Dernière mise à jour le 01/06/2026
SUNITINIB TEVA 25 mg, gélule
Indications thérapeutiques
SUNITINIB TEVA contient la substance active sunitinib, qui est un inhibiteur des protéines kinase. Il est utilisé dans le traitement des cancers, en diminuant l’activité d’un groupe de protéines spécifiques impliquées dans la croissance et la dissémination des cellules cancéreuses.
SUNITINIB TEVA est utilisé chez les adultes dans le traitement des cancers suivants :
· Tumeur stromale gastro-intestinale (GIST), un type de cancer de l’estomac et de l’intestin lorsque l’imatinib (un autre médicament traitant le cancer) ne marche plus ou lorsque vous ne pouvez plus prendre l’imatinib.
· Cancer du rein métastatique (MRCC), un type de cancer du rein qui s’est disséminé dans d’autres parties du corps.
· Tumeur neuroendocrine du pancréas (pNET) (tumeur des cellules sécrétrices d’hormones du pancréas) qui a progressé et ne peut être enlevé par chirurgie.
Si vous vous posez des questions sur le mode d’action de SUNITINIB TEVA ou voulez savoir pourquoi ce médicament vous a été prescrit, veuillez consulter votre médecin.
Présentations
> 28 plaquette(s) PVC (ACLAR RX) polytrifluorochloroéthylène PVC-Aluminium de 1 gélule(s)
Code CIP : 34009 301 385 4 0
Déclaration de commercialisation : 25/01/2022
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 928,62 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 929,64 €
- Taux de remboursement :100 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : TEVA BV
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription hospitalière
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 674 184 7
ANSM - Mis à jour le : 12/07/2024
SUNITINIB TEVA 25 mg, gélule
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Sunitinib................................................................................................................................ 25 mg
Pour une gélule.
Pour la liste complète des excipients, voir rubrique 6.1.
Gélule en gélatine dure constituée d’une coiffe opaque orange clair sur laquelle est imprimé « 25 » à l’encre noire, et d’un corps opaque orange moyen. Chaque gélule de taille 3 (longueur fermée totale d’environ 15,8 mm) contient une poudre granulée orange.
4.1. Indications thérapeutiques
Tumeur stromale gastro-intestinale (GIST)
SUNITINIB TEVA est indiqué dans le traitement des tumeurs stromales gastro-intestinales (GIST) malignes non résécables et/ou métastatiques chez l’adulte, après échec d’un traitement par imatinib dû à une résistance ou à une intolérance.
Cancer du rein métastatique (MRCC)
SUNITINIB TEVA est indiqué dans le traitement des cancers du rein avancés/métastatiques (MRCC) chez l’adulte.
Tumeur neuroendocrine du pancréas (pNET)
SUNITINIB TEVA est indiqué dans le traitement des tumeurs neuroendocrines du pancréas (pNET) non résécables ou métastatiques, bien différenciées, avec progression de la maladie chez l’adulte.
4.2. Posologie et mode d'administration
Le traitement par sunitinib doit être instauré par un médecin ayant l’expérience de l’administration des agents anticancéreux.
Dans les GIST et les MRCC, la dose de SUNITINIB TEVA recommandée est de 50 mg, par voie orale, à raison d’une prise quotidienne pendant 4 semaines consécutives, suivie d’une fenêtre thérapeutique de 2 semaines (Schéma posologique 4/2), correspondant à un cycle complet de 6 semaines.
Dans les pNET, la dose de SUNITINIB TEVA recommandée est de 37,5 mg, par voie orale, à raison d’une prise quotidienne, sans fenêtre thérapeutique préétablie.
Ajustements de doses
Tolérance et innocuité
Dans les GIST et les MRCC, des ajustements de doses par paliers de 12,5 mg pourront être effectués en fonction de la tolérance individuelle au traitement. La dose journalière ne devra pas excéder 75 mg ni être inférieure à 25 mg.
Dans les pNET, des ajustements de doses par paliers de 12,5 mg pourront être effectués en fonction de la tolérance individuelle au traitement. La dose maximale administrée au cours de l’étude de phase III pNET était de 50 mg par jour.
Des interruptions de doses pourront être envisagées selon la tolérance individuelle au traitement.
Inhibiteurs/inducteurs du CYP3A4
L’administration concomitante de sunitinib et d’inducteurs puissants du CYP3A4 tels que la rifampicine devra être évitée (voir rubriques 4.4 et 4.5). Si cela n’est pas possible, la dose de sunitinib administrée pourra être augmentée par paliers de 12,5 mg (jusqu’à 87,5 mg par jour pour les GIST et les MRCC ou 62,5 mg par jour pour les pNET) sous étroite surveillance de la tolérance.
L’administration concomitante de sunitinib et d’inhibiteurs puissants du CYP3A4 tels que le kétoconazole devra être évitée (voir rubriques 4.4 et 4.5). Si cela n’est pas possible, la dose de sunitinib pourra être diminuée jusqu’à une dose minimale de 37,5 mg par jour pour les GIST et les MRCC ou 25 mg par jour pour les pNET, sous étroite surveillance de la tolérance.
Le choix d’un traitement médicamenteux alternatif concomitant ayant peu ou pas d’effet sur l’induction ou l’inhibition du CYP3A4 devra être envisagé.
Populations particulières
Population pédiatrique
La sécurité et l’efficacité du sunitinib n’ont pas été établies chez les patients de moins de 18 ans. Les données actuellement disponibles sont décrites aux rubriques 4.8, 5.1 et 5.2. En revanche, aucune recommandation sur la posologie ne peut être donnée.
Personnes âgées
Environ un tiers des patients ayant participé aux études cliniques qui ont reçu le sunitinib étaient âgés de 65 ans ou plus. Aucune différence significative relative à la tolérance ou à l’efficacité n’a été observée par rapport à des patients plus jeunes.
Altération de la fonction hépatique
Aucun ajustement de la dose initiale du sunitinib n’est recommandé chez les patients présentant une insuffisance hépatique légère ou modérée (classe A et B de Child-Pugh). Le sunitinib n’a pas été étudié chez les patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh) et par conséquent son utilisation chez les patients présentant une insuffisance hépatique sévère ne peut pas être recommandée (voir rubrique 5.2).
Altération de la fonction rénale
Aucun ajustement de la dose initiale de sunitinib n’est exigé chez les patients présentant une altération de la fonction rénale (modérée à sévère) ou présentant une insuffisance rénale terminale (IRT) sous hémodialyse. De tels ajustements doivent être fonction de la sécurité et de la tolérance individuelle (voir rubrique 5.2).
Mode d’administration
SUNITINIB TEVA est destiné à une administration orale. SUNITINIB TEVA peut être pris au cours ou en dehors d’un repas.
Si une dose est oubliée, le patient ne doit pas prendre de dose supplémentaire, mais il doit prendre la dose habituellement prescrite, le jour suivant.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
L’administration concomitante avec des inducteurs puissants du CYP3A4 doit être évitée, car elle peut conduire à une diminution des concentrations plasmatiques du sunitinib (voir rubriques 4.2 et 4.5).
L’administration concomitante avec des inhibiteurs puissants du CYP3A4 doit être évitée, car elle peut conduire à une augmentation des concentrations plasmatiques du sunitinib (voir rubriques 4.2 et 4.5).
Affections de la peau et du tissu
Les patients doivent également être avertis qu’une dépigmentation de la peau ou des cheveux peut survenir pendant le traitement par sunitinib. D’autres effets dermatologiques peuvent se produire, tels qu’une sécheresse, un épaississement ou un craquellement de la peau, l’apparition de vésicules ou des éruptions sur la paume des mains ou la plante des pieds.
Les réactions rapportées ci-dessus n’étaient pas cumulatives, elles ont généralement été réversibles et en général n’ont pas nécessité l’interruption du traitement. Des cas de pyoderma gangrenosum, généralement réversibles après l’interruption du sunitinib, ont été rapportés. Des cas de réactions cutanées sévères ont été signalés, notamment des cas d’érythème polymorphe (EP), des cas suggérant un syndrome de Stevens-Johnson (SSJ) et une nécrolyse épidermique toxique (NET), dont certains ont été d’issue fatale. En présence de signes ou de symptômes de SSJ, de NET ou d’EP (p. ex. rash cutané évolutif souvent accompagné d’ampoules ou de lésions des muqueuses), le traitement par sunitinib doit être interrompu. Si le diagnostic de SSJ ou de NET est confirmé, le traitement ne doit pas être réintroduit. Dans certains cas de suspicion d’EP, la réintroduction du sunitinib à dose plus faible après la disparition de la réaction a été bien tolérée ; certains de ces patients ont également reçu un traitement concomitant par corticostéroïdes ou antihistaminiques (voir rubrique 4.8).
Hémorragies et saignements tumoraux
Les événements hémorragiques, dont certains ont été d’issue fatale, rapportés lors des études cliniques sur le sunitinib et pendant la surveillance après la commercialisation sont des hémorragies gastro-intestinales, respiratoires, du tractus urinaire et cérébrales (voir rubrique 4.8).
Les évaluations de routine de ces épisodes hémorragiques doivent comprendre une numération formule sanguine et un examen physique.
L’épistaxis a été l’effet indésirable hémorragique le plus fréquemment rapporté, celui-ci étant survenu chez environ la moitié des patients présentant des tumeurs solides et ayant eu des événements hémorragiques. Certains de ces épistaxis ont été sévères, mais très rarement fatals.
Des cas d’hémorragies tumorales, parfois associés à des nécroses tumorales, ont été rapportés ; certains de ces événements hémorragiques ont été fatals.
Des hémorragies tumorales sont susceptibles d’apparaître de façon soudaine et, dans les cas de tumeurs pulmonaires, peuvent se présenter sous forme d’hémoptysies ou d’hémorragies pulmonaires sévères mettant en jeu le pronostic vital. Des cas d’hémorragie pulmonaire, dont certaines d’issue fatale, ont été observés au cours des essais cliniques et ont été rapportés après la mise sur le marché chez des patients traités par sunitinib pour MRCC, GIST et cancer du poumon. L’utilisation de SUNITINIB TEVA n’est pas autorisée chez les patients atteints de cancer du poumon.
Les patients recevant un traitement anticoagulant concomitant (par exemple, warfarine ou acénocoumarol) pourront être surveillés de façon périodique en procédant à des numérations des cellules sanguines complètes (plaquettes), des tests de facteurs de coagulation (TP/INR) et des examens physiques.
Affections gastro-intestinales
Diarrhée, nausée/vomissement, douleur abdominale, dyspepsie et stomatite/douleur buccale ont été les effets indésirables gastro-intestinaux les plus fréquemment rapportés ; des cas d’œsophagite ont également été rapportés (voir rubrique 4.8).
La prise en charge symptomatique des effets indésirables gastro-intestinaux peut consister en un traitement par des médicaments aux propriétés anti-émétiques, anti-diarrhéiques ou anti-acides.
Des complications gastro-intestinales graves parfois mortelles, incluant une perforation gastro-intestinale, ont été rapportées chez des patients présentant des tumeurs malignes intra-abdominales et traités avec sunitinib.
Hypertension
De l’hypertension a été signalée en lien avec le sunitinib y compris l’hypertension sévère (> 200 mmHg de systolique ou 110 mmHg de diastolique). L’hypertension doit être dépistée et si nécessaire traitée de façon appropriée. Une interruption temporaire du traitement est recommandée chez les patients atteints d’hypertension sévère non contrôlée médicalement. Le traitement peut être repris dès que l’hypertension est correctement contrôlée (voir rubrique 4.8).
Affections hématologiques
Des diminutions du nombre absolu des neutrophiles et des diminutions du nombre de plaquettes ont été rapportées en lien avec le sunitinib (voir rubrique 4.8). Les événements rapportés ci-dessus n’étaient pas cumulatifs, et en général ont été réversibles et n’ont pas nécessité l’interruption du traitement. Aucun de ces événements pendant les études de phases III n’a été d’issue fatale, mais de rares événements hématologiques d’issue fatale, incluant des hémorragies associées à des thrombocytopénies et des infections neutropéniques, ont été rapportés lors de la surveillance après la commercialisation.
Des anémies ont été observées aussi bien précocement que tardivement au cours du traitement avec le sunitinib.
Une numération formule sanguine devra être effectuée au début de chaque cycle de traitement par le sunitinib (voir rubrique 4.8).
Affections cardiaques
Des événements cardiovasculaires, incluant insuffisance cardiaque, cardiomyopathie, diminution de la fraction d’éjection ventriculaire gauche inférieure à la limite inférieure de la normale, myocardite, ischémie myocardique et infarctus du myocarde, dont certains ont été d’issue fatale, ont été rapportés chez les patients traités par sunitinib. Ces données suggèrent une augmentation du risque de cardiomyopathie avec le sunitinib. A l’exception des effets spécifiques du médicament, aucun facteur de risque spécifique supplémentaire de survenue des cardiomyopathies induites par le sunitinib n’a été identifié chez les patients traités. Utiliser sunitinib avec précaution chez les patients à risque ou ayant des antécédents de ces événements (voir rubrique 4.8).
Les patients ayant présenté des événements cardiaques dans les 12 mois précédant l’administration de sunitinib, tels qu’infarctus du myocarde (y compris angor sévère ou instable), pontage artériel coronarien ou périphérique par greffe, insuffisance cardiaque congestive (ICC) symptomatique, accident vasculaire cérébral ou épisode ischémique transitoire, ou embolie pulmonaire, ont été exclus de toutes les études cliniques de sunitinib. On ne sait pas si les patients atteints de ces pathologies concomitantes seraient à plus haut risque de développer une dysfonction ventriculaire gauche liée au sunitinib.
Il est conseillé aux médecins de bien apprécier ce risque par rapport aux bénéfices escomptés du traitement. L’apparition de signes cliniques ou symptômes d’ICC chez les patients devra être soigneusement surveillée au cours du traitement par sunitinib, en particulier chez des patients ayant des facteurs de risque cardiaques et/ou des antécédents de maladie coronarienne. Des évaluations initiales et périodiques de la FEVG devront être envisagées chez les patients traités par sunitinib. Chez les patients sans facteurs de risques cardiaques, une évaluation initiale de la fraction d’éjection devra être envisagée.
En cas de manifestations cliniques d’ICC, il est recommandé d’interrompre l’administration de sunitinib. L’administration de sunitinib devra également être interrompue, et/ou les doses réduites, chez les patients sans signe clinique d’ICC mais dont la fraction d’éjection a diminué de 20 % par rapport à la valeur initiale et est inférieure à 50 %.
Allongement de l’intervalle QT
Un allongement de l’intervalle QT et des torsades de pointes ont été observés chez les patients exposés au sunitinib. L’allongement de l’intervalle QT peut entraîner une augmentation du risque d’arythmies ventriculaires y compris une torsade de pointes.
Le sunitinib devra être utilisé avec précaution chez les patients ayant déjà présenté un allongement de l’intervalle QT, ainsi que chez ceux qui prennent des antiarythmiques, ou des médicaments susceptibles d’allonger l’intervalle QT ou présentant une pathologie cardiaque préexistante, une bradycardie ou des troubles électrolytiques. L’administration concomitante de sunitinib et d’inhibiteurs puissants du CYP3A4 devra être limitée en raison de la possible augmentation des concentrations plasmatiques de sunitinib (voir rubriques 4.2, 4.5 et 4.8).
Evénements thromboemboliques veineux
Des événements thromboemboliques veineux liés au traitement ont été rapportés chez les patients ayant reçu du sunitinib, y compris des thromboses veineuses profondes et une embolie pulmonaire (voir rubrique 4.8).
Des cas d’embolie pulmonaire avec une issue fatale ont été observés lors de la surveillance après la commercialisation.
Evènements thromboemboliques artériels
Des cas d’évènements thromboemboliques artériels, parfois fatals, ont été rapportés chez des patients traités par le sunitinib. Les évènements les plus fréquents comportaient accidents vasculaires cérébraux, accidents ischémiques transitoires et infarctus cérébraux. Les facteurs de risque associés aux évènements thromboemboliques artériels, en plus de la pathologie maligne sous-jacente et de l’âge ≥ 65 ans, étaient l’hypertension artérielle, le diabète de type II et des antécédents de maladies thromboemboliques.
Anévrismes et dissections artérielles
L’utilisation d’inhibiteurs des voies du VEGF chez les patients souffrant ou non d’hypertension peut favoriser la formation d’anévrismes et/ou de dissections artérielles. Avant l’initiation du sunitinib, le risque doit être soigneusement évalué chez les patients ayant des facteurs de risque tels que l’hypertension ou des antécédents d’anévrisme.
Microangiopathie thrombotique (MAT)
Le diagnostic de MAT, y compris de purpura thrombotique thrombocytopénique (PTT) et le syndrome hémolytique urémique (SHU), parfois entraînant une atteinte rénale ou une issue fatale, doit être considéré en cas de survenue d’une anémie hémolytique, d’une thrombocytopénie, d’une fatigue, de manifestations neurologiques fluctuantes, d’une insuffisance rénale et d’une fièvre. Le traitement par sunitinib doit être interrompu chez les patients qui développent une MAT et un traitement doit être aussitôt instauré. Une régression des effets de la MAT a été observée après l’arrêt du traitement (voir rubrique 4.8).
Dysfonction thyroïdienne
Une évaluation par tests biologiques de la fonction thyroïdienne préalable au traitement par sunitinib est recommandée chez tous les patients. Les patients atteints d’hypothyroïdie ou d’hyperthyroïdie préexistante devront être traités conformément à la pratique médicale standard. Au cours du traitement par sunitinib, une surveillance de routine de la fonction thyroïdienne devra être effectuée tous les 3 mois. De plus, les signes et symptômes de dysfonction thyroïdienne devront être étroitement surveillés chez les patients au cours du traitement et les patients développant des signes et/ou des symptômes évocateurs d’une dysfonction thyroïdienne devront bénéficier de tests biologiques de la fonction thyroïdienne tel que cliniquement indiqué. Les patients développant une dysfonction thyroïdienne devront être traités conformément à la pratique médicale standard.
L’hypothyroïdie a été observée aussi bien en début de traitement que tardivement au cours du traitement par sunitinib (voir rubrique 4.8).
Pancréatite
Des augmentations sériques des lipases et des amylases ont été observées chez des patients présentant diverses formes de tumeurs solides et ayant reçu du sunitinib. Les augmentations sériques des lipases ont été transitoires et généralement non associées à des signes ou symptômes de pancréatite chez des patients présentant diverses formes de tumeurs solides (voir rubrique 4.8).
Des cas graves de pancréatite, dont certains d’issue fatale, ont été rapportés.
En présence de symptômes de pancréatite, le traitement par sunitinib devrait être arrêté et les patients devront bénéficier d’un suivi médical approprié.
Hépatotoxicité
Une hépatotoxicité a été observée chez des patients traités par le sunitinib. Des cas d’insuffisance hépatique, dont certains d’issue fatale, ont été observés chez < 1 % des patients présentant une tumeur solide et traités par le sunitinib. Surveiller les tests de la fonction hépatique (taux d’alanine aminotransférase (ALAT), d’aspartate aminotransférase (ASAT), de bilirubine) avant l’initiation du traitement, au cours de chaque cycle de traitement, et en cas de symptômes cliniques. En présence de signes ou symptômes d’insuffisance hépatique, le traitement par sunitinib doit être arrêté et un traitement approprié doit être mis en place (voir rubrique 4.8).
Fonction rénale
Des cas d’altération de la fonction rénale, d’insuffisance rénale, et/ou d’insuffisance rénale aigüe, dont certains d’issue fatale, ont été rapportés (voir rubrique 4.8).
En plus du carcinome rénal (RCC) sous-jacent, les facteurs de risque associés à l’altération de la fonction rénale/l’insuffisance rénale chez les patients traités par le sunitinib étaient les suivants : patients plus âgés, diabète de type II, altération de la fonction rénale sous-jacente, insuffisance cardiaque, hypertension, sepsis, déshydratation/hypovolémie et rhabdomyolyse.
La sécurité des patients présentant une protéinurie modérée à sévère, poursuivant le traitement par sunitinib, n’a pas été systématiquement évaluée.
Des cas de protéinuries et de rares cas de syndrome néphrotique ont été rapportés. Il est recommandé de pratiquer une analyse urinaire avant l’initiation du traitement et de surveiller l’apparition ou l’aggravation d’une protéinurie. Le traitement par sunitinib doit être arrêté chez les patients présentant un syndrome néphrotique.
Fistule
Si la formation d’une fistule se produit, le traitement par sunitinib doit être interrompu. Peu d’informations sont disponibles sur la poursuite de l’utilisation de sunitinib chez des patients présentant des fistules (voir rubrique 4.8).
Troubles de la cicatrisation des plaies
Des cas de troubles de la cicatrisation des plaies ont été rapportés au cours du traitement par le sunitinib.
Aucune étude clinique formelle de l’effet du sunitinib sur la cicatrisation des plaies n’a été menée. Par précaution, une interruption temporaire du traitement est recommandée chez les patients devant subir une intervention chirurgicale majeure. L’expérience clinique concernant le délai de réintroduction du traitement après une intervention chirurgicale majeure est limitée. Aussi, la décision de reprendre le traitement par sunitinib après une intervention chirurgicale majeure doit se baser sur l’appréciation clinique du rétablissement après la chirurgie.
Ostéonécrose de la mâchoire
Des cas d’ostéonécrose de la mâchoire ont été rapportés chez des patients traités par sunitinib. La majorité de ces cas ont été rapportés chez des patients ayant reçu antérieurement ou de façon concomitante un traitement par des bisphosphonates par voie intraveineuse, pour lesquels l’ostéonécrose de la mâchoire est un risque identifié. La prudence est donc de rigueur chez les patients traités par sunitinib en cas d’administration antérieure ou concomitante de bisphosphonates par voie intraveineuse.
Les interventions dentaires invasives sont également un facteur de risque identifié. Avant d’instaurer un traitement par sunitinib, un examen dentaire et des soins dentaires préventifs appropriés doivent être envisagés. Les interventions dentaires invasives doivent être évitées autant que possible chez les patients qui ont reçu précédemment ou qui reçoivent des bisphosphonates par voie intraveineuse (voir rubrique 4.8).
Hypersensibilité/angio-œdème
Si un angio-œdème dû à de l’hypersensibilité se produit, le traitement par sunitinib doit être interrompu et le traitement médical standard doit être appliqué (voir rubrique 4.8).
Crises convulsives
Dans les études cliniques et suite à la surveillance après la commercialisation du sunitinib, des crises convulsives ont été rapportées. Les patients atteints de crises convulsives et présentant des signes/symptômes suggérant un syndrome de leucoencéphalopathie postérieure réversible (SLPR), tels qu’hypertension, céphalées, baisse de vigilance ou des facultés mentales, perte de la vision, et notamment cécité corticale, devront être surveillés et traités pour leur hypertension. Il est recommandé d’interrompre temporairement la prise de sunitinib ; après normalisation de l’état du patient, le traitement pourra être repris selon l’avis du médecin (voir rubrique 4.8).
Syndrome de lyse tumorale (SLT)
Des cas de SLT, certains fatals, ont été rarement observés au cours des essais cliniques et ont été rapportés après la mise sur le marché chez les patients traités par sunitinib. Les facteurs de risque de SLT incluent : un volume tumoral élevé, une insuffisance rénale chronique pré-existante, une oligurie, une déshydratation, une hypotension et des urines acides. Ces patients doivent faire l’objet d’une surveillance étroite et être traités tel que cliniquement indiqué, et une hydratation en prophylaxie doit être envisagée.
Infections
Des infections sévères, avec ou sans neutropénie, dont certaines d’issue fatale, ont été rapportées. Des cas peu fréquents de fasciite nécrosante, y compris du périnée, parfois d’issue fatale, ont été rapportés (voir rubrique 4.8).
Le traitement par sunitinib doit être interrompu chez les patients qui présentent une fasciite nécrosante et un traitement approprié doit être instauré immédiatement.
Hypoglycémie
Des diminutions de la glycémie, dans certains cas accompagnées de symptômes cliniques et nécessitant une hospitalisation en raison d’une perte de connaissance, ont été rapportées au cours du traitement par sunitinib. En cas d’hypoglycémie symptomatique, le traitement par sunitinib doit être interrompu temporairement. La glycémie des patients diabétiques doit être régulièrement surveillée afin de déterminer si un ajustement de la posologie du traitement antidiabétique est nécessaire pour limiter le risque d’hypoglycémie (voir rubrique 4.8).
Encéphalopathie hyperammoniémique
Une encéphalopathie hyperammoniémique a été observée avec sunitinib (voir rubrique 4.8). Le taux d’ammoniaque doit être mesuré et une prise en charge clinique appropriée doit être mise en place chez les patients présentant une léthargie inexpliquée ou des changements dans leur état mental.
Excipient(s)
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les études d’interaction n’ont été réalisées que chez l’adulte.
Médicaments pouvant augmenter les concentrations plasmatiques de sunitinib
Effet des inhibiteurs du CYP3A4
Chez les volontaires sains, l’administration concomitante d’une dose unique de sunitinib et d’un puissant inhibiteur du CYP3A4, le kétoconazole, a provoqué une élévation de la valeur de la concentration maximale (Cmax) et de la valeur de l’aire sous la courbe (ASC0-¥) de la combinaison [sunitinib + métabolite principal] de 49 % et 51 % respectivement.
L’administration concomitante de sunitinib et d’inhibiteurs puissants du CYP3A4 (tels que le ritonavir, l’itraconazole, l’érythromycine, la clarithromycine, le jus de pamplemousse) peut augmenter les concentrations de sunitinib.
L’association de sunitinib avec des inhibiteurs du CYP3A4 devra donc être évitée, ou l’utilisation d’un autre traitement pris de façon concomitante et présentant un potentiel inhibiteur du CYP3A4 minimal ou nul devra être envisagée.
Si cela n’est pas possible, la dose de sunitinib pourra être réduite jusqu’à une dose minimale journalière de 37,5 mg pour les GIST et MRCC ou de 25 mg pour les pNET, sous surveillance étroite de la tolérance (voir rubrique 4.2).
Effet des inhibiteurs de la protéine de résistance au cancer du sein (BCRP)
Les données cliniques disponibles concernant l’interaction entre le sunitinib et les inhibiteurs de la BCRP sont limitées et une possible interaction entre le sunitinib et d’autres inhibiteurs de la BCRP ne peut être exclue (voir rubrique 5.2).
Médicaments pouvant diminuer des concentrations plasmatiques de sunitinib
Effet des inducteurs du CYP3A4
Chez les volontaires sains, l’administration concomitante d’une dose unique de sunitinib et d’un inducteur du CYP3A4, la rifampicine, a provoqué une diminution de la valeur de la Cmax et de la valeur de l’ASC0-¥ de la combinaison [sunitinib + métabolite principal] de 23 % et 46 % respectivement.
L’administration concomitante de sunitinib et d’inducteurs puissants du CYP3A4 (tels que la dexaméthasone, la phénytoïne, la carbamazépine, la rifampicine, le phénobarbital ou des préparations à base de plante contenant de l’Hypericum perforatum (millepertuis)) peut diminuer les concentrations de sunitinib. L’association de sunitinib avec des inducteurs du CYP3A4 devra donc être évitée, ou le choix d’un autre traitement concomitant ayant un potentiel inducteur sur le CYP3A4 nul ou réduit devra être envisagé.
Si cela n’est pas possible, la dose de sunitinib pourra être augmentée par paliers de 12,5 mg (jusqu’à une dose maximale journalière de 87,5 mg pour les GIST et les MRCC ou de 62,5 mg par jour pour les pNET), sous surveillance étroite de la tolérance (voir rubrique 4.2).
4.6. Fertilité, grossesse et allaitement
Contraception chez les femmes et les hommes
Il est conseillé aux femmes en âge de procréer d’utiliser une méthode efficace de contraception et d’éviter d’être enceintes au cours du traitement par SUNITINIB TEVA.
Grossesse
On ne dispose d’aucune étude chez la femme enceinte utilisant du sunitinib. Les études animales ont mis en évidence une toxicité sur la reproduction, comprenant des malformations fœtales (voir rubrique 5.3). SUNITINIB TEVA ne doit pas être utilisé pendant la grossesse ou chez des femmes n’utilisant pas de méthode de contraception efficace, à moins que les bénéfices escomptés ne justifient le risque potentiel pour le fœtus. Si SUNITINIB TEVA est utilisé pendant la grossesse ou si la patiente tombe enceinte en cours de traitement par SUNITINIB TEVA, elle devra être avertie des risques potentiels pour le fœtus.
Allaitement
Le sunitinib et/ou ses métabolites sont excrétés dans le lait du rat femelle. On ne sait pas si le sunitinib ou son métabolite actif principal est excrété dans le lait maternel de la femme. Dans la mesure où les substances actives sont généralement excrétées dans le lait maternel et où il existe un risque potentiel d’événements indésirables graves chez le nouveau-né allaité, les femmes ne doivent pas allaiter pendant un traitement par SUNITINIB TEVA.
Fertilité
Selon des données non cliniques, la fertilité des hommes et des femmes pourrait être affectée par le traitement par le sunitinib (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables les plus graves associés au sunitinib, dont certains d’issue fatale, sont l’insuffisance rénale, l’insuffisance cardiaque, l’embolie pulmonaire, la perforation gastro-intestinale et les hémorragies (par exemple, hémorragie des voies respiratoires, gastro-intestinale, tumorale, des voies urinaires ou cérébrale). Les effets indésirables de tout grade les plus fréquents (survenus chez des patients au cours des essais d’enregistrement RCC, GIST et pNET) ont inclus la diminution de l’appétit, les troubles du goût, l’hypertension, la fatigue, les troubles gastro-intestinaux (tels que diarrhée, nausées, stomatite, dyspepsie et vomissements), la décoloration de la peau et le syndrome d’érythrodysesthésie palmo-plantaire. Ces symptômes peuvent diminuer avec la poursuite du traitement. Une hypothyroïdie peut se développer en cours de traitement. Des troubles hématologiques (tels que neutropénie, thrombocytopénie et anémie) sont parmi les effets indésirables le plus fréquemment rapportés avec le médicament.
Les événements d’issue fatale autres que ceux qui sont énumérés dans la rubrique 4.4 ci-dessus ou dans la rubrique 4.8 ci-dessous, considérés comme pouvant être liés au sunitinib, ont été : défaillance multi-viscérale, coagulation intravasculaire disséminée, hémorragie péritonéale, insuffisance surrénalienne, pneumothorax, choc et mort subite.
Liste tabulée des effets indésirables
Les effets indésirables rapportés chez des patients atteints de GIST, MRCC et pNET dans les données fusionnées de 7 115 patients sont listés ci-dessous, classés par organe, par fréquence et par grade de sévérité (NCI-CTCAE). Les effets indésirables rapportés dans les études cliniques après la commercialisation sont également inclus. Pour chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Les fréquences des événements sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1 - Effets indésirables rapportés dans les essais cliniques
|
Classe de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Fréquence indéterminée |
|
Infections et infestations |
|
Infections viralesa Infections respiratoiresb,* Abcèsc,* Infections fongiquesd Infections des voies urinaires Infections cutanéese Sepsisf,* |
Fasciite nécrosante* Infections bactériennesg |
|
|
|
Affections hématologiques et du système lymphatique |
Neutropénie Thrombocytopénie Anémie Leucopénie |
Lymphopénie |
Pancytopénie |
Microangiopathie thrombotiqueh,* |
|
|
Affections du système immunitaire |
|
|
Hypersensibilité |
Angio-œdème |
|
|
Affections endocriniennes |
Hypothyroïdie |
|
Hyperthyroïdie |
Thyroïdite |
|
|
Troubles du métabolisme et de la nutrition |
Diminution de l’appétiti |
Déshydratation Hypoglycémie |
|
Syndrome de lyse tumorale* |
|
|
Affections psychiatriques |
Insomnie |
Dépression |
|
|
|
|
Affections du système nerveux |
Sensation vertigineuse Céphalées Troubles du goûtj |
Neuropathie périphérique Paresthésie Hypoesthésie Hyperesthésie |
Hémorragie cérébrale* Accident vasculaire cérébral* Accident ischémique transitoire |
Syndrome d’encéphalopathie postérieure réversible* |
Encéphalopathie hyperammoniémique |
|
Affections oculaires |
|
Œdème périorbitaire Œdème des paupières Augmentation des sécrétions lacrymales |
|
|
|
|
Affections cardiaques |
|
Ischémie myocardiquek,* Fraction d’éjection ventriculaire diminuéel |
Insuffisance cardiaque congestive Infarctus du myocardem,* Insuffisance cardiaque* Cardiomyopathie* Épanchement péricardique Allongement de l’intervalle QT sur l’électrocardio-gramme |
Insuffisance ventriculaire gauche* Torsade de pointes |
|
|
Affections vasculaires |
Hypertension |
Thrombose veineuse profonde Bouffée de chaleur Bouffée vasomotrice |
Hémorragie tumorale* |
|
Anévrismes et dissections artérielles* |
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée Epistaxis Toux |
Embolie pulmonaire* Épanchement pleural* Hémoptysie Dyspnée d’effort Douleur oropharyngéen Congestion nasale Sécheresse nasale |
Hémorragie pulmonaire* Insuffisance respiratoire* |
|
|
|
Affections gastro-intestinales |
Stomatiteo Douleur abdominalep Vomissements Diarrhée Dyspepsie Nausées Constipation |
Reflux gastro-œsophagien Dysphagie Hémorragie gastro-intestinale* Œsophagite* Distension abdominale Gêne abdominale Hémorragie rectale Saignement gingival Ulcération buccale Proctalgie Chéilite Hémorroïdes Glossodynie Douleur buccale Sécheresse de la bouche Flatulence Gêne buccale Eructation |
Perforation gastro-intestinaleq,* Pancréatite Fistule anale Coliter Colite ischémiquer |
|
|
|
Affections hépatobiliaires |
|
|
Insuffisance hépatique* Cholécystites,* Fonction hépatique anormale |
Hépatite |
|
|
Affections de la peau et du tissu sous-cutané |
Décoloration de la peaut Syndrome d’érythrodysesthésie palmo-plantaire Rashu Modification de la couleur des cheveux Sécheresse de la peau |
Exfoliation cutanée Réaction cutanéev Eczéma Ampoules Erythème Alopécie Acné Prurit Hyperpigmentation de la peau Lésion cutanée Hyperkératose Dermatite Altération des onglesw |
|
Érythème polymorphe* Syndrome de Stevens-Johnson* Pyoderma gangrenosum Nécrolyse épidermique toxique* |
|
|
Affections musculo-squelettiques et systémiques |
Douleurs des extrémités Arthralgie Mal de dos |
Douleur musculo-squelettique Spasmes musculaires Myalgie Faiblesse musculaire |
Ostéonécrose de la mâchoire Fistule* |
Rhabdomyolyse* Myopathie |
|
|
Affections du rein et des voies urinaires |
|
Insuffisance rénale* Insuffisance rénale aiguë* Chromaturie Protéinurie |
Hémorragie des voies urinaires |
Syndrome néphrotique |
|
|
Troubles généraux et anomalies au site d’administration |
Inflammation des muqueuses Fatiguex Œdèmey Pyrexie |
Douleur thoracique Douleur Syndrome pseudo-grippal Frissons |
Trouble de la cicatrisation |
|
|
|
Investigations |
|
Perte de poids Diminution des globules blancs Elévation de la lipase Diminution des plaquettes Diminution du taux d’hémoglobine Élévation de l’amylasez Élévation de l’aspartate aminotransférase Élévation de l’alanine aminotransférase Élévation de la créatinine sérique Augmentation de la tension artérielle Elévation de l’uricémie |
Élévation de la créatinine phosphokinase sérique Augmentation de la concentration de thyrotropine (TSH) |
|
|
* Dont évènements d’issue fatale
Les termes suivants ont été combinés :
a Rhinopharyngite et herpès buccal
b Bronchite, infection des voies respiratoires inférieures, pneumonie et infection des voies respiratoires
c Abcès, abcès de membre, abcès anal, abcès gingival, abcès hépatique, abcès pancréatique, abcès du périnée, abcès périrectal, abcès rectal, abcès sous-cutané et abcès dentaire
d Candidose œsophagienne et candidose orale
e Cellulite et infection cutanée
f Sepsis et choc septique
g Abcès abdominal, sepsis abdominal, diverticulite et ostéomyélite
h Microangiopathie thrombotique, purpura thrombotique thrombocytopénique, et syndrome hémolytique urémique
i Diminution de l’appétit et anorexie
j Dysgueusie, agueusie et trouble du goût
k Syndrome coronaire aigu, angine de poitrine, angor instable, occlusion de l’artère coronaire, et ischémie myocardique
l Fraction d’éjection ventriculaire diminuée/anormale
m Infarctus du myocarde aigu, infarctus du myocarde, et infarctus du myocarde silencieux
n Douleur oro-pharyngée et pharyngo-laryngée
o Stomatite et stomatite aphteuse
p Douleur abdominale, douleur abdominale basse, douleur abdominale haute
q Perforation gastro-intestinale et perforation intestinale
r Colite et colite ischémique
s Cholécystite et cholécystite alithiasique
t Coloration jaune de la peau, décoloration de la peau et troubles de la pigmentation
u Dermatite psoriasiforme, rash avec exfoliation, rash, éruption érythémateuse, éruption folliculaire, rash généralisé, éruption maculeuse, éruption maculopapuleuse, éruption papuleuse et rash prurigineux
v Réaction cutanée et trouble de la peau
w Altération des ongles et modification de la couleur des ongles
x Fatigue et asthénie
y Œdème du visage, œdème et œdème périphérique
z Amylase et élévation de l’amylase
Description des effets indésirables sélectionnés
Infections et infestations
Des cas graves d’infection (avec ou sans neutropénie associée), y compris des cas d’issue fatale, ont été rapportés. Des cas de fasciite nécrosante, y compris du périnée, parfois d’issue fatale, ont été rapportés (voir également rubrique 4.4).
Affections hématologiques et du système lymphatique
Des diminutions du nombre absolu des neutrophiles de grade 3 et 4, respectivement, ont été rapportées chez 10 % et 1,7 % des patients de l’étude de phase III portant sur des patients atteints de GIST, chez 16 % et 1,6 % des patients de l’étude de phase III portant sur des patients atteints de MRCC et chez 13 % et 2,4 % des patients de l’étude de phase III portant sur des patients atteints de pNET. Des diminutions du nombre de plaquettes de grade 3 et 4, respectivement, ont été rapportées chez 3,7 % et 0,4 % des patients de l’étude de phase III portant sur des patients atteints de GIST, chez 8,2 % et 1,1 % des patients de l’étude de phase III portant sur des patients atteints de MRCC et chez 3,7 % et 1,2 % des patients de l’étude de phase III portant sur des patients atteints de pNET (voir rubrique 4.4).
Au cours d’une étude de phase III portant sur des patients atteints de GIST, des épisodes hémorragiques ont été rapportés chez 18 % des patients recevant le sunitinib, comparativement à 17 % des patients recevant le placebo. Parmi les patients atteints de MRCC non prétraités recevant le sunitinib, 39 % ont présenté des épisodes hémorragiques, comparativement à 11 % des patients recevant l’interféron-alpha (IFN-α). Dix-sept patients (4,5 %) recevant du sunitinib et 5 (1,7 %) des patients recevant l’IFN-α ont présenté des épisodes hémorragiques de grade supérieur ou égal à 3. Parmi les patients atteints de MRCC recevant du sunitinib après l’échec d’un traitement à base de cytokine, 26 % ont présenté des épisodes hémorragiques.
Au cours de l’étude de phase III portant sur des patients atteints de pNET, des épisodes hémorragiques (hors épistaxis), sont survenus chez 21,7 % des patients traités par le sunitinib comparativement à 9,85 % des patients ayant reçu un placebo (voir rubrique 4.4).
Dans les études cliniques, des hémorragies tumorales ont été rapportées chez environ 2 % des patients atteints de GIST.
Affections du système immunitaire
Des réactions d’hypersensibilité ont été rapportées, incluant des angio-œdèmes (voir rubrique 4.4).
Affections endocriniennes
Une hypothyroïdie a été rapportée comme un effet indésirable chez 7 patients (4 %) traités par sunitinib inclus dans l’une ou l’autre des deux études portant sur des patients atteints de MRCC après l’échec d’un traitement à base de cytokine ; chez 61 patients (16 %) recevant le sunitinib et chez 3 patients (< 1 %) recevant l’IFN-α dans l’étude portant sur des patients atteints de MRCC non prétraités.
De plus, une élévation de l’hormone stimulant la thyroïde (TSH) a été rapportée chez 4 patients (2 %) présentant un MRCC après échec d’un traitement à base de cytokine. Globalement, 7 % des patients atteints de MRCC ont présenté des signes cliniques ou biologiques d’hypothyroïdie sous traitement. Une hypothyroïdie acquise a été rapportée chez 6,2 % des patients atteints de GIST sous sunitinib versus 1 % sous placebo. Dans l’étude de phase III portant sur des patients atteints de pNET, une hypothyroïdie a été rapportée chez 6 patients (7,2 %) traités par sunitinib, et chez un patient (1,2 %) sous placebo.
Une surveillance prospective de la fonction thyroïdienne a été réalisée dans deux études menées chez des patients atteints d'un cancer du sein. Le sunitinib n’est pas indiqué chez les patients atteints d’un cancer du sein. Dans l'une des études, une hypothyroïdie a été rapportée chez 15 patients (13,6 %) traités par sunitinib et chez 3 patients (2,9 %) recevant un traitement médical standard. Une élévation du taux sanguin de TSH a été rapportée chez 1 patient (0,9 %) traité par sunitinib et chez aucun des patients recevant un traitement médical standard. Aucun cas d'hyperthyroïdie n'a été observé chez les patients traités par sunitinib et 1 patient (1,0 %) recevant un traitement médical standard a présenté une hyperthyroïdie. Dans l'autre étude, une hypothyroïdie a été rapportée chez 31 patients (13 %) traités par sunitinib et 2 patients (0,8 %) traités par capécitabine. Une élévation du taux sanguin de TSH a été rapportée chez 12 patients (5,0 %) traités par sunitinib et chez aucun des patients traités par capécitabine. Une hyperthyroïdie a été rapportée chez 4 patients (1,7 %) traités par sunitinib et chez aucun des patients traités par capécitabine. Une diminution du taux sanguin de TSH a été rapportée chez 3 patients (1,3 %) traités par sunitinib et chez aucun des patients traités par capécitabine. Une augmentation du taux de T4 a été rapportée chez 2 patients (0,8 %) traités par sunitinib et chez 1 patient (0,4 %) traité par capécitabine. Une augmentation du taux de T3 a été rapportée chez 1 patient (0,8 %) traité par sunitinib et chez aucun des patients traités par capécitabine. Tous les événements relatifs à la fonction thyroïdienne rapportés étaient de grade 1 ou 2 (voir rubrique 4.4).
Troubles du métabolisme et de la nutrition
Une incidence plus élevée des évènements d’hypoglycémie a été rapportée chez les patients atteints de pNET comparativement aux patients atteints de MRCC et GIST. Néanmoins, la plupart de ces évènements indésirables, observés dans les études cliniques, n’ont pas été considérés reliés au traitement expérimental (voir rubrique 4.4).
Affections du système nerveux
Au cours des études cliniques sur le sunitinib et de l’expérience après la commercialisation, de rares cas (< 1 %), dont certains d’issue fatale, de patients présentant des crises convulsives et des signes radiologiques de SLPR ont été rapportés. Des crises convulsives ont été observées chez certains patients, avec ou sans signes radiologiques de métastases cérébrales (voir rubrique 4.4).
Affections cardiaques
Dans les essais cliniques, une diminution de la fraction d’éjection du ventricule gauche (FEVG) supérieure ou égale à 20 %, et en dessous de la limite inférieure de la normale, est survenue chez environ 2 % des patients atteints de GIST, chez 4 % des patients atteints de MRCC traités par sunitinib après échec d’un traitement à base de cytokine, et chez 2 % des patients atteints de GIST recevant un placebo. Ces diminutions de la FEVG ne semblent pas avoir été évolutives et se sont souvent améliorées avec la poursuite du traitement.
Au cours de l’étude portant sur des patients ayant un MRCC non prétraité, 27 % des patients recevant le sunitinib et 15 % des patients recevant l’IFN-α, ont présenté une valeur de la FEVG inférieure à la limite inférieure de la normale. Une insuffisance cardiaque congestive (ICC) a été diagnostiquée chez deux patients (< 1 %) ayant reçu du sunitinib. Chez les patients atteints de GIST, des cas « d’insuffisance cardiaque », « d’insuffisance cardiaque congestive » ou « d’insuffisance ventriculaire gauche » ont été rapportés chez 1,2 % des patients traités par sunitinib et 1 % des patients recevant un placebo. Au cours de l’étude pivot de phase III chez des patients atteints de GIST (N = 312), des effets cardiaques d’issue fatale liés au traitement sont survenus chez 1 % des patients de chaque bras de l’étude (c’est-à-dire bras sunitinib et bras placebo). Au cours d’une étude de phase II chez des patients atteints de MRCC réfractaire au traitement par cytokine, un infarctus du myocarde d’issue fatale lié au traitement a été rapporté chez 0,9 % des patients et, dans l’étude de phase III chez des patients atteints de MRCC non prétraités, des événements cardiaques d’issue fatale ont été rapportés chez 0,6 % des patients du bras IFN-α et aucun événement cardiaque d’issue fatale n’a été rapporté chez les patients du bras sunitinib. Dans l’étude de phase III portant sur des patients atteints de pNET, un patient (1 %), traité par le sunitinib, a présenté une insuffisance cardiaque fatale liée au traitement.
Affections vasculaires
Hypertension
L’hypertension a été un effet indésirable très fréquemment rapporté lors des essais cliniques. Les doses de sunitinib ont été réduites ou son administration temporairement suspendue chez environ 2,7 % des patients qui ont présenté une hypertension. Le sunitinib n’a été définitivement arrêté chez aucun de ces patients. Une hypertension sévère (pression systolique > 200 mmHg ou pression diastolique > 110 mmHg) est survenue chez 4,7 % des patients présentant des tumeurs solides. Une hypertension a été rapportée chez environ 33,9 % des patients atteints de MRCC non prétraités qui recevaient du sunitinib et chez 3,6 % de ceux qui recevaient l’IFN-α. Une hypertension sévère est survenue chez 12 % des patients non prétraités recevant du sunitinib et chez < 1 % des patients recevant l’IFN-α. Une hypertension a été rapportée chez 26,5 % des patients traités par sunitinib dans l’étude de phase III portant sur des patients atteints de pNET, comparativement à 4,9 % des patients ayant reçu le placebo. Une hypertension sévère est survenue chez 10 % des patients atteints de pNET traités par le sunitinib et chez 3 % des patients sous placebo.
Événements thromboemboliques veineux
Des événements thromboemboliques veineux liés au traitement ont été rapportés chez environ 1,0 % des patients présentant une tumeur solide qui ont reçu sunitinib lors des essais cliniques, y compris GIST et RCC.
Dans une étude de phase III portant sur des patients atteints de GIST, des événements thromboemboliques veineux sont survenus chez sept patients (3 %) recevant le sunitinib ; 5 de ces 7 patients ont eu des thromboses veineuses profondes (TVP) de grade 3, et 2 de ces patients des TVP de grade 1 ou 2. Quatre de ces 7 patients ont vu leur traitement interrompu après la première observation de TVP.
Des événements thromboemboliques veineux ont été rapportés chez treize patients (3 %) atteints de MRCC non prétraités recevant le sunitinib dans une étude de phase III, et chez 4 patients (2 %) inclus dans les deux études portant sur les patients atteints de MRCC après échec d’un traitement à base de cytokine. Neuf de ces patients ont présenté des embolies pulmonaires : 1 de grade 2 et 8 de grade 4. Huit de ces patients ont présenté une TVP : 1 de grade 1, 2 de grade 2, 4 de grade 3 et 1 de grade 4. Une embolie pulmonaire observée chez un patient atteint de MRCC dans l’étude sunitinib après échec d’un traitement à base de cytokine a nécessité une interruption de traitement.
Des événements thromboemboliques veineux sont survenus chez 6 patients (2 %) atteints de MRCC non prétraités recevant l’IFN-α : 1 patient (< 1 %) a présenté une TVP de grade 3 et 5 patients (1 %) des embolies pulmonaires, toutes de grade 4.
Dans l’étude de phase III portant sur des patients atteints de pNET, des événements thromboemboliques veineux ont été rapportés chez 1 patient (1,2 %) du bras sunitinib et 5 patients (6,1 %) du bras placebo. Deux de ces patients sous placebo présentaient une TVP, 1 de grade 2 et 1 de grade 3.
Aucun cas d’issue fatale n’a été rapporté dans les études d’enregistrement GIST, MRCC et pNET. Des cas d’issue fatale ont été observés depuis la mise sur le marché.
Des cas d’embolie pulmonaire ont été observés chez environ 3,1 % des patients atteints de GIST et chez environ 1,2 % des patients atteints de MRCC ayant reçu sunitinib dans les études de phase III. Aucune embolie pulmonaire n’a été rapportée chez les patients atteints de pNET ayant reçu du sunitinib dans l’étude de phase III. Des cas rares d’issue fatale ont été observés depuis la mise sur le marché.
Les patients ayant présenté une embolie pulmonaire au cours des 12 mois précédents ont été exclus des études cliniques de sunitinib.
Chez les patients qui ont reçu le sunitinib dans les études d’enregistrement de phase III, des événements pulmonaires (c.-à-d. dyspnée, épanchement pleural, embolie pulmonaire ou œdème pulmonaire) ont été rapportés chez environ 17,8 % des patients atteints de GIST, chez environ 26,7 % des patients atteints de MRCC et chez 12 % des patients atteints de pNET.
Environ 22,2 % des patients ayant des tumeurs solides, y compris GIST et MRCC, qui ont reçu du sunitinib dans les essais cliniques ont présenté des événements pulmonaires.
Affections gastro-intestinales
Les cas de pancréatite ont été peu fréquemment (< 1 %) observés chez les patients atteints de MRCC ou de GIST traités par sunitinib. Dans l’étude de phase III portant sur des patients atteints de pNET, aucun cas de pancréatite liée au traitement n’a été rapporté (voir rubrique 4.4).
Une hémorragie gastro-intestinale d’issue fatale a été rapportée chez 0,98 % des patients recevant le placebo au cours de l’étude GIST de phase III.
Affections hépatobiliaires
Des troubles de la fonction hépatique ont été rapportés et peuvent inclure des anomalies des tests de la fonction hépatique, des hépatites ou des insuffisances hépatiques (voir rubrique 4.4).
Affections de la peau et du tissu sous-cutané
Des cas de pyoderma gangrenosum, généralement réversibles après l’interruption du sunitinib, ont été rapportés (voir également rubrique 4.4).
Affections musculo-squelettiques et systémiques
Des cas de myopathie et/ou de rhabdomyolyse, certains associés à une insuffisance rénale aiguë, ont été rapportés. Les patients présentant des signes ou des symptômes de toxicité musculaire devront être traités conformément à la pratique médicale courante (voir rubrique 4.4).
Des cas de formation de fistule ont été rapportés, parfois associés à une nécrose et une régression tumorales, et dans certains cas à une issue fatale (voir rubrique 4.4).
Des cas d’ostéonécrose de la mâchoire ont été rapportés chez les patients traités par sunitinib, la plupart étant apparues chez des patients ayant des facteurs de risque identifiés pour l’ostéonécrose de la mâchoire, en particulier l’exposition aux bisphosphonates par voie intraveineuse et/ou un antécédent de pathologie dentaire nécessitant une intervention dentaire invasive (voir également rubrique 4.4).
Investigations
Les données des études non cliniques (in vitro et in vivo) menées avec des doses supérieures à la dose recommandée chez l’homme indiquent que le sunitinib peut inhiber le processus de repolarisation du potentiel d’action cardiaque (par exemple, allongement de l’intervalle QT).
Des allongements de l'intervalle QTc de plus de 500 ms se sont produits chez 0,5 % des 450 patients présentant une tumeur solide et des modifications de plus de 60 ms ont été observées par rapport aux valeurs initiales chez 1,1 % de ces patients. Ces deux paramètres sont considérés comme des modifications potentiellement significatives. A des concentrations d’environ deux fois les concentrations thérapeutiques, le sunitinib a montré une prolongation de l’intervalle QTcF (correction de Fridericia).
L’allongement de l’intervalle QTc a été étudié dans un essai clinique, chez 24 patients âgés de 20 à 87 ans, présentant des cancers à un stade avancé. Les résultats de cette étude ont montré que le sunitinib a eu un effet sur l’intervalle QTc (défini comme une modification moyenne, ajustée pour les valeurs sous placebo, supérieure à 10 ms avec une limite supérieure de l’IC à 90 % supérieure à 15 ms) aux concentrations thérapeutiques (jour 3) en utilisant la méthode de correction des valeurs initiales en cours de journée, et aux concentrations supérieures aux concentrations thérapeutiques (jour 9) en utilisant les deux méthodes de correction des valeurs initiales. Aucun patient n’a présenté de valeur du QTc supérieure à 500 ms.
Bien qu’un effet sur l’intervalle QTcF ait été observé 24 heures après la prise du jour 3 (c’est-à-dire à la concentration plasmatique thérapeutique attendue après l’administration de la dose initiale recommandée de 50 mg) avec la méthode de correction des valeurs initiales en cours de journée, la pertinence clinique de cette observation n’est pas claire.
Les évaluations d’ECG complètes, répétées, pratiquées lors des expositions thérapeutiques ou lors d’expositions supérieures ont montré qu’aucun patient des populations évaluable ou en intention de traiter (ITT) n’avait développé un allongement de l’intervalle QTc considéré comme sévère (c’est-à-dire supérieur ou égal au grade 3 des Critères de terminologie communs des effets indésirables [CTCAE] version 3.0).
Aux concentrations plasmatiques thérapeutiques, la modification maximale moyenne de l’intervalle QTcF (correction de Fridericia) par rapport aux valeurs initiales a été de 9 ms (IC à 90 % : 15,1 ms). A des concentrations d’environ deux fois les concentrations thérapeutiques, la modification maximale de l’intervalle QTcF par rapport aux valeurs initiales a été de 15,4 ms (IC à 90 % : 22,4 ms). Après administration de moxifloxacine (400 mg), utilisé comme témoin positif, la modification maximale moyenne de l’intervalle QTcF par rapport aux valeurs initiales a été de 5,6 ms. Aucun sujet n’a présenté d’allongement de l’intervalle QTc supérieur au grade 2 (CTCAE version 3.0) (voir rubrique 4.4).
La sécurité d’emploi à long terme dans le MRCC
La sécurité d’emploi à long terme du sunitinib chez les patients atteints de MRCC a été analysée au cours de 9 études cliniques terminées, menées dans le traitement de première intention du MRCC réfractaire au bévacizumab et aux cytokines chez 5 739 patients parmi lesquels 807 (14 %) ont été traités pendant une période ≥ 2 ans pouvant aller jusqu’à 6 ans. Chez les 807 patients ayant reçu un traitement par sunitinib à long terme, la majorité des effets indésirables liés au traitement sont apparus initialement au cours des 6 premiers mois à 1 an puis ont été stables ou leur fréquence a diminué au fil du temps à l’exception de l’hypothyroïdie qui a progressivement augmenté au fil du temps, de nouveaux cas survenant pendant toute la période de 6 ans. Un traitement prolongé par sunitinib n’a pas été associé à de nouveaux types d’effets indésirables liés au traitement.
Population pédiatrique
Le profil de sécurité du sunitinib a été établi à partir d’une étude de phase I, en escalade de doses, d’une étude de phase II en ouvert, d’une étude de phase I/II à bras unique et de publications décrites ci-dessous.
Une étude de phase I, en escalade de doses avec du sunitinib par voie orale, a été menée chez 35 patients pédiatriques, dont 30 patients pédiatriques (âgés de 3 à 17 ans) et 5 jeunes adultes (âgés de 18 à 21 ans) atteints de tumeurs solides réfractaires. La majorité des patients avait un diagnostic principal de tumeur cérébrale. Tous les patients participant à l’étude ont présenté des effets indésirables au médicament ; la plupart de ces effets étaient sévères (grade de toxicité ≥ 3) et incluaient une cardiotoxicité. Les effets indésirables les plus fréquents étaient la toxicité gastro-intestinale (GI), la neutropénie, la fatigue et l’élévation des taux d’ALAT. Le risque d’effets indésirables cardiaques induits par le médicament semblait supérieur chez les patients pédiatriques ayant été précédemment exposés à une irradiation cardiaque ou aux anthracyclines, en comparaison aux patients pédiatriques sans exposition antérieure. Chez ces patients pédiatriques sans exposition antérieure aux anthracyclines ou à une irradiation cardiaque, la dose maximale tolérée (DMT) a été identifiée (voir rubrique 5.1).
Une étude de phase II en ouvert a été menée chez 29 patients, dont 27 patients pédiatriques (âgés de 3 à 16 ans) et 2 jeunes adultes (âgés de 18 à 19 ans) atteints de gliome ou d’épendymome récidivant/évolutif/réfractaire de haut grade (HGG). Aucun effet indésirable de grade 5 n’a été rapporté dans les deux groupes. Les événements indésirables liés au traitement les plus fréquents (≥ 10 %) ont été une diminution de la numération des neutrophiles (6 patients [20,7 %]) et une hémorragie intracrânienne (3 patients [10,3 %]).
Une étude de phase I/II à bras unique a été menée chez 6 patients pédiatriques (âgés de 13 à 16 ans) atteints de GIST non résécables avancées. Les effets indésirables les plus fréquents ont été la diarrhée, les nausées, la diminution de la numération leucocytaire, la neutropénie et les céphalées chacun chez 3 patients (50,0 %) principalement de grade de sévérité 1 ou 2. Quatre patients sur 6 (66,7 %) ont présenté des événements indésirables liés au traitement de grade 3 ou 4 (hypophosphatémie, neutropénie et thrombocytopénie de grade 3 chacune chez 1 patient et neutropénie de grade 4 chez 1 patient). Aucun effet indésirable grave (EIG) ou effet indésirable de grade 5 n’a été rapporté au cours de cette étude. Dans l’étude clinique et dans les publications, le profil de sécurité correspondait au profil de sécurité connu chez les adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Dans la mesure où il n’existe pas d’antidote spécifique au surdosage par sunitinib, le traitement devra consister en des mesures habituelles de traitement symptomatique. Si cela s’avère nécessaire, la substance active non absorbée peut être éliminée par des vomissements provoqués ou par lavage gastrique. Des cas de surdosage ont été rapportés, certains cas ont été associés à des effets indésirables cohérents avec le profil de tolérance connu du sunitinib.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le sunitinib inhibe plusieurs récepteurs à tyrosine kinase (RTK) impliqués dans la croissance tumorale, la néoangiogenèse pathologique et la progression métastatique du cancer. Le sunitinib a été identifié comme un inhibiteur des récepteurs du facteur de croissance plaquettaire (PDGFRα et PDGFRβ), des récepteurs du facteur de croissance endothélial vasculaire (VEGFR1, VEGFR2 et VEGFR3), du récepteur du facteur de cellule souche (KIT), du récepteur Fms-like tyrosine kinase-3 (FLT3), du récepteur du facteur stimulant la formation de colonies (CSF-1R) et du récepteur du facteur neurotrophique de la lignée gliale (RET). Les tests biochimiques et cellulaires ont montré que le principal métabolite du sunitinib présentait le même pouvoir inhibiteur que le sunitinib.
Efficacité et sécurité clinique
La sécurité et l’efficacité clinique du sunitinib ont été étudiées lors du traitement de patients présentant un GIST résistant à l’imatinib (patients dont la tumeur a progressé pendant ou après le traitement par l’imatinib) ou n’ayant pas toléré ce médicament (patients ayant présenté une toxicité significative pendant le traitement par l’imatinib, ayant empêché sa continuation), chez les patients atteints d’un MRCC et chez les patients atteints de pNET non résécables.
Les données d’efficacité reposent sur le temps jusqu’à progression tumorale (Time to Tumour Progression TTP) et sur l’allongement de la durée de survie pour les patients atteints de GIST, sur la survie sans progression (SSP) pour les patients atteints de MRCC non prétraités, sur les taux de réponse objective (TRO) pour les patients atteints de MRCC après échec d’un traitement à base de cytokine et sur la SSP pour les patients atteints de pNET.
Tumeurs stromales gastro-intestinales
Une première étude en ouvert, en escalade de doses, a été menée chez des patients atteints de GIST après échec d’un traitement par l’imatinib (dose médiane journalière maximale : 800 mg) dû à une résistance ou à une intolérance. Quatre-vingt-dix-sept patients ont été inclus pour recevoir différentes doses et schémas posologiques ; 55 patients ont reçu 50 mg selon le Schéma posologique recommandé de 4 semaines de traitement suivi de 2 semaines sans traitement (« Schéma posologique 4/2 »).
Dans cette étude, le TTP médian était de 34,0 semaines (IC à 95 % : 22,0 ; 46,0).
Une étude randomisée de phase III, en double aveugle, contrôlée contre placebo, a été menée chez des patients atteints de GIST intolérants à un traitement par l’imatinib ou ayant présenté une progression de la maladie pendant ou après le traitement (dose médiane journalière maximale : 800 mg). Trois cent douze patients ont été randomisés (selon un ratio de 2:1) pour recevoir soit 50 mg de sunitinib, soit un placebo, administré par voie orale à raison d’une prise par jour selon un Schéma posologique 4/2, jusqu’à progression de la maladie ou sortie de l’étude pour une autre raison (207 patients ont reçu du sunitinib et 105 ont reçu un placebo). Le critère d’efficacité principal était le TTP, défini par le temps écoulé entre la randomisation et la première confirmation d’une progression tumorale.
Au moment de l’analyse intermédiaire pré-définie, le TTP médian sous sunitinib était de 28,9 semaines (IC à 95 % : 21,3 ; 34,1) selon l’évaluation de l’investigateur et de 27,3 semaines (IC à 95 % : 16,0 ; 32,1) selon l’évaluation du Comité de revue indépendant, ce qui est statistiquement significativement plus long que le TTP observé sous placebo : 5,1 semaines (IC à 95 % : 4,4 ; 10,1) selon l’évaluation de l’investigateur et 6,4 semaines (IC à 95 % : 4,4 ; 10,0) selon l’évaluation du Comité de revue indépendant. La survie globale (SG) était statistiquement en faveur du sunitinib [rapport de risque (RR) : 0,491 (IC à 95 % : 0,290 ; 0,831)]. Le risque de mortalité était deux fois plus grand pour les patients dans le bras placebo comparé à celui des patients dans le bras sunitinib.
Après l’analyse intermédiaire de l’efficacité et de la sécurité des patients, selon la recommandation du Comité de surveillance et de suivi (Data Safety Monitoring Board DSMB) indépendant, une levée d’aveugle a été réalisée et les patients du bras placebo se sont vus offrir le traitement par sunitinib en ouvert.
Au total, 255 patients ont reçu du sunitinib dans la phase de traitement en ouvert de l’étude, dont 99 patients qui étaient initialement traités par placebo.
L’analyse des critères primaires et secondaires dans la partie en ouvert de l’étude a reconfirmé les résultats obtenus au moment de l’analyse intermédiaire, tel que montré dans le tableau 2 :
Tableau 2 - Résumé des critères d’efficacité GIST (population ITT)
|
|
Traitement en double-aveuglea |
||||
|
Médiane (IC à 95 %) |
Rapport de risque |
Groupe initialement traité par placebob |
|||
|
Critère |
Sunitinib |
Placebo |
(IC à 95 %) |
Valeur de p |
|
|
Primaire |
|
|
|
|
|
|
TTP (en semaines) |
|
|
|
|
|
|
Intermédiaire |
27,3 (16,0 ; 32,1) |
6,4 (4,4 ; 10,0) |
0,329 (0,233 ; 0,466) |
< 0,001 |
- |
|
Final |
26,6 (16,0 ; 32,1) |
6,4 (4,4 ; 10,0) |
0,339 (0,244 ; 0,472) |
< 0,001 |
10,4 (4,3 ; 22,0) |
|
Secondaire |
|
|
|
|
|
|
SSP (en semaines)c |
|
|
|
|
|
|
Intermédiaire |
24,1 (11,1 ; 28,3) |
6,0 (4,4 ; 9,9) |
0,333 (0,238 ; 0,467) |
< 0,001 |
- |
|
Final |
22,9 (10,9 ; 28,0) |
6,0 (4,4 ; 9,7) |
0,347 (0,253 ; 0,475) |
< 0,001 |
- |
|
TRO (%)d |
|
|
|
|
|
|
Intermédiaire |
6,8 (3,7 ; 11,1) |
0 (-) |
NA |
0,006 |
- |
|
Final |
6,6 (3,8 ; 10,5) |
0 (-) |
NA |
0,004 |
10,1 (5,0 ; 17,8) |
|
SG (en semaines)e |
|
|
|
|
|
|
Intermédiaire |
- |
- |
0,491 (0,290 ; 0,831) |
0,007 |
- |
|
Final |
72,7 (61,3 ; 83,0) |
64,9 (45,7 ; 96,0) |
0,876 (0,679 ; 1,129) |
0,306 |
- |
|
Abréviations : IC = intervalle de confiance ; ITT = en intention de traiter ; NA = non applicable ; TRO = taux de réponse objective ; SG = survie globale ; SSP = survie sans progression ; TTP = temps jusqu’à progression tumorale aLes résultats du traitement en double-aveugle sont donnés pour la population ITT et en utilisant les mesures du radiologue centralisé, si approprié. bRésultats d’efficacité pour les 99 sujets qui ont changé de traitement du placebo vers le sunitinib après la levée d’aveugle. L’état initial a été repris au moment du changement de traitement et les analyses d’efficacité sont basées sur les évaluations des investigateurs. cLes chiffres intermédiaires de SSP ont été mis à jour suite à un recalcul des données d’origine. dLes résultats pour le TRO sont donnés en pourcentages de sujets ayant montré une réponse confirmée avec un IC à 95 %. eLa médiane n’est pas atteinte car les données ne sont pas encore matures. |
|||||
La survie globale médiane dans la population ITT était de 72,7 semaines et 64,9 semaines (RR : 0,876 ; IC à 95 % : 0,679 – 1,129 ; p = 0,306), dans les bras sunitinib et placebo respectivement. Dans cette analyse, le bras placebo incluait les patients randomisés au placebo qui ont reçu par la suite un traitement par sunitinib en ouvert.
Cancer du rein métastatique non prétraité
Une étude randomisée de phase III, multicentrique, internationale évaluant l’efficacité et la tolérance du sunitinib versus interféron IFN-α chez des patients atteints d’un MRCC non prétraités a été menée. Sept cent cinquante patients ont été randomisés en deux groupes de traitement selon un ratio de 1/1, à savoir un groupe recevant du sunitinib par cycles consécutifs de 6 semaines consistant en l’administration de 50 mg par jour de sunitinib par voie orale pendant 4 semaines, suivie de 2 semaines de fenêtre thérapeutique (Schéma 4/2), et un groupe recevant l’IFN-α en injection sous-cutanée de 3 millions d’unités (MU) la 1re semaine, 6 MU la 2e semaine et 9 MU la 3e semaine et ensuite, chaque semaine suivante, 3 injections effectuées à des jours non consécutifs.
La durée médiane de traitement était de 11,1 mois (durée allant de 0,4 à 46,1 mois) pour le traitement par sunitinib et de 4,1 mois (durée allant de 0,1 à 45,6 mois) pour le traitement par IFN-α. Des évènements indésirables graves liés au traitement ont été rapportés chez 23,7 % des patients recevant du sunitinib et chez 6,9 % des patients recevant l’IFN-α. Cependant, les taux d’arrêt du traitement liés aux évènements indésirables étaient de 20 % pour le sunitinib et de 23 % pour de l’IFN-α. Les interruptions de traitement ont eu lieu chez 202 patients (54 %) traités par sunitinib et 141 patients (39 %) traités par IFN-α. Des réductions de doses ont eu lieu chez 194 patients (52 %) traités par sunitinib et chez 98 patients (27 %) traités par IFN-α. Les patients ont été traités jusqu’à progression de la maladie ou jusqu’à la sortie de l’étude. Le critère principal d’efficacité était la SSP. Une analyse intermédiaire prévue dans l’étude a montré un avantage statistiquement significatif pour le sunitinib comparé à l’IFN-α, dans cette étude, la valeur médiane de la SSP était de 47,3 semaines dans le groupe traité par sunitinib et de 22,0 semaines dans le groupe traité par IFN-α ; le RR était égal à 0,415 (IC à 95 % : 0,320 ; 0,539 ; valeur de p < 0,001). Les autres critères étudiés regroupaient le TRO, la SG et la tolérance. L’évaluation radiologique principale a été abandonnée après l’atteinte du critère principal. Lors de l’analyse finale, le taux de réponse objective tel que déterminé par les investigateurs était de 46 % (IC à 95 % : 41 % ; 51 %) pour le bras sunitinib et de 12,0 % (IC à 95 % : 9 % ; 16 %) pour le bras IFN-α (p < 0,001).
Le traitement par sunitinib a été associé à une durée de survie plus longue comparée à l’IFN-α. La survie globale médiane était de 114,6 semaines pour le bras sunitinib (IC à 95 % : 100,1 ; 142,9) et de 94,9 semaines pour le bras IFN-α (IC à 95 % : 77,7 ; 117,0) avec un rapport de risque de 0,821 (IC à 95 % : 0,673 ; 1,001 ; p = 0,0510 par le test du log-rank non stratifié).
La survie sans progression et la survie globale, observées dans la population ITT, et telles que déterminées par l’évaluation clinique radiologique principale, sont résumées dans le tableau 3 :
Tableau 3 - Résumé des critères d’efficacité mRCC non prétraité (population ITT)
|
Résumé de la survie sans progression |
Sunitinib (N = 375) |
IFN-α (N = 375) |
|
Sujets qui n’ont pas progressé ou qui ne sont pas décédés [n (%)] |
161 (42,9) |
176 (46,9) |
|
Sujets identifiés comme ayant progressé ou étant décédés [n (%)] |
214 (57,1) |
199 (53,1) |
|
SSP (en semaines) |
|
|
|
Quartile (IC à 95 %) |
|
|
|
25 % |
22,7 (18,0 ; 34,0) |
10,0 (7,3 ; 10,3) |
|
50 % |
48,3 (46,4 ; 58,3) |
22,1 (17,1 ; 24,0) |
|
75 % |
84,3 (72,9 ; 95,1) |
58,1 (45,6 ; 82,1) |
|
Analyse non stratifiée |
|
|
|
Rapport de risque (sunitinib versus IFN-α) |
0,5268 |
|
|
IC à 95 % pour le rapport de risque |
(0,4316 ; 0,6430) |
|
|
Valeur de pa |
< 0,0001 |
|
|
Résumé de la survie globale |
|
|
|
Sujets non identifiés comme décédés [n (%)] |
185 (49,3) |
175 (46,7) |
|
Sujets identifiés décédés [n (%)] |
190 (50,7) |
200 (53,3) |
|
Survie globale (en semaines) |
|
|
|
Quartile (IC à 95 %) |
|
|
|
25 % |
56,6 (48,7 ; 68,4) |
41,7 (32,6 ; 51,6) |
|
50 % |
114,6 (100,1 ; 142,9) |
94,9 (77,7 ; 117,0) |
|
75 % |
NA (NA ; NA) |
NA (NA ; NA) |
|
Analyse non stratifiée |
|
|
|
Rapport de risque (sunitinib versus IFN-α) |
0,8209 |
|
|
IC à 95 % pour le rapport de risque |
(0,6730 ; 1,0013) |
|
|
Valeur de pa |
0,0510 |
|
Abréviations : IC = intervalle de confiance ; INF-α = interféron-alpha ; ITT = en intention de traiter ; N= nombre de patients ; NA = non applicable ; SG = survie globale ; SSP = survie sans progression.
a A partir du test du log-rank bilatéral.
Cancer du rein métastatique après échec d’un traitement par cytokine
Une étude de phase II a été conduite chez des patients après échec d’un traitement préalable par cytokine à base d’interleukine-2 ou d’IFN-α. Soixante-trois patients ont reçu 50 mg de sunitinib par voie orale, à raison d’une prise par jour pendant 4 semaines consécutives, suivies d’une fenêtre thérapeutique de 2 semaines, correspondant à un cycle complet de 6 semaines (Schéma posologique 4/2). Le critère d’efficacité principal était le TRO calculé d’après les critères d’évaluation des réponses tumorales relatives aux tumeurs solides (RECIST).
Dans cette étude, le taux de réponse objective était de 36,5 % (IC à 95 % : 24,7 ; 49,6 %) et le TTP de 37,7 semaines (IC à 95 % : 24,0 ; 46,4).
Une étude confirmatoire multicentrique, en ouvert, non contrôlée, évaluant l’efficacité et la tolérance du sunitinib, a été menée chez des patients présentant un cancer du rein métastatique après échec d’un précédent traitement par cytokine. 106 patients ont reçu au moins une dose de 50 mg de sunitinib selon le Schéma 4/2.
Le principal critère d’efficacité de cette étude était le TRO. Les critères d’évaluation secondaires comprenaient le TTP, la durée de la réponse (DR) et la SG.
Dans cette étude, le TRO était de 35,8 % (IC à 95 % : 26,8 % – 47,5 %) ; les valeurs médianes de la durée de la réponse et de la survie globale n’avaient pas encore été atteintes.
Tumeurs neuroendocrines du pancréas
Une étude support de phase II, en ouvert, multicentrique, a évalué l’efficacité et la tolérance du sunitinib en monothérapie à une dose journalière de 50 mg selon le Schéma 4/2 chez des patients atteints de pNET non résécables. Dans une cohorte de 66 patients atteints d’une tumeur des îlots de Langerhans, le taux de réponse, critère principal, était de 17 %.
Une étude pivot de phase III, multicentrique, internationale, randomisée, en double aveugle, portant sur le sunitinib en monothérapie, le groupe contrôle étant sous placebo, a été menée chez des patients atteints de pNET non résécables.
Les patients devaient avoir présenté une progression documentée, sur la base des critères RECIST, dans les 12 derniers mois et ont été randomisés (1/1) pour recevoir soit une dose journalière de 37,5 mg de sunitinib sans fenêtre thérapeutique préétablie (N = 86) soit le placebo (N = 85).
L’objectif principal était de comparer la SSP des patients recevant le sunitinib versus les patients recevant le placebo. D’autres critères comprenaient la SG, le TRO, les PRO et la tolérance.
Les données démographiques étaient comparables entre le groupe sunitinib et le groupe placebo. De plus, 49 % des patients recevant le sunitinib avaient une tumeur non fonctionnelle comparativement à 52 % des patients recevant le placebo, et 92 % des patients dans les deux bras présentaient des métastases hépatiques.
L’utilisation d’analogues de la somatostatine a été autorisée dans l’étude.
Un total de 66 % des patients sous sunitinib avaient reçu une thérapie par voie systémique antérieure comparativement à 72 % des patients sous placebo. De plus, 24 % des patients du groupe sunitinib avaient reçu des analogues de la somatostatine contre 22 % des patients du groupe placebo.
Un avantage cliniquement significatif pour la SSP évaluée par les investigateurs a été observé avec le sunitinib par rapport au placebo. La SSP médiane était de 11,4 mois pour le bras sunitinib comparé à 5,5 mois pour le bras placebo [rapport de risque : 0,418 (IC à 95 % : 0,263 - 0,662) valeur de p = 0,0001]. Des résultats similaires ont été observés lorsque l’évaluation de la réponse tumorale basée sur l’application des critères RECIST pour des mesures tumorales faites par l’investigateur était utilisée pour déterminer la progression de la maladie, voir tableau 4. Un rapport de risque en faveur du sunitinib a été observé dans tous les sous-groupes de patients, déterminés sur leurs caractéristiques à l’entrée dans l’étude, y compris une analyse par nombre de traitements systémiques antérieurs. Un total de 29 patients dans le bras sunitinib et 24 dans le bras placebo n’avaient reçu aucun traitement systémique antérieur ; parmi ces patients, le risque relatif pour la SSP était de 0,365 (IC à 95 % : 0,156 - 0,857), p = 0,0156. De la même manière, parmi les 57 patients dans le bras sunitinib (dont 28 ayant reçu 1 thérapie systémique antérieure et 29 ayant reçu 2 thérapies systémiques antérieures ou plus), et parmi les 61 patients dans le bras placebo (dont 25 patients ayant reçu 1 thérapie systémique antérieure et 36 ayant reçu 2 thérapies systémiques antérieures ou plus), le risque relatif pour la SSP était de 0,456 (IC à 95 % : 0,264 - 0,787), p = 0,0036.
Une analyse de sensibilité de la SSP a été menée au cours de laquelle la progression était basée sur les mesures tumorales rapportées par l’investigateur et au cours de laquelle tous les patients censurés pour toute raison autre que l’arrêt de l’étude, ont été considérés comme ayant présenté un évènement de SSP. Cette analyse a donné une estimation prudente de l’effet du traitement par le sunitinib et a conforté l’analyse primaire, en démontrant un risque relatif de 0,507 (IC à 95 % : 0,350 - 0,733), p = 0,000193. L’étude pivot dans les tumeurs neuroendocrines du pancréas a été terminée prématurément selon les recommandations du Comité de surveillance (Drug Monitoring Committee) indépendant, et le critère principal a été basé sur l’évaluation de l’investigateur, ce qui a pu impacter les estimations de l’effet du traitement.
Afin d’éliminer un biais dans l’évaluation de la SSP par l’investigateur, une BICR des scanners a été réalisée. Cette revue a confirmé l’évaluation de la SSP par l’investigateur, voir tableau 4.
Tableau 4 – Résultats d’efficacité de l’étude de phase III pNET
|
Paramètres d’efficacité |
Sunitinib (N = 86) |
Placebo (N = 85) |
Rapport de risque (IC à 95 %) |
Valeur de p |
|
Evaluation de la survie sans progression [médiane, mois (IC à 95 %)] par l’investigateur |
11,4 (7,4 ; 19,8) |
5,5 (3,6 ; 7,4) |
0,418 (0,263 ; 0,662) |
0,0001a |
|
Survie sans progression [médiane, mois (IC à 95 %)] par l’évaluation de la réponse tumorale basée sur l’application des critères RECIST à l’évaluation tumorale par l’investigateur |
12,6 (7,4 ; 16,9) |
5,4 (3,5 ; 6,0) |
0,401 (0,252 ; 0,640) |
0,000066a |
|
Survie sans progression [médiane, mois (IC à 95 %)] par la revue en aveugle, centralisée et indépendante de l’évaluation de la tumeur |
12,6 (11,1 ; 20,6) |
5,8 (3,8 ; 7,2) |
0,315 (0,181 ; 0,546) |
0,000015a |
|
Survie globale [5 ans de suivi] [médiane, mois (IC à 95 %)] |
38,6 (25,6 ; 56,4) |
29,1 (16,4 ; 36,8) |
0,730 (0,504 ; 1,057) |
0,0940a |
|
Taux de réponse objective [% (IC à 95 %)] |
9,3 (3,2 ; 15,4) |
0 |
NA |
0,0066b |
Abréviations : IC = intervalle de confiance ; N = nombre de patients ; NA = non applicable ; pNET = tumeur neuroendocrine du pancréas ; RECIST = critères d’évaluation de la réponse dans les tumeurs solides.
a Test du log-rank bilatéral non stratifié
b Test exact de Fisher
Figure 1 – Courbe de la SSP de Kaplan-Meier dans l’étude de phase III pNET
|
|
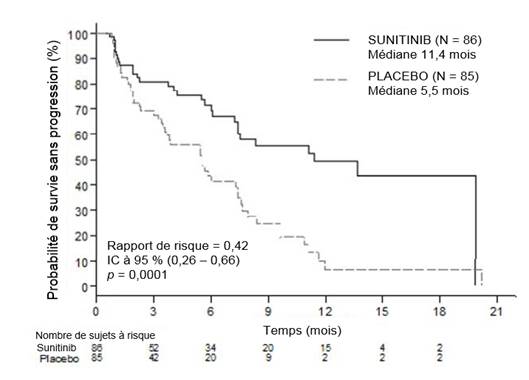
Abréviations : IC = intervalle de confiance ; N = nombre de patients ; SSP = survie sans progression ; pNET = tumeurs neuroendocrines du pancréas.
Les données de survie globale n’étaient pas matures au moment de l’arrêt de l’étude [20,6 mois (IC à 95 % ; 20,6 ; NR) pour le bras sunitinib par rapport au NR (IC à 95 % ; 15,5 ; NR) pour le bras placebo, rapport de risque : 0,409 (IC à 95 % : 0,187 ; 0,894), p = 0,0204]. Il y a eu 9 décès dans le bras sunitinib et 21 décès dans le bras placebo.
En cas de progression de la maladie, une levée d’aveugle était effectuée et les patients sous placebo avaient la possibilité de prendre du sunitinib dans une étude d’extension séparée, en ouvert. En conséquence de l’arrêt prématuré de l’étude, une levée d’aveugle a été effectuée pour les patients restants dans celle-ci et ils ont eu la possibilité de prendre du sunitinib en ouvert dans une étude d’extension. Un total de 59 patients sur 85 (69,4 %) du bras placebo a eu un cross over par sunitinib en ouvert à la suite de la progression de la maladie ou de la levée de l’aveugle au moment de l’arrêt de l’étude. La survie globale, observée après 5 ans de suivi dans l’étude d’extension, a donné un rapport de risque de 0,730 (IC à 95 % ; 0,504 ; 1,057).
Les résultats du questionnaire de l’organisation européenne pour la recherche et le traitement du cancer et de la qualité de vie (EORTC QLQ-C30) ont démontré que la qualité de vie globale liée à la santé et les 5 domaines fonctionnels (physique, rôle, cognitif, émotionnel et social) ont été maintenus pour les patients recevant le sunitinib en comparaison avec le placebo avec des effets indésirables symptomatiques limités.
Une étude de phase IV internationale, multicentrique, en ouvert, à bras unique, évaluant l’efficacité et la tolérance du sunitinib a été menée chez des patients atteints de pNET non résécables, bien différenciées, avancées/métastatiques.
Cent-six patients (61 dans la cohorte des patients non prétraités et 45 dans la cohorte de la ligne suivante) ont reçu un traitement oral par du sunitinib 37,5 mg une fois par jour selon un schéma d’administration quotidienne en continu.
La SSP (survie sans progression) médiane évaluée par l’investigateur a été de 13,2 mois dans la population générale (IC à 95 % : 10,9 ; 16,7) et dans la cohorte des patients non prétraités (IC à 95 % : 7,4 ; 16,8).
Population pédiatrique
L’expérience en matière d’utilisation du sunitinib chez les patients pédiatriques reste limitée (voir rubrique 4.2).
Une étude de phase I en escalade de doses avec du sunitinib par voie orale, a été menée chez 35 patients, dont 30 patients pédiatriques (âgés de 3 à 17 ans) et 5 jeunes adultes (âgés de 18 à 21 ans) atteints de tumeurs solides réfractaires, La plupart d’entre eux ont présenté un diagnostic principal de tumeur cérébrale. Une cardiotoxicité dose-dépendante a été observée lors de la première partie de l’étude qui a été modifiée en conséquence afin d’exclure les patients soumis à une exposition antérieure à des traitements potentiellement cardiotoxiques (y compris les anthracyclines) ou à une irradiation cardiaque. Lors de la seconde partie de l'étude incluant les patients soumis à un traitement anticancéreux préalable, mais sans facteurs de risque de cardiotoxicité, le sunitinib était généralement toléré et cliniquement gérable à la dose de 15 mg/m2/jour (DMT) selon le Schéma 4/2. Aucun des patients n’a obtenu une réponse complète ou partielle. Une stabilisation de la maladie a été observée chez 6 patients (17 %). Un patient atteint de GIST a été inscrit à un niveau de dose de 15 mg/m2, sans aucune preuve d’efficacité. Les effets indésirables observés ont été globalement similaires à ceux observés chez les adultes (voir rubrique 4.8).
Une étude de phase II en ouvert a été menée chez 29 patients, dont 27 patients pédiatriques (âgés de 3 à 16 ans) et 2 jeunes adultes (âgés de 18 à 19 ans) atteints de HGG ou d’épendymome. L’étude a été clôturée au moment de l’analyse intermédiaire prévue en raison de l’absence de contrôle de la maladie. La SSP médiane a été de 2,3 mois dans le groupe HGG et de 2,7 mois dans le groupe épendymome. La SG médiane a été de 5,1 mois dans le groupe HGG et de 12,3 mois dans le groupe épendymome. Les événements indésirables liés au traitement les plus fréquents (≥ 10 %) rapportés chez les patients des deux groupes combinés ont été une diminution de la numération des neutrophiles (6 patients [20,7 %]) et une hémorragie intracrânienne (3 patients [10,3 %]) (voir rubrique 4.8).
Les résultats d’une étude de phase I/II portant sur le sunitinib administré par voie orale à 6 patients pédiatriques atteints de GIST et âgés de 13 à 16 ans ayant reçu du sunitinib selon le Schéma 4/2, à des doses allant de 15 mg/m2 par jour à 30 mg/m2 par jour, et les données publiées disponibles (20 patients pédiatriques ou jeunes adultes atteints de GIST) ont indiqué que le traitement par sunitinib a permis une stabilisation de la maladie chez 18 des 26 patients (69,2 %), soit après un échec ou une intolérance à l’imatinib (16 patients présentant une maladie stable sur 21), soit de novo/après une intervention chirurgicale (2 patients présentant une maladie stable sur 5). Au cours de cette étude de phase I/II, une maladie stable et une progression de la maladie ont été observées chacune chez 3 patients sur 6 (1 patient a reçu le traitement néo-adjuvant et 1 patient a reçu l’imatinib adjuvant, respectivement). Au cours de cette même étude, 4 patients sur 6 (66,7 %) ont présenté des événements indésirables liés au traitement de grade 3-4 (hypophosphatémie, neutropénie et thrombocytopénie de grade 3 chacune chez 1 patient et neutropénie de grade 4 chez 1 patient). De plus, les effets indésirables de grade 3 survenus chez 5 patients suivants sont rapportés dans les publications : fatigue (2), effets indésirables gastro-intestinaux (notamment diarrhée) (2), effets indésirables hématologiques (notamment anémie) (2), cholécystite (1), hyperthyroïdie (1) et mucite (1).
Une analyse de la population de pharmacocinétique (PK) et de pharmacocinétique/ pharmacodynamique (PK/PD) a été menée en vue d’extrapoler la PK et les principaux paramètres de sécurité et d’efficacité du sunitinib chez les patients pédiatriques atteints de GIST (âgés de 6 à 17 ans). Cette analyse a été basée sur les données recueillies auprès des adultes atteints de GIST ou de tumeurs solides, et auprès de patients pédiatriques atteints de tumeurs solides. Selon les analyses par modélisation, un jeune âge et une petite corpulence ne semblent pas avoir d’effets négatifs sur la sécurité et l’efficacité des réponses à l’exposition plasmatique au sunitinib. Il ne semble pas que le jeune âge ou la petite corpulence ait des répercussions négatives sur le rapport bénéfice/risque du sunitinib qui est dû principalement à son exposition plasmatique.
L'EMA a accordé une dérogation à l'obligation de soumettre les résultats d’études réalisées avec le produit de référence contenant du sunitinib dans tous les sous-groupes de la population pédiatrique dans le traitement du carcinome rénal ou du bassinet (à l'exclusion du néphroblastome, de la néphroblastomatose, du sarcome à cellule claire, du néphrome mésoblastique, du carcinome médullaire rénal et de la tumeur rhabdoïde du rein) (voir rubrique 4.2).
L'EMA a accordé une dérogation à l'obligation de soumettre les résultats d’études réalisées avec le produit de référence contenant du sunitinib dans tous les sous-groupes de la population pédiatrique dans le traitement des tumeurs neuroendocrines gastro-entéro-pancréatiques (à l'exclusion des neuroblastomes, neuroganglioblastomes et phéochromocytomes) (voir rubrique 4.2).
5.2. Propriétés pharmacocinétiques
La PK du sunitinib a été étudiée chez 135 volontaires sains et 266 patients présentant une tumeur solide. Les données PK étaient similaires dans toutes les populations de patients présentant des tumeurs solides et chez les volontaires sains.
Dans l’éventail de doses comprises entre 25 et 100 mg, l’aire sous la courbe de concentration plasmatique en fonction du temps (ASC) et la Cmax augmentent de façon proportionnelle en fonction de la dose. Sous l’effet répété des doses journalières, le sunitinib s’accumule et sa concentration est multipliée par 3 à 4, et celle de son principal métabolite actif par 7 à 10. Les concentrations à l’équilibre du sunitinib et de son principal métabolite actif sont atteintes en 10 à 14 jours. Au 14e jour, les concentrations plasmatiques cumulées du sunitinib et de son métabolite actif sont comprises entre 62,9 et 101 ng/mL et correspondent aux concentrations cibles, anticipées d’après les résultats des études précliniques, qui inhibent in vitro la phosphorylation du récepteur et provoquent l’arrêt ou le ralentissement de la croissance tumorale in vivo. Le principal métabolite actif représente 23 % à 37 % de l’exposition au médicament. Aucune modification significative de la PK du sunitinib ou de son principal métabolite actif n’a été observée lors de l’administration de doses journalières répétées, ni lors de cycles répétés avec les schémas posologiques testés.
Absorption
Les Cmax de sunitinib sont généralement atteintes entre 6 à 12 heures (temps nécessaire pour atteindre la concentration maximale Tmax) après l’administration orale de sunitinib.
La biodisponibilité de sunitinib n’est pas affectée par la prise de nourriture.
Distribution
In vitro, la liaison du sunitinib et de son principal métabolite actif aux protéines plasmatiques humaines est respectivement de 95 % et 90 % et ne semble pas dépendre de la concentration. Le volume de distribution apparent du sunitinib (Vd) est important – 2230 L, ce qui indique une distribution tissulaire.
Interactions métaboliques
Les valeurs de Ki calculées in vitro pour toutes les isoformes du cytochrome P450 (CYP) testées (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 et CYP4A9/11) indiquent qu’il est peu probable que le sunitinib et son principal métabolite actif induisent le métabolisme d’autres substances actives susceptibles d’être métabolisés par ces enzymes, de manière cliniquement significative.
Biotransformation
Le sunitinib est principalement métabolisé par le CYP3A4, isoforme du CYP, qui produit son principal métabolite actif, le deséthyl de sunitinib, lequel est ensuite de nouveau métabolisé par la même isoenzyme.
L’administration concomitante de sunitinib et d’inducteurs puissants ou d’inhibiteurs du CYP3A4 doit être évitée en raison d’une altération possible des niveaux plasmatiques de sunitinib (voir rubriques 4.4 et 4.5).
Elimination
L’excrétion se fait principalement par les selles (61 %) ; seulement 16 % de la dose de sunitinib administrée est éliminée par voie rénale sous la forme de substance active inchangée ou de ses métabolites. Le sunitinib et son principal métabolite actif représentent la plus grande partie des composés retrouvés dans le plasma, l’urine et les selles, soit respectivement 91,5 %, 86,4 % et 73,8 % de la radioactivité mesurée sur des échantillons groupés. Des métabolites mineurs ont été identifiés dans les urines et les selles, mais n’ont généralement pas été retrouvés dans le plasma. Les valeurs de la clairance orale totale (CL/F) étaient comprises entre 34 et 62 L/h. Après administration orale chez des volontaires sains, les demi-vies d’élimination du sunitinib et de son principal métabolite actif déséthylé sont respectivement comprises entre environ 40 et 60 heures, et 80 et 110 heures.
Administration concomitante avec des médicaments inhibiteurs de la BCRP
In vitro, le sunitinib est un substrat du transporteur d’efflux BCRP. Dans l’étude A6181038, l’administration concomitante de géfitinib, un inhibiteur de la BCRP, n’a pas eu d’effet cliniquement significatif sur la Cmax et l’ASC du sunitinib ou du médicament total (sunitinib + métabolite) (voir rubrique 4.5). Cette étude était multicentrique, en ouvert, de phase I/II visant à évaluer la sécurité d’emploi/la tolérance, la dose maximale tolérée et l’activité antitumorale du sunitinib en association avec le géfitinib chez des sujets atteints de MRCC. La PK du géfitinib (250 mg par jour) et du sunitinib (37,5 mg [cohorte 1, n = 4] ou 50 mg [cohorte 2, n = 7] par jour selon un schéma posologique de 4 semaines/2 semaines) en cas d’administration concomitante a été évaluée en tant qu’objectif secondaire de l’étude. Les modifications des paramètres PK du sunitinib n’ont pas été cliniquement significatives et n’ont indiqué aucune interaction médicamenteuse. Cependant, compte tenu du nombre relativement faible de sujets (c.-à-d., N = 7 + 4) et de la variabilité inter-patient modérée à élevée au niveau des paramètres PK, la prudence est requise lors de l’interprétation des résultats d’interaction médicamenteuse pharmacocinétique issus de cette étude.
Populations particulières
Insuffisance hépatique
Le sunitinib et son métabolite primaire sont essentiellement métabolisés par le foie. Chez les patients atteints d’une insuffisance hépatique légère ou modérée (classes A et B de Child-Pugh) et chez les patients ayant une fonction hépatique normale les concentrations systémiques étaient similaires, après l’administration d’une dose unique de sunitinib. Le sunitinib n’a pas été étudié chez les patients atteints d’une insuffisance hépatique sévère (classe C de Child-Pugh).
Les patients dont les taux d’ALAT ou d’ASAT étaient supérieurs à 2,5 × LSN (limite supérieure de la normale) ou supérieurs à 5,0 × LSN en présence de métastases hépatiques, ont été exclus des études chez les patients atteints d’un cancer.
Insuffisance rénale
Les analyses PK de population ont indiqué que la clairance apparente de sunitinib (CL/F) n’était pas affectée par la clairance de la créatinine (CLcr) dans les fourchettes de valeurs évaluées (42– 347 mL/min).
Chez les patients présentant une altération sévère de la fonction rénale (CLcr < 30 mL/min) et chez les patients ayant une fonction rénale normale (CLcr > 80 mL/min), les concentrations plasmatiques étaient similaires après l’administration d’une dose unique de sunitinib. Bien que le sunitinib et son métabolite principal ne soient pas éliminés par hémodialyse chez les patients présentant une IRT, les concentrations plasmatiques totales étaient inférieures de 47 % pour le sunitinib et de 31 % pour son métabolite principal par rapport aux patients ayant une fonction rénale normale.
Poids, indice de performance
Les analyses PK de population de données démographiques indiquent qu’aucun ajustement de la dose initiale n’est nécessaire en fonction du poids, ou de l’indice de performance Eastern Cooperative Oncology Group (ECOG).
Sexe
Les données disponibles indiquent que les femmes sont susceptibles de présenter une clairance apparente (CL/F) du sunitinib de 30 % inférieure à celles des hommes : néanmoins cette différence ne nécessite pas de procéder à des ajustements de la dose initiale.
Population pédiatrique
L’expérience en matière d’utilisation du sunitinib chez les patients pédiatriques reste limitée (voir rubrique 4.2). Les analyses de population de PK sur un ensemble de données regroupées provenant de patients adultes atteints de GIST et de tumeurs solides et de patients pédiatriques atteints de tumeurs solides ont été finalisées. Les analyses de modélisation des covariables par étapes ont été réalisées pour évaluer l’effet de l’âge et de la corpulence (poids corporel total ou surface corporelle) ainsi que d’autres co-variables sur les paramètres pharmacocinétiques importants pour le sunitinib et son métabolite actif. Parmi les co-variables testées, liées à l’âge et à la corpulence, l’âge était une co-variable significative sur la clairance apparente du sunitinib (plus le patient pédiatrique était jeune, plus la clairance apparente était faible). De façon similaire, la surface corporelle était une co-variable significative pour la clairance apparente du métabolite actif (plus la surface corporelle est faible, plus la clairance apparente est faible).
De plus, d’après une analyse PK de population intégrée de données groupées provenant de 3 études pédiatriques (2 études pédiatriques sur les tumeurs solides et 1 étude pédiatrique sur les GIST ; âges : de 6 à 11 ans et de 12 à 17 ans), la surface corporelle (SC) initiale était une co-variable significative de la clairance apparente du sunitinib et de son métabolite actif. D’après cette analyse, une dose d’environ 20 mg/m2 par jour (intervalle de SC : 1,10– 1,87 m2) chez les patients pédiatriques devrait entraîner une exposition plasmatique au sunitinib et à son métabolite actif comparable (entre 75 et 125 % de l’ASC) à celle des adultes atteints de GIST ayant reçu 50 mg de sunitinib par jour selon le Schéma 4/2 (ASC de 1 233 ng.h/mL). Dans les études pédiatriques, la dose initiale de sunitinib était de 15 mg/m2, laquelle, chez les patients pédiatriques atteints de GIST, a augmenté à 22,5 mg/m2, puis à 30 mg/m2 (sans dépasser la dose totale de 50 mg/jour) en fonction de la sécurité et de la tolérance individuelles. En outre, selon la documentation publiée sur les patients pédiatriques atteints de GIST, la dose initiale calculée variait de 16,6 mg/m2 à 36 mg/m2 et pouvait atteindre des doses aussi élevées que 40,4 mg/m2 (sans dépasser la dose totale de 50 mg/jour).
5.3. Données de sécurité préclinique
D’autres effets ont été observés dans d’autres études, et notamment un allongement de l’intervalle QTc, une réduction de la fraction d’éjection (FEVG), une atrophie tubulaire des testicules, une augmentation du volume de la matrice mésengiale du rein, des hémorragies du tractus gastro-intestinal et de la muqueuse buccale et une hypertrophie des cellules de l’hypophyse antérieure. Les modifications de l’utérus (atrophie de l’endomètre) et des cartilages de conjugaison (épaississement ou dysplasie des cartilages) sont considérées comme liées à l’action pharmacologique du sunitinib. La plupart de ces effets ont été réversibles et ont disparu 2 à 6 semaines après l’arrêt du traitement.
Génotoxicité
Le potentiel génotoxique du sunitinib a été évalué in vitro et in vivo. Le sunitinib n’est pas mutagène chez des bactéries testées avec activation métabolique en utilisant des cellules hépatiques de rats. Le sunitinib n’induit pas d’aberrations chromosomiques dans des lymphocytes périphériques humains in vitro.
Une polyploïdie (anomalie du nombre de chromosomes) a été observée dans des lymphocytes périphériques humains in vitro, avec ou sans activation métabolique. Le sunitinib n’est pas clastogène sur la moelle osseuse de rat in vivo. Le principal métabolite actif n’a pas fait l’objet d’une évaluation du potentiel génotoxique.
Carcinogénicité
Au cours d’une étude de recherche de dose menée pendant un mois par gavage oral (0, 10, 25, 75 ou 200 mg/kg/jour) en dosage journalier en continu chez des souris transgéniques rasH2, des carcinomes et des hyperplasies des glandes de Brunner du duodénum ont été observés à la dose testée la plus élevée (200 mg/kg/jour).
Une étude de carcinogénicité de 6 mois par gavage oral (0, 8, 25, 75 [réduit à 50] mg/kg/jour), en dosage journalier a été menée chez les souris transgéniques rasH2. Des carcinomes gastro-duodénaux, une élévation de l’incidence des hémangiosarcomes sous-jacents, et/ou une hyperplasie de la muqueuse gastrique ont été rapportés à des doses ≥ 25 mg/kg/jour après une durée de traitement de 1 ou 6 mois (≥ 7,3 fois l’ASC chez les patients ayant reçu la dose recommandée journalière [DRJ]).
Au cours d’une étude de carcinogénicité de 2 ans chez le rat (0 ; 0,33 ; 1 ou 3 mg/kg/jour), l’administration de sunitinib par cycles de 28 jours suivis d’une fenêtre thérapeutique de 7 jours a résulté en l’augmentation de l’incidence des phéochromocytomes et des hyperplasies de la médullosurénale chez les rats mâles ayant reçu 3 mg/kg/jour après > 1 an d’administration (≥ 7,8 fois l’ASC chez les patients ayant reçu la DRJ).
Des carcinomes des glandes de Brunner sont apparus dans le duodénum à des doses ≥ 1 mg/kg/jour chez les femelles et à 3 mg/kg/jour chez les mâles, et des hyperplasies des cellules de la muqueuse étaient visibles dans la partie glandulaire de l’estomac à 3 mg/kg/jour chez les mâles, qui sont apparus à ≥ 0,9, 7,8 et 7,8 fois l’ASC chez les patients ayant reçu la DRJ, respectivement. La pertinence chez l’homme des résultats de néoplasie observés chez la souris (transgénique rasH2) et des études de carcinogénicité chez le rat après un traitement par sunitinib n’est pas claire.
Toxicité sur la reproduction et sur le développement du fœtus
Aucun effet sur la fertilité mâle ou femelle n’a été observé dans les études de toxicité sur la reproduction. Cependant, dans des études de toxicité à doses répétées menées chez des rats et des singes, des effets sur la fertilité femelle ont été observés, avec une atrésie folliculaire, une dégénérescence du corps jaune, des modifications de l’endomètre et une diminution du poids de l’utérus et des ovaires, à des expositions systémiques cliniquement pertinentes. Chez le rat, des effets sur la fertilité mâle ont été observés, avec une atrophie tubulaire des testicules, une réduction du nombre de spermatozoïdes dans l’épididyme, une déplétion colloïdale dans la prostate et les vésicules séminales, à des expositions plasmatiques correspondant à 25 fois l’exposition systémique chez l’homme.
Chez le rat, la mortalité embryo-fœtale a été mise en évidence à travers une réduction significative du nombre de fœtus vivants, une augmentation du nombre de résorptions fœtales, une augmentation de la perte post-implantatoire et une perte de toute la portée chez 8 des 28 femelles gravides, à des expositions plasmatiques correspondant à 5,5 fois l’exposition systémique chez l’homme. Chez le lapin, les diminutions de poids de l’utérus gravide et du nombre de fœtus vivants étaient dues à l’augmentation du nombre de résorptions et de l’incidence des pertes post-implantatoires et à la perte de toute la portée chez 4 des 6 femelles gravides exposées à des doses correspondant à 3 fois l’exposition systémique chez l’homme.
Le traitement des rates par sunitinib, au cours de la période de l’organogenèse, a provoqué des effets sur le développement fœtal à ≥ 5 mg/kg/jour, consistant en une augmentation de l’incidence des malformations squelettiques du fœtus, principalement caractérisées par un retard de l’ossification des vertèbres thoraciques et/ou lombaires, survenus chez le rat à des expositions plasmatiques correspondant à 5,5 fois l’exposition systémique chez l’homme. Chez le lapin, ces effets consistaient en une augmentation de l’incidence des fentes labiales à des expositions plasmatiques environ équivalentes à celles utilisées en pratique clinique, et en une augmentation des fentes labiales et palatines à des expositions correspondant à 2,7 fois l’exposition systémique chez l’homme.
Le sunitinib (0,3 ; 1,0 ; 3,0 mg/kg/jour) a été évalué dans une étude de développement pré- et postnatal chez des rates gravides. Les gains de poids corporel maternel ont été diminués au cours de la gestation et de la lactation à des doses ≥ 1 mg/kg/jour mais aucune toxicité de la reproduction maternelle n’a été observée jusqu’à 3 mg/kg/jour (exposition estimée à ≥ 2,3 fois l’ASC chez les patients ayant reçu la DRJ). Une diminution du poids corporel de la progéniture a été observée au cours des périodes pré-sevrage et post-sevrage à la dose de 3 mg/kg/jour. Aucune toxicité du développement n’a été observée à 1 mg/kg/jour (exposition environ ≥ 0,9 fois l’ASC chez les patients ayant reçu la DRJ).
Mannitol, povidone K-25, croscarmellose sodique, stéarate de magnésium.
Enveloppe de la gélule
Gélatine, dioxyde de titane (E171), oxyde de fer jaune (E172), oxyde de fer rouge (E172).
Encre d’impression
Gomme laque, oxyde de fer noir (E172), propylène glycol, solution d'ammoniaque concentrée, hydroxyde de potassium.
3 ans
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30 °C.
A conserver dans l’emballage extérieur d’origine, à l’abri de l’humidité.
6.5. Nature et contenu de l'emballage extérieur
Flacon en polyéthylène haute densité (PEHD) muni d’un bouchon en polypropylène (PP) avec fermeture de sécurité enfant contenant 30 gélules.
Plaquette en PVC/Aclar/PVC//Aluminium.
Boîtes de 28 et 30 gélules sous plaquettes ou de 28 × 1 et 30 × 1 gélules sous plaquettes unitaires.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
SWENSWEG 5
2031 GA HAARLEM
PAYS-BAS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 385 1 9 : Comprimés en flacon (PEHD) avec bouchon de sécurité-enfant (PP). Boîte de 30
· 34009 301 385 2 6 : Comprimés sous plaquettes (PVC/Aclar/PVC//Aluminium). Boîte de 28
· 34009 301 385 3 3 : Comprimés sous plaquettes (PVC/Aclar/PVC//Aluminium). Boîte de 30
· 34009 301 385 4 0 : Comprimés sous plaquettes unitaires (PVC/Aclar/PVC//Aluminium). Boîte de 28x1
· 34009 301 385 5 7 : Comprimés sous plaquettes unitaires (PVC/Aclar/PVC//Aluminium). Boîte de 30x1
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.
Médicament à prescription réservée aux spécialistes et services de cancérologie, hématologie et oncologie médicale.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 12/07/2024
Sunitinib
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que SUNITINIB TEVA 25 mg, gélule et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre SUNITINIB TEVA 25 mg, gélule ?
3. Comment prendre SUNITINIB TEVA 25 mg, gélule ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver SUNITINIB TEVA 25 mg, gélule ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE SUNITINIB TEVA 25 mg, gélule ET DANS QUELS CAS EST-IL UTILISE ?
SUNITINIB TEVA contient la substance active sunitinib, qui est un inhibiteur des protéines kinase. Il est utilisé dans le traitement des cancers, en diminuant l’activité d’un groupe de protéines spécifiques impliquées dans la croissance et la dissémination des cellules cancéreuses.
SUNITINIB TEVA est utilisé chez les adultes dans le traitement des cancers suivants :
· Tumeur stromale gastro-intestinale (GIST), un type de cancer de l’estomac et de l’intestin lorsque l’imatinib (un autre médicament traitant le cancer) ne marche plus ou lorsque vous ne pouvez plus prendre l’imatinib.
· Cancer du rein métastatique (MRCC), un type de cancer du rein qui s’est disséminé dans d’autres parties du corps.
· Tumeur neuroendocrine du pancréas (pNET) (tumeur des cellules sécrétrices d’hormones du pancréas) qui a progressé et ne peut être enlevé par chirurgie.
Si vous vous posez des questions sur le mode d’action de SUNITINIB TEVA ou voulez savoir pourquoi ce médicament vous a été prescrit, veuillez consulter votre médecin.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE SUNITINIB TEVA 25 mg, gélule ?
Ne prenez jamais SUNITINIB TEVA 25 mg, gélule :
· si vous êtes allergique au sunitinib ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin avant de prendre SUNITINIB TEVA :
· Si vous présentez une tension artérielle élevée. SUNITINIB TEVA peut augmenter la tension artérielle. Votre médecin pourra vérifier votre tension artérielle au cours de votre traitement par SUNITINIB TEVA, et vous pourrez être traité par des médicaments pour abaisser la tension artérielle si nécessaire.
· Si vous avez ou avez eu une maladie du sang, des problèmes de saignement ou de bleus. Le traitement par SUNITINIB TEVA peut conduire à un risque plus élevé de saignement ou à des modifications dans le nombre de certaines cellules du sang qui pourraient conduire à une anémie ou modifier la capacité de votre sang à coaguler. Si vous prenez de la warfarine ou de l’acénocoumarole, des médicaments qui fluidifient le sang pour éviter la formation de caillots sanguins, le risque de saignement pourrait être plus élevé. Informez votre médecin si vous avez un quelconque saignement au cours de votre traitement par SUNITINIB TEVA.
· Si vous avez des problèmes cardiaques. SUNITINIB TEVA peut causer des problèmes au niveau du cœur. Informez votre médecin si vous vous sentez très fatigué, si vous manquez de souffle, ou si vous avez des gonflements des pieds et des chevilles.
· Si vous présentez des modifications anormales du rythme cardiaque. SUNITINIB TEVA peut causer des anomalies du rythme cardiaque. Votre médecin peut se procurer des électrocardiogrammes pour évaluer ces problèmes pendant votre traitement par SUNITINIB TEVA. Informez votre médecin si vous vous sentez étourdi, si vous vous évanouissez, ou avez des battements du cœur anormaux au cours de votre traitement par SUNITINIB TEVA.
· Si vous avez eu récemment un problème de caillots sanguins dans vos veines et/ou dans vos artères (types de vaisseaux sanguins), tels qu’un accident vasculaire cérébral, une crise cardiaque, une embolie ou une thrombose. Informez votre médecin immédiatement si vous présentez des symptômes tels qu’une douleur ou une oppression à la poitrine, des douleurs dans les bras, le dos, le cou ou la mâchoire, un essoufflement, un engourdissement ou une faiblesse d’un côté du corps, des troubles de la parole, des maux de tête ou des étourdissements au cours de votre traitement par SUNITINIB TEVA.
· Si vous souffrez ou avez souffert d’un anévrisme (élargissement et affaiblissement de la paroi d’un vaisseau sanguin) ou d’une déchirure dans la paroi d’un vaisseau sanguin.
· Si vous souffrez ou avez souffert d’une lésion des plus petits vaisseaux sanguins appelée microangiopathie thrombotique (MAT). Informez votre médecin si vous présentez de la fièvre, une fatigue, des bleus, un saignement, un gonflement, une confusion, une perte de vision et des crises convulsives.
· Si vous avez des problèmes de thyroïde. SUNITINIB TEVA peut causer des problèmes de thyroïde. Informez votre médecin si vous êtes fatigué plus facilement, si vous avez généralement plus froid que les autres personnes ou si votre voix devient plus grave alors que vous prenez SUNITINIB TEVA. Votre fonction thyroïdienne devrait être vérifiée avant votre traitement par SUNITINIB TEVA et régulièrement pendant votre traitement. Si votre thyroïde ne produit pas suffisamment d’hormone, vous pourrez être traité par une hormone thyroïdienne de substitution.
· Si vous avez ou avez eu des troubles pancréatiques ou de la vésicule biliaire. Informez votre médecin si l’un des signes et symptômes suivants se développe : douleur dans la zone de l’estomac (haut de l’abdomen), nausées, vomissements et fièvre. Ils pourraient être dus à une inflammation du pancréas ou de la vésicule biliaire.
· Si vous avez ou avez eu des problèmes au foie. Informez votre médecin si l’un des signes et symptômes de problème au foie suivants se développe pendant votre traitement par SUNITINIB TEVA : démangeaisons, yeux ou peau jaune, urines foncées, douleur ou inconfort au niveau de la zone supérieure droite de votre estomac. Votre médecin devra faire des tests sanguins pour vérifier votre fonction hépatique avant et pendant votre traitement avec SUNITINIB TEVA, et en cas d’indication clinique.
· Si vous avez ou avez eu des problèmes aux reins. Votre médecin surveillera votre fonction rénale.
· Si vous devez subir une opération chirurgicale ou si vous avez eu une opération récemment. SUNITINIB TEVA pourrait modifier la façon dont vos plaies cicatrisent. En général, vous arrêterez SUNITINIB TEVA si vous devez subir une opération. Votre médecin décidera quand reprendre SUNITINIB TEVA.
· On peut vous recommander de faire un bilan dentaire avant de commencer votre traitement par SUNITINIB TEVA
o Si vous avez ou avez eu des douleurs au niveau de la bouche, des dents et/ou de la mâchoire, un gonflement ou une irritation dans la bouche, un engourdissement ou une sensation de lourdeur dans la mâchoire, ou la perte d’une dent, informez-en votre médecin et votre dentiste immédiatement.
o Si vous devez subir un traitement dentaire invasif ou une chirurgie dentaire, informez votre dentiste que vous êtes traité par SUNITINIB TEVA, en particulier si vous recevez également ou avez reçu des bisphosphonates par voie intraveineuse. Les bisphosphonates sont des médicaments utilisés pour prévenir les complications osseuses, qui peuvent vous avoir été prescrits pour une autre maladie.
· Si vous avez ou avez eu des affections de la peau et du tissu sous-cutané. Un « pyoderma gangrenosum » (ulcération douloureuse de la peau) ou une « fasciite nécrosante » (infection de la peau/des tissus mous se propageant rapidement et pouvant être mortelle) peut apparaître lorsque vous prenez ce médicament. Contactez immédiatement votre médecin si vous présentez des symptômes d’infection autour d’une plaie cutanée, notamment de la fièvre, une douleur, une rougeur, un gonflement ou un écoulement de pus ou de sang. Cet effet est généralement réversible après l’arrêt du sunitinib. Des éruptions cutanées sévères (syndrome de Stevens-Johnson, nécrolyse épidermique toxique, érythème polymorphe) ont été signalées avec l’utilisation du sunitinib, apparaissant initialement sous la forme de taches rougeâtres en forme de « cibles » ou de plaques circulaires présentant fréquemment des ampoules centrales sur le tronc. L’éruption cutanée peut évoluer en l’apparition d’ampoules sur tout le corps ou en une exfoliation généralisée de la peau et peut mettre en jeu le pronostic vital. Si vous présentez une éruption cutanée ou ces symptômes cutanés, consultez immédiatement un médecin.
· Si vous avez ou avez eu des crises convulsives. Informez votre médecin dès que possible si avez une élévation de la tension artérielle, des maux de tête ou une perte de la vue.
· Si vous avez du diabète. La glycémie des patients diabétiques doit être régulièrement surveillée afin de déterminer si un ajustement de la posologie du traitement antidiabétique est nécessaire pour limiter le risque d’hypoglycémie. Prévenez votre médecin dès que possible si vous présentez des signes et symptômes d’hypoglycémie (fatigue, palpitations, transpiration, faim et perte de connaissance).
Enfants et adolescents
L’utilisation de SUNITINIB TEVA n’est pas recommandée chez les personnes âgées de moins de 18 ans.
Autres médicaments et SUNITINIB TEVA 25 mg, gélule
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance.
Certains médicaments peuvent affecter le niveau de SUNITINIB TEVA dans votre organisme. Vous devez informer votre médecin si vous prenez des médicaments contenant les substances actives suivantes :
· le kétoconazole, l’itraconazole - utilisés pour traiter les infections fongiques
· l’érythromycine, la clarithromycine, la rifampicine - utilisées pour traiter les infections
· le ritonavir - utilisé pour traiter le VIH
· la dexaméthasone - un corticostéroïde utilisé dans diverses maladies (telles que les troubles allergiques/respiratoires ou les maladies de la peau)
· la phénytoïne, la carbamazépine, le phénobarbital - utilisés pour traiter l’épilepsie et d’autres maladies neurologiques
· des préparations à base de plantes contenant du millepertuis (Hypericum perforatum) - utilisées pour traiter la dépression et l’anxiété.
SUNITINIB TEVA 25 mg, gélule avec des aliments et boissons
Vous devez éviter de boire du jus de pamplemousse durant votre traitement par SUNITINIB TEVA.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Si vous êtes susceptible d’être enceinte, vous devez utiliser une méthode de contraception fiable pendant le traitement par SUNITINIB TEVA.
Si vous allaitez, signalez-le à votre médecin. Vous ne devez pas allaiter pendant le traitement par SUNITINIB TEVA.
Conduite de véhicules et utilisation de machines
Si vous ressentez des étourdissements ou êtes anormalement fatigué(e), soyez très vigilant(e) en conduisant ou utilisant des machines.
SUNITINIB TEVA 25 mg, gélule contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE SUNITINIB TEVA 25 mg, gélule ?
Votre médecin vous prescrira une dose qui convient à votre cas, selon le type de cancer à traiter. Si vous êtes traité pour :
· une tumeur maligne stromale gastro-intestinale ou un MRCC, la dose usuelle de traitement est de 50 mg par jour, pris pendant une période de 28 jours (4 semaines), suivie d’une période de repos thérapeutique de 14 jours (2 semaines sans traitement), par cycles de traitement de 6 semaines ;
· une tumeur neuroendocrine du pancréas, la dose usuelle est de 37,5 mg, une fois par jour, sans période de repos thérapeutique.
Votre médecin déterminera la dose que vous devez prendre et si vous devez arrêter votre traitement par SUNITINIB TEVA et quand l’arrêter.
SUNITINIB TEVA peut être pris accompagné ou non de nourriture.
Si vous avez pris plus de SUNITINIB TEVA 25 mg, gélule que vous n’auriez dû
Si vous avez accidentellement pris trop de gélules, avertissez-en immédiatement votre médecin. Vous pourriez avoir besoin de certains soins médicaux.
Si vous oubliez de prendre SUNITINIB TEVA 25 mg, gélule
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre SUNITINIB TEVA 25 mg, gélule
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Vous devez immédiatement contacter votre médecin si vous ressentez l’un des effets indésirables graves suivants (voir également Quelles sont les informations à connaître avant de prendre SUNITINIB TEVA 25 mg, gélule ?) :
Problèmes cardiaques. Avertissez votre médecin si vous vous sentez très fatigué, si vous manquez de souffle, ou si vous avez des gonflements des pieds et des chevilles. Ce pourrait être les symptômes de problèmes cardiaques, notamment une insuffisance cardiaque et des problèmes au muscle du cœur (cardiomyopathie).
Problèmes aux poumons ou respiratoires. Avertissez votre médecin si vous développez de la toux, des douleurs à la poitrine, un accès soudain d’essoufflement ou une toux sanglante. Ce pourrait être les symptômes d’une maladie appelée embolie pulmonaire qui apparaît lorsque des caillots sanguins vont se loger dans vos poumons.
Problèmes aux reins. Avertissez votre médecin si vous ressentez un changement dans la fréquence ou une absence d’émission d’urine qui peuvent être les symptômes d’une insuffisance rénale.
Saignements. Avertissez votre médecin si vous avez l’un des symptômes suivants ou des problèmes de saignement importants au cours du traitement par SUNITINIB TEVA : estomac (abdomen) douloureux ou gonflé, vomissements de sang, selles noires, collantes, présence de sang dans les urines, mal de tête, ou modification de votre état mental, toux sanglante ou crachat sanglant en provenance des poumons ou des voies respiratoires.
Destruction tumorale entraînant un trou dans l’intestin. Avertissez votre médecin si vous présentez des douleurs abdominales sévères, de la fièvre, des nausées, des vomissements, du sang dans les selles ou des changements dans les mouvements habituels de l’intestin.
Les autres effets indésirables avec SUNITINIB TEVA peuvent inclure :
Très fréquent : pouvant affecter plus de 1 personne sur 10
· Diminution du nombre de plaquettes, de globules rouges et/ou de globules blancs (par exemple neutrophiles).
· Essoufflement.
· Elévation de la tension artérielle.
· Fatigue extrême, perte de la force.
· Gonflement dû à du liquide sous la peau et autour des yeux, éruption cutanée allergique profonde.
· Irritation, douleur ou inflammation buccale, sécheresse buccale, altération du goût, maux d’estomac, nausées, vomissements, diarrhée, constipation, douleur/gonflement abdominal, perte/diminution de l’appétit.
· Baisse de l’activité thyroïdienne (hypothyroïdie).
· Sensations vertigineuses.
· Mal de tête.
· Saignement de nez.
· Douleurs dans le dos, dans les articulations.
· Douleur dans les bras et les jambes.
· Coloration jaune de la peau/modification de la couleur de la peau, hyperpigmentation de la peau, modification de la couleur des cheveux, éruption cutanée des paumes des mains et plantes des pieds, éruption cutanée, sécheresse de la peau.
· Toux.
· Fièvre.
· Difficulté à s’endormir.
Fréquent : pouvant affecter jusqu’à 1 personne sur 10
· Caillots sanguins dans les vaisseaux sanguins.
· Insuffisance de l’apport de sang au muscle du cœur due à une obstruction ou une constriction des artères coronaires.
· Douleur dans le thorax.
· Diminution de la quantité de sang pompée par le cœur.
· Rétention de fluides, y compris autour des poumons.
· Infections.
· Complication d’infection grave (infection présente dans la circulation sanguine) qui peut entraîner des lésions des tissus, une défaillance des organes et le décès.
· Diminution du taux de sucre dans le sang (voir rubrique 2).
· Perte de protéines dans les urines, conduisant parfois à des gonflements.
· Syndrome pseudo-grippal.
· Tests sanguins anormaux, notamment pour les enzymes du pancréas et du foie.
· Taux élevé d'acide urique dans le sang.
· Hémorroïdes, douleur au rectum, saignement des gencives, difficulté ou incapacité à avaler.
· Sensation de brûlure ou de douleur sur la langue, inflammation au niveau du tube digestif, excès de gaz dans l’estomac ou l’intestin.
· Perte de poids.
· Douleurs musculo-squelettiques (douleur dans les muscles et les os), faiblesse musculaire, fatigue musculaire, douleur dans les muscles, spasmes musculaires.
· Sècheresse nasale, nez bouché.
· Augmentation de la sécrétion lacrymale.
· Sensations anormales au niveau de la peau, démangeaisons, inflammation et écaillement de la peau, cloques, acné, décoloration des ongles, perte de cheveux.
· Sensations anormales dans les extrémités.
· Sensibilité anormalement élevée ou diminuée, en particulier au toucher.
· Brûlures d’estomac.
· Déshydratation.
· Bouffées de chaleur.
· Coloration anormale des urines.
· Dépression.
· Frissons.
Peu fréquent : pouvant affecter jusqu’à 1 personne sur 100
· Infection des tissus mous menaçant le pronostic vital, y compris dans la région ano-génitale (voir rubrique 2).
· Accident vasculaire cérébral.
· Crise cardiaque provoquée par une interruption ou une diminution de l’apport de sang au cœur.
· Changement dans l’activité électrique du cœur ou rythme anormal du cœur.
· Liquide autour du cœur (épanchement péricardique).
· Insuffisance hépatique.
· Douleur dans l’estomac (abdomen) en raison d’une inflammation du pancréas.
· Destruction tumorale entraînant un trou dans l’intestin (perforation).
· Inflammation (gonflement et rougeur) de la vésicule biliaire associée ou non à des calculs biliaires.
· Formation d’une communication anormale (conduit) entre une cavité du corps et une autre cavité du corps ou bien de la peau.
· Douleur au niveau de la bouche, des dents et/ou de la mâchoire, gonflement ou irritation dans la bouche, engourdissement ou sensation de lourdeur dans la mâchoire, ou déchaussement d’une dent. Ce pourrait être les signes et symptômes d’un endommagement de l’os de la mâchoire (ostéonécrose), voir rubrique 2.
· Surproduction d’hormones par la thyroïde, ce qui augmente la consommation d’énergie par le corps au repos.
· Problème de cicatrisation des plaies après une chirurgie.
· Augmentation du taux sanguin d’une enzyme (la créatinine-phosphokinase) produite par les muscles.
· Réaction excessive à un allergène incluant rhume des foins, rash cutané, démangeaisons, urticaire, gonflement de certaines parties du corps et difficulté à respirer.
· Inflammation du colon (colite, colite ischémique).
Rare : pouvant affecter jusqu’à 1 personne sur 1 000
· Réaction sévère de la peau et/ou des muqueuses (syndrome de Stevens-Johnson, nécrolyse épidermique toxique, érythème polymorphe).
· Syndrome de lyse tumorale (SLT) – Le SLT correspond à un groupe de complications métaboliques qui peuvent apparaître au cours du traitement contre le cancer. Ces complications sont dues aux produits de décomposition des cellules cancéreuses détruites et peuvent inclure : nausées, essoufflement, battements du cœur irréguliers, crampes musculaires, crise convulsive, urines troubles, et fatigue associés à des résultats de test sanguins anormaux (taux sanguins élevés de potassium, d’acide urique et de phosphore et taux sanguin diminué de calcium) qui peuvent entraîner des modifications de la fonction rénale et une insuffisance rénale aiguë.
· Dégradation anormale des muscles pouvant entraîner des problèmes rénaux (rhabdomyolyse).
· Modifications anormales au niveau du cerveau, pouvant provoquer un ensemble de symptômes incluant des maux de tête, une confusion, des convulsions et une perte de la vision (syndrome de leucoencéphalopathie postérieure réversible).
· Ulcération douloureuse de la peau (pyoderma gangrenosum).
· Inflammation du foie (hépatite).
· Inflammation de la glande thyroïde.
· Lésion des plus petits vaisseaux sanguins appelée microangiopathie thrombotique (MAT).
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
· Elargissement et affaiblissement de la paroi d’un vaisseau sanguin ou déchirure dans la paroi d’un vaisseau sanguin (anévrismes et dissections artérielles).
· Manque d’énergie, confusion, somnolence, perte de connaissance/coma – ces symptômes peuvent être des signes de toxicité cérébrale causée par un taux sanguin élevé d’ammoniaque (encéphalopathie hyperammoniémique).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER SUNITINIB TEVA 25 mg, gélule ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte, le flacon et la plaquette après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 30 °C. A conserver dans l’emballage extérieur d’origine, à l’abri de l’humidité.
N’utilisez pas ce médicament si vous remarquez que la boîte est endommagée ou présente des signes d’altération.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient SUNITINIB TEVA 25 mg, gélule
· La substance active est :
Sunitinib.......................................................................................................................... 25 mg
Pour une gélule.
· Les autres composants sont :
Contenu de la gélule : mannitol, povidone K-25, croscarmellose sodique, stéarate de magnésium.
Enveloppe de la gélule : gélatine, dioxyde de titane (E171), oxyde de fer jaune (E172), oxyde de fer rouge (E172).
Encre d’impression : gomme laque, oxyde de fer noir (E172), propylène glycol, solution d'ammoniaque concentrée, hydroxyde de potassium.
Qu’est-ce que SUNITINIB TEVA 25 mg, gélule et contenu de l’emballage extérieur
Gélule en gélatine dure constituée d’une coiffe opaque orange clair sur laquelle est imprimé « 25 » à l’encre noire, et d’un corps opaque orange moyen. Chaque gélule de taille 3 (longueur fermée totale d’environ 15,8 mm) contient une poudre granulée orange.
SUNITINIB TEVA est disponible en flacons PEHD blancs de 30 gélules, en plaquettes de 28 ou 30 gélules ou en plaquettes unitaires de 28 × 1 ou 30 × 1 gélules.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
SWENSWEG 5
2031 GA HAARLEM
PAYS-BAS
Exploitant de l’autorisation de mise sur le marché
100-110, ESPLANADE DU GENERAL DE GAULLE
92931 PARIS LA DEFENSE CEDEX
TEVA OPERATIONS POLAND SP. Z.O.O
UL. MOGILSKA 80
31-546 KRAKOW
POLOGNE
OU
MERCKLE GMBH
LUDWIG-MERCKLE-STRASSE 3
89143 BLAUBEUREN
ALLEMAGNE
OU
PLIVA HRVATSKA D.O.O (PLIVA CROATIA LTD.)
PRILAZ BARUNA FILIPOVICA 25
10000 ZAGREB
CROATIE
OU
ACTAVIS INTERNATIONAL LTD.
4 SQAQ TAL-GIDI OFF VALLETTA ROAD
LUQA LQA 6000
MALTE
OU
BALKANPHARMA-DUPNITSA AD
3 SAMOKOVSKO SHOSSE STR.
DUPNITSA, 2600
BULGARIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).