Dernière mise à jour le 01/06/2026
NEXPLANON 68 mg, implant pour usage sous-cutané
Indications thérapeutiques
NEXPLANON est un implant contraceptif préchargé dans un applicateur jetable. La sécurité et l’efficacité ont été établies chez les femmes entre 18 et 40 ans. L’implant est un petit bâtonnet en plastique, flexible, souple, de 4 cm de long et 2 mm de diamètre, qui contient 68 milligrammes d’une substance active, l’étonogestrel. L’applicateur permet au professionnel de santé d’insérer l’implant juste sous la peau au niveau de votre bras. L’étonogestrel est une hormone féminine synthétique proche de la progestérone. Une petite quantité d’étonogestrel est libérée en continu dans la circulation sanguine. Le bâtonnet lui-même est composé de copolymère éthylène/acétate de vinyle, une matière plastique qui ne se dissout pas dans le corps. Il contient également une petite quantité de sulphate de baryum qui le rend visible aux rayons X.
NEXPLANON est utilisé pour empêcher la survenue d’une grossesse.
Comment fonctionne NEXPLANON
L’implant est inséré juste sous la peau. Le composé actif, l’étonogestrel, a une double action :
· Il empêche la libération d’un ovocyte par les ovaires.
· Il entraîne des modifications de la glaire cervicale qui rendent difficile le passage des spermatozoïdes vers l’utérus.
En conséquence, NEXPLANON vous protège de la survenue d’une éventuelle grossesse pendant une période de trois ans, mais si vous êtes en surpoids, le professionnel de santé qui vous prescrit NEXPLANON peut vous conseiller de remplacer l’implant plus tôt. NEXPLANON fait partie des nombreux moyens de contraception. Une autre méthode de contraception fréquemment utilisée est la pilule combinée. A l’inverse des pilules combinées, NEXPLANON peut être utilisé par les femmes qui ne peuvent pas, ou ne veulent pas utiliser d’estrogènes.
Lorsque vous utilisez NEXPLANON, vous n'avez pas à vous rappeler de prendre un comprimé tous les jours. C’est l’une des raisons pour lesquelles NEXPLANON est très fiable (plus de 99 % d’efficacité). Si, dans de rares cas, NEXPLANON n’est pas inséré correctement ou n’est pas inséré du tout, vous pouvez ne pas être protégée de la survenue d’une éventuelle grossesse. Lors de l’utilisation de NEXPLANON, vos règles peuvent être modifiées et devenir absentes, irrégulières, rares, fréquentes, prolongées ou rarement abondantes. Le profil de saignement que vous observerez au cours des trois premiers mois est généralement prédictif de votre futur profil de saignement. Des règles douloureuses peuvent s’améliorer.
Vous pouvez arrêter d’utiliser NEXPLANON à tout moment (voir le paragraphe « Si vous voulez arrêter d’utiliser NEXPLANON »).
Présentations
> plaquette(s) thermoformée(s) PETG (polyéthylène téréphtalate glycol) de 1 implant avec applicateur acrylonitrile butadiène styrène avec aiguille acier inoxydable
Code CIP : 351 544-3 ou 34009 351 544 3 9
Déclaration de commercialisation : 02/05/2001
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 96,09 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 97,11 €
- Taux de remboursement :65%
Documents de bon usage du médicament
- Contraception chez la femme en post-partum
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception d’urgence
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception chez l’homme
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Stérilisation à visée contraceptive chez l’homme et chez la femme
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception chez la femme adulte et de l'adolescente en âge de procréer (hors post-partum et post-IVG)
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception chez la femme à risque cardio-vasculaire
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception : prescriptions et conseils aux femmes
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception hormonale orale : dispensation en officine
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception d’urgence : dispensation en officine
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception estroprogestative transdermique ou vaginale : dispensation en officine
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception chez la femme après une interruption volontaire de grossesse (IVG)
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Méthodes contraceptives : Focus sur les méthodes les plus efficaces disponibles
Auteur : Haute autorité de santé
Type : Evaluation des technologies de santé
Date de mise à jour :Novembre 2017
- Contraceptifs oraux estroprogestatifs : préférez les «pilules» de 1re ou 2e génération
Auteur : Haute autorité de santé
Type : Fiche Bon Usage du Médicament
Date de mise à jour :Janvier 2013
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 17/12/2025 | Réévaluation suite à résultats étude post-inscript | Le service médical rendu par NEXPLANON 68 mg (étonogestrel), implant pour usage sous-cutané, reste important dans l’indication de l’AMM. |
| Important | Avis du 16/09/2015 | Renouvellement d'inscription (CT) | Le service médical rendu par NEXPLANON reste important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 17/12/2025 | Réévaluation suite à résultats étude post-inscript | Compte tenu : • de l’efficacité bien établie de la contraception avec l’implant sous-cutané à base du progestatif étonogestrel, qui est le métabolite actif du désogestrel, • que l’ANSM a considéré qu’au plan pharmacologique, le risque de méningiome observé avec la pilule orale progestative au désogestrel 75 Mug, s’applique également à l’implant contraceptif NEXPLANON, l’étonogestrel étant le métabolite actif du désogestrel, • du profil de tolérance du désogestrel avec un sur-risque identifié de méningiome dans une étude pharmaco-épidémiologique EPI-PHARE sur une cohorte française réalisée à partir du SNDS sur le risque de méningiome intracrânien associé à l’utilisation prolongée de la contraception orale à base de désogestrel (OR = 1,25 [1,10-1,42], et de l’augmentation du risque avec la durée du traitement et avec l’âge de la femme, le risque étant accru à partir de l’âge de 45 ans, • de l’amplitude du risque de méningiome associé au désogestrel 75 Mug très inférieure à celle retrouvée lors de l’étude de l’utilisation prolongée d’autres progestatifs utilisés à fortes doses comme les acétates de cyprotérone, chlormadinone, nomégestrol, médroxyprogestérone, la médrogestone et la promégestone, la Commission considère que NEXPLANON 68 mg (étonogestrel), implant pour usage sous-cutané, n’apporte pas d’amélioration du service médical rendu (ASMR V) dans la stratégie thérapeutique actuelle qui comprend les comparateurs pertinents (cf. paragraphe 5.2). |
ANSM - Mis à jour le : 23/09/2025
NEXPLANON 68 mg, implant pour usage sous-cutané
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
NEXPLANON est un implant flexible, purement progestatif, non biodégradable, radio-opaque, préchargé dans un applicateur stérile, jetable.
Etonogestrel......................................................................................................................... 68 mg
Pour un implant.
Le taux de libération de l’étonogestrel est approximativement de 60˗70 µg/jour en 5 à 6 semaines puis diminue pour atteindre approximativement 35-45 µg/jour à la fin de la première année, environ 30- 40 µg/jour à la fin de la deuxième année et environ 25-30 µg/jour à la fin de la troisième année. La conception de l’applicateur permet une utilisation avec une seule main et facilite une insertion sous-cutanée correcte de l’implant.
Pour la liste complète des excipients, voir rubrique 6.1.
Implant pour usage sous-cutané.
Bâtonnet flexible, souple, blanc à blanc cassé, non biodégradable, radio-opaque, de 4 cm de long et 2 mm de diamètre.
4.1. Indications thérapeutiques
La sécurité et l’efficacité ont été établies chez les femmes entre 18 et 40 ans.
4.2. Posologie et mode d'administration
1 implant, qui peut être laissé en place pendant 3 ans.
Population pédiatrique
La sécurité et l’efficacité de NEXPLANON n’ont pas été étudiées chez les adolescentes de moins de 18 ans.
Mode d’administration
Toute grossesse doit être exclue avant l’insertion de NEXPLANON.
Il est fortement recommandé que NEXPLANON soit inséré et retiré uniquement par des professionnels de santé ayant été formés à l’utilisation de l’applicateur de NEXPLANON et aux techniques d’insertion et de retrait de l’implant NEXPLANON et le cas échéant, qu’une supervision soit demandée lors de l’insertion ou du retrait de l’implant.
Avant d’insérer l’implant, lire et suivre attentivement les instructions pour l'insertion et le retrait de l'implant dans la rubrique 4.2. « Comment insérer NEXPLANON » et « Comment retirer NEXPLANON ».
Des vidéos illustrant l’insertion et le retrait de l’implant sont disponibles en ligne www.nexplanonvideos.eu Veuillez contacter ORGANON France si vous avez des questions (tél. 01.57.77.32.00).
En cas de doute sur les étapes nécessaires pour insérer et/ou retirer NEXPLANON en toute sécurité, ne tentez pas la procédure de pose et/ou de retrait de l’implant.
Comment utiliser NEXPLANON
NEXPLANON est un contraceptif hormonal d’action prolongée. Un seul implant est inséré en sous-cutané et il peut être laissé en place pendant trois ans. Ne pas retirer l’implant plus de trois ans après la date d’insertion. L’utilisatrice doit être informée qu’elle peut demander le retrait de l’implant à n’importe quel moment. Les professionnels de santé habilités à prescrire NEXPLANON devront envisager de remplacer l’implant plus tôt chez les femmes en surpoids (voir rubrique 4.4.). Après le retrait de l’implant, l’insertion immédiate d’un autre implant assurera la continuité de la protection contraceptive. Si le souhait de la femme est de ne pas continuer avec NEXPLANON, une autre méthode contraceptive doit lui être conseillée pour éviter une grossesse.
L’étui de NEXPLANON contient une Carte d’Alerte Patiente destinée à la patiente mentionnant le numéro de lot de l'implant. Il est demandé au professionnel de santé effectuant l'insertion de noter la date d'insertion, le bras où l'implant est inséré et le jour prévu du retrait sur la Carte d’Alerte Patiente. Les patientes doivent être informées de conserver leur Carte d’Alerte Patiente dans un endroit sûr et de montrer leur carte lors de toute visite relative à l’usage de leur implant. La Carte d’Alerte Patiente contient également des instructions pour que la patiente palpe délicatement de manière occasionnelle l’implant afin d’être certaine qu’elle connaisse sa localisation. Les patientes doivent être informées de contacter leur médecin dès que possible si à tout moment elles ne sentent plus leur implant à la palpation. L’étui comprend également des étiquettes adhésives destinées au dossier médical mentionnant le numéro de lot. Cette information doit être incluse dans le dossier médical électronique de la patiente, si utilisé.
La réussite de l’utilisation et du retrait de NEXPLANON repose sur une insertion sous-cutanée de l’implant réalisée correctement et avec précaution conformément aux instructions.
· Si l’implant n’est pas inséré conformément aux instructions, et pas le jour adapté, ceci peut entraîner une grossesse non désirée (voir rubrique 4.2. « Comment insérer NEXPLANON » et rubrique « Quand insérer NEXPLANON »).
· Un implant inséré plus profondément qu’en sous-cutané (insertion profonde) peut ne pas être palpable et la localisation et/ou le retrait peuvent être difficiles (voir rubrique 4.2. « Comment retirer NEXPLANON » et rubrique 4.4).
L’implant NEXPLANON doit être inséré à la face interne du bras non dominant en sous-cutané, JUSTE SOUS LA PEAU. Le site d’insertion est en regard du triceps, à environ 8 à 10 cm de l’épicondyle médial de l’humérus et 3 à 5 cm postérieur au (sous le) sillon (gouttière) qui sépare le biceps du triceps. Cet emplacement est destiné à éviter les principaux vaisseaux sanguins et nerfs se trouvant dans et autour du sillon (voir Figures 2a, 2b et 2c).
Immédiatement après l’insertion, la présence de l’implant doit être vérifiée par palpation. Si l’implant ne peut pas être palpé ou s’il y a un doute sur sa présence, voir rubrique 4.2. « Comment insérer NEXPLANON», sous-rubrique « Si l’implant n’est pas palpable après l’insertion ».
Quand insérer NEXPLANON
IMPORTANT : Exclure toute grossesse avant l’insertion de l’implant.
Le moment choisi pour l’insertion dépend de la situation contraceptive récente de la femme, comme suit :
Absence préalable de contraception hormonale utilisée au cours du mois précédent
L’implant doit être inséré entre le 1er jour (premier jour des menstruations) et le 5ème jour du cycle menstruel, même si la femme saigne toujours.
Si l’insertion a lieu au moment recommandé, une contraception complémentaire n'est pas nécessaire. Si l’insertion a lieu à un autre moment que celui recommandé pour l’insertion, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue.
Passage d’une méthode contraceptive hormonale à NEXPLANON
Relais d’un contraceptif hormonal combiné (ex : contraceptif oral combiné (COC), anneau vaginal ou patch transdermique)
L’implant doit être inséré de préférence le lendemain de la prise du dernier comprimé actif (le dernier comprimé contenant les substances actives) de son précédent contraceptif oral combiné ou le jour du retrait de l’anneau vaginal ou du patch transdermique.
Au plus tard, l’implant doit être inséré le lendemain de l’intervalle habituel sans comprimé, sans anneau, sans patch ou de la prise de comprimés placebo de son précédent contraceptif hormonal combiné, quand la prochaine prise/insertion/application aurait dû avoir lieu. Toutes les méthodes contraceptives (patch transdermique, anneau vaginal) peuvent ne pas être commercialisées dans tous les pays.
Si l’insertion a lieu au moment recommandé, une contraception complémentaire n'est pas nécessaire. Si l’insertion a lieu à un autre moment que celui recommandé pour l’insertion, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue.
Relais d’une méthode purement progestative (ex : pilule progestative, injection, implant ou système intra-utérin (SIU) libérant un progestatif)
Comme plusieurs types de méthodes purement progestatives existent, l’insertion de l’implant doit se faire comme suit :
· Contraceptifs injectables : insérer l’implant le jour prévu pour l’injection suivante.
· Pilule progestative : la femme peut passer de la pilule purement progestative à NEXPLANON n’importe quel jour du mois. L’implant doit être inséré dans les 24 heures suivant la prise du dernier comprimé.
· Implant/Système intra-utérin (SIU) : insérer l’implant le jour du retrait du précédent implant ou du SIU.
Si l’insertion a lieu au moment recommandé, une contraception complémentaire n'est pas nécessaire. Si l’insertion a lieu à un autre moment que celui recommandé pour l’insertion, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue.
Après un avortement ou une fausse couche
L’implant peut être inséré directement après un avortement ou une fausse couche.
· Premier trimestre : Si l’insertion a lieu sous 5 jours, une contraception complémentaire n’est pas nécessaire.
· Deuxième trimestre : Si l’insertion a lieu sous 21 jours, une contraception complémentaire n’est pas nécessaire.
Si l’insertion a lieu après le moment recommandé pour l’insertion, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue avant l’insertion.
En post-partum
L’implant peut être inséré immédiatement après l’accouchement à la fois chez une patiente qui allaite et une patiente qui n’allaite pas, en considérant les bénéfices et les risques individuels pour la patiente.
· Si l’insertion a lieu sous 21 jours, une contraception complémentaire n'est pas nécessaire.
· Si l’insertion a lieu plus de 21 jours après l’accouchement, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue avant l’insertion.
Comment insérer NEXPLANON
La réussite de l’utilisation et du retrait de NEXPLANON repose sur une insertion sous-cutanée de l’implant dans le bras non dominant réalisée correctement et avec précaution conformément aux instructions. Le professionnel de santé ayant effectué l'insertion ainsi que la patiente doivent être capables de palper l’implant sous la peau de la femme après insertion.
L’implant doit être inséré à la face interne du bras non dominant en sous-cutané, juste sous la peau.
· Un implant inséré plus profondément qu’en sous-cutané (insertion profonde) peut ne pas être palpable et la localisation et/ou le retrait peuvent être difficiles (voir rubrique 4.2. « Comment retirer NEXPLANON » et rubrique 4.4).
· Si l’implant est inséré profondément, une lésion nerveuse ou vasculaire peut se produire. Des insertions profondes ou incorrectes ont été associées à une paresthésie (due à une lésion nerveuse) et à une migration de l’implant (due à une insertion dans le muscle ou dans le fascia), et dans de rares cas, à une insertion intravasculaire.
L’insertion de NEXPLANON doit être effectuée dans des conditions d’asepsie et uniquement par un professionnel de santé habilité à prescrire NEXPLANON et familiarisé avec la technique. L’insertion de l’implant doit être réalisée uniquement avec l’applicateur préchargé.
Procédure d’insertion
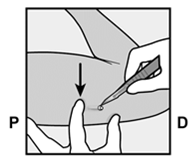
Pour s’assurer que l’implant est inséré juste sous la peau, le professionnel de santé doit se placer de manière à voir le mouvement de l’aiguille en regardant l’applicateur par le côté et non par le dessus du bras. Par cette vue de côté, le site d’insertion et le mouvement de l’aiguille juste sous la peau pourront être bien visualisés.
À des fins d’illustration, les figures représentent la face interne du bras gauche.
|
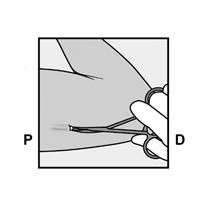
· Demandez à la patiente de s’allonger sur le dos sur la table d’examen avec son bras non dominant plié au niveau du coude et tourné vers l’extérieur, ainsi, sa main est placée sous la tête (ou le plus près possible) (Figure 1). |
|
|
· Identifiez le site d’insertion, qui se situe à la face interne du bras non dominant. Le site d’insertion est en regard du triceps, à environ 8 à 10 cm de l’épicondyle médial de l’humérus et 3 à 5 cm postérieur au (sous le) sillon (gouttière) qui sépare le biceps du triceps (Figures 2a, 2b et 2c). Cet emplacement est destiné à éviter les principaux vaisseaux sanguins et nerfs se trouvant dans et autour du sillon. S’il n’est pas possible d’insérer l’implant à cet endroit (par exemple, chez une femme avec des bras minces), il devra de toute façon être inséré en arrière et aussi loin que possible du sillon. |
|
|
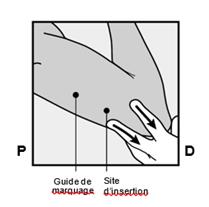
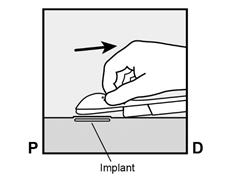
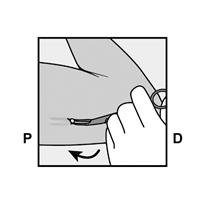
· Faites deux repères avec un marqueur chirurgical : un premier point, pour repérer l’endroit où l’implant sera inséré, et un second point, à 5 centimètres au-dessus (en direction de l’épaule) du premier repère (Figure 2a et 2b). Ce second repère (guide de marquage) servira plus tard de guide pour la direction pendant l’insertion. |
|
|
|
|
|
Figure 2a P, proximal (en direction de l’épaule) D, distal (en direction du coude) |
Figure 2b |
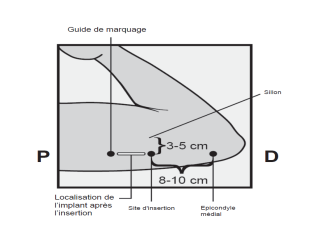
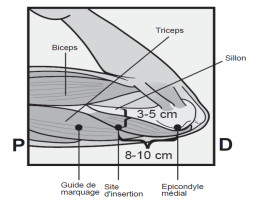
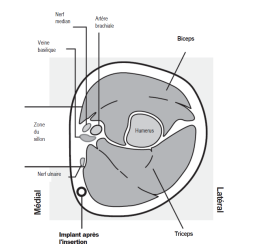
Figure 2c
Coupe transversale du bras gauche,
vu du coude
Médial (face interne du bras)
Latéral (face externe du bras)
· Après avoir marqué le bras, confirmez que le site est correctement localisé à la face interne du bras.
· Nettoyez la peau depuis le site d’insertion jusqu’au guide de marquage avec une solution antiseptique.
· Anesthésiez la zone d’insertion (par exemple, avec un anesthésique en spray ou en injectant 2 ml de lidocaïne à 1 % juste sous la peau le long du tunnel d’insertion prévu).
· Sortez de son emballage l’applicateur NEXPLANON préchargé stérile jetable contenant l’implant. Avant utilisation, inspecter visuellement l’emballage pour s’assurer qu’il n’est pas endommagé (i.e. emballage tordu, perforé ). Si l’emballage présente une altération visible, qui pourrait compromettre la stérilité, l’applicateur ne doit pas être utilisé.
|
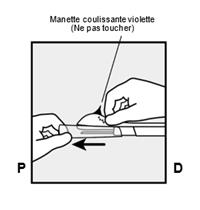
· Tenez l’applicateur juste au-dessus de l’aiguille au niveau de la zone striée. Retirez le capuchon protecteur transparent de l’aiguille en le faisant glisser horizontalement, dans le sens de la flèche (Figure 3). Si le capuchon ne se retire pas facilement, l’applicateur ne doit pas être utilisé. Vous devez voir l’implant blanc en regardant dans la pointe de l’aiguille. Ne touchez pas la manette coulissante violette avant d’avoir entièrement inséré l’aiguille sous la peau, car cela rétracterait l’aiguille et libérerait prématurément l’implant de l’applicateur. · Si la manette coulissante violette est libérée prématurément, recommencez la procédure d’insertion avec un nouvel applicateur. |
|
|
|
Figure 3
|
|
· Avec votre main libre, tendez la peau autour du site d’insertion vers le coude (Figure 4). |
|
|
|
Figure 4 |
|
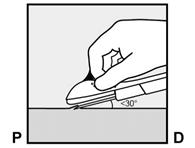
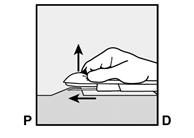
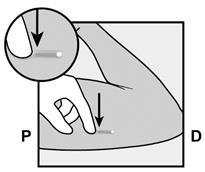
· L’implant doit être inséré en sous-cutané, juste sous la peau (voir rubrique 4.4). Pour s’assurer que l’implant est inséré juste sous la peau, vous devez vous placer de manière à voir le mouvement de l’aiguille en regardant l’applicateur par le côté et non par le dessus du bras. Par cette vue de côté, vous pouvez clairement voir le site d’insertion et le mouvement de l’aiguille juste sous la peau (Figure 6). · Piquez la peau avec la pointe de l’aiguille légèrement inclinée selon un angle inférieur à 30° (Figure 5a). |
|
|
|
Figure 5a |
|
· Insérez l’aiguille jusqu’à ce que le biseau (ouverture inclinée de la pointe) soit juste sous la peau (et pas plus loin) (Figure 5b). Si vous avez inséré l’aiguille plus en profondeur que le biseau, retirez l’aiguille jusqu’à ce que seul le biseau soit sous la peau. |
|
|
|
Figure 5b |
|
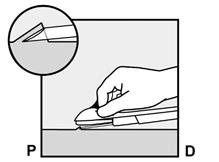
· Abaissez l’applicateur en position presque horizontale. Pour faciliter le positionnement sous-cutané, soulevez la peau avec l’aiguille, tout en la faisant glisser sur toute sa longueur (Figure 6). Vous pouvez ressentir une légère résistance mais n’exercez pas de force excessive. Si l’aiguille n’est pas entièrement insérée, l’implant ne sera pas correctement inséré. Si la pointe de l’aiguille sort de la peau avant que l’aiguille soit entièrement insérée, l’aiguille doit être retirée et replacée en sous-cutané pour terminer la procédure d’insertion. |
|
|
|
Figure 6 |
|
· Maintenez l’applicateur dans la même position avec l’aiguille insérée sur toute sa longueur (Figure 7). Si nécessaire, vous pouvez utiliser votre main libre pour immobiliser l’applicateur. |
|
|
|
Figure 7 |
|
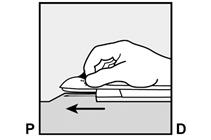
· Déverrouillez la manette coulissante violette en la poussant légèrement vers le bas (Figure 8a). Déplacez la manette coulissante complètement en arrière jusqu’à la butée. Ne déplacez pas ( |
|
|
|
Figure 8a |
|
|
|
|
|
Figure 8b |
|
|
Figure 8c |
|
Si l’applicateur n’est pas maintenu dans la même position au cours de la procédure d’insertion ou si la manette coulissante violette n’est pas complètement tirée en arrière jusqu’à la butée, l’implant ne sera pas correctement inséré et il pourrait sortir du site d’insertion. Si l’implant sort du site d’insertion, retirez l’implant et effectuez une nouvelle procédure d’insertion au niveau du même site d’insertion en utilisant un nouvel applicateur. Ne renfoncez pas l’implant ressorti dans l’incision. · Appliquez un petit pansement adhésif sur le site d’insertion
|
|
|
· Vérifiez toujours la présence de l’implant dans le bras de la patiente par palpation immédiatement après l’insertion. En palpant les deux extrémités de l’implant, vous devez pouvoir confirmer la présence du bâtonnet de 4 cm (Figure 9). Voir rubrique ci-dessous « Si l’implant n’est pas palpable après l’insertion ». |
|
|
|
Figure 9 |
|
· Demandez à la patiente de palper elle-même l’implant. · Appliquez une compresse stérile avec un bandage compressif pour minimiser le risque d’ecchymose. La patiente peut retirer le bandage compressif au bout de 24 heures et le petit pansement adhésif au bout de 3 à 5 jours. · Complétez la Carte d’Alerte Patiente et remettez-la à la patiente en lui demandant de la conserver. Complétez également les étiquettes adhésives et collez-les dans le dossier médical de la patiente. Si des dossiers médicaux électroniques sont utilisés, les informations figurant sur l’étiquette adhésive doivent être enregistrées. · L’applicateur est à usage unique et doit être correctement éliminé, conformément aux réglementations nationales d’élimination des déchets biologiques. |
|
Si l’implant n’est pas palpable après l’insertion
Si vous ne pouvez pas palper l’implant ou si vous doutez de sa présence, l’implant peut ne pas avoir été inséré ou il peut avoir été inséré profondément :
· Vérifiez l’applicateur. L’aiguille doit être complètement rétractée et seul le bout violet de l’obturateur doit être visible.
· Utilisez d’autres méthodes pour confirmer sa présence. Etant donné la nature radio-opaque de l’implant, les méthodes de localisation adaptées sont la radiographie bidimensionnelle et la tomodensitométrie (TDM). L’échographie avec sonde linéaire à haute fréquence (10 MHz ou plus) ou l’imagerie par résonance magnétique (IRM) peuvent être utilisées. Si l’implant ne peut pas être trouvé avec ces méthodes d’imagerie, il est recommandé de vérifier la présence de l’implant en mesurant le taux d’étonogestrel dans un échantillon de sang prélevé chez la patiente. Dans ce cas, contactez ORGANON France qui vous communiquera le protocole approprié.
· La patiente doit utiliser une méthode contraceptive non hormonale tant que la présence de l’implant n’est pas confirmée.
· Les implants insérés trop profondément doivent être localisés et retirés dès que possible pour éviter tout risque de migration (voir rubrique 4.4).
Comment retirer NEXPLANON
Le retrait de l’implant doit être effectué uniquement dans des conditions d’asepsie et par un professionnel de santé familiarisé avec la technique de retrait. Si vous n’êtes pas familiarisé avec la technique de retrait, contactez ORGANON France pour plus d’informations.
Avant de débuter la procédure de retrait, le professionnel de santé effectuant le retrait doit localiser l’implant. Vérifiez la localisation exacte de l’implant dans le bras par palpation.
Si l’implant n’est pas palpable, consultez la Carte d’Alerte Patiente ou le dossier médical pour vérifier le bras porteur de l’implant. Si l’implant ne peut pas être palpé, il pourrait être localisé en profondeur ou avoir migré. Envisagez qu’il puisse se situer à proximité de vaisseaux et de nerfs. Le retrait des implants non palpables doit uniquement être effectué par un professionnel de santé ayant l’expérience du retrait des implants insérés profondément et familiarisé avec la localisation de l’implant et l’anatomie du bras. Contactez ORGANON France pour plus d’informations.
Voir la rubrique ci-dessous « Localisation et retrait d’un implant non palpable » si l’implant ne peut pas être palpé.
Procédure de retrait d’un implant palpable
À des fins d’illustration, les figures représentent la face interne du bras gauche.
|
· Demandez à la patiente de s’allonger sur le dos sur la table. Le bras doit être placé avec le coude plié et la main sous la tête (ou le plus près possible) (Voir Figure 10) |
Figure 10 |
|
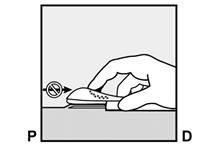
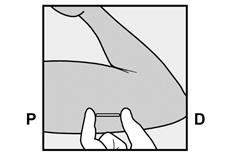
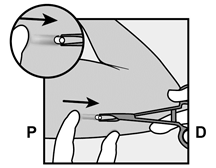
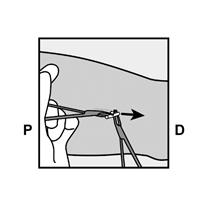
· Localisez l’implant par palpation. Appuyez sur l’extrémité de l’implant la plus proche de l’épaule (Figure 11) pour l’immobiliser ; un renflement devrait apparaître indiquant l’extrémité de l’implant la plus proche du coude. Si l’extrémité n’apparaît pas, le retrait de l’implant pourrait être difficile et il devra être effectué par un professionnel de santé familiarisé avec le retrait des implants profonds. Contactez ORGANON France pour plus d’informations. · Marquez l’extrémité distale (l’extrémité la plus proche du coude), par exemple, avec un marqueur chirurgical. · Nettoyez le site avec une solution antiseptique. |
Figure 11 P, proximal (en direction de l’épaule) ; D, distal (en direction du coude) |
|
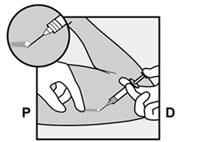
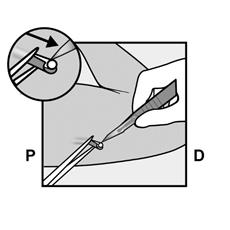
· Anesthésiez le site, par exemple, avec 0,5 à 1 ml de lidocaïne à 1 % à l’endroit de l’incision (Figure 12). Veillez à injecter l’anesthésique local sous l’implant pour que l’implant reste près de la surface de la peau. L’injection d’un anesthésique local au-dessus de l’implant peut rendre le retrait plus difficile. |
|
|
|
Figure 12 |
|
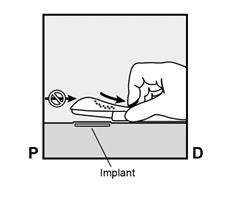
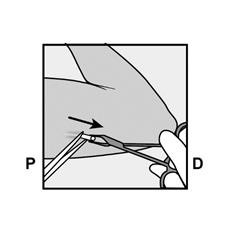
· Appuyez sur l’extrémité de l’implant la plus proche de l’épaule (Figure 13) pour l’immobiliser tout au long de la procédure de retrait. En partant au-dessus de l’extrémité de l’implant la plus proche du coude, faire une incision longitudinale (parallèle à l’implant) de 2 mm vers le coude. Veillez à ne pas couper l’extrémité de l’implant. |
|
|
|
Figure 13 |
|
· L’extrémité de l’implant doit sortir de l’incision. Si ce n’est pas le cas, poussez doucement l’implant vers l’incision jusqu’à ce que l’extrémité soit visible. Saisissez l’implant avec une pince et si possible, retirez l’implant (Figure 14). Si nécessaire, retirez doucement le tissu adhérent de l’extrémité de l’implant par dissection mousse. Si l’extrémité de l’implant n’est pas exposée après la dissection mousse, pratiquez une incision dans la gaine tissulaire et ensuite retirez l’implant avec une pince (Figures 15 et 16). |
|
|
|
Figure 14 |

|
|
|
|
Figure 15 |
Figure 16 |
|
· Si l’extrémité de l’implant n’est pas visible au niveau de l’incision, insérez doucement une pince (de préférence une pince mosquito courbe, avec les extrémités dirigées vers le haut) superficiellement dans l’incision (Figure 17). · Saisissez délicatement l’implant puis, tournez la pince avec votre autre main (Figure 18). · Avec une seconde pince, disséquez soigneusement le tissu autour de l’implant et saisissez l’implant (Figure 19). L’implant peut alors être retiré. · Si l’implant ne peut pas être saisi, arrêtez la procédure de retrait et orientez la patiente vers un professionnel de santé familiarisé avec les retraits complexes ou contactez ORGANON France. |
|
|
|
|
|
|
Figure 17 |
Figure 18 |
Figure 19 |
|
· Vérifiez que la totalité du bâtonnet, qui mesure 4 cm de long, a été retiré en le mesurant. Des cas d’implants cassés dans le bras des patientes ont été rapportés. Dans certains cas, une difficulté de retrait de l’implant cassé a été signalée. Si un implant incomplet (moins de 4 cm) est retiré, le morceau restant devra être retiré en suivant les instructions de cette rubrique. · Si la femme souhaite continuer à utiliser NEXPLANON, un nouvel implant peut être inséré immédiatement après le retrait du précédent implant en utilisant la même incision à condition que la localisation du site soit correcte (voir rubrique 4.2 « Comment remplacer NEXPLANON »). · Après avoir retiré l’implant, fermez l’incision avec un pansement adhésif stérile. · Appliquez une compresse stérile avec un bandage compressif pour minimiser le risque d’ecchymose. La patiente peut retirer le bandage compressif au bout de 24 heures et le pansement adhésif stérile au bout de 3 à 5 jours. |
||
Localisation et retrait d’un implant non palpable
Il y a eu des rapports occasionnels de migration de l’implant ; habituellement il s’agit d’un mouvement mineur par rapport à la position initiale (voir aussi rubrique 4.4.) mais cela peut conduire à un implant non palpable à l’endroit où il avait été placé. Un implant qui a été inséré profondément ou qui a migré peut ne pas être palpable et c’est pourquoi des procédures d’imagerie, telles que décrites ci-dessous, peuvent s’avérer nécessaires pour la localisation.
Un implant non palpable doit toujours être localisé avant d’essayer de le retirer. Etant donné la nature radio-opaque de l’implant, les méthodes adaptées pour la localisation comprennent la radiographie bidimensionnelle et la tomodensitométrie (TDM), L’échographie avec sonde linéaire à haute fréquence (10 MHz ou plus) ou l’imagerie par résonance magnétique (IRM) peuvent être utilisées. Une fois l’implant localisé dans le bras, l’implant doit être retiré par un professionnel de santé ayant l’expérience du retrait des implants insérés profondément et familiarisé avec l’anatomie du bras. L’utilisation du guidage échographique pendant le retrait doit être envisagée.
Si l’implant ne peut pas être trouvé dans le bras après des tentatives exhaustives de localisation, envisagez d’appliquer les techniques d’imagerie au niveau du thorax, car des cas extrêmement rares de migration dans le système vasculaire pulmonaire ont été rapportés. Si l’implant est localisé dans le thorax, des interventions chirurgicales ou endovasculaires peuvent être nécessaires pour le retrait ; des professionnels de santé familiarisés avec l’anatomie thoracique doivent être consultés.
Si, à tout moment, ces méthodes d’imagerie échouent à localiser l’implant, la détermination du taux sanguin d’étonogestrel peut être utilisée pour vérifier la présence de l’implant. Veuillez contacter ORGANON France pour plus de conseils.
Si l’implant a migré dans le bras, le retrait peut nécessiter une intervention chirurgicale mineure avec une incision plus grande ou une intervention chirurgicale en salle d’opération. Le retrait d’implants insérés profondément doit être effectué avec précaution afin d’aider à éviter toute lésion des structures nerveuses ou vasculaires profondes dans le bras.
Les implants non palpables et insérés profondément doivent être retirés par un professionnel de santé familiarisé avec l’anatomie du bras et le retrait des implants insérés profondément.
Une chirurgie exploratoire sans connaissance de la localisation exacte de l’implant est fortement déconseillée.
Veuillez contacter ORGANON France pour plus de conseils.
Comment remplacer NEXPLANON
Un remplacement immédiat peut être réalisé après le retrait du précédent implant et la procédure est semblable à la procédure d’insertion décrite dans la rubrique 4.2 « Comment insérer NEXPLANON ».
Le nouvel implant peut être inséré dans le même bras et par l’incision effectuée pour retirer le précédent implant à condition que la localisation du site soit correcte, c’est-à-dire 8 à 10 cm de l’épicondyle médial de l’humérus et 3 à 5 cm postérieur au (sous le) sillon (voir rubrique 4.2 « Comment insérer NEXPLANON »). Si la même incision est utilisée pour insérer le nouvel implant, anesthésiez le site d’insertion en injectant un anesthésique, par exemple 2 ml de lidocaïne à 1 %, juste sous la peau à partir de l’incision de retrait et le long du « canal d’insertion » et suivez les étapes suivantes des instructions d’insertion.
· Accident thromboembolique veineux évolutif.
· Tumeurs malignes, connues ou suspectées, sensibles aux stéroïdes sexuels.
· Présence ou antécédent de tumeurs du foie (bénigne ou maligne).
· Présence ou antécédent d’affection hépatique sévère tant que les paramètres de la fonction hépatique ne se sont pas normalisés.
· Hémorragies génitales non diagnostiquées.
· Hypersensibilité à la substance active ou à l’un des excipients listés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Cancer du sein :
Le risque de cancer du sein augmente généralement avec l’âge. Pendant l’utilisation de contraceptifs oraux (combinés) [CO], le risque de diagnostic d’un cancer du sein est légèrement augmenté. L’augmentation de ce risque disparaît progressivement au cours des 10 ans suivant l’arrêt de l’utilisation de CO et n’est pas liée à la durée d’utilisation, mais à l’âge de la femme au moment de l’utilisation du CO. Le nombre attendu de cas diagnostiqués pour 10 000 femmes qui utilisent des CO combinés (jusqu’à 10 ans après l’arrêt) comparativement à celles ne les ayant jamais utilisés pendant la même période a été calculé pour les groupes d’âge respectifs comme étant : 4,5/4 (16‑19 ans), 17,5/16 (20‑24 ans), 48,7/44 (25‑29 ans), 110/100 (30‑34 ans), 180/160 (35‑39 ans) et 260/230 (40‑44 ans). Le risque chez les utilisatrices de méthodes contraceptives uniquement progestatives est peut-être du même ordre que celui associé aux CO combinés. Cependant, pour ces méthodes, la preuve est moins concluante. Comparativement au risque de cancer du sein au cours d’une vie, l’augmentation du risque associé aux CO est faible. Les cas de cancers du sein diagnostiqués chez les utilisatrices de CO tendent à être moins avancés que chez les non‑utilisatrices de CO. L’augmentation du risque observé chez les utilisatrices de CO peut être liée à un diagnostic plus précoce, aux effets biologiques des CO ou à l’association des deux.
Troubles hépatiques :
En cas de troubles hépatiques aigus ou chroniques, la femme doit être adressée à un spécialiste pour examen et conseils.
Evènements thrombotiques et vasculaires :
Des études épidémiologiques ont associé l’utilisation des contraceptifs oraux combinés (estrogène + progestatif) à une augmentation de l’incidence d’accidents thromboemboliques veineux (TEV, thrombose veineuse profonde et embolie pulmonaire) et d’accidents thromboemboliques artériels (TEA, infarctus du myocarde et accident vasculaire cérébral ischémique). La pertinence clinique de ces résultats pour l’étonogestrel (le métabolite actif du désogestrel), utilisé comme contraceptif purement progestatif en l’absence d’un composant estrogénique, n’est pas connue.
Des données épidémiologiques limitées ne suggèrent pas d’augmentation du risque de TEV ou de TEA chez les femmes utilisant l’implant ; cependant, des TEV et des TEA ont été rapportés depuis la commercialisation chez les femmes utilisant des implants à l’étonogestrel.
Il est recommandé d’évaluer les facteurs de risque connus pour augmenter le risque de TEV et de TEA. Les femmes ayant des antécédents d’accidents thromboemboliques doivent être averties de la possibilité d’une récidive. L’implant doit être retiré en cas de thrombose. Le retrait de l’implant doit aussi être considéré en cas d’immobilisation de longue durée liée à une intervention chirurgicale ou à une maladie.
Pression artérielle élevée :
Si une hypertension artérielle prolongée se développe pendant l’utilisation de NEXPLANON, ou si une augmentation significative de la pression artérielle ne répond pas convenablement à une thérapeutique antihypertensive, l’utilisation de NEXPLANON doit être arrêtée.
Effet sur le métabolisme des glucides :
L’utilisation de contraceptifs contenant des progestatifs peut avoir un effet sur la résistance périphérique à l’insuline et sur la tolérance au glucose. Par conséquent, les femmes diabétiques doivent être attentivement suivies au cours des premiers mois d’utilisation de NEXPLANON.
Chloasma :
Un chloasma peut occasionnellement apparaître, notamment chez les femmes ayant des antécédents de chloasma gravidique. Les femmes ayant des prédispositions au chloasma doivent éviter de s’exposer au soleil ou aux ultraviolets au cours de l’utilisation de NEXPLANON.
Poids corporel :
L’effet contraceptif de NEXPLANON est lié aux concentrations plasmatiques d’étonogestrel, qui sont inversement proportionnelles au poids corporel, et qui diminuent avec le temps après l’insertion. L’expérience clinique chez les femmes en surpoids au cours de la 3ème année d’utilisation est limitée. Il ne peut donc être exclu que l’effet contraceptif chez ces femmes au cours de la 3ème année d’utilisation puisse être inférieur à celui observé chez les femmes de poids normal. Les professionnels de santé habilités à prescrire NEXPLANON doivent donc envisager de remplacer plus tôt l’implant chez les femmes en surpoids.
Complications d’insertion :
Des cas de migration de l’implant dans le bras depuis le site d’insertion ont été rapportés, pouvant être liés à une insertion profonde (voir rubrique 4.2. « Comment insérer NEXPLANON ») ou à des pressions extérieures (ex : manipulation de l’implant ou sports de contact). Depuis la commercialisation, de rares cas d’implants localisés dans les vaisseaux du bras et dans l’artère pulmonaire ont également été rapportés, pouvant être liés à des insertions profondes ou une insertion intravasculaire. Dans les cas où l’implant a migré dans le bras depuis le site d’insertion, la localisation de l’implant peut être rendue plus difficile et le retrait peut nécessiter une intervention chirurgicale mineure avec une incision plus large ou une intervention chirurgicale dans une salle d’opération. Dans les cas où l’implant a migré dans l’artère pulmonaire, une intervention endovasculaire ou chirurgicale peut être nécessaire pour le retrait (voir rubrique 4.2. « Comment retirer NEXPLANON »). Si, à tout moment, l’implant ne peut être palpé, il doit être localisé et le retrait est recommandé dès que médicalement approprié. Si l’implant n’est pas retiré, son effet contraceptif et le risque d’effets indésirables liés au progestatif pourront persister au-delà de la durée désirée par la femme.
Une expulsion peut survenir en particulier si l’implant n’a pas été inséré conformément aux instructions données à la rubrique 4.2. « Comment insérer NEXPLANON », ou à la suite d’une réaction inflammatoire locale.
Kystes ovariens :
Avec tous les contraceptifs hormonaux faiblement dosés, le développement folliculaire persiste et occasionnellement un follicule peut continuer à croître dépassant la taille qu’il atteindrait au cours d’un cycle normal. En général, ces follicules hypertrophiés disparaissent spontanément. Ils sont souvent asymptomatiques ; dans certains cas, ils sont associés à de légères douleurs abdominales. Ils nécessitent rarement une intervention chirurgicale.
Grossesses extra-utérines :
Avec les pilules uniquement progestatives traditionnelles, la protection vis-à-vis des grossesses extra‑utérines n’est pas aussi bonne qu’avec les CO combinés, en raison de la survenue fréquente d’ovulations au cours de leur utilisation. Bien que NEXPLANON inhibe l’ovulation, une grossesse extra‑utérine doit être envisagée au cours d’un diagnostic différentiel si la femme présente une aménorrhée ou des douleurs abdominales.
Troubles psychiatriques :
L’état dépressif et la dépression sont des effets indésirables bien connus liés à l’utilisation de contraceptifs hormonaux (voir rubrique 4.8). La dépression peut être grave et constitue un facteur de risque bien connu de comportement suicidaire et de suicide. Il convient de conseiller aux femmes de contacter leur médecin en cas de changements d’humeur et de symptômes dépressifs, y compris peu de temps après le début du traitement.
Autres événements :
Les événements suivants ont été rapportés à la fois lors d’une grossesse et lors de l’utilisation de stéroïdes sexuels, mais l’imputabilité de ces manifestations à l’utilisation de progestatifs n’a pas été démontrée : ictère et/ou prurit lié à une cholestase ; formation de lithiase biliaire ; porphyrie ; lupus érythémateux disséminé ; syndrome hémolytique et urémique ; chorée de Sydenham ; herpès gestationnel ; perte d’audition liée à une otosclérose et angiœdème (héréditaire).
Examen / consultation médical(e)
Avant l’insertion ou la réinsertion de NEXPLANON, un interrogatoire médical complet (incluant les antécédents médicaux familiaux) doit être fait et une grossesse doit être exclue. La pression artérielle doit être mesurée et un examen physique doit être réalisé, avec recherche des contre-indications (voir rubrique 4.3.) et des mises en garde (voir rubrique 4.4.). Il est recommandé que la femme revienne pour un contrôle médical trois mois après l’insertion de NEXPLANON.
Au cours de ce contrôle, le professionnel de santé devra mesurer la pression artérielle et vérifier si la femme a des questions, des plaintes ou si des effets indésirables sont apparus. La fréquence et la nature des examens médicaux réguliers futurs doivent être adaptées à chaque femme, en fonction de l’avis médical. L’implant doit être palpé à chaque visite de contrôle. La patiente doit être informée de contacter son médecin dès que possible si elle ne sent plus son implant à la palpation à tout moment entre chaque visite de contrôle.
Les femmes seront informées que NEXPLANON ne protège pas du VIH (SIDA) ni des autres infections sexuellement transmissibles.
Diminution de l’efficacité avec des traitements concomitants
L’efficacité de NEXPLANON peut être réduite lors de l’utilisation de traitements concomitants qui diminuent la concentration plasmatique de l’étonogestrel (voir rubrique 4.5.).
Changements du profil de saignement vaginal
Pendant l’utilisation de NEXPLANON, il est probable que les femmes observent des changements de leur profil de saignement vaginal qui seront imprédictibles. Ceux-ci peuvent inclure l’apparition de saignements vaginaux irréguliers (absents, moins fréquents, plus fréquents ou continus) et des changements de l’intensité des saignements (réduits ou augmentés) ou de leur durée. Des aménorrhées ont été rapportées chez environ 1 femme sur 5 tandis que chez d’autres femmes (1 femme sur 5), il a été rapporté des saignements fréquents et/ou prolongés. Chez beaucoup de femmes, le profil de saignement observé au cours des trois premiers mois est généralement prédictif du futur profil de saignement.
Une information, des conseils et l’utilisation d’un calendrier des saignements peuvent améliorer l’adhésion de la femme à son profil de saignement. L’évaluation des saignements vaginaux doit être faite au cas par cas et peut inclure un examen visant à éliminer une pathologie gynécologique ou une grossesse.
Implant cassé ou plié in situ
Des cas d’implants cassés ou pliés qui peuvent être dus à des pressions extérieures exercées dans le bras des patientes ont été rapportés. Des cas de migration dans le bras d'un fragment d'implant cassé ont également été rapportés. Sur la base des données in vitro, lorsque l’implant est cassé ou plié, le taux de diffusion de l’étonogestrel peut être légèrement augmenté.
Il n’est pas attendu d’effet cliniquement significatif avec ce changement.
Cependant, lorsque l’implant est cassé, il doit être retiré, et il est important de l’enlever dans son intégralité. Se référer à la rubrique 4.2 concernant les procédures de retrait de l’implant (à la fois palpable ou non palpable).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Effets d’autres médicaments sur NEXPLANON
Des interactions peuvent se produire avec des médicaments inducteurs des enzymes microsomales, ce qui peut conduire à une augmentation de la clairance des hormones sexuelles et peut entraîner des hémorragies de privation et/ou un échec de l’effet contraceptif.
Conduite à tenir
L’induction enzymatique peut être observée après seulement quelques jours de traitement. L’induction enzymatique maximale est généralement observée en quelques semaines. Après l’arrêt du traitement, l’induction enzymatique peut perdurer pendant environ 4 semaines.
Les femmes traitées par des médicaments inducteurs des enzymes hépatiques ou des produits à base de plantes doivent être averties que l’efficacité de NEXPLANON peut être réduite. Le retrait de l’implant n’est pas nécessaire, mais il est conseillé aux femmes d’utiliser une méthode contraceptive non hormonale supplémentaire pendant la durée du traitement concomitant et pendant 28 jours après l’arrêt de celui-ci afin d’obtenir une protection maximale.
Les interactions suivantes ont été rapportées dans la littérature (principalement avec les contraceptifs combinés mais occasionnellement aussi avec les contraceptifs purement progestatifs, y compris NEXPLANON) :
Substances augmentant la clairance des contraceptifs hormonaux (diminution de l’efficacité des contraceptifs hormonaux par induction enzymatique), par ex :
Barbituriques, bosentan, carbamazépine, phénytoïne, primidone, rifampicine et traitements anti-VIH/VHC tels que l’éfavirenz, le bocéprévir, la névirapine et potentiellement aussi le felbamate, la griséofulvine, l’oxcarbazépine, le topiramate et les produits contenant du millepertuis (Hypericum perforatum).
Substances ayant des effets variables sur la clairance des contraceptifs hormonaux
Lors de l’administration concomitante avec des contraceptifs hormonaux, de nombreuses associations d’inhibiteurs de la protéase du VIH et d’inhibiteurs non nucléosidiques de la transcriptase inverse, y compris des associations avec des inhibiteurs du VHC, peuvent augmenter ou diminuer les concentrations plasmatiques des progestatifs, y compris l’étonogestrel. Dans certains cas, l’impact de ces modifications peut être cliniquement significatif.
Par conséquent, le résumé des caractéristiques du produit des traitements concomitants VIH/VHC doit être consulté afin d’identifier les interactions potentielles ainsi que toute recommandation s’y rapportant. En cas de doute, les femmes traitées par un inhibiteur de la protéase ou par un inhibiteur non nucléosidique de la transcriptase inverse doivent utiliser une méthode contraceptive barrière supplémentaire.
Substances diminuant la clairance des contraceptifs hormonaux (inhibiteurs enzymatiques)
L’administration concomitante d’inhibiteurs puissants du CYP3A4 (par ex. kétoconazole, itraconazole, clarithromycine) ou modérés (par ex. fluconazole, diltiazem, érythromycine) peut augmenter les concentrations sériques des progestatifs, y compris l’étonogestrel.
Effets de NEXPLANON sur d’autres médicaments
Les contraceptifs hormonaux peuvent influer sur le métabolisme de certaines autres substances actives. Par conséquent, les concentrations plasmatiques et tissulaires peuvent être soit augmentées (ex : ciclosporine), soit diminuées (ex : lamotrigine).
Tests biologiques
Les données obtenues avec les CO combinés ont montré que les stéroïdes contraceptifs peuvent influencer les résultats de certains tests biologiques, dont les paramètres biochimiques hépatiques, thyroïdiens, surrénaliens et rénaux, les taux sériques de protéines (porteuses), comme la globuline se liant aux corticostéroïdes (corticosteroid binding globulin) et les fractions lipidiques/lipoprotéiniques, les paramètres du métabolisme des hydrates de carbone et les paramètres de la coagulation et de la fibrinolyse. Les modifications restent généralement dans les valeurs normales. Il n’est pas établi si ceci s’applique aussi aux contraceptifs purement progestatifs.
4.6. Fertilité, grossesse et allaitement
Grossesse
NEXPLANON n’est pas indiqué pendant la grossesse. Si une grossesse survient au cours de l’utilisation de NEXPLANON, l’implant doit être retiré. Les études chez l’animal ont montré que des doses très élevées de substances progestatives peuvent causer une masculinisation des fœtus féminins. De vastes études épidémiologiques n’ont mis en évidence ni d’augmentation du risque de malformations chez les enfants nés de femmes ayant utilisé des CO avant leur grossesse, ni d’effet tératogène lors de l’utilisation de CO par inadvertance au cours de la grossesse. Bien que cela soit probablement le cas pour tous les CO, il n’est pas démontré qu’il en soit de même pour NEXPLANON.
Les données de pharmacovigilance de différents médicaments contenant de l’étonogestrel et du désogestrel (l’étonogestrel est un métabolite du désogestrel) n’indiquent pas non plus de risque augmenté.
Des données cliniques indiquent que NEXPLANON ne modifie pas la production ou la qualité (concentrations en protéines, lactose ou lipides) du lait maternel. Cependant, de faibles quantités d’étonogestrel sont excrétées dans le lait maternel.
En se basant sur une ingestion moyenne quotidienne de lait de 150 ml/kg, la dose moyenne d’étonogestrel reçue par l’enfant est estimée à 27 ng/kg/jour après un mois. Ceci correspond approximativement à 2,2 % de la dose quotidienne maternelle moyenne ajustée au poids et approximativement à 0,2 % de la dose maternelle quotidienne estimée en valeur absolue. Par la suite, la concentration en étonogestrel dans le lait diminue avec le temps pendant la période d’allaitement.
Des données à long terme, limitées, sont disponibles chez 38 enfants, dont les mères ont commencé à utiliser l’implant entre la 4ème et la 8ème semaine après l’accouchement. Ils ont été allaités pendant une période moyenne de 14 mois et un suivi a été effectué jusqu’à l’âge de 36 mois. L’évaluation de la croissance, du développement physique et psychomoteur n’a montré aucune différence par rapport aux enfants allaités dont les mères avaient un DIU (n = 33). Néanmoins, le développement et la croissance de l’enfant devront être suivis avec précaution. En se basant sur les données disponibles, NEXPLANON peut être utilisé pendant l’allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Pendant l’utilisation de NEXPLANON, il est probable que les femmes observent des changements de leur profil de saignement vaginal qui seront imprédictibles. Ceux‑ci peuvent inclure l’apparition de saignements vaginaux irréguliers (absents, moins fréquents, plus fréquents ou continus) et des changements de l’intensité des saignements (réduits ou augmentés) ou de leur durée. Des aménorrhées ont été rapportées chez environ 1 femme sur 5 tandis que chez d’autres femmes (1 femme sur 5), il a été rapporté des saignements fréquents et/ou prolongés. Occasionnellement, des saignements abondants ont été rapportés. Lors des essais cliniques, les changements du profil de saignement ont été la raison la plus fréquente d’arrêt du traitement (environ 11 %). Chez beaucoup de femmes, le profil de saignement observé au cours des trois premiers mois est généralement prédictif du futur profil de saignement.
Les effets indésirables rapportés au cours des essais cliniques et possiblement liés à l’utilisation de NEXPLANON ont été listés dans le tableau ci-dessous :
|
Classe de systèmes d’organes |
Réactions indésirables en terme MedDRA1 |
||
|
Très fréquent (≥1/10) |
Fréquent (≥1/100 à <1/10) |
Peu fréquent (≥1/1000 à <1/100) |
|
|
Infections et infestations |
Infection vaginale |
|
Pharyngite, rhinite, infection des voies urinaires |
|
Affections du système immunitaire |
|
|
Hypersensibilité |
|
Affections du métabolisme et de la nutrition |
|
Augmentation de l’appétit |
|
|
Affections psychiatriques |
|
Instabilité émotionnelle, humeur dépressive, nervosité, diminution de la libido |
Anxiété, insomnie |
|
Affections du système nerveux |
Céphalées |
Etourdissements |
Migraine, somnolence |
|
Affections vasculaires |
|
Bouffées de chaleur |
|
|
Affections gastro‑intestinales |
|
Douleur abdominale, nausée, flatulences |
Vomissements, constipation, diarrhée |
|
Affections de la peau et du tissu sous-cutané |
Acné |
Alopécie |
Hypertrichose, rash, prurit |
|
Affections musculo‑squelettiques et systémiques |
|
|
Dorsalgies, arthralgies, myalgies, douleurs musculo-squelettiques |
|
Affections du rein et des voies urinaires |
|
|
Dysurie |
|
Affections des organes de reproduction et du sein |
Tensions mammaires, mastodynie, règles irrégulières |
Dysménorrhée, kyste ovarien |
Pertes vaginales, gêne vulvovaginale, galactorrhée, augmentation du volume des seins, prurit génital |
|
Troubles généraux et anomalies au site d’administration |
|
Douleur au site d’insertion, réaction au site d’insertion, fatigue, symptômes pseudo‑grippaux, douleur |
Pyrexie, œdème |
|
Investigations |
Prise de poids |
Perte de poids |
|
1 les termes MedDRA (version 10.1) les plus appropriés pour décrire certaines réactions indésirables ont été repris. Les synonymes et les conditions apparentées ne sont pas mentionnés mais doivent aussi être pris en compte.
Au cours de la surveillance post-commercialisation, une augmentation de la pression artérielle cliniquement significative a été observée dans de rares cas. De plus, une hypertension intracrânienne idiopathique a été rapportée. Des cas de séborrhée ont aussi été rapportés. Des réactions anaphylactiques, une urticaire, un angio-œdème, une aggravation d'un angio‑œdème et/ou une aggravation d’un œdème angioneurotique héréditaire peuvent survenir.
Les effets indésirables suivants ont été rapportés en lien avec les procédures d’insertion ou de retrait de l’implant :
L’insertion ou le retrait de l’implant peuvent entraîner des ecchymoses, incluant un hématome dans certains cas, une légère irritation locale, des douleurs ou des démangeaisons.
L'insertion de l’implant peut entrainer des réactions vasovagales (telles que de l’hypotension, sensations vertigineuses, ou syncope).
Une fibrose au site d'insertion peut se produire, une cicatrice peut se former ou un abcès peut se développer. Des paresthésies ou des sensations pseudo‑paresthésiques peuvent survenir. Une expulsion ou une migration de l’implant y compris, rarement, dans la paroi thoracique, ont été rapportées. Dans de rares cas, des implants ont été trouvés dans le système vasculaire y compris l’artère pulmonaire.
Dans certains cas d’implants trouvés dans l’artère pulmonaire, une douleur thoracique et/ou des troubles respiratoires (tels que dyspnée, toux, hémoptysie) ont été rapportées ; d’autres cas ont été rapportés comme asymptomatiques (voir rubrique 4.4.). Si les instructions ne sont pas suivies (voir rubrique 4.2.), des insertions incorrectes, des difficultés de localisation et de retrait de l’implant peuvent se produire. Une intervention chirurgicale peut être nécessaire pour le retrait de l’implant.
Dans de rare cas, des grossesses extra-utérines ont été rapportées (voir rubrique 4.4.).
Chez les femmes utilisant des contraceptifs (oraux combinés), un certain nombre d’effets indésirables (graves) ont été rapportés. Ils comprennent : des accidents thromboemboliques veineux, des accidents thromboemboliques artériels, des tumeurs hormono‑dépendantes (ex : tumeurs hépatiques, cancer du sein) et des chloasmas, plusieurs d’entre eux sont détaillés dans la rubrique 4.4.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Un implant doit toujours être retiré avant d’en insérer un nouveau. Aucune donnée n’est disponible sur un surdosage avec l’étonogestrel. Il n’a pas été rapporté d’effet délétère grave lié à un surdosage avec les contraceptifs en général.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L’implant NEXPLANON est un implant non biodégradable, radio-opaque, contenant de l’étonogestrel destiné à un usage sous-cutané, préchargé dans un applicateur stérile, jetable. L’étonogestrel est le métabolite biologiquement actif du désogestrel, un progestatif largement utilisé dans les CO. Sa structure est dérivée de la 19‑nortestostérone et il se lie avec une haute affinité aux récepteurs de la progestérone dans les organes cibles. L’effet contraceptif de l’étonogestrel est principalement dû à une inhibition de l’ovulation. Il n’a pas été observé d’ovulation durant les deux premières années d’utilisation de l’implant et rarement durant la troisième année. En plus de l’inhibition de l’ovulation, l’étonogestrel entraîne aussi des modifications de la glaire cervicale, qui gênent le passage des spermatozoïdes.
Efficacité et sécurité clinique
Les essais cliniques ont été conduits chez des femmes entre 18 et 40 ans. Bien qu’aucune comparaison directe n’ait été faite, l’efficacité contraceptive est au moins comparable à celle des CO combinés. Lors des études cliniques, aucune grossesse n’a été observé pour les 35 057 cycles d’exposition ; l’indice de Pearl étudié est de 0,00 (intervalle de confiance à 95 % : 0,00 – 0,14). Cependant, il faut savoir qu’en pratique aucune méthode ne peut être considérée comme efficace à 100 %. Ce taux élevé d’efficacité contraceptive est obtenu, entre autres, parce que l’action contraceptive de NEXPLANON est indépendante de l’observance d’un traitement par la femme elle-même. L’action contraceptive de l’étonogestrel est réversible, ce qui se manifeste par un retour rapide à un cycle menstruel normal après le retrait de l’implant. Bien que l’étonogestrel inhibe l’ovulation, l’activité ovarienne n’est pas complètement supprimée. Les concentrations moyennes en estradiol restent supérieures au taux observé en phase folliculaire précoce. Dans une étude sur 2 ans, au cours de laquelle la densité minérale osseuse de 44 utilisatrices a été comparée à celle d’un groupe témoin de 29 femmes utilisant un DIU, aucun effet indésirable relatif à la masse osseuse n’a été observé. Aucun effet cliniquement significatif sur le métabolisme des lipides n’a été observé. L’utilisation de contraceptifs contenant un progestatif peut avoir un effet sur la résistance à l’insuline et la tolérance au glucose. Des essais cliniques additionnels ont indiqué que les utilisatrices de NEXPLANON auraient souvent des menstruations moins douloureuses (dysménorrhées).
5.2. Propriétés pharmacocinétiques
Après l’insertion de l’implant, l’étonogestrel est rapidement absorbé dans la circulation. Les concentrations permettant l’inhibition de l’ovulation sont atteintes en 1 jour. Les concentrations sériques maximales (entre 472 et 1270 pg/ml) sont atteintes en 1 à 13 jours. Le taux de libération de l’implant diminue avec le temps.
En conséquence, les concentrations sériques diminuent rapidement au cours des premiers mois. A la fin de la première année, une concentration moyenne d’approximativement 200 pg/ml (entre 150‑261 pg/ml) est observée, celle-ci diminue lentement jusqu’à 156 pg/ml (entre 111‑202 pg/ml) à la fin de la troisième année. Les variations observées dans les concentrations sériques peuvent être, en partie, attribuées aux différences de poids corporel.
Distribution
L’étonogestrel est lié à 95,5‑99 % aux protéines sériques, principalement à l’albumine et avec une importance moindre à la protéine porteuse des stéroïdes sexuels (SHBG). Le volume central de distribution et le volume total de distribution sont respectivement de 27 L et de 220 L, et ils ne se modifient quasiment pas au cours de l’utilisation de NEXPLANON.
Biotransformation
L’étonogestrel subit une hydroxylation et une réduction. Les métabolites sont conjugués aux sulphates et aux glucuronides. Des études sur l’animal montrent que la circulation entérohépatique ne contribue probablement pas à l’activité progestative de l’étonogestrel.
Élimination
Après une administration intraveineuse d’étonogestrel, la demi‑vie d’élimination moyenne est approximativement de 25 heures et la clairance sérique est approximativement de 7,5 L/heure. La clairance et la demi‑vie d’élimination restent constantes pendant la période de traitement. L’excrétion de l’étonogestrel et de ses métabolites, sous forme de stéroïdes libres ou sous forme conjuguée, est urinaire ou fécale (avec un ratio de 1,5/1). Après insertion chez une femme qui allaite, l’étonogestrel est excrété dans le lait maternel avec un ratio lait/sérum de 0,44/0,50 au cours des quatre premiers mois. Chez la femme qui allaite, la dose moyenne d’étonogestrel ingérée par l’enfant est d’environ 0,2 % de la dose maternelle quotidienne estimée en valeur absolue (2,2 % lorsque les valeurs sont normalisées par kg de poids corporel). Les concentrations diminuent de façon progressive et statistiquement significative au cours du temps.
5.3. Données de sécurité préclinique
Noyau :
· copolymère éthylène / acétate de vinyle (28 % d’acétate de vinyle, 43 mg)
· sulphate de baryum (15 mg)
· stéarate de magnésium (0,1 mg).
Enveloppe :
Copolymère éthylène / acétate de vinyle (15 % d’acétate de vinyle, 15 mg).
5 ans.
NEXPLANON ne doit pas être inséré après la date de péremption mentionnée sur le conditionnement extérieur.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de conditions particulières de conservation.
A conserver dans le conditionnement primaire d’origine.
6.5. Nature et contenu de l'emballage extérieur
La plaquette thermoformée contient un implant (4 cm de long et 2 mm de diamètre) qui est préchargé dans l’aiguille en acier inoxydable d’un applicateur stérile, prêt à l’emploi, jetable. L’applicateur contenant l’implant est conditionné sous plaquette thermoformée en polyéthylène téréphtalate glycol (PETG) transparent scellée avec un opercule en polyéthylène haute densité (PEHD). Le contenu de la plaquette thermoformée est stérile à moins que le conditionnement soit endommagé ou ouvert.
Boîte de 1 ou de 5 implants.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Voir rubrique 4.2.
L’applicateur est à usage unique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
176 RUE MONTMARTRE
75002 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 351 544 3 9 : 1 implant préchargé dans l’aiguille (acier inoxydable) d’un applicateur (acrylonitrile butadiène styrène) sous plaquette thermoformée (polyéthylène téréphtalate glycol). Boîte de 1 implant.
· 34009 587 005 9 0 : 1 implant préchargé dans l’aiguille (acier inoxydable) d’un applicateur (acrylonitrile butadiène styrène) sous plaquette thermoformée (polyéthylène téréphtalate glycol). Boîte de 5 implants
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Informations importantes
Les informations importantes disponibles pour ce médicament sont les suivantes :
- Migration de l'implant contraceptif Nexplanon dans l'artère pulmonaire : bilan et nouvelles recommandations
- Contraception et risque de méningiome : nouvelles recommandations
- Implant contraceptif Nexplanon : renforcement des mesures de réduction du risque de migration notamment dans l'artère pulmonaire - Point d'Informationn
ANSM - Mis à jour le : 23/09/2025
NEXPLANON 68 mg, implant pour usage sous cutané
Etonogestrel
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre sage-femme.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre sage-femme. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
Votre professionnel de santé vous remettra une Carte d’Alerte Patiente contenant des informations importantes que vous avez besoin de connaître. Conservez la carte dans un endroit sûr et montrez la à votre professionnel de santé à chaque visite relative à l’utilisation de votre implant.
1. Qu'est-ce que NEXPLANON 68 mg, implant pour usage sous cutané et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser NEXPLANON 68 mg, implant pour usage sous cutané ?
3. Comment utiliser NEXPLANON 68 mg, implant pour usage sous cutané ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver NEXPLANON 68 mg, implant pour usage sous cutané ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE NEXPLANON 68 mg, implant pour usage sous cutané ET DANS QUELS CAS EST-IL UTILISE ?
NEXPLANON est un implant contraceptif préchargé dans un applicateur jetable. La sécurité et l’efficacité ont été établies chez les femmes entre 18 et 40 ans. L’implant est un petit bâtonnet en plastique, flexible, souple, de 4 cm de long et 2 mm de diamètre, qui contient 68 milligrammes d’une substance active, l’étonogestrel. L’applicateur permet au professionnel de santé d’insérer l’implant juste sous la peau au niveau de votre bras. L’étonogestrel est une hormone féminine synthétique proche de la progestérone. Une petite quantité d’étonogestrel est libérée en continu dans la circulation sanguine. Le bâtonnet lui-même est composé de copolymère éthylène/acétate de vinyle, une matière plastique qui ne se dissout pas dans le corps. Il contient également une petite quantité de sulphate de baryum qui le rend visible aux rayons X.
NEXPLANON est utilisé pour empêcher la survenue d’une grossesse.
Comment fonctionne NEXPLANON
L’implant est inséré juste sous la peau. Le composé actif, l’étonogestrel, a une double action :
· Il empêche la libération d’un ovocyte par les ovaires.
· Il entraîne des modifications de la glaire cervicale qui rendent difficile le passage des spermatozoïdes vers l’utérus.
En conséquence, NEXPLANON vous protège de la survenue d’une éventuelle grossesse pendant une période de trois ans, mais si vous êtes en surpoids, le professionnel de santé qui vous prescrit NEXPLANON peut vous conseiller de remplacer l’implant plus tôt. NEXPLANON fait partie des nombreux moyens de contraception. Une autre méthode de contraception fréquemment utilisée est la pilule combinée. A l’inverse des pilules combinées, NEXPLANON peut être utilisé par les femmes qui ne peuvent pas, ou ne veulent pas utiliser d’estrogènes.
Lorsque vous utilisez NEXPLANON, vous n'avez pas à vous rappeler de prendre un comprimé tous les jours. C’est l’une des raisons pour lesquelles NEXPLANON est très fiable (plus de 99 % d’efficacité). Si, dans de rares cas, NEXPLANON n’est pas inséré correctement ou n’est pas inséré du tout, vous pouvez ne pas être protégée de la survenue d’une éventuelle grossesse. Lors de l’utilisation de NEXPLANON, vos règles peuvent être modifiées et devenir absentes, irrégulières, rares, fréquentes, prolongées ou rarement abondantes. Le profil de saignement que vous observerez au cours des trois premiers mois est généralement prédictif de votre futur profil de saignement. Des règles douloureuses peuvent s’améliorer.
Vous pouvez arrêter d’utiliser NEXPLANON à tout moment (voir le paragraphe « Si vous voulez arrêter d’utiliser NEXPLANON »).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER NEXPLANON 68 mg, implant pour usage sous cutané ?
N'utilisez jamais NEXPLANON 68 mg, implant pour usage sous-cutané :
N'utilisez jamais NEXPLANON si vous êtes dans l’une des situations énumérées ci-dessous. Si vous êtes concernée par l'une de ces situations, parlez-en au professionnel de santé qui vous prescrit NEXPLANON avant l'insertion de NEXPLANON. Il pourra vous conseiller une méthode contraceptive non hormonale.
· si vous êtes allergique (hypersensible) à l’étonogestrel ou à l’un des autres composants contenus dans ce médicament (mentionnés à la rubrique 6).
· si vous avez une thrombose. Une thrombose consiste en la formation d’un caillot de sang dans un vaisseau sanguin [par exemple, dans les jambes (phlébite) ou dans les poumons (embolie pulmonaire)].
· si vous avez ou avez eu un ictère (jaunissement de la peau), une maladie grave du foie (lorsque le foie ne fonctionne pas normalement) ou une tumeur du foie.
· si vous avez (ou avez eu) ou si l’on suspecte chez vous un cancer du sein ou des organes génitaux.
· si vous avez des saignements vaginaux inexpliqués.
Si l’une de ces situations apparaît pour la première fois lors de l’utilisation de NEXPLANON, consultez immédiatement un médecin ou le professionnel de santé qui vous prescrit NEXPLANON.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou sage-femme avant d’utiliser NEXPLANON.
Si NEXPLANON est utilisé dans l’une des situations énumérées ci-dessous, il se peut qu’il soit nécessaire de vous placer sous étroite surveillance. Le professionnel de santé qui vous prescrit NEXPLANON vous donnera les explications nécessaires. Si vous êtes concernée par l’une de ces situations, parlez-en à un médecin ou au professionnel de santé qui vous prescrit NEXPLANON avant l’insertion de NEXPLANON. De même, si l’une de ces situations se développe ou s’aggrave lors de l’utilisation de NEXPLANON, vous devez également en parler à un médecin ou au professionnel de santé qui vous prescrit NEXPLANON.
· vous avez eu un cancer du sein ;
· vous avez ou avez eu une maladie du foie ;
· vous avez déjà eu une thrombose ;
· vous êtes diabétique ;
· vous êtes en surpoids ;
· vous souffrez d’épilepsie ;
· vous souffrez de tuberculose ;
· vous avez une tension artérielle élevée ;
· vous avez ou avez eu un chloasma (taches de pigmentation de couleur jaune-brun sur la peau, en particulier sur le visage) ; si c’est votre cas, évitez de vous exposer trop au soleil ou aux ultra-violets.
Situations graves possibles
Cancer
Les informations présentées ci-dessous ont été obtenues dans des études chez des femmes qui prenaient quotidiennement une contraception orale combinée contenant deux hormones féminines différentes (« la pilule »). Il n’est pas établi que ces observations soient également applicables aux femmes qui utilisent une contraception hormonale différente, telle que les implants contenant uniquement un progestatif.
Des cancers du sein ont été diagnostiqués un peu plus fréquemment chez les femmes utilisant les pilules orales combinées, mais il n’est pas établi si cela est dû au traitement. Par exemple, il est possible que les cancers soient diagnostiqués plus fréquemment chez les femmes sous pilules combinées, car elles font l’objet d’un suivi plus rapproché par le professionnel de santé qui leur prescrit NEXPLANON. Le risque de développer un cancer du sein diminue progressivement après l'arrêt de la pilule combinée. Il est important de vérifier régulièrement vos seins et vous devez contacter un médecin si vous ressentez une grosseur dans vos seins. Vous devez aussi informer le professionnel de santé qui vous prescrit NEXPLANON si une personne de votre famille proche a ou a déjà eu un cancer du sein.
Dans de rares cas, des tumeurs bénignes du foie et plus rarement encore, des tumeurs malignes du foie ont été rapportées chez des femmes utilisant la pilule. Si vous avez des douleurs abdominales sévères, vous devez contacter immédiatement un médecin.
Thrombose
Un caillot de sang dans une veine (appelé « thrombose veineuse ») peut boucher cette veine. Cela peut se produire dans les veines de la jambe (phlébite), du poumon (embolie pulmonaire) ou d'autres organes. Un caillot sanguin dans une artère (appelé « thrombose artérielle ») peut boucher l’artère et provoquer, par exemple, une crise cardiaque, ou un accident vasculaire dans le cerveau.
Chez une femme, l’utilisation d’un contraceptif hormonal combiné augmente le risque de développer de tels caillots par rapport à une femme qui ne prend pas de contraceptif hormonal combiné. Le risque n'est pas aussi élevé que le risque de développer un caillot sanguin pendant la grossesse. Ce risque avec une méthode uniquement progestative, comme NEXPLANON, est estimé plus faible que celui constaté chez les utilisatrices de pilules qui contiennent également des estrogènes. Des formations de caillots sanguins, telles qu’embolie pulmonaire, thrombose veineuse profonde, crises cardiaques et accidents vasculaires cérébraux, ont été rapportées chez des femmes utilisant des implants à l’étonogestrel ; cependant, les données disponibles ne suggèrent pas d’augmentation du risque de ces événements chez les femmes utilisant l’implant.
Si vous remarquez soudainement des signes possibles de thrombose, vous devez contacter immédiatement votre médecin (voir le paragraphe « Quand devez‑vous contacter un médecin ou le professionnel de santé qui vous prescrit NEXPLANON ? »).
Autres situations
Changement du profil de saignement
Comme avec tous les contraceptifs purement progestatifs, le profil de saignement peut changer lors de l’utilisation de NEXPLANON. Vous pouvez présenter une modification de leur fréquence (absents, moins fréquents, plus fréquents ou continus), de leur intensité (réduits ou augmentés) ou de leur durée. L’absence de saignement a été rapportée chez environ 1 femme sur 5 tandis que chez d’autres femmes (1 femme sur 5), il a été rapporté des saignements fréquents et/ou prolongés. Occasionnellement, des saignements abondants ont été rapportés. Lors des essais cliniques, les changements du profil de saignement ont été la raison la plus fréquente d’arrêt du traitement (environ 11 %). Le profil de saignement que vous observerez au cours des trois premiers mois est généralement prédictif de votre futur profil de saignement.
Un changement du profil de saignement ne signifie pas que NEXPLANON ne vous convient pas ou ne vous procure pas une protection contraceptive. En général, vous n'avez pas besoin de prendre de mesures. Vous devez consulter le professionnel de santé qui vous a prescrit NEXPLANON si vos règles deviennent abondantes ou prolongées.
Evènements liés à l'insertion et au retrait
L'implant peut se déplacer de son site d'insertion initial dans le bras, s’il est incorrectement inséré ou sous l’action de pressions extérieures (ex : manipulation de l'implant ou sports de contact). Dans de rares cas, des implants ont été trouvés dans les vaisseaux sanguins du bras ou dans l’artère pulmonaire (vaisseau sanguin situé dans le poumon).
Dans les cas où l’implant a migré de son site d’insertion initial, la localisation de l’implant peut être rendue plus difficile et le retrait peut nécessiter une incision plus large ou un retrait par chirurgie à l’hôpital. Si l’implant ne peut pas être localisé dans le bras, votre professionnel de santé peut utiliser la radiographie ou d’autres méthodes d’imagerie au niveau du thorax. Si l’implant est localisé dans le thorax, une chirurgie peut être nécessaire.
Si l’implant ne peut être localisé et s’il n’existe aucune preuve qu’il ait été expulsé, son effet contraceptif et le risque d’effets indésirables liés au progestatif pourront persister plus longtemps que ce que vous souhaiteriez.
Si, à tout moment, l’implant ne peut être palpé, vous devez contacter votre médecin dès que possible.
Troubles psychiatriques
Certaines femmes qui utilisent des contraceptifs hormonaux dont NEXPLANON ont fait état d’une dépression ou d’un état dépressif. La dépression peut être grave et peut parfois donner lieu à des idées suicidaires. Si vous présentez des changements d’humeur et des symptômes dépressifs, sollicitez les conseils de votre médecin dès que possible.
Kystes ovariens
Pendant l’utilisation de tous les contraceptifs hormonaux faiblement dosés, de petits sacs remplis de liquide peuvent se développer dans les ovaires. Ce sont des kystes ovariens. Ils disparaissent généralement d'eux‑mêmes. Parfois, ils peuvent provoquer une légère douleur abdominale. Rarement, ils peuvent conduire à des problèmes plus graves.
Implant cassé ou plié
Si l’implant se casse ou se plie dans votre bras, son fonctionnement ne devrait pas être affecté. Une cassure ou une courbure peuvent survenir en raison de pressions extérieures. L'implant cassé peut se déplacer du site d'insertion. Si vous avez des questions, contactez votre professionnel de santé.
Autres médicaments et NEXPLANON 68 mg, implant pour usage sous-cutané
|
Informez toujours votre médecin des médicaments et des produits à base de plantes que vous utilisez déjà. Informez également tout autre médecin ou dentiste (ou pharmacien) qui vous prescrit un autre médicament que vous utilisez NEXPLANON. Ils pourront vous dire si vous avez besoin de prendre des précautions contraceptives supplémentaires (par exemple des préservatifs) et, si c’est le cas, pendant combien de temps ou si un autre médicament dont vous avez besoin doit être changé. |
Certains médicaments
· peuvent avoir une influence sur les taux sanguins de NEXPLANON
· peuvent le rendre moins efficace pour prévenir une grossesse
· peuvent provoquer des saignements inattendus.
Cela inclut des médicaments utilisés pour le traitement
· de l'épilepsie (par exemple, primidone, phénytoïne, barbituriques, carbamazépine, oxcarbazépine, topiramate, felbamate),
· de la tuberculose (par exemple, rifampicine),
· des infections par le VIH (par exemple, nelfinavir, névirapine, éfavirenz),
· de l’infection par le VHC (par exemple, bocéprévir, télaprévir),
· d’autres maladies infectieuses (par exemple, griséofulvine),
· d’une pression sanguine élevée dans les vaisseaux sanguins situés au niveau des poumons (bosentan),
· des états dépressifs (produits à base de plantes contenant du millepertuis (Hypericum perforatum)).
NEXPLANON peut avoir une influence sur l’effet d’autres médicaments, par exemple
· les médicaments contenant de la ciclosporine
· l’anti-épileptique lamotrigine (cela pourrait entraîner une augmentation de la fréquence des crises d’épilepsie).
Demandez conseil à votre médecin ou votre pharmacien avant d’utiliser tout médicament.
NEXPLANON 68 mg, implant pour usage sous-cutané avec des aliments et boissons
Il n’y a pas de donnée indiquant un effet des aliments et des boissons sur l’utilisation de NEXPLANON.
Grossesse et allaitement
Vous ne devez pas utiliser NEXPLANON si vous êtes enceinte, ou si vous pensez l’être. En cas de doute, vous devez effectuer un test de grossesse avant de commencer à utiliser NEXPLANON.
NEXPLANON peut être utilisé au cours de l’allaitement. Bien qu’une petite quantité de substance active de NEXPLANON passe dans le lait maternel, il n’y a d’effet ni sur la production ou la qualité du lait maternel, ni sur la croissance ou le développement de l’enfant.
Si vous allaitez, demandez conseil à votre médecin avant de prendre ce médicament.
Enfants et adolescents
La sécurité d’emploi et l’efficacité de NEXPLANON n’ont pas été étudiées chez les adolescentes de moins de 18 ans.
Conduite de véhicules et utilisation de machines
Il n’y a pas de donnée indiquant un possible effet sur la vigilance et la concentration lors de l’utilisation de NEXPLANON.
Quand devez-vous contacter un médecin ou le professionnel de santé qui vous prescrit NEXPLANON 68 mg, implant pour usage sous-cutané ?
Examens médicaux réguliers
Avant l’insertion de NEXPLANON, le professionnel de santé qui vous prescrit NEXPLANON vous posera quelques questions sur vos antécédents médicaux et ceux de votre famille. Il mesurera aussi votre tension artérielle et en fonction de votre situation personnelle, il pourra également pratiquer d’autres tests. Quand vous utilisez NEXPLANON, il vous demandera de revenir le voir pour des examens médicaux réguliers après l’insertion de l’implant. La fréquence et la nature de ces examens médicaux dépendront de votre situation personnelle. Votre professionnel de santé doit palper l’implant à chaque visite de contrôle.
Contactez un médecin dès que possible si :
· vous remarquez des changements de votre état de santé, en particulier relatifs aux situations mentionnées dans cette notice (voir les paragraphes : « N’utilisez jamais NEXPLANON 68 mg, implant pour usage sous-cutané » et « Avertissements et précautions », sans oublier ce qui concerne votre famille proche) ;
· vous remarquez des signes possibles d’une thrombose tels qu’une douleur sévère ou un gonflement dans l'une de vos jambes, des douleurs inexpliquées dans la poitrine, un essoufflement, une toux inhabituelle, surtout si vous crachez du sang ;
· vous avez brutalement d’importants maux d'estomac ou si votre teint devient jaune ;
· vous sentez une grosseur dans un sein (voir également le paragraphe « Cancer ») ;
· vous ressentez une douleur soudaine ou importante au niveau du bas-ventre ou de l'estomac ;
· vous avez des saignements vaginaux inhabituellement abondants ;
· vous devez être immobilisée (par exemple, confinée au lit) ou devez être opérée (consultez votre médecin au moins quatre semaines avant) ;
· vous pensez être enceinte ;
· l’implant n’est pas palpable après l’insertion ou à tout moment.
NEXPLANON 68 mg, implant pour usage sous-cutané contient
Sans objet.
3. COMMENT UTILISER NEXPLANON 68 mg, implant pour usage sous cutané ?
Comment l’utiliser
NEXPLANON doit être inséré et retiré uniquement par un professionnel de santé familiarisé avec les procédures telles que décrites au verso de cette notice. Il décidera en accord avec vous quelle sera la période la plus adaptée pour l'insertion. Cela dépend de votre situation personnelle (par exemple, de la méthode contraceptive que vous utilisez actuellement). A moins que vous utilisiez une autre méthode contraceptive hormonale, l'insertion doit être effectuée entre le 1er et le 5ème jour de votre cycle menstruel naturel afin d’exclure une grossesse.
Si l’implant est inséré après le 5ème jour des règles, vous devez utiliser une méthode contraceptive supplémentaire (par exemple, un préservatif) pendant les 7 premiers jours suivant l’insertion.
Avant l'insertion ou le retrait de NEXPLANON, le professionnel de santé vous fera une anesthésie locale. NEXPLANON est inséré directement sous la peau, à la face interne de votre bras non dominant (le bras avec lequel vous n’écrivez pas). Une description des procédures d'insertion et de retrait de NEXPLANON est présentée à la fin de la notice.
L’implant doit être palpable après l’insertion
A la fin de la procédure d’insertion, le professionnel de santé qui a inséré NEXPLANON vous demandera de palper l’implant (de sentir l’implant sous votre peau). Un implant correctement inséré doit être facilement palpable à la fois par le professionnel de santé et par vous-même, et vous devez être capable de sentir les deux extrémités entre votre pouce et un doigt. Votre attention doit être attirée sur le fait que la présence de l’implant ne peut pas être vérifiée à 100 % par palpation. Si l’implant ne peut être palpé immédiatement après l’insertion, ou à tout moment, l’implant peut ne pas avoir été inséré, avoir été inséré profondément, ou avoir migré du site où il a été inséré.
Ainsi, il est important de palper doucement et occasionnellement l’implant afin d’être sûre que vous connaissiez sa localisation. Si, à tout moment, vous ne sentez pas l’implant à la palpation, contactez votre médecin dès que possible.
Au moindre doute, vous devez utiliser une méthode contraceptive mécanique (ex : préservatif) jusqu'à ce que le professionnel de santé et vous-même soyez absolument sûrs que l'implant a été inséré. Le professionnel de santé peut recourir à la radiographie, l'échographie ou l'imagerie par résonance magnétique, ou à une prise de sang, pour garantir la présence de l'implant dans votre bras. Si l’implant ne peut être localisé dans le bras après une recherche approfondie, votre professionnel de santé pourra utiliser la radiographie ou d’autres méthodes d’imagerie au niveau de votre thorax. Une fois que le professionnel de santé aura localisé l’implant qui n’était pas palpable, celui-ci devra être retiré.
NEXPLANON 68 mg, implant pour usage sous-cutané doit être retiré ou remplacé au plus tard trois ans après l'insertion.
Carte d’Alerte Patiente
Pour vous aider à vous rappeler quand et où a été inséré NEXPLANON, et quand il doit être retiré au plus tard, le professionnel de santé qui a inséré NEXPLANON vous remettra une Carte d’Alerte Patiente qui contient ces informations. La Carte d’Alerte Patiente contient également des instructions pour palper doucement et occasionnellement l’implant pour être sûre que vous connaissez sa localisation. Si, à tout moment, vous ne sentez pas l’implant à la palpation, contactez votre médecin dès que possible. Conservez cette carte dans un endroit sûr ! Montrez la Carte d’Alerte Patiente au professionnel de santé à chaque visite relative à l’utilisation de votre implant.
Dans le cas où vous désireriez remplacer NEXPLANON, un nouvel implant peut être inséré immédiatement après le retrait de l'ancien implant. Le nouvel implant peut être inséré dans le même bras et au même endroit que l’implant précédent à condition que la localisation du site soit correcte. Le professionnel de santé qui vous prescrit NEXPLANON vous conseillera.
Si vous avez utilisé plus de NEXPLANON 68 mg, implant pour usage sous-cutané que vous n’auriez dû
Sans objet.
Si vous oubliez d’utiliser NEXPLANON 68 mg, implant pour usage sous-cutané
Si vous arrêtez d’utiliser NEXPLANON 68 mg, implant pour usage sous-cutané
Vous pouvez demander à votre professionnel de santé de retirer l'implant à tout moment.
Si l'implant ne peut pas être localisé par palpation, le professionnel de santé réalisera une radiographie, une échographie ou une imagerie par résonance magnétique pour localiser l’implant. En fonction de la position exacte de l'implant, le retrait peut être difficile et peut nécessiter une chirurgie.
Si vous ne voulez pas être enceinte après le retrait de NEXPLANON, interrogez votre professionnel de santé sur les autres méthodes de contraception fiables.
Si vous arrêtez d'utiliser NEXPLANON parce que vous souhaitez être enceinte, il est généralement recommandé que vous attendiez d’avoir un cycle naturel avant d'essayer d’avoir un enfant. Le calcul de la date de la naissance en sera facilité.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Des saignements menstruels peuvent survenir à intervalles irréguliers lors de l’utilisation de NEXPLANON. Il peut juste s’agir de petites pertes qui peuvent ne pas nécessiter de protection hygiénique, ou de saignements plus abondants, qui ressemblent plutôt à des règles peu abondantes et qui eux, nécessitent une protection hygiénique. Vous pouvez aussi n’avoir aucun saignement. Les saignements irréguliers ne signifient pas que l’action contraceptive de NEXPLANON est diminuée. En général, vous n’avez pas besoin de prendre de mesures. Cependant, si les saignements sont abondants ou prolongés, consultez le professionnel de santé qui vous prescrit NEXPLANON.
Les effets indésirables graves sont décrits dans les paragraphes de la rubrique 2. « Cancer » et « Thrombose ». Lisez ces paragraphes pour plus d'informations et demandez conseil au professionnel de santé qui vous prescrit NEXPLANON si nécessaire.
Les effets indésirables suivants ont été rapportés :
|
Très fréquemment (peut affecter plus d’une personne sur 10) |
Fréquemment (peut affecter jusqu’à une personne sur 10) |
Peu fréquemment (peut affecter jusqu’à une personne sur 100) |
|
Acné, maux de tête, prise de poids, sensibilité et douleur au niveau des seins, saignements irréguliers, infection vaginale. |
Chute de cheveux, étourdissements, humeur dépressive, instabilité émotionnelle, nervosité, diminution des pulsions sexuelles, augmentation de l'appétit, douleurs abdominales, nausées, gaz dans l’estomac et l’intestin, règles douloureuses, perte de poids, symptômes pseudo-grippaux, douleur, fatigue, bouffées de chaleur, douleur au site d'insertion de l'implant, réaction au site d’insertion de l’implant, kyste ovarien. |
Démangeaisons, démangeaisons génitales, éruption cutanée, augmentation de la pousse des poils, migraine, anxiété, insomnie, somnolence, diarrhée, vomissements, constipation, infection urinaire, inconfort vaginal (par exemple, sécrétion vaginale), augmentation du volume des seins, écoulement mammaire, douleurs au niveau du dos, fièvre, œdème, miction urinaire difficile ou douloureuse, réactions allergiques, inflammation et douleur de la gorge, rhinite, douleurs articulaires, douleurs musculaires, douleurs osseuses. |
En dehors de ces effets indésirables, il a été occasionnellement observé une augmentation de la tension artérielle. Une augmentation de pression dans la tête (hypertension intracrânienne bégnine) avec des signes tels qu’un mal de tête persistant, accompagné de nausées, de vomissements et d’une modification de la vue, y compris une vision floue a été rapportée. Il a également été observé des cas de peau grasse. Vous devez consulter immédiatement un médecin si vous ressentez les symptômes d’une réaction allergique sévère, tels qu’un gonflement du visage, de la langue ou de la gorge ; des difficultés à avaler ; ou une urticaire et des difficultés à respirer.
L'insertion ou le retrait de NEXPLANON peut entraîner des ecchymoses (graves dans certains cas), une douleur, un gonflement ou des démangeaisons ainsi que dans de rares cas, une infection. Une cicatrice peut se former ou un abcès peut se développer au niveau du site d'insertion.En raison de l’insertion de l’implant, une sensation de faiblesse, peut survenir. Un engourdissement ou une sensation d'engourdissement (diminution de la sensibilité) peut apparaître. L'expulsion ou la migration de l’implant peut survenir, notamment s’il n'a pas été inséré correctement. Dans de rares cas, des implants ont été localisés dans un vaisseau sanguin, y compris un vaisseau sanguin dans le poumon, ce qui peut être associé à un essoufflement et/ou une toux avec ou sans saignement. Une intervention chirurgicale peut être nécessaire pour retirer l’implant.
Des caillots de sang dans une veine (appelés « thrombose veineuse ») ou dans une artère (appelés « thrombose artérielle ») ont été rapportés chez des femmes utilisant un implant à l’étonogestrel. Un caillot de sang dans une veine peut boucher cette veine. Cela peut se produire dans les veines de la jambe (thrombose veineuse profonde), du poumon (embolie pulmonaire) ou d'autres organes. Un caillot sanguin dans une artère peut boucher l’artère et provoquer une crise cardiaque, ou un accident vasculaire dans le cerveau.
Si vous manifestez un effet indésirable quelconque, parlez-en à votre médecin, pharmacien ou infirmier(ère). Ceci inclut tout effet indésirable qui n’est pas listé dans cette notice.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre sage-femme. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER NEXPLANON 68 mg, implant pour usage sous cutané ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la plaquette ou sur l’emballage.
A conserver dans le conditionnement primaire d’origine.
Ce médicament ne nécessite pas de conditions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient NEXPLANON 68 mg, implant pour usage sous cutané
Chaque applicateur contient un implant dont :
· La substance active est :
Etonogestrel ................................................................................................................... 68 mg
· Les autres composants sont :
Copolymère éthylène / acétate de vinyle, sulphate de baryum, stéarate de magnésium.
Qu’est-ce que NEXPLANON 68 mg, implant pour usage sous cutané et contenu de l’emballage extérieur
NEXPLANON est un contraceptif hormonal sous-cutané d’action prolongée. Il s’agit d’un implant purement progestatif, radio-opaque, préchargé dans un applicateur innovant, prêt à l’emploi, facile à utiliser, jetable. L’implant de couleur blanc cassé mesure 4 cm de long et 2 mm de diamètre et contient de l’étonogestrel et du sulphate de baryum. L’applicateur a été conçu pour faciliter l’insertion de l’implant juste sous la peau de la face interne de votre bras (non dominant). L’implant doit être inséré et retiré par un professionnel de santé familiarisé avec les procédures.
Pour faciliter le retrait, il est nécessaire que l’implant soit inséré juste sous la peau (voir le verso de la notice). Une anesthésie locale doit être pratiquée avant d’insérer ou de retirer l’implant. Le risque de complications est faible si les instructions fournies sont respectées.
Boîte de 1 implant ou de 5 implants.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
176 RUE MONTMARTRE
75002 PARIS
Exploitant de l’autorisation de mise sur le marché
ORGANON FRANCE
176 RUE MONTMARTRE
75002 PARIS
P.O. BOX 20
5340 BH OSS
PAYS-BAS
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen et au Royaume-Unis (Irlande du Nord) sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Ces pictogrammes sont uniquement destinés à illustrer les procédures d’insertion et de retrait pour la patiente qui recevra l’implant.
Note : Les procédures exactes d’insertion et de retrait de NEXPLANON par un professionnel de santé qualifié sont décrites dans le résumé des caractéristiques du produit ainsi que dans la rubrique « Les informations suivantes sont destinées uniquement aux professionnels de santé » au verso de cette notice.
Comment est inséré NEXPLANON
|
· L'insertion de NEXPLANON doit être effectuée uniquement par un professionnel de santé familiarisé avec la technique. · Pour faciliter l'insertion de l’implant, vous devez vous allonger sur le dos, avec votre bras plié au niveau du coude et votre main sous votre tête (ou le plus près possible). |
|
|
|
· L’implant sera inséré à la face interne de votre bras non dominant (le bras avec lequel vous n'écrivez pas). · Le site d'insertion sera indiqué sur la peau et le site est désinfecté et anesthésié. · La peau est tendue et l’aiguille est insérée, directement sous la peau. Une fois que l’extrémité est dans la peau, l’aiguille est entièrement insérée selon un mouvement parallèle à la peau. |
|
P, proximal (en direction de l’épaule) ; D, distal (en direction du coude) · La manette coulissante violette est déverrouillée afin de rétracter l’aiguille. L'implant restera dans le bras lors du retrait de l’aiguille.
· La présence de l’implant doit être vérifiée en le touchant (par palpation) immédiatement après l’insertion. Un implant correctement inséré doit être facilement senti entre le pouce et un doigt à la fois par le professionnel de santé et vous‑même. Votre attention doit être attirée sur le fait que la présence de NEXPLANON ne peut pas être vérifiée à 100 % par palpation.
· Si l’implant ne peut pas être palpé ou s’il y a un doute sur sa présence, d’autres méthodes doivent être utilisées pour confirmer la présence de l’implant. · Une fois que le professionnel de santé a localisé l’implant qui n’était pas palpable, celui-ci doit être retiré. · Tant que la présence de l’implant n’a pas été confirmée, vous pouvez ne pas être protégée contre une grossesse et une méthode contraceptive mécanique (ex : préservatif) doit être utilisée. · Un petit pansement adhésif sera placé sur le site d’insertion et un bandage compressif sera appliqué pour minimiser le risque d’ecchymose. Vous pourrez retirer le bandage compressif au bout de 24 heures et le petit pansement adhésif placé sur le site d’insertion après 3 à 5 jours. · Après l’insertion de l’implant, le professionnel de santé qui a inséré NEXPLANON vous donnera une Carte d’Alerte Patiente mentionnant le site d’insertion, la date d’insertion et la date limite à laquelle l’implant doit être retiré ou remplacé. Rangez-la dans un endroit sûr, car les informations sur la carte pourront faciliter le retrait ultérieur. |
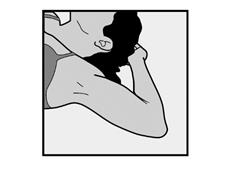
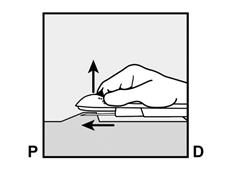
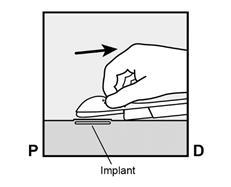
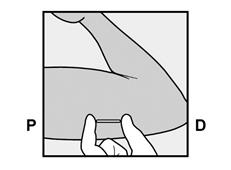
Comment doit être retiré NEXPLANON
|
· L’implant doit être retiré uniquement par un professionnel de santé familiarisé avec la technique. · L’implant est retiré à votre demande ou - au plus tard - trois ans après l’insertion. · La localisation du site d’insertion de l’implant est indiquée sur la Carte d’Alerte Patiente. · Le professionnel de santé localisera l'implant. Si l’implant ne peut pas être localisé, le professionnel de santé peut recourir à la radiographie, la tomodensitométrie, l’échographie ou l’imagerie par résonnance magnétique. ·
|
|||||||||
|
|
|
||||||||
|
|
|
||||||||
|
|
|
||||||||
|
· Parfois, l’implant peut être entouré de tissu fibreux. Dans ce cas, une petite incision devra être faite dans le tissu avant que l’implant puisse être retiré. · Si vous voulez que le professionnel de santé qui a retiré NEXPLANON remplace NEXPLANON par un autre implant, le nouvel implant peut être inséré par la même incision, à condition que la localisation du site soit correcte. · L'incision sera fermée par un pansement adhésif stérile. · Un bandage compressif sera appliqué pour minimiser le risque d’ecchymose. Vous pourrez retirer le bandage compressif au bout de 24 heures et le pansement adhésif stérile placé sur le site d’insertion après 3 à 5 jours. |
|||||||||
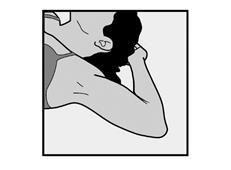
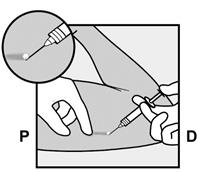
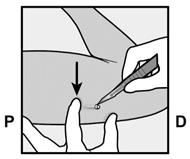
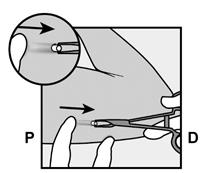
Les informations suivantes sont destinées uniquement aux professionnels de santé.
Quand insérer NEXPLANON
IMPORTANT : Exclure toute grossesse avant l’insertion de l’implant.
Le moment choisi pour l’insertion dépend de la situation contraceptive récente de la femme, comme suit :
Absence préalable de contraception hormonale utilisée au cours du mois précédent
L’implant doit être inséré entre le 1er jour (premier jour des menstruations) et le 5ème jour du cycle menstruel, même si la femme saigne toujours.
Si l’insertion a lieu au moment recommandé, une contraception complémentaire n'est pas nécessaire. Si l’insertion a lieu à un autre moment que celui recommandé pour l’insertion, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue.
Passage d’une méthode contraceptive hormonale à NEXPLANON 68 mg, implant pour usage sous-cutané
Relais d’un contraceptif hormonal combiné (ex : contraceptif oral combiné (COC), anneau vaginal ou patch transdermique)
L’implant doit être inséré de préférence le lendemain de la prise du dernier comprimé actif (le dernier comprimé contenant les substances actives) de son précédent contraceptif oral combiné ou le jour du retrait de l’anneau vaginal ou du patch transdermique. Au plus tard, l’implant doit être inséré le lendemain de l’intervalle habituel sans comprimé, sans anneau, sans patch ou de la prise de comprimés placebo de son précédent contraceptif hormonal combiné, quand la prochaine prise/insertion/application aurait dû avoir lieu. Toutes les méthodes contraceptives (patch transdermique, anneau vaginal) peuvent ne pas être commercialisées dans tous les pays.
Si l’insertion a lieu au moment recommandé, une contraception complémentaire n'est pas nécessaire. Si l’insertion a lieu à un autre moment que celui recommandé pour l’insertion, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue.
Relais d’une méthode purement progestative (ex : pilule progestative, injection, implant ou système intra-utérin (SIU) libérant un progestatif)
Comme plusieurs types de méthodes purement progestatives existent, l’insertion de l’implant doit se faire comme suit :
· Contraceptifs injectables : insérer l’implant le jour prévu pour l’injection suivante.
· Pilule progestative : la femme peut passer de la pilule purement progestative à NEXPLANON n’importe quel jour du mois. L’implant doit être inséré dans les 24 heures suivant la prise du dernier comprimé.
· Implant/Système intra-utérin (SIU) : insérer l’implant le jour du retrait du précédent implant ou du SIU.
Si l’insertion a lieu au moment recommandé, une contraception complémentaire n'est pas nécessaire. Si l’insertion a lieu à un autre moment que celui recommandé pour l’insertion, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue.
Après un avortement ou une fausse couche
L’implant peut être inséré immédiatement après un avortement ou une fausse couche.
· Premier trimestre : Si l’insertion a lieu sous 5 jours, une contraception complémentaire n’est pas nécessaire.
· Deuxième trimestre : Si l’insertion a lieu sous 21 jours, une contraception complémentaire n’est pas nécessaire.
Si l’insertion a lieu après le moment recommandé pour l’insertion, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue avant l’insertion.
En post-partum
L’implant peut être inséré immédiatement après l’accouchement à la fois chez une patiente qui allaite et une patiente qui n’allaite pas, en considérant les bénéfices et les risques individuels pour la patiente.
· Si l’insertion a lieu sous 21 jours, une contraception complémentaire n'est pas nécessaire.
· Si l’insertion a lieu plus de 21 jours après l’accouchement, la patiente devra être avertie qu’elle doit utiliser une méthode contraceptive non hormonale pendant les 7 jours suivant l’insertion. Si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue avant l’insertion.
Comment insérer NEXPLANON 68 mg, implant pour usage sous-cutané
La réussite de l’utilisation et du retrait de NEXPLANON repose sur une insertion sous-cutanée de l’implant dans le bras non dominant réalisée correctement et avec précaution conformément aux instructions. Le professionnel de santé qui a inséré NEXPLANON ainsi que la patiente doivent être capables de palper l’implant sous la peau de la femme après insertion.
L’implant doit être inséré à la face interne du bras non dominant en sous-cutané, juste sous la peau.
· Un implant inséré plus profondément qu’en sous-cutané (insertion profonde) peut ne pas être palpable et la localisation et/ou le retrait peuvent être difficiles (voir rubrique 4.2. « Comment retirer NEXPLANON » et rubrique 4.4. du RCP).
· Si l’implant est inséré profondément, une lésion nerveuse ou vasculaire peut se produire. Des insertions profondes ou incorrectes ont été associées à une paresthésie (due à une lésion nerveuse) et à une migration de l’implant (due à une insertion dans le muscle ou dans le fascia), et dans de rares cas, à une insertion intravasculaire.
L’insertion de NEXPLANON doit être effectuée dans des conditions d’asepsie et uniquement par un professionnel de santé familiarisé avec la technique. L’insertion de l’implant doit être réalisée uniquement avec l’applicateur préchargé.
Procédure d’insertion
Pour s’assurer que l’implant est inséré juste sous la peau, le professionnel de santé doit se placer de manière à voir le mouvement de l’aiguille en regardant l’applicateur par le côté et non par le dessus du bras. Par cette vue de côté, le site d’insertion et le mouvement de l’aiguille juste sous la peau pourront être bien visualisés.
À des fins d’illustration, les figures représentent la face interne du bras gauche.
|
· Demandez à la patiente de s’allonger sur le dos sur la table d’examen avec son bras non dominant plié au niveau du coude et tourné vers l’extérieur, ainsi, sa main est placée sous sa tête (ou le plus près possible) (Figure 1). |
Figure 1 |
||||||||||||||||||||||
|
· Identifiez le site d’insertion, qui se situe à la face interne du bras non dominant. Le site d’insertion est en regard du triceps, à environ 8 à 10 cm de l’épicondyle médial de l’humérus et 3 à 5 cm postérieur au (sous le) sillon (gouttière) qui sépare le biceps du triceps (Figures 2a, 2b et 2c). Cet emplacement est destiné à éviter les principaux vaisseaux sanguins et les nerfs se trouvant dans et autour du sillon. S’il n’est pas possible d’insérer l’implant à cet endroit (par exemple, chez une femme avec des bras minces), il devra de toute façon être inséré en arrière et aussi loin que possible du sillon. · Faites deux repères avec un marqueur chirurgical : un premier point, pour repérer l’endroit où l’implant sera inséré, et un second point, à 5 centimètres au-dessus (en direction de l’épaule) du premier repère (Figures 2a et 2b). Ce second repère (guide de marquage) servira plus tard de guide pour la direction pendant l’insertion.
|
|
||||||||||||||||||||||
|
· Après avoir marqué le bras, confirmez que le site est correctement localisé à la face interne du bras. · Nettoyez la peau depuis le site d’insertion jusqu’au guide de marquage avec une solution antiseptique. · Anesthésiez la zone d’insertion (par exemple, avec un anesthésique en spray ou en injectant 2 ml de lidocaïne à 1 % juste sous la peau le long du tunnel d’insertion prévu). · Sortez de son emballage l’applicateur NEXPLANON préchargé stérile jetable contenant l’implant. Avant utilisation, inspecter visuellement l’emballage pour s’assurer qu’il n’est pas endommagé (i.e. emballage tordu, perforé ). Si l’emballage présente une altération visible, qui pourrait compromettre la stérilité, l’applicateur ne doit pas être utilisé.
|
|||||||||||||||||||||||
|
· Tenez l’applicateur juste au-dessus de l’aiguille au niveau de la zone striée. Retirez le capuchon protecteur transparent de l’aiguille en le faisant glisser horizontalement, dans le sens de la flèche (Figure 3). Si le capuchon ne se retire pas facilement, l’applicateur ne doit pas être utilisé. Vous devez voir l’implant blanc en regardant dans la pointe de l’aiguille. Ne touchez pas la manette coulissante violette avant d’avoir entièrement inséré l’aiguille sous la peau, car cela rétracterait l’aiguille prématurément et libérerait l’implant de l’applicateur. · Si la manette coulissante violette est libérée prématurément, recommencez la procédure d’insertion avec un nouvel applicateur.
|
Figure 3 |
|
|||||||||||||||||||||
|
Avec votre main libre, tendez la peau autour du site d’insertion vers le coude (Figure 4). |
Figure 4 |
|
|||||||||||||||||||||
|
· L’implant doit être inséré en sous-cutanée, juste sous la peau (voir rubrique 4.4). Pour s’assurer que l’implant est inséré juste sous la peau, vous devez vous placer de manière à voir le mouvement de l’aiguille en regardant l’applicateur par le côté et non par le dessus du bras. Par cette vue de côté, vous pouvez clairement voir le site d’insertion et le mouvement de l’aiguille juste sous la peau (Figure 6). · Piquez la peau avec la pointe de l’aiguille, légèrement inclinée selon un angle inférieur à 30° (Figure 5a).
|
Figure 5a
|
|
|||||||||||||||||||||
|
|
|
|
|||||||||||||||||||||
|
· Insérez l’aiguille jusqu’à ce que le biseau (ouverture inclinée de la pointe) soit juste sous la peau (et pas plus loin) (Figure 5b). Si vous avez inséré l’aiguille plus en profondeur que le biseau, retirez l’aiguille jusqu’à ce que seul le biseau soit sous la peau. |
Figure 5b |
|
|||||||||||||||||||||
|
· Abaissez l’applicateur en position presque horizontale. Pour faciliter le positionnement sous-cutané, soulevez la peau avec l’aiguille, tout en la faisant glisser sur toute sa longueur (Figure 6). Vous pouvez ressentir une légère résistance mais n’exercez pas de force excessive. Si l’aiguille n’est pas entièrement insérée, l’implant ne sera pas correctement inséré. Si la pointe de l’aiguille sort de la peau avant que l’aiguille soit entièrement insérée, l’aiguille doit être retirée et replacée en sous-cutané pour terminer la procédure d’insertion.
|
Figure 6 |
|
|||||||||||||||||||||
|
· Maintenez l’applicateur dans la même position avec l’aiguille insérée sur toute sa longueur (Figure 7). Si nécessaire, vous pouvez utiliser votre main libre pour immobiliser l’applicateur.
|
Figure 7 |
|
|||||||||||||||||||||
|
· Déverrouillez la manette coulissante violette en la poussant légèrement vers le bas (Figure 8a). Déplacez la manette coulissante complètement en arrière jusqu’à la butée.
Ne déplacez pas (
|
Figure 8a |
|
|||||||||||||||||||||
|
Figure 8b |
Figure 8c |
|
|||||||||||||||||||||
|
|
|
|
|||||||||||||||||||||
|
Si l’applicateur n’est pas maintenu dans la même position au cours de la procédure d’insertion ou si la manette coulissante violette n’est pas complètement tirée en arrière jusqu’à la butée, l’implant ne sera pas correctement inséré et il pourrait sortir du site d’insertion. Si l’implant sort du site d’insertion, retirez l’implant et effectuez une nouvelle procédure d’insertion au niveau du même site d’insertion en utilisant un nouvel applicateur. Ne renfoncez pas l’implant ressorti dans l’incision. |
|
||||||||||||||||||||||
|
· Appliquez un petit pansement adhésif sur le site d’insertion.
· Vérifiez toujours la présence de l’implant dans le bras de la patiente par palpation immédiatement après l’insertion. En palpant les deux extrémités de l’implant, vous devez pouvoir confirmer la présence du bâtonnet de 4 cm (Figure 9). Voir la rubrique ci-dessous « Si l’implant n’est pas palpable après l’insertion ». |
Figure 9 |
|
|||||||||||||||||||||
|
· Demandez à la patiente de palper elle-même l’implant. · Appliquez une compresse stérile avec un bandage compressif pour minimiser le risque d’ecchymose. La patiente peut retirer le bandage compressif au bout de 24 heures et le petit pansement adhésif au bout de 3 à 5 jours. |
|
||||||||||||||||||||||
|
· Complétez la Carte d’Alerte Patiente et remettez-la à la patiente en lui demandant de la conserver. Complétez également les étiquettes adhésives et collez-les dans le dossier médical de la patiente. Si des dossiers médicaux électroniques sont utilisés, les informations figurant sur l’étiquette adhésive doivent être enregistrées. |
|
||||||||||||||||||||||
|
· L’applicateur est à usage unique et doit être correctement éliminé, conformément aux réglementations nationales d’élimination des déchets biologiques. |
|
||||||||||||||||||||||
|
Si l’implant n’est pas palpable après l’insertion Si vous ne pouvez pas palper l’implant ou si vous doutez de sa présence, l’implant peut ne pas avoir été inséré ou il peut avoir été inséré profondément : · Vérifiez l’applicateur. L’aiguille doit être complètement rétractée et seul le bout violet de l’obturateur doit être visible. · Utilisez d’autres méthodes pour confirmer sa présence. Etant donné la nature radio-opaque de l’implant, les méthodes de localisation adaptées sont la radiographie bidimensionnelle et la tomodensitométrie (TDM). L’échographie avec sonde linéaire à haute fréquence (10 MHz ou plus) ou l’imagerie par résonance magnétique (IRM) peuvent être utilisées. Si l’implant ne peut pas être trouvé avec ces méthodes d’imagerie, il est recommandé de vérifier la présence de l’implant en mesurant le taux d’étonogestrel dans un échantillon de sang prélevé chez la patiente. Dans ce cas, contactez ORGANON France qui vous communiquera le protocole approprié. · La patiente doit utiliser une méthode contraceptive non hormonale doit être utilisée tant que la présence de l’implant n’est pas confirmée. · Les implants insérés trop profondément doivent être localisés et retirés dès que possible pour éviter tout risque de migration (voir rubrique 4.4. du RCP). |
|
||||||||||||||||||||||
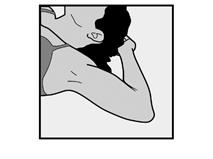
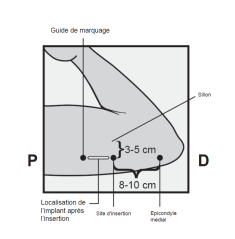
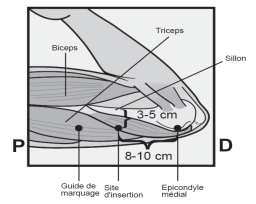
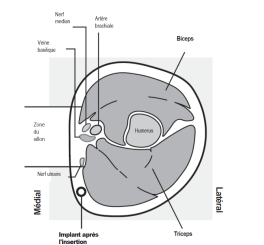
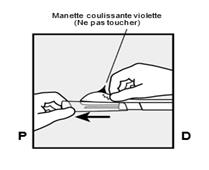
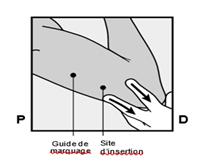
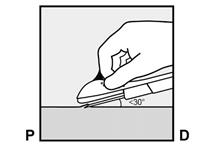
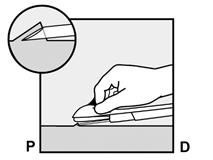
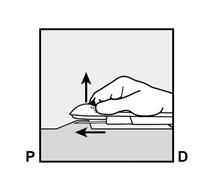
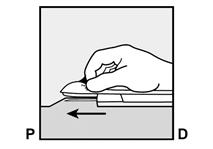
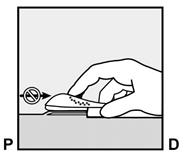
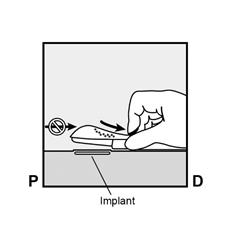
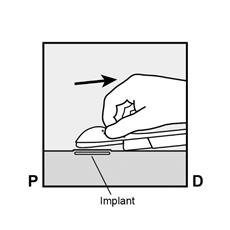
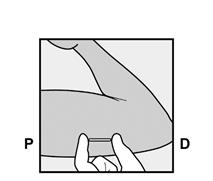
Comment retirer NEXPLANON
Le retrait de l’implant doit être effectué uniquement dans des conditions d’asepsie et par un professionnel de santé familiarisé avec la technique de retrait. Si vous n’êtes pas familiarisé avec la technique de retrait, contactez ORGANON France pour plus d’informations.
Avant de débuter la procédure de retrait, le professionnel de santé doit localiser l’implant. Vérifiez la localisation exacte de l’implant dans le bras par palpation.
Si l’implant n’est pas palpable, consultez la Carte d’Alerte Patiente ou le dossier médical pour vérifier le bras porteur de l’implant. Si l’implant ne peut pas être palpé, il pourrait être localisé en profondeur ou avoir migré. Envisagez qu’il puisse se situer à proximité de vaisseaux et de nerfs. Le retrait des implants non palpables doit uniquement être effectué par un professionnel de santé ayant l’expérience du retrait des implants insérés profondément et familiarisé avec la localisation de l’implant et l’anatomie du bras. Contactez ORGANON France pour plus d’informations.
Voir la rubrique ci-dessous « Localisation et retrait d’un implant non palpable » si l’implant ne peut pas être palpé.
Procédure de retrait d’un implant palpable
A des fins d’illustration, les figures représentent la face interne du bras gauche.
|
· Demandez à la patiente de s’allonger sur le dos sur la table. Le bras doit être placé avec le coude plié et la main sous la tête (ou le plus près possible) (Voir Figure 10) · |
Figure 10
|
||
|
· Localisez l’implant par palpation. Appuyez sur l’extrémité de l’implant la plus proche de l’épaule (Figure 11) pour l’immobiliser ; un renflement devrait apparaître indiquant l’extrémité de l’implant la plus proche du coude. Si l’extrémité n’apparaît pas, le retrait de l’implant pourrait être difficile et il devra être effectué par un professionnel de santé familiarisé avec le retrait des implants profonds. Contactez ORGANON France pour plus d’informations. · Marquez l’extrémité distale (l’extrémité la plus proche du coude), par exemple, avec un marqueur chirurgical. · Nettoyez le site avec une solution antiseptique. |
Figure 11 P, proximal (en direction de l’épaule) ; D, distal (en direction du coude) |
||
|
· Anesthésiez le site, par exemple, avec 0,5 à 1 ml de lidocaïne à 1 % à l’endroit de l’incision (Figure 12). Veillez à injecter l’anesthésique local sous l’implant pour que l’implant reste près de la surface de la peau. L’injection d’un anesthésique local au-dessus de l’implant peut rendre le retrait plus difficile. |
Figure 12 |
||
|
· Appuyez sur l’extrémité de l’implant la plus proche de l’épaule (Figure 13) pour l’immobiliser tout au long de la procédure de retrait. En partant au-dessus de l’extrémité de l’implant la plus proche du coude, faire une incision longitudinale (parallèle à l’implant) de 2 mm vers le coude. Veillez à ne pas couper l’extrémité de l’implant. |
Figure 13 |
||
|
· L’extrémité de l’implant doit sortir de l’incision. Si ce n’est pas le cas, poussez doucement l’implant vers l’incision jusqu’à ce que l’extrémité soit visible. Saisissez l’implant avec une pince et retirez si possible l’implant (Figure 14). · Si nécessaire, retirez doucement le tissu adhérent de l’extrémité de l’implant par dissection mousse. Si l’extrémité de l’implant n’est pas exposée après la dissection mousse, pratiquez une incision dans la gaine tissulaire et ensuite retirez l’implant avec une pince (Figures 15 et 16). |
Figure 14 |
||
|
Figure 15 |
Figure 16 |
||
|
· Si l’extrémité de l’implant n’est pas visible au niveau de l’incision, insérez doucement une pince (de préférence une pince mosquito courbe, avec les extrémités dirigées vers le haut) superficiellement dans l’incision (Figure 17). · Saisissez délicatement l’implant puis, tournez la pince avec votre autre main (Figure 18). · Avec une seconde pince, disséquez soigneusement le tissu autour de l’implant et saisissez l’implant (Figure 19). L’implant peut alors être retiré. · Si l’implant ne peut être saisi, arrêtez la procédure de retrait et orientez la patiente vers un professionnel de santé familiarisé avec les retraits complexes ou contactez ORGANON France. |
|||
|
|
|
|
|
|
Figure 17 Figure 18
Figure 19 · Vérifiez que la totalité du bâtonnet, qui mesure 4 cm de long, a été retiré en le mesurant. Des cas d’implants cassés dans le bras des patientes ont été rapportés. Dans certains cas, une difficulté de retrait de l’implant cassé a été signalée. Si un implant incomplet (moins de 4 cm) est retiré, le morceau restant devra être retiré en suivant les instructions de cette rubrique. · Si la femme souhaite continuer à utiliser NEXPLANON, un nouvel implant peut être inséré immédiatement après le retrait du précédent implant en utilisant la même incision à condition que la localisation du site soit correcte (voir paragraphe « Comment remplacer NEXPLANON »). |
|||
|
· Après avoir retiré l’implant, fermez l’incision avec un pansement adhésif stérile. |
|||
|
· Appliquez une compresse stérile avec un bandage compressif pour minimiser le risque d’ecchymose. La patiente peut retirer le bandage compressif au bout de 24 heures et le pansement adhésif stérile au bout de 3 à 5 jours. |
|||

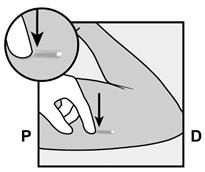
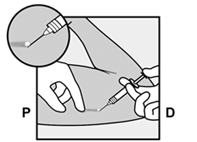
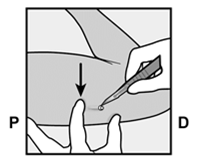
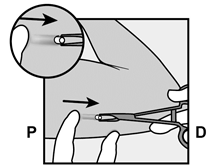
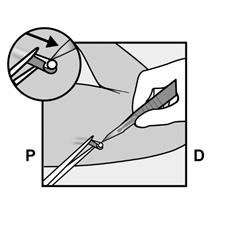
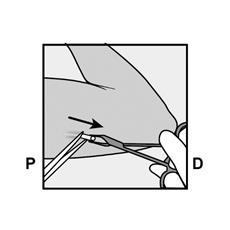
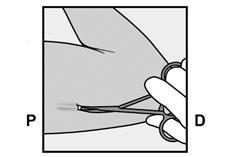
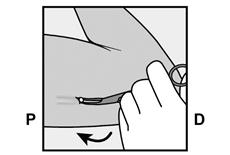
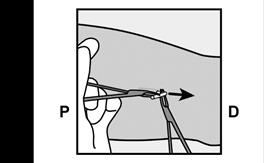
Localisation et retrait d’un implant non palpable
Il y a eu des rapports occasionnels de migration de l’implant ; habituellement il s’agit d’un mouvement mineur par rapport à la position initiale (voir aussi rubrique 4.4. du RCP) mais cela peut conduire à un implant non palpable à l’endroit où il avait été placé. Un implant qui a été inséré profondément ou qui a migré peut ne pas être palpable et c’est pourquoi des procédures d’imagerie, telles que décrites ci-dessous, peuvent s’avérer nécessaires pour la localisation.
Un implant non palpable doit toujours être localisé avant d’essayer de le retirer. Etant donné la nature radio-opaque de l’implant, les méthodes adaptées pour la localisation comprennent la radiographie bidimensionnelle et la tomodensitométrie (TDM). L’échographie avec sonde linéaire à haute fréquence (10 MHz ou plus) ou l’imagerie par résonance magnétique (IRM) peuvent être utilisées. Une fois l’implant localisé dans le bras, l’implant doit être retiré par un professionnel de santé ayant l’expérience du retrait des implants insérés profondément et familiarisé avec l’anatomie du bras. L’utilisation du guidage échographique pendant le retrait doit être envisagée.
Si l’implant ne peut pas être trouvé dans le bras après des tentatives exhaustives de localisation, envisagez d’appliquer les techniques d’imagerie au niveau du thorax, car des cas extrêmement rares de migration dans le système vasculaire pulmonaire ont été rapportés. Si l’implant est localisé dans le thorax, des interventions chirurgicales ou endovasculaires peuvent être nécessaires pour le retrait ; des professionnels de santé familiarisés avec l’anatomie thoracique doivent être consultés.
Si, à tout moment, ces méthodes d’imagerie échouent à localiser l’implant, la détermination du taux sanguin d’étonogestrel peut être utilisée pour vérifier la présence de l’implant. Veuillez contacter ORGANON France pour plus de conseils.
Si l’implant a migré dans le bras, le retrait peut nécessiter une intervention chirurgicale mineure avec une incision plus grande ou une intervention chirurgicale en salle d’opération. Le retrait d’implants insérés profondément doit être effectué avec précaution afin d’aider à éviter toute lésion des structures nerveuses ou vasculaires profondes dans le bras.
Les implants non palpables et insérés profondément doivent être retirés par un professionnel de santé familiarisé avec l’anatomie du bras et le retrait des implants insérés profondément.
Une chirurgie exploratoire sans connaissance de la localisation exacte de l’implant est fortement déconseillée.
Veuillez contacter ORGANON France pour plus de conseils.
Comment remplacer NEXPLANON
Un remplacement immédiat peut être réalisé après le retrait du précédent implant et la procédure est semblable à la procédure d’insertion décrite dans le paragraphe « Comment insérer NEXPLANON ».
Le nouvel implant peut être inséré dans le même bras et par l’incision effectuée pour retirer le précédent implant à condition que la localisation du site soit correcte, c’est-à-dire 8 à 10 cm de l’épicondyle médial de l’humérus et 3 à 5 cm postérieur au (sous le) sillon (voir rubrique 4.2 « Comment insérer NEXPLANON » du RCP). Si la même incision est utilisée pour insérer le nouvel implant, anesthésiez le site d’insertion en injectant un anesthésique, par exemple 2 ml de lidocaïne à 1 %, juste sous la peau à partir de l’incision de retrait et le long du « canal d’insertion » et suivez les étapes suivantes des instructions d’insertion.
Carte d’Alerte Patiente et Etiquettes adhésives :
Carte d’Alerte Patiente
Recto
NEXPLANON 68 mg, implant pour usage sous-cutané
étonogestrel
Information importante :
Occasionnellement, palpez doucement l’implant pour être sûre que vous connaissez sa localisation. Si, à tout moment, vous ne sentez pas l’implant à la palpation, contactez votre médecin dès que possible.
La propriétaire de cette carte est porteuse d’un implant contraceptif sous‑cutané uniquement progestatif.
L’implant est situé à la face interne du bras.
En cas d’accident, n’essayez pas de retirer l’implant.
NEXPLANON est visible aux rayons X.
Pour plus d’informations, veuillez contacter :
ORGANON France
176 RUE MONTMARTRE
75002 PARIS
Information médicale : 01 57 77 32 00
CONSERVEZ CETTE CARTE DANS UN ENDROIT SUR !
Verso
Renseignements cliniques / Nom et ville du professionnel de santé ayant inséré l'implant
Lot
Nom de la patiente
Date d’insertion
Date prévue de retrait
Bras * Gauche * Droit
Etiquettes adhésives
Etiquette n°1
NEXPLANON 68 mg, implant pour usage sous-cutané
étonogestrel
Lot
Pour le dossier du professionnel de santé
Nom de la patiente :
Date d’insertion :
Date prévue de retrait :
Bras : * Gauche * Droit
N’oubliez pas de vérifier la présence de l’implant par palpation.
N’oubliez pas de donner à la patiente la notice, la carte et le sticker lui étant destinés.
Etiquette n°2
NEXPLANON 68 mg, implant pour usage sous-cutané
étonogestrel
Lot
Pour le dossier patient.
Nom de la patiente :
Date d’insertion :
Date prévue de retrait :
Bras : * Gauche * Droit