Dernière mise à jour le 29/06/2026
EFFERALGANMED 250 mg, comprimé dispersible
Indications thérapeutiques
Classe pharmacothérapeutique : Autres analgésiques et antipyrétiques - code ATC: N02BE01
Ce médicament est un analgésique et un antipyrétique. Il contient du paracétamol.
Il est indiqué dans le traitement symptomatique des douleurs d'intensité légère à modérée et/ou fièvre, telles que maux de tête, états grippaux, douleurs dentaires, courbatures ou règles douloureuses.
Ce médicament est réservé aux ENFANTS pesant de 13 à 50 kg (environ 2 à 15 ans). Voir rubrique 3. "Comment prendre EFFERALGANMED 250 mg, comprimé dispersible ? "
Présentations
> 12 plaquette(s) polyamide PVC-Aluminium aluminium polytéréphtalate (PET) de 1 comprimé(s)
Code CIP : 346 365-7 ou 34009 346 365 7 8
Déclaration de commercialisation : 18/01/2021
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 1,31 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 2,33 €
- Taux de remboursement :65 %
Documents de bon usage du médicament
- Prise en charge de la fièvre chez l’enfant
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Octobre 2016
- Prise en charge médicamenteuse de la douleur chez l'enfant : alternatives à la codéine
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Février 2016
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 20/03/2020 | Inscription (CT) | Le service médical rendu par EFFERALGANMED 250mg, comprimé dispersible, est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 20/03/2020 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à aux présentations déjà inscrites. |
ANSM - Mis à jour le : 11/03/2025
EFFERALGANMED 250 mg, comprimé dispersible
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
(sous forme de cristaux de paracétamol enrobés).
Pour un comprimé dispersible.
Excipient à effet notoire: chaque comprimé contient 30 mg d'aspartam (E951).
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé blanc, rond, biconvexe avec une dépression centrale concave et ayant une odeur caractéristique de banane.
4.1. Indications thérapeutiques
Traitement symptomatique des douleurs d'intensité légère à modérée et/ou des états fébriles.
4.2. Posologie et mode d'administration
Cette présentation est réservée à l’enfant de 13 kg à 50 kg (environ 2 à 15 ans).
Chez l’enfant, il est impératif de respecter les posologies définies en fonction du poids de l’enfant et donc de choisir une présentation adaptée. Les âges approximatifs en fonction du poids sont donnés à titre d’information.
La posologie usuelle de paracétamol recommandée est d’environ 60 mg/kg/jour, à répartir en 4 ou 6 prises, soit environ 15 mg/kg toutes les 6 heures ou 10 mg/kg toutes les 4 heures.
· Pour les enfants ayant un poids de 13 à 20 kg (environ 2 à 7 ans), la posologie est de 1 comprimé par prise, à renouveler si besoin au bout de 6 heures, sans dépasser 4 comprimés par jour.
· Pour les enfants ayant un poids de 21 à 25 kg (environ 6 à 10 ans), la posologie est de 1 comprimé par prise, à renouveler si besoin au bout de 4 heures, sans dépasser 6 comprimés par jour.
· Pour les enfants ayant un poids de 26 à 40 kg (environ 8 à 13 ans), la posologie est de 2 comprimés par prise, à renouveler si besoin au bout de 6 heures, sans dépasser 8 comprimés par jour.
· Pour les enfants ayant un poids de 41 à 50 kg (environ 12 à 15 ans), la posologie est de 2 comprimés par prise, à renouveler si besoin au bout de 4 heures, sans dépasser 12 comprimés par jour.
Doses maximales recommandées :
La dose totale de paracétamol ne doit pas dépasser 80 mg/kg/jour chez l’enfant de moins de 37 kg et 3 g par jour chez le grand enfant au-delà de 38 kg (voir rubrique 4.9).
Fréquence d’administration
Chez l’enfant, les prises doivent être régulièrement espacées, y compris la nuit, de préférence de 6 heures, et d’au moins 4 heures.
Situations cliniques particulières :
Insuffisance de la fonction hépatique :
Chez les patients présentant une insuffisance hépatique ou un syndrome de Gilbert, la dose doit être réduite ou l'intervalle entre 2 prises doit être augmenté.
Sauf prescription médicale, la dose quotidienne ne doit pas dépasser 2g.
Insuffisance rénale
Chez les patients présentant une insuffisance rénale, la dose doit être réduite et l'intervalle minimal entre deux administrations doit être augmenté, conformément au tableau suivant :
|
Filtration glomérulaire |
Dose |
|
≥ 50 ml/min |
Toutes les 4 heures |
|
10-50 ml/min |
Toutes les 6 heures |
|
< 10 ml/min |
Toutes les 8 heures |
La dose quotidienne de paracétamol ne doit pas dépasser 60 mg/kg/jour (jusqu'à 2 g par jour) dans les situations suivantes, sauf avis contraire d'un médecin (voir rubrique 4.4) :
· Poids inférieur à 50kg chez l'adulte
· Alcoolisme chronique
· Déshydratation
· Malnutrition chronique.
Mode d'administration
Voie orale.
Chez les enfants de moins de 6 ans, les comprimés sont à dissoudre dans une cuillère remplie d’eau ou de lait (pas de jus de fruit en raison du risque d’amertume), avant d’être administrés à l’enfant.
Chez les enfants de plus de 6 ans, les comprimés peuvent être sucés, ils fondent très rapidement dans la bouche au contact de la salive.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Phénylcétonurie (liée à la présence d’aspartam).
· Insuffisance hépatocellulaire sévère.
4.4. Mises en garde spéciales et précautions d'emploi
Ne pas dépasser la dose prescrite.
L'utilisation prolongée de ce médicament, sauf sous contrôle médical, peut être dangereuse.
Ce produit ne doit être utilisé qu'en cas de nécessité absolue.
Des doses supérieures à celles recommandées peuvent entraîner des risques hépatiques graves. Le traitement avec l'antidote doit être administré le plus rapidement possible (voir rubrique 4.9).
Pour éviter un risque de surdosage, il est recommandé aux patients de ne pas prendre simultanément d'autres produits contenant du paracétamol.
La dose totale de paracétamol ne doit pas dépasser 80 mg/kg/jour chez l’enfant de moins de 37 kg et 3 g par jour chez l’enfant pesant 38 kg ou plus (Voir rubrique 4.9).
La prise de comprimé dispersible n’est pas recommandée chez l’enfant de moins de 6 ans car elle peut entraîner une fausse-route.
Cette spécialité contient 30 mg d'aspartam par comprimé. L’aspartam est une source de phénylalanine. Il peut être nocif si vous êtes atteint de phénylcétonurie (PCU), une maladie génétique rare dans laquelle la phénylalanine s'accumule parce que le corps ne peut pas l'éliminer correctement.
Il n’existe aucune donnée clinique ou non clinique concernant l’utilisation de l’aspartam chez les enfants âgés de moins de 12 semaines.
Précautions d'emploi
Hépatotoxicité
Des cas d'hépatotoxicité induite par le paracétamol ont été rapportés chez des patients présentant (un ou plusieurs) facteur(s) de risque.
Le paracétamol doit être administré avec prudence chez les patients présentant des facteurs de risque et notamment :
· Adultes pesant moins de 50 kg (voir rubrique 4.2).
· Insuffisance hépatocellulaire légère à modérée (voir rubrique 4.2) (à noter : le paracétamol est contre-indiqué en cas d'insuffisance hépatocellulaire sévère).
· Insuffisance rénale (voir rubrique 4.2).
· Syndrome de Gilbert (ictère familial non hémolytique) (voir rubrique 4.2).
· Alcoolisme chronique (voir rubrique 4.2).
· Malnutrition chronique (faibles réserves de glutathion hépatique) (voir rubrique 4.2).
· Déshydratation (voir rubrique 4.2).
· Prise concomitante de médicaments hépatotoxiques.
Les doses de paracétamol doivent être revues à intervalles cliniquement appropriés et les patients doivent être surveillés pour détecter l'émergence de nouveaux facteurs de risque d'hépatotoxicité, qui peuvent justifier une adaptation de la posologie.
Si une hépatite virale aiguë est diagnostiquée, le traitement doit être arrêté.
Chez l’enfant traité par 60 mg/kg/jour de paracétamol, l’association d’un autre antipyrétique n’est justifiée qu’en cas d’inefficacité.
En cas de fièvre élevée, de survenue de signe de surinfection ou de persistance des symptômes au-delà de 3 jours, une réévaluation du traitement doit être faite.
Des cas d’acidose métabolique à trou anionique élevé (AMTAE) due à une acidose pyroglutamique ont été rapportés chez les patients atteints d’une maladie grave telle qu’une insuffisance rénale sévère et un sepsis, ou chez les patients souffrant de malnutrition et d'autres sources de déficit en glutathion (par exemple, alcoolisme chronique) qui ont été traités par du paracétamol à une dose thérapeutique pendant une période prolongée ou par une association de paracétamol et de flucloxacilline. En cas de suspicion d’AMTAE due à une acidose pyroglutamique, il est recommandé d’arrêter immédiatement le paracétamol et d’effectuer une surveillance étroite.
La mesure de la 5-oxoproline urinaire peut être utile pour identifier l’acidose pyroglutamique comme cause sous-jacente de l’AMTAE chez les patients présentant de multiples facteurs de risque
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
· Le probénécide diminue presque de moitié la clairance du paracétamol en inhibant sa conjugaison avec l'acide glucuronique. Une réduction de la dose de paracétamol doit être conseillée en cas de traitement concomitant avec le probénécide.
· Le salicylamide peut prolonger la demi-vie d'élimination du paracétamol.
· La prise concomitante de substances inductrices des enzymes (notamment la carbamazépine, le phénobarbital, la phénytoïne, la primidone, la rifampicine, le millepertuis, etc.) ou de substances potentiellement hépatotoxiques demande une attention particulière (voir rubrique 4.9).
· Métoclopramide et dompéridone : accélèrent l'absorption du paracétamol.
· Cholestyramine : réduit l'absorption du paracétamol.
· L’utilisation concomitante de paracétamol (4 g par jour au moins pendant 4 jours) avec des anticoagulants oraux peut entraîner de légères variations des valeurs de l'INR, s'accompagnant d'une aggravation des risques de saignements. Dans ce cas, une surveillance accrue des valeurs de l’INR devra être menée pendant la durée de l’association et après.
· Des précautions doivent être prises lorsque le paracétamol est utilisé en même temps que la flucloxacilline, car une prise concomitante a été associée à une acidose métabolique à trou anionique élevé due à une acidose pyroglutamique, en particulier chez les patients présentant des facteurs de risque (voir rubrique 4.4).
Interactions avec les examens paracliniques :
La prise de paracétamol peut fausser le dosage de l’acide urique réalisé par la méthode à l’acide phosphotungstique, et le dosage de la glycémie réalisé par la méthode au glucose oxydase-peroxydase.
4.6. Fertilité, grossesse et allaitement
Grossesse
Une vaste quantité de données portant sur les femmes enceintes démontrent l’absence de toute malformation ou de toute toxicité fœtale/néonatale. Les études épidémiologiques consacrées au neurodéveloppement des enfants exposés au paracétamol in utero produisent des résultats non concluants. Si cela s’avère nécessaire d’un point de vue clinique, le paracétamol peut être utilisé pendant la grossesse; cependant, il devra être utilisé à la dose efficace la plus faible, pendant la durée la plus courte possible et à la fréquence la plus réduite possible.
Allaitement
Après administration par voie orale, le paracétamol est excrété dans le lait maternel en petites quantités. Aucun effet indésirable chez le nourrisson allaité n'a été signalé. A doses thérapeutiques, l'administration de ce médicament est possible pendant l'allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
|
Rare (³1/10 000, < 1/1 000) |
Très rares (<1/10 000) |
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) |
|
|
Affections hématologiques et du système lymphatique |
|
|
Thrombopénie, leucopénie, neutropénie (cas isolés). |
|
Affections du système immunitaire |
|
Hypersensibilité (allant de simples rashs cutanés ou urticaire jusqu'au choc anaphylactique nécessitant l'arrêt du traitement) |
|
|
Troubles du métabolisme et de la nutrition |
|
|
Acidose métabolique à trou anionique élevé |
|
Affections hépatobiliaires |
Augmentation des transaminases hépatiques |
|
Diminution ou augmentation des enzymes hépatiques (en particulier dans la population présentant des facteurs de risque) (voir rubrique 4.4). |
|
Affections de la peau et du tissu sous-cutané |
|
De très rares cas de réactions cutanées graves ont été rapportés. |
|
Description des effets indésirables sélectionnés
Acidose métabolique à trou anionique élevé
Des cas d’acidose métabolique à trou anionique élevé due à une acidose pyroglutamique ont été observés chez des patients présentant des facteurs de risque et prenant du paracétamol (voir rubrique 4.4). Une acidose pyroglutamique peut survenir chez ces patients en raison des faibles taux de glutathion.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement.social-sante.gouv.fr
Les symptômes apparaissent généralement dans les 24 premières heures et sont caractérisés par : nausées, vomissements, anorexie, pâleur et douleurs abdominales.
Le surdosage, à partir de 7,5 g de paracétamol en une seule prise chez l'adulte ou à partir de 140 mg/kg de poids corporel en une seule prise chez l’enfant, provoque une cytolyse hépatique susceptible d'aboutir à une nécrose complète et irréversible, se traduisant par une insuffisance hépatocellulaire, une acidose métabolique et une encéphalopathie pouvant aller jusqu'au coma et à la mort. Simultanément, on observe une augmentation des transaminases hépatiques (ASAT, ALAT), de la lacto-déshydrogénase, de la bilirubine et une diminution du taux de prothrombine pouvant apparaître 12 à 48 heures après administration.
Les premiers symptômes cliniques de l’atteinte hépatique se manifestent en général 2 jours après le surdosage pour atteindre un maximum après 4 à 6 jours.
Une insuffisance rénale aiguë avec une nécrose tubulaire aiguë peut survenir, même en l'absence d’atteintes hépatiques sévères. Les autres symptômes non-hépatiques qui ont été signalés suite à un surdosage de paracétamol sont des anomalies myocardiques et des pancréatites.
Conduite d'urgence :
· Transfert immédiat en milieu hospitalier, même en l'absence de symptômes précoces significatifs.
· Analyse sanguine afin de déterminer la concentration plasmatique initiale de paracétamol.
· Lavage gastrique.
· Administration par voie I.V. (ou par voie orale, si possible) de l'antidote N-acétylcystéine, si possible avant la dixième heure suivant l'ingestion. La N-acétylcystéine peut encore protéger à un moindre degré 10 heures après et jusqu'à 48 heures après le surdosage, mais dans ces cas un traitement plus long est nécessaire.
· Un traitement symptomatique sera institué.
· La méthionine par voie orale peut être utilisée à la place de la N-acétylcystéine. Dans ce cas, l'administration doit être aussi précoce que possible et dans tous les cas dans les dix heures qui suivent ce surdosage.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : AUTRES ANALGESIQUES ET ANTIPYRETIQUES, code ATC : N02BE01
Le mécanisme précis des propriétés analgésique et antipyrétique du paracétamol reste encore à établir, il pourrait impliquer des actions centrales et périphériques.
5.2. Propriétés pharmacocinétiques
L'absorption du paracétamol par voie orale est complète et rapide.
Les concentrations plasmatiques maximales sont atteintes 30 à 60 minutes après ingestion.
Distribution
Le paracétamol est distribué rapidement dans tous les tissus. Les concentrations sont comparables dans le sang, la salive et le plasma. La liaison aux protéines plasmatiques est faible.
Métabolisme
Le paracétamol est essentiellement métabolisé au niveau du foie, selon deux voies principales : conjugaison à l’acide glycuronique et conjugaison à l’acide sulfurique. Cette dernière voie est rapidement saturable aux posologies supérieures aux doses thérapeutiques. Une voie mineure, catalysée par le cytochrome P 450 (principalement CYP2E1), conduit à la formation d'un intermédiaire réactif, la N-acétyl benzoquinone imine, qui, dans les conditions normales d'utilisation, est rapidement détoxifiée par le glutathion réduit et éliminée dans les urines après conjugaison à la cystéine et à l'acide mercapturique. En revanche, lors d'intoxications massives, la quantité de ce métabolite toxique est augmentée.
Elimination
L'élimination est essentiellement urinaire. 90 % de la dose ingérée est éliminée par les reins en 24 heures, principalement en glucuronide (60 à 80 %) et en sulfate-conjugué (20 à 30 %). Moins de 5 % est éliminé sous forme inchangée.
La demi-vie d'élimination est d'environ 2 heures.
Insuffisance rénale
En cas d’insuffisance rénale sévère (clairance de la créatinine inférieure à 10 ml/min), l’élimination du paracétamol et de ses métabolites est retardée.
Sujet âgé
La capacité de conjugaison n'est pas modifiée.
5.3. Données de sécurité préclinique
Des études complémentaires n’ont pas mis en évidence de risque génotoxique du paracétamol aux doses thérapeutiques (doses non-toxiques).
Des études animales (rats et souris) à long terme n'ont pas mis en évidence d'effet cancérigène du paracétamol à des doses non-hépatoxiques.
Le paracétamol traverse le placenta.
Les études effectuées chez l'animal et les données recueillies lors de l’utilisation chez l’homme n'ont pas mis en évidence jusqu'à ce jour de données sur la toxicité de reproduction.
Aucune étude conventionnelle s’appuyant sur les normes actuellement admises pour évaluer la toxicité pour la reproduction et le développement n’est disponible.
Cristaux de paracétamol enrobé :
Copolymère basique de méthacrylate de butyle, dispersion de polyacrylate à 30 %, silice hydrophobe colloïdale.
Matrice du comprimé:
Mannitol (granulé, poudre), crospovidone, aspartam (E951), arôme banane, stéarate de magnésium.
6.4. Précautions particulières de conservation
Pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Comprimés sous plaquettes, unidoses, perforées, sécurisées et avec opercule détachable: Polyamide/PVC/Aluminium - Aluminium/PET
Boîte de 12 ou 24.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Sans objet.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
194 Bureaux de La Colline, Bâtiment D
92213 Saint Cloud cedex
France
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Médicament non soumis à prescription médicale.
ANSM - Mis à jour le : 11/03/2025
EFFERALGANMED 250 mg, comprimé dispersible
Paracétamol
Veuillez lire attentivement cette notice avant de prendre ce médicamentcar elle contient des informations importantes pour vous.
Vous devez toujours prendre ce médicament en suivant scrupuleusement les informations fournies dans cette notice ou par votre médecin ou votre pharmacien.
· Gardez cette notice, vous pourriez avoir besoin de la relire.
· Adressez-vous à votre pharmacien pour tout conseil ou information.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien après 3 jours.
1. Qu'est-ce que EFFERALGANMED 250 mg, comprimé dispersible et dans quel cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre EFFERALGANMED 250 mg, comprimé dispersible ?
3. Comment prendre EFFERALGANMED 250 mg, comprimé dispersible ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver EFFERALGANMED 250 mg, comprimé dispersible ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE EFFERALGANMED 250 mg, comprimé dispersible ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Autres analgésiques et antipyrétiques - code ATC: N02BE01
Ce médicament est un analgésique et un antipyrétique. Il contient du paracétamol.
Il est indiqué dans le traitement symptomatique des douleurs d'intensité légère à modérée et/ou fièvre, telles que maux de tête, états grippaux, douleurs dentaires, courbatures ou règles douloureuses.
Ce médicament est réservé aux ENFANTS pesant de 13 à 50 kg (environ 2 à 15 ans). Voir rubrique 3. "Comment prendre EFFERALGANMED 250 mg, comprimé dispersible ? "
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE EFFERALGANMED 250 mg, comprimé dispersible ?
Ne prenez jamais EFFERALGANMED 250 mg comprimé dispersible
· Si vous êtes allergique à la substance active ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· En cas de maladie grave du foie,
· En cas de phénylcétonurie (maladie héréditaire dépistée à la naissance), en raison de la présence d'aspartam.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre EFFERALGANMED 250 mg, comprimé dispersible :
· si vous pesez moins de 50 kg (pour un adulte),
· en cas de maladie du foie,
· en cas d'alcoolisme chronique,
· en cas de malnutrition chronique,
· en cas de déshydratation,
· en cas de maladie des reins
· si vous utilisez tout autre médicament affectant la fonction hépatique.
Pendant le traitement par EFFERALGANMED 250 mg, comprimé dispersible, informez immédiatement votre médecin : si vous avez des maladies graves, y compris une insuffisance rénale grave ou un sepsis (lorsque des bactéries et leurs toxines circulent dans le sang, entraînant des lésions au niveau des organes), ou si vous êtes atteint de malnutrition, d’alcoolisme chronique ou si vous prenez également de la flucloxacilline (un antibiotique). Une affection grave appelée acidose métabolique (une anomalie du sang et des fluides) a été signalée chez les patients qui prennent régulièrement du paracétamol pendant une période prolongée ou qui prennent du paracétamol en association avec de la flucloxacilline. Les symptômes de l’acidose métabolique peuvent inclure : de graves difficultés respiratoires avec une respiration rapide et profonde, une somnolence, une envie de vomir (nausée) et des vomissements.
Vous devrez peut-être réduire la quantité de paracétamol que vous prenez ou éviter complètement d'utiliser ce produit.
En cas d'hépatite virale aiguë, arrêtez votre traitement et consultez votre médecin.
Ce médicament contient du paracétamol. Afin d'éviter un risque de surdosage, vous ne devez pas prendre d'autres produits contenant du paracétamol en même temps que ce médicament.
Enfants
La dose totale de paracétamol ne doit pas dépasser 80 mg/kg/ par jour chez l’enfant de moins de 37 kg et 3 g par jour chez le grand enfant au-delà de 38 kg (voir rubrique "Si vous avez pris plus de EFFERALGANMED 250 mg, comprimé dispersible que vous n’auriez dû").
La prise de comprimé dispersible n’est pas recommandée chez l’enfant de moins de 6 ans car elle peut entraîner une fausse-route.
Chez l’enfant traité par 60 mg/kg/jour de paracétamol, l’association d’un autre antipyrétique n’est justifiée qu’en cas d’inefficacité.
Ce médicament contient un arôme banane et peut être attrayant pour les enfants. Tenir hors de la vue et de la portée des enfants.
Autres médicaments et EFFERALGANMED 250 mg, comprimé dispersible
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Informez toujours votre médecin ou votre pharmacien si vous prenez l'un des médicaments suivants avant de commencer le traitement par EFFERALGANMED 250 mg, comprimé dispersible :
· probénécide (utilisé pour traiter la goutte) ;
· salicylamide (utilisé pour traiter la fièvre) ;
· médicaments inducteurs enzymatiques, notamment la carbamazépine, le phénobarbital, la phénytoïne, la primidone (utilisée pour traiter l'épilepsie), la rifampicine (antibactérien), le millepertuis (herbe médicinale utilisée pour le traitement de la dépression légère à modérée), etc. ;
· métoclopramide et dompéridone (utilisés pour traiter et prévenir les nausées et les vomissements) ;
· cholestyramine (utilisée pour traiter les concentrations élevées de lipides (graisses) dans le sang) ;
· anticoagulants, notamment la warfarine (utilisée pour empêcher la coagulation du sang).
Grossesse et Allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Au besoin, EFFERALGANMED 250 mg, comprimé dispersible peut être utilisé pendant la grossesse. Vous devez utiliser la dose la plus faible possible qui permette de soulager la douleur et/ou la fièvre et la prendre pendant la durée la plus courte possible.
Contactez votre médecin si la douleur et/ou la fièvre ne diminuent pas ou si vous devez prendre le médicament plus fréquemment.
EFFERALGANMED 250 mg, comprimé dispersible contient de l’aspartam
Ce médicament contient 30 mg d’aspartam par dose.
L’aspartam contient une source de phénylalanine. Peut être dangereux pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de phénylalanine ne pouvant être éliminée correctement.
Analyses biologiques
Si vous devez subir des analyses biologiques destinées à vérifier vos concentrations sanguines de glucose ou d'acide urique, informez le médecin de votre traitement par EFFERALGANMED 250 mg, comprimé dispersible.
3. COMMENT PRENDRE EFFERALGANMED 250 mg, comprimé dispersible ?
Ce médicament est réservé aux ENFANTS pesant de 13 à 50 kg (environ 2 à 15 ans).
Chez les enfants de moins de 6 ans, les comprimés sont à dissoudre dans une cuillère remplie d’eau ou de lait (pas de jus de fruit en raison du risque d’amertume), avant d’être administrés à l’enfant.
Chez les enfants de plus de 6 ans, les comprimés peuvent être sucés, ils fondent très rapidement dans la bouche au contact de la salive.
Méthode d’administration
Instructions d’ouverture :
Ce médicament est disponible sous plaquettes thermoformées, unidoses, perforées, sécurisées et avec opercule détachable:
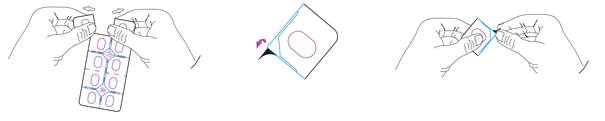 |
Retirer une seule dose en déchirant le long de la ligne perforée de la plaquette thermoformée et retirer l’opercule de la plaquette thermoformée pour retirer le comprimé.
Posologie
Chez l'enfant, il est impératif de respecter la posologie définie sur la base du poids corporel, et par conséquent d'utiliser une présentation adaptée. Les âges approximatifs en fonction du poids sont donnés à titre d’information.
La dose quotidienne recommandée de paracétamol est approximativement de 60 mg/kg par jour, à répartir en 4 ou 6 prises quotidiennes, c'est-à-dire approximativement 15 mg/kg toutes les 6 heures ou 10 mg/kg toutes les 4 heures.
· Pour les enfants ayant un poids de 13 à 20 kg (environ 2 à 7 ans), la dose recommandée est de 1 comprimé par prise, à renouveler si besoin au bout de 6 heures, sans dépasser 4 comprimés par jour.
· Pour les enfants ayant un poids de 21 à 25 kg (environ 6 à 10 ans), la dose recommandée est de 1 comprimé par prise, à renouveler si besoin au bout de 4 heures, sans dépasser 6 comprimés par jour.
· Pour les enfants ayant un poids de 26 à 40 kg (environ 8 à 13 ans), la dose recommandée est de 2 comprimés par prise, à renouveler si besoin au bout de 6 heures, sans dépasser 8 comprimés par jour.
· Pour les enfants ayant un poids de 41 à 50 kg (environ 12 à 15 ans), la dose recommandée est de 2 comprimés par prise, à renouveler si besoin au bout de 4 heures, sans dépasser 12 comprimés par jour.
Populations particulières :
Chez les patients présentant une insuffisance de la fonction hépatique ou rénale ou un syndrome de Gilbert, le médecin adaptera la dose.
Chez les patients ayant une insuffisance rénale sévère, l'intervalle entre deux prises sera au minimum de 8 heures.
Parlez à votre médecin ou à votre pharmacien avant de prendre EFFERALGANMED 250 mg dans les situations suivantes :
· Poids inférieur à 50 kg chez l'adulte
· Alcoolisme chronique
· Déshydratation
· Malnutrition chronique.
Si vous avez l'impression que l'effet de EFFERALGANMED 250 mg est trop fort ou trop faible, parlez-en à votre médecin ou votre pharmacien.
Respectez la posologie afin d'éviter la douleur ou une récidive de la fièvre.
Chez l’enfant, les prises doivent être régulièrement espacées, y compris la nuit, de préférence à intervalles de 6 heures, ou à intervalles minimum de 4 heures.
Vous devez consulter un médecin si vos symptômes s'aggravent ou ne s'améliorent pas au bout de 3 jours.
Si vous avez pris plus de EFFERALGANMED 250 mg, comprimé dispersible que vous n’auriez dû :
Arrêtez le traitement et consultez immédiatement un médecin.
Les symptômes de surdosage apparaissent généralement dans les premières 24 heures et sont caractérisés par : nausées, vomissements, perte d'appétit, pâleur et douleurs abdominales.
Consulter immédiatement un médecin en cas de surdosage, même si le patient se sent bien, à cause du risque de lésions hépatiques tardives, graves et irréversibles.
Si vous oubliez de prendre EFFERALGANMED 250 mg, comprimé dispersible :
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament est susceptible d'avoir des effets indésirables, bien que tout le monde n’y soit pas sujet.
Le médicament peut notamment entraîner les effets indésirables suivants :
Effets indésirables rares (peuvent affecter jusqu’à 1 personne sur 1000):
Augmentation du taux d'enzymes hépatiques (transaminases).
Effets indésirables très rares (peuvent affecter jusqu’à 1 personne sur 10000):
Réactions allergiques : rash ou éruption cutanée avec boules d'œdème prurigineuses, gonflement du visage ou de la langue, essoufflement ou difficultés respiratoires susceptibles d'être provoqués par une réaction allergique grave. Si vous présentez une réaction allergique grave, interrompez le traitement par EFFERALGANMED 250 mg et consultez immédiatement un médecin.
Réactions cutanées : de très rares cas de réactions cutanées graves ont été rapportés.
Effets indésirables de fréquence indéterminée (ne peut pas être estimée à partir des données disponibles)
Diminution ou augmentation des enzymes hépatiques (dommages causés au foie).
Troubles sanguins : diminution du nombre de plaquettes entraînant des saignements de nez ou des gencives, et diminution du nombre de globules blancs entraînant une sensibilité accrue aux infections.
Une affection grave qui peut rendre le sang plus acide (appelée acidose métabolique), chez les patients atteints d’une maladie grave et utilisant du paracétamol (voir rubrique 2).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER EFFERALGANMED 250 mg, comprimé dispersible ?
Tenir hors de la vue et de la portée des enfants.
Ne pas utiliser ce médicament après la date de péremption mentionnée sur la plaquette thermoformée et la boîte.
La date d’expiration fait référence au dernier jour du mois.
Pas de précautions particulières de conservation.
Ne pas utiliser ce médicament si vous remarquez des signes visibles de dégradation.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient EFFERALGANMED 250 mg, comprimé dispersible
· La substance active est : paracétamol
Chaque comprimé contient 250 mg de paracétamol (sous forme de cristaux de paracétamol enrobés).
· Les autres composants sont :
Copolymère basique de méthacrylate de butyle, dispersion de polyacrylate à 30 %, silice hydrophobe colloïdale, mannitol (granulé, poudre), crospovidone, aspartam (E951), arôme banane, stéarate de magnésium.
Qu’est-ce que EFFERALGANMED 250 mg, comprimé dispersible et contenu de l’emballage extérieur ?
EFFERALGANMED 250 mg se présente sous forme de comprimé dispersible blanc, rond, biconvexe avec une dépression centrale concave et ayant une odeur caractéristique de banane.
Conditionnements: boîtes de 12 ou 24 comprimés en plaquettes, unidoses, perforées, sécurisées et avec opercule détachable.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
194 Bureaux de La Colline, Bâtiment D
92213 Saint Cloud cedex
France
Exploitant de l’autorisation de mise sur le marché
UPSA SAS
3 rue joseph monier
92500 rueil malmaison
france
Z.I. de Saint-Arnoult
28170 CHATEAUNEUF-EN-THYMERAIS
france
ETHYPHARM
CHEMIN DE LA POUDRIERE
76120 GRAND QUEVILLY
france
DELPHARM NOVARA SrL
VIA CROSA, 86
28065 CERANO (NO)
ITALIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[À compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Conseil d’éducation sanitaire :
QUE FAIRE EN CAS DE FIEVRE :
La température normale du corps est variable d'un individu à l'autre et est comprise entre 36°5C et 37°5C. Une élévation de température au-delà de 38°C peut être considérée comme une fièvre.
Ce médicament est destiné à l'adulte et à l'enfant pesant de 13 à 50 kg (environ 2 à 15 ans).
Si les troubles que la fièvre entraîne sont trop gênants, vous pouvez prendre ce médicament qui contient du paracétamol en respectant les posologies indiquées.
Pour éviter tout risque de déshydratation, pensez à boire fréquemment.
Avec ce médicament, la fièvre doit baisser rapidement. Néanmoins :
· si d'autres signes inhabituels apparaissent
· si la fièvre persiste plus de 3 jours ou si elle s'aggrave,
· si les maux de tête deviennent violents, ou en cas de vomissements.
CONSULTEZ IMMEDIATEMENT VOTRE MEDECIN.
QUE FAIRE EN CAS DE DOULEURS :
L'intensité de la perception de la douleur et la capacité à lui résister varient d'une personne à l'autre.
· S'il n'y a pas d'amélioration au bout de 5 jours de traitement,
· Si la douleur est violente, inattendue et survient de façon brutale (notamment une douleur forte dans la poitrine) et/ou au contraire revient régulièrement,
· Si elle s'accompagne d'autres signes comme un état de malaise général, de la fièvre, un gonflement inhabituel de la zone douloureuse, une diminution de la force dans un membre,
· Si elle vous réveille la nuit,
CONSULTEZ IMMEDIATEMENT VOTRE MEDECIN.