les adultes. La dose et/ ou la fréquence d'administration pour chaque individu doivent toujours être fondées sur l’efficacité clinique et sur l’activité en FXIII.
Population âgée
La posologie et mode d'administration chez les personnes âgées (> 65 ans) n'a pas été documenté dans des études cliniques.
Mode d’administration :
Instructions générales
· La solution doit être claire ou légèrement opalescente. Après filtration/prélèvement (voir ci-dessous) le produit reconstitué doit être inspecté visuellement afin de mettre en évidence la présence de particules ou un changement de coloration avant administration.
· La reconstitution et le prélèvement doivent être faits sous conditions aseptiques.
· N’utilisez pas de solutions troubles ou contenant des résidus (dépôts/particules).
Reconstitution
Amener le solvant à température ambiante. Retirer les capuchons protecteurs des flacons de produit et de solvant et nettoyer les bouchons avec une solution antiseptique puis laisser sécher avant l’ouverture de l’emballage du Mix2vial.
|
|
1. Ouvrir l’emballage du dispositif Mix2Vial en retirant l’opercule. N'enlevez pas le Mix2Vial de l’emballage. |
|
|
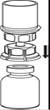
2. Placer le flacon de solvant sur une surface plane et propre et le maintenir fermement. En tenant le dispositif Mix2Vial à travers son emballage, pousser l’extrémité bleue tout droit à travers le bouchon du flacon de solvant. |
|
|
3. Retirer avec précaution l’emballage du dispositif Mix2Vial en tenant les bords et en tirant verticalement vers le haut. Bien s’assurer que vous avez seulement retiré l’emballage et que le dispositif Mix2Vial est bien resté en place. |
|
|
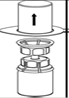
4. Poser le flacon de produit sur une surface plane et rigide. Retourner l’ensemble flacon de diluant-dispositif Mix2Vial et pousser la partie transparente de l’adaptateur tout droit à travers le bouchon du flacon de produit. Le solvant coule automatiquement dans le flacon de poudre. |
|
|
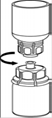
5. En maintenant la partie produit reconstitué d’une main et la partie solvant de l’autre, séparer les flacons en dévissant dans le sens opposé des aiguilles d’une montre le dispositif Mix2Vial. Jeter le flacon de solvant avec la partie bleue attachée de l’adaptateur Mix2Vial. |
|
|
6. Agiter délicatement le flacon de produit avec la partie transparente de l’adaptateur attaché dessus jusqu’à ce que la substance soit totalement dissoute. Ne pas secouer. |
|
|
7. Remplir d’air une seringue stérile vide. Tout en maintenant verticalement le flacon de produit reconstitué, connecter la seringue au Luer Lock du dispositif Mix2Vial en vissant dans le sens des aiguilles d’une montre. Injecter l’air dans le flacon de produit. |
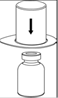
Prélèvement et administration
|
|
8. Tout en maintenant le piston de la seringue appuyé, retourner l’ensemble et prélever la solution dans la seringue en tirant lentement sur le piston. |
|
|
9. Une fois la solution transférée dans la seringue, tenir le corps de la seringue fermement (en maintenant le piston de la seringue dirigé vers le bas) et déconnecter la partie transparente de l’adaptateur Mix2Vial de la seringue en vissant dans le sens opposé des aiguilles d’une montre. |
Veiller à ce que du sang ne pénètre pas dans la seringue remplie de produit en raison d'un risque de coagulation dans la seringue et d'administration de caillots de fibrine au patient.
Dans le cas où des volumes importants de Fibrogammin sont nécessaires, il est possible d’utiliser plusieurs flacons de Fibrogammin pour une seule perfusion via un dispositif de perfusion disponible dans le commerce.
La solution de Fibrogammin ne doit pas être diluée.
La solution reconstituée doit être administrée par la ligne d’injection/de perfusion fournie par injection intraveineuse lente, sans dépasser la vitesse de perfusion recommandée de 4 ml par minute.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Si vous avez utilisé plus de FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/ perfusion que vous n’auriez dû :
Les conséquences d’un surdosage sont inconnues.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Dans de rares cas, une augmentation de la température corporelle a été observée.
Tableau récapitulatif des effets indésirables :
Le tableau présenté ci-dessous est établi conformément à la classification des systèmes d’organes MedDRA (classes de systèmes d’organes et termes préconisés). Les fréquences ont été estimées d’après la convention suivante : très fréquent (≥ 1/10), fréquent (≥1/100 à <1/10), peu fréquent (≥1/1000 à <1/100), rare (≥1/10 000 à <1/1000), très rare (<1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classification MedDRA Classe de système d’organes |
Effets indésirables |
Frequence |
|
Affections du système immunitaire |
Réactions allergiques anaphylactiques (telles que urticaire, urticaire généralisée, rash, hypotension, dyspnée) |
Rare |
|
Développement d’inhibiteurs du FXIII |
Très rare |
|
|
Troubles généraux et administration au site administration |
Augmentation de la température corporelle |
Rare |
Si une réaction allergique-anaphylactique survient, l’administration de FIBROGAMMIN doit être immédiatement interrompue (par arrêt de l’injection) et un traitement approprié doit être initié. Les standards habituels de traitement de l’état de choc doivent être suivis.
Population pédiatrique
Le profil de sécurité pour les patients pédiatriques n'est pas différent de celle des adultes dans les études cliniques.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage et le flacon après EXP.
La date de péremption fait référence au dernier jour de ce mois.
FIBROGAMMIN doit être conservé au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler. Conserver le flacon dans l’emballage extérieur à l’abri de la lumière.
La stabilité chimique et physique a été démontrée pendant 24 h à une température ne dépassant pas 25°C. D’un point de vue microbiologique, le produit doit être utilisé immédiatement. S’il n’est pas administré immédiatement, la durée de conservation ne doit pas dépasser 4 heures à une température ne dépassant pas 25°C. Ne pas réfrigérer ou congeler de solution reconstituée.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient FIBROGAMMIN 62,5 UI/ml, poudre et solvant pour solution injectable/perfusion
· La substance active est :
le facteur XIII de coagulation humain 62,5 UI/ml. Flacons contenant 250 UI et 1 250 UI de facteur XIII de coagulation humain.
· Les autres composants sont :
l’albumine humaine, le glucose, le chlorure de sodium et l’hydroxyde de sodium.
Fibrogammin est un concentré purifié de facteur XIII de coagulation humain. Il est extrait du plasma humain,
Présentations
Etui de 250 UI
· 1 flacon de poudre
· 1 flacon avec 4 ml d’eau pour préparation injectable
· 1 dispositif de transfert avec filtre 20/20 (Mix2Vial)
· Set d'administration (boîte intérieure) :
· 1 seringue jetable de 5 ml
· 1 nécessaire de ponction veineuse
· 2 tampons alcoolisés
· 1 pansement non stérile
Etui de 1250 UI
· 1 flacon de poudre
· 1 flacon avec 20 ml d’eau pour préparation injectable
· 1 dispositif de transfert avec filtre 20/20 (Mix2Vial)
· Set d'administration (boîte intérieure) :
· 1 seringue jetable de 20 ml
· 1 nécessaire de ponction veineuse
· 2 tampons alcoolisés
· 1 pansement non stérile
Toutes les présentations peuvent ne pas être commercialisées.
Fibrogammin se présente sous forme de poudre blanche et d’un solvant pour solution injectable/perfusion (250 UI en flacon / 4 ml en flacon, et 1 250 UI en flacon / 20 ml en flacon avec un dispositif de transfert 20/20 avec filtre (Mix2Vial) et un set d’administration – boîte de 1).
Titulaire de l’autorisation de mise sur le marché
EMIL-VON-BEHRING-STRASSE 76
35041 MARBURG
ALLEMAGNE
Exploitant de l’autorisation de mise sur le marché
CSL BEHRING SA
Carré Suffren
31-35 Rue de la Fédération
75015 PARIS
EMIL-VON-BEHRING-STRASSE 76
35041 MARBURG
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Posologie
1 ml correspond à 62,5 UI et 100 UI est équivalent à 1,6 ml, respectivement.
Important :
La quantité à administrer et la fréquence d’administration doivent toujours être déterminées en fonction de l’efficacité clinique dans chaque cas individuel.
Dosage
La posologie doit être individualisée en fonction du poids du patient, des valeurs du laboratoire et de l’état clinique du patient.
Schéma posologique pour la prophylaxie de routine dans le traitement de la carence en facteur XIII congénitale
Dose initiale
· 40 unités internationales (UI) par kilogramme de poids corporel.
· La vitesse d'injection ne doit pas dépasser 4 ml par minute.
Doses ultérieures :
· La posologie doit être guidée par le plus bas niveau récent de l'activité en facteur XIII, avec une administration tous les 28 jours (4 semaines) pour maintenir un niveau bas d'activité en FXIII d'environ 5 à 20%.
· Les ajustements posologiques recommandés de ± 5 UI par kg doivent être fondés sur les niveaux d'activité en FXIII comme indiqué dans le tableau 1 et l'état clinique du patient.
· Ces ajustements doivent être fondés sur la base d'une analyse sensible spécifique de détermination du taux de facteur XIII. Un exemple d’ajustements de la dose à l'aide du test standard d'activité Berichrom FXIII est décrit dans le tableau 1 ci-dessous.
Tableau 1 : Ajustement de la dose par l’utilisation du test standard Berichrom FXIII
|
Niveau d’activité du facteur XIII (%) |
Ajustement de posologie |
|
Niveau <5% |
Augmentation de 5 unités par kg |
|
Niveau entre 5% et 20% |
Pas d’ajustement |
|
2 mesures >20% |
Diminution de 5 unités par kg |
|
1 mesure >25% |
Diminution de 5 unités par kg |
L’activité exprimée en unités est déterminée en utilisant le test d'activité Berichrom FXIII, par rapport à la norme internationale en vigueur pour le facteur XIII de coagulation plasmatique. Par conséquent, une unité est égale à une unité internationale.
Prophylaxie avant intervention chirurgicale :
Après la dernière dose prophylactique de routine du patient, si une intervention chirurgicale est prévue :
· Entre 21 et 28 jours plus tard : administrer la dose prophylactique complète au patient immédiatement avant la chirurgie et la dose prophylactique suivante 28 jours plus tard.
· Entre 8 et 21 jours plus tard : une dose supplémentaire (totale ou partielle) peut être administrée avant la chirurgie. La dose doit être fondée sur le niveau d'activité en FXIII du patient, de son état clinique et doit être ajusté en fonction de la demi-vie de Fibrogammin.
· Dans les 7 jours suivant la dernière dose : une administration supplémentaire peut ne pas être nécessaire.
Les ajustements de posologie peuvent être différents de ces recommandations et doivent être individualisées en fonction des niveaux d'activité du FXIII et de l'état clinique du patient. Tous les patients doivent être étroitement surveillés pendant et après la chirurgie.
Ainsi, il est recommandé de contrôler l'augmentation de FXIII-activité avec un dosage du FXIII Dans le cas d'une intervention chirurgicale majeure et d’hémorragies graves, le but est d'obtenir près des valeurs normales (70% à 140%).
Mode d’administration
Reconstituer le produit comme décrit au paragraphe « Reconstitution » (rubrique 3. « Mode d’administration »). Avant administration, la préparation doit être amenée à la température ambiante ou corporelle. Injecter ou perfuser le produit avec le set d’administration fourni, par voie intraveineuse, lentement, à un débit confortable pour le patient. Le débit d’injection ou de perfusion ne doit pas dépasser 4 ml par minute.
Surveiller le patient à la recherche d’une éventuelle réaction immédiate. En cas de survenue d’une réaction paraissant liée à l’administration de FIBROGAMMIN, le débit de perfusion devra être ralenti ou la perfusion interrompue, selon l’état clinique du patient.
Mises en garde spéciales et précautions d’emploi
Chez les patients présentant des antécédents d’allergies au produit (associés à des symptômes tels que : urticaire généralisé, rash, chute de la pression sanguine, dyspnée), des antihistaminiques et des corticoïdes peuvent être administrés en prophylaxie.
En cas de thromboses récentes, des précautions doivent être prises en raison de l’effet stabilisant de la fibrine.
Après des traitements répétés par Fibrogammin, en l’absence de réponse clinique ou si le taux de facteur XIII n’atteint pas le niveau prévu, il est indispensable de rechercher la présence d’inhibiteur du facteur XIII (anticorps neutralisant le facteur XIII). Il faut alors démontrer la présence dans le plasma et le titrer à l’aide de tests biologiques appropriés.
Remarque à l’attention des patients sous régime hyposodé :
FIBROGAMMIN contient de 124,4 à 195,4 mg (5,41 à 8,50 mmol) de sodium par dose (40 UI/ kg de poids corporel – pour une moyenne de 70 kg), si une dose journalière recommandée (2800 UI= 44,8 ml) est administrée. Ceci est à prendre en compte chez les patients suivant un régime hyposodé strict.
Interactions avec d’autres médicaments et autres formes d’interaction
Aucune interaction du facteur XIII de coagulation humain avec d’autres médicaments n’est connue.