Dernière mise à jour le 03/08/2026
ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique anticholinergiques - code ATC : R03BB01
Qu’est-ce que ATROVENT 20 microgrammes/dose ?
ATROVENT 20 microgrammes/dose appartient à une famille de médicaments appelée les anticholinergiques.
Il agit en augmentant le diamètre des bronches.
Dans quels cas est-il utilisé ?
Votre médecin vous a prescrit ce médicament car vous avez besoin de recevoir cette substance active afin de traiter :
· les symptômes d’une crise d’asthme,
· ou des difficultés pour respirer provoquées par une maladie au long cours encombrant vos bronches et vos poumons (broncho-pneumopathie chronique obstructive).
Pour prendre ce médicament, vous utiliserez un flacon sous pression qui vous permettra de prendre ce médicament en utilisant votre respiration (également appelée voie inhalée).
Si vous avez une crise d’asthme aiguë ou des difficultés importantes pour respirer
Si vous avez une crise d’asthme aiguë ou une crise au cours de laquelle vous avez une difficulté importante pour respirer, ce médicament ne suffit pas à lui tout seul.
Vous devrez alors utiliser en complément un autre médicament par voie inhalée qui augmentera également le diamètre de vos bronches. Cet autre médicament contiendra une substance active appartenant à une famille de médicaments appelée les bêta2 mimétiques dont l’action est rapide et de courte durée. Un soulagement de la crise doit être observé rapidement après la prise de ces deux médicaments. En cas d’échec de cette association, consultez immédiatement votre médecin.
Présentations
> 1 flacon(s) pressurisé(s) acier de 200 doses avec valve(s) avec embout(s) buccal(aux)
Code CIP : 360 797-8 ou 34009 360 797 8 6
Déclaration d'arrêt de commercialisation : 08/10/2025
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Faible | Avis du 31/01/2007 | Inscription (CT) | Le service médical rendu par cette spécialité est faible dans le traitement symptomatique de la crise d'asthme et des exacerbations au cours de la maladie asthmatique ou de BPCO, en complément d'un bêta-2 mimétique d'action rapide et de courte durée par voie inhalée. |
| Important | Avis du 31/01/2007 | Inscription (CT) | Le service médical rendu par cette spécialité est important dans le traitement symptomatique continu du bronchospasme réversible de la bronchopneumopathie chronique obstructive. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 31/01/2007 | Inscription (CT) | ATROVENT 20 µg/dose solution pour inhalation en flacon pressurisé avec HFa n'apporte pas d'amélioration du service médical rendu (ASMR V) par rapport à la formulation actuellement commercialisée d'ATROVENT 20 µg/dose solution pour inhalation en flacon pressurisé avec CFC. |
ANSM - Mis à jour le : 01/08/2022
ATROVENT20 microgrammes/dose, solution pour inhalation en flacon pressurisé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Bromure d’ipratropium anhydre.......................................................................... 20,00 microgrammes
Sous forme de bromure d’ipratropium monohydraté
Pour une dose.
Ce médicament contient 8,415 mg d’alcool (éthanol) par dose.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution pour inhalation en flacon pressurisé.
Liquide limpide, incolore et dépourvu de particules en suspension.
4.1. Indications thérapeutiques
Traitement symptomatique des exacerbations au cours de la maladie asthmatique ou de la bronchopneumopathie chronique obstructive, en complément d'un bêta2 mimétique d'action rapide et de courte durée par voie inhalée.
Traitement symptomatique continu du bronchospasme réversible au cours de la bronchopneumopathie chronique obstructive.
4.2. Posologie et mode d'administration
La posologie doit être adaptée aux besoins individuels et le traitement doit être administré aux patients sous surveillance médicale. Il est conseillé de ne pas dépasser la dose quotidienne recommandée, que ce soit pendant la phase aiguë de traitement ou d’entretien.
Si le traitement n’apporte pas d’amélioration significative ou si l’état clinique du patient s’aggrave, il convient de demander conseil à un médecin afin d’établir un nouveau plan de traitement. Le patient doit être informé qu’en cas de dyspnée aiguë ou présentant une aggravation rapide, un médecin doit être consulté immédiatement.
Les posologies recommandées sont les suivantes:
Traitement de la crise d'asthme et des exacerbations en association à un bêta2 mimétique d'action rapide et de courte durée par voie inhalée : dès les premiers symptômes, inhaler 1 à 2 bouffées. Cette dose est généralement suffisante. En cas de persistance des symptômes, elle peut être renouvelée quelques minutes plus tard.
Traitement symptomatique continu de la bronchopneumopathie chronique obstructive : inhalation de 1 à 2 bouffées 2 à 4 fois par jour réparties régulièrement dans la journée.
La dose quotidienne ne doit habituellement pas dépasser 16 bouffées par 24 heures.
Mode d’administration
Veuillez lire attentivement les instructions d’utilisation pour une administration correcte.
Inhalation par distributeur avec embout buccal.
Pour une utilisation correcte, il est souhaitable que le médecin s’assure du bon usage de l’appareil par le patient.
En cas de mise en évidence chez le patient d'une mauvaise synchronisation main/poumon empêchant la coordination des mouvements inspiration/déclenchement de l'appareil, l'utilisation d'une chambre d'inhalation est indiquée.
Lors de la première utilisation du flacon pressurisé, appuyer deux fois sur l’embout buccal après avoir ôté le capuchon protecteur, et cela sans inhaler les deux bouffées expulsées.
Si le flacon pressurisé n'a pas été utilisé pendant trois jours, réamorcer la valve en appuyant une fois sur l’embout buccal, sans inhaler la bouffée expulsée.
|
1. Après avoir enlevé le capuchon le patient devra : |
|
|
2. Expirer profondément |
|
|
3. Présenter l’embout buccal à l’entrée de la bouche, le fond de la cartouche métallique dirigé vers le haut. |
|
|
4. Commencer à inspirer et presser sur la cartouche métallique tout en continuant à inspirer lentement et profondément. Retirer l’embout buccal et retenir sa respiration pendant au moins 10 secondes. L’embout buccal de l’appareil de propulsion doit, par mesure d’hygiène, être nettoyé après emploi. |
|
|
5. Replacer le capuchon protecteur après utilisation. |
|
|
6. Lorsque l’inhalateur n’a pas été utilisé pendant trois jours, la valve doit être actionnée une fois. |
|
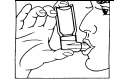
|
Le récipient n’étant pas transparent, il n’est pas possible de voir lorsqu’il est vide. L’inhalateur délivrera 200 bouffées (200 doses indiquées sur l’étiquette). Lorsque les 200 doses ont été utilisées (en général, après 3 semaines d’utilisation comme recommandé), la cartouche peut sembler contenir encore une petite quantité de liquide. L’inhalateur doit toutefois être remplacé afin d’être certain d’obtenir la bonne quantité délivrée de médicament à chaque utilisation.
|
|
|
|
|
|
Nettoyer l’embout buccal de votre inhalateur au moins une fois par semaine. Il est important de maintenir l’embout buccal de l’inhalateur propre, afin d’éviter l’accumulation de médicament qui peut bloquer le pulvérisateur. Pour le nettoyage, commencer par retirer le capuchon antipoussière, puis retirer la cartouche de l’embout buccal. Rincer l’embout buccal abondamment à l’eau chaude jusqu’à ce qu’aucune accumulation de médicament ni de poussière ne soit visible. |
|
|
Une fois le nettoyage terminé, secouer l’embout buccal et laisser-le sécher à l’air sans utiliser de système de chauffage. Lorsque l’embout buccal est sec, replacer la cartouche et le capuchon antipoussière. |
|
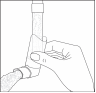
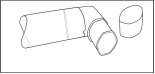
L’embout buccal en plastique est spécifiquement conçu pour être utilisé avec l’aérosol doseur ATROVENT, solution pour inhalation en flacon pressurisé afin de garantir l’administration de la quantité correcte de médicament. L’embout buccal ne doit jamais être utilisé avec un autre aérosol doseur et l’aérosol doseur ATROVENT® ne doit jamais être utilisé avec un embout buccal autre que celui fourni avec le produit. Le récipient est sous pression et ne doit en aucun cas être ouvert de force ni exposé à des températures supérieures à 50 °C.
4.4. Mises en garde spéciales et précautions d'emploi
L’action bronchodilatatrice de l’ipratropium est moins puissante que celle des bêta2 mimétiques par voie inhalée. Il convient en cas de crise d’asthme de ne pas l’utiliser en première intention ou seul et par conséquent de bien rappeler au patient d’avoir recours aux bronchodilatateurs bêta2 mimétiques par voie inhalée de courte durée d’action pour traiter les symptômes aigus.
Informer le patient qu’une consultation médicale immédiate est nécessaire si, en cas de crise d’asthme, le soulagement habituellement obtenu n’est pas rapidement observé après inhalation de ces traitements.
Si un patient développe en quelques jours une augmentation rapide de sa consommation en bronchodilatateurs bêta2 mimétiques à action rapide et de courte durée par voie inhalée, on doit craindre (surtout si les valeurs du débitmètre de pointe s’abaissent et/ou deviennent très irrégulières) une décompensation de sa maladie et la possibilité d’une évolution vers un état de mal asthmatique. Le médecin devra donc prévenir le patient de la nécessité dans ce cas, d’une consultation immédiate, sans avoir au préalable, dépassé les doses maximales prescrites. La conduite thérapeutique devra alors être réévaluée.
Chez les patients asthmatiques adultes, l’association à un traitement anti-inflammatoire continu doit être envisagée dès qu’il est nécessaire de recourir plus de 1 fois par semaine aux bêta2 mimétiques. Le patient doit, dans ce cas, être averti que l’amélioration de son état clinique ne doit pas conduire à une modification de son traitement, en particulier à l’arrêt de la corticothérapie par voie inhalée sans avis médical.
En raison de son activité anticholinergique, la projection accidentelle d’ipratropium dans l’œil provoque une mydriase par effet parasympatholytique. Les patients prédisposés à un risque de glaucome par fermeture de l’angle devront se protéger des risques de projections intraoculaires de ce médicament.
L’apparition de signes de glaucome par fermeture de l’angle (douleur ou gêne oculaire, vision trouble, perception d’images colorées, rougeur conjonctivale, congestion de la cornée) nécessite l’interruption du traitement et un avis médical spécialisé immédiat.
Comme avec les autres médicaments inhalés, ATROVENT peut entrainer un bronchospasme paradoxal pouvant mettre en jeu le pronostic vital. Si un bronchospasme paradoxal survient, ATROVENT doit être arrêté immédiatement et un autre traitement doit être instauré.
Précautions d’emploi
En cas d’infection bronchique ou de bronchorrhée abondante, un traitement approprié est nécessaire afin de favoriser la diffusion optimale du produit dans les voies respiratoires.
Le traitement par le bromure d’ipratropium doit être prescrit avec prudence chez les personnes âgées, notamment chez les sujets masculins présentant des antécédents d’obstruction des voies urinaires (par exemple, un adénome prostatique ou une obstruction urétrale).
Le bromure d’ipratropium sera utilisé avec prudence chez les patients atteints de mucoviscidose plus souvent sujets à des troubles de la motilité gastro-intestinale.
Veillez à éviter la pénétration du brumisat dans les yeux. L’aérosol doseur étant utilisé par l’intermédiaire d’un embout buccal et commandé manuellement, le risque de pénétration de brumisat dans les yeux est limité.
Excipients
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Association à prendre en compte
+ Médicaments atropiniques
L’administration concomitante chronique d’ATROVENT avec d’autres médicaments anticholinergiques n’a pas été étudiée et n’est donc pas recommandée.
Il faut prendre en compte le fait que les substances atropiniques peuvent additionner leurs effets indésirables et entraîner plus facilement une rétention urinaire, une poussée aiguë de glaucome, une constipation, une sécheresse de la bouche, etc
Les divers médicaments atropiniques sont représentés par les antidépresseurs imipraminiques, la plupart des antihistaminiques H1 atropiniques, les antiparkinsoniens anticholinergiques, les antispasmodiques atropiniques, le disopyramide, les neuroleptiques phénothiaziniques ainsi que la clozapine.
+ Médicaments anticholinestérasiques
Risque de moindre efficacité de l’anticholinestérasique par antagonisme des récepteurs de l’acétylcholine par l’atropine
+ Morphiniques
Risque important d’akinésie colique, avec constipation sévère
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études chez l'animal n'ont pas mis en évidence d'effet tératogène. En l’absence d’effet tératogène chez l’animal, un effet malformatif dans l’espèce humaine n’est pas attendu. En effet, à ce jour, les substances responsables de malformations dans l’espèce humaine se sont révélées tératogènes chez l’animal au cours d’études bien conduites sur deux espèces.
En clinique, aucun effet malformatif ou fœtotoxique particulier n’est apparu à ce jour. Toutefois, le suivi de grossesses exposées au bromure d’ipratropium est insuffisant pour exclure tout risque.
En conséquence, l’utilisation du bromure d’ipratropium ne doit être envisagée au cours de la grossesse que si nécessaire. En cas d’administration en fin de grossesse, tenir compte des répercussions possibles pour le nouveau-né des propriétés atropiniques de cette molécule.
En l’absence de données sur le passage dans le lait de l’ipratropium, et compte tenu de ses propriétés atropiniques son utilisation est déconseillée durant l’allaitement sauf nécessité absolue.
Fertilité
Les études précliniques réalisées avec le bromure d’ipratropium n’ont pas mis en évidence d’effets néfastes sur la fertilité (voir rubrique 5.3). Les données cliniques sur la fertilité ne sont pas disponibles pour le bromure d’ipratropium.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés.
Les patients doivent être informés du risque de troubles de l’accommodation visuelle, de mydriase, de vision trouble et de possibles sensations vertigineuses au cours du traitement par ATROVENT. Par conséquent, il convient de faire preuve de prudence lors de la conduite d’un véhicule ou de l’utilisation d’une machine.
Beaucoup d’effets indésirables listés peuvent être attribués aux propriétés anticholinergiques d’ATROVENT.
Comme tous les traitements par inhalation, ATROVENT peut engendrer des symptômes d'irritation locale. Résumé du profil de sécurité d’emploi
Les effets indésirables les plus fréquemment signalés lors des essais cliniques étaient les céphalées, les irritations de la gorge, la toux, la sécheresse de la bouche, les troubles de la motilité gastro-intestinale (constipation, diarrhée et vomissement), les nausées et les vertiges.
Tableau résumé des effets indésirables
Les effets indésirables suivants ont été rapportés dans les essais cliniques menés avec ATROVENT et de l’expérience acquise depuis la commercialisation.
Les effets indésirables ont été classés en fonction de leur fréquence en utilisant la classification suivante :
Très fréquent (³ 1/10) ; fréquent (³ 1/100, < 1/10) ; peu fréquent (³ 1/1000, < 1/100) ; rare (³ 1/10000, < 1/1000) ; très rare (< 1/10000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Terme usuel MedDRA |
Fréquence |
|
Affections du système immunitaire Hypersensibilité Réaction anaphylactique |
Peu fréquent Peu fréquent |
|
Affections du système nerveux Céphalées Sensations vertigineuses |
Fréquent Fréquent |
|
Affections oculaires Glaucome par fermeture de l’angle Augmentation de la pression intraoculaire Mydriase Douleur oculaire Œdème cornéen Hyperémie conjonctivale Vision de halo Vision trouble Troubles de l’accommodation visuelle |
Peu fréquent Peu fréquent Peu fréquent Peu fréquent Peu fréquent Peu fréquent Peu fréquent Peu fréquent Rare |
|
Troubles cardiaques Tachycardie supraventriculaire Palpitations Fibrillation auriculaire Tachycardie |
Peu fréquent Peu fréquent Rare Rare |
|
Affections respiratoires, thoraciques et médiastinales Toux Irritation pharyngée Bronchospasme Bronchospasme Laryngospasme Œdème pharyngé Sécheresse de la gorge |
Fréquent Fréquent Peu fréquent Peu fréquent Peu fréquent Peu fréquent Peu fréquent |
|
Affections gastro-intestinales Sécheresse de la bouche Nausées Troubles de la motilité gastro-intestinale Constipation Vomissements Diarrhées Œdème buccal Stomatite |
Fréquent Fréquent Fréquent Peu fréquent Peu fréquent Peu fréquent Peu fréquent Peu fréquent |
|
Affections de la peau et du tissu sous-cutané Rashs cutanés Prurit Œdème de Quincke Urticaire |
Peu fréquent Peu fréquent Peu fréquent Rare |
|
Affections du rein et des voies urinaires Rétention urinaire |
Peu fréquent |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
L'emploi à des doses très supérieures aux doses recommandées est le reflet d'une aggravation de l'affection respiratoire nécessitant une consultation médicale rapide pour réévaluation thérapeutique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : anticholinergiques, code ATC : R03BB01.
Ce médicament est un bronchodilatateur anticholinergique par voie inhalée.
Administré par voie inhalée, le bromure d'ipratropium exerce une action compétitive préférentielle au niveau des récepteurs cholinergiques du muscle lisse bronchique entraînant par effet parasympatholytique une relaxation de celui-ci et une bronchodilatation. Son effet bronchodilatateur est moins puissant que celui des bêta2 mimétiques par voie inhalée.
L'action bronchospasmolytique apparaît rapidement (environ 3 minutes) et persiste pendant 4 à 6 heures.
Les anticholinergiques préviennent l’augmentation de la concentration intracellulaire de Ca++ provoquée par l’interaction de l’acétylcholine avec les récepteurs muscariniques du muscle lisse bronchique. La libération de Ca++ a pour médiateur le système de second messager constitué d’IP3 (inositol triphosphate) et de DAG (diacylglycérol).
La bronchodilatation faisant suite à l’inhalation d’ATROVENT (bromure d’ipratropium) est principalement locale et spécifique au poumon, et non systémique.
5.2. Propriétés pharmacocinétiques
La quantité absorbée après administration par voie inhalée est minime et les taux sériques faibles correspondraient à l’absorption intestinale partielle de la fraction déglutie très faiblement absorbée par voie digestive. Après administration orale, cette absorption peut être évaluée entre 15 et 30 % de la quantité administrée. La demi-vie d’élimination est de l’ordre de 3,5 à 4 heures. L’excrétion se fait essentiellement par voie urinaire.
Après administration par voie inhalée, le passage de la barrière hémato-encéphalique est très faible.
L’excrétion rénale cumulée (0 à 24 heures) du composé parent est d’environ 46 % d’une dose administrée par voie intraveineuse, de moins de 1 % d’une dose orale et environ 3 à 13 % d’une dose inhalée. Sur la base de ces données, la biodisponibilité systémique totale de doses orales et inhalées de bromure d’ipratropium est estimée à respectivement 2 % et 7 à 28 %.
De ce fait, les portions de dose de bromure d’ipratropium avalées ne contribuent pas de manière importante à l’exposition systémique.
Distribution
Les paramètres cinétiques décrivant le sort de l’ipratropium ont été calculés à partir des concentrations plasmatiques après administration par voie intraveineuse. Une baisse biphasique rapide des concentrations plasmatiques est observée. Le volume de distribution apparent à l’équilibre (Vdss) est d’environ 176 L (environ 2,4 L/kg). Le médicament se lie de manière minimale (moins de 20 %) aux protéines plasmatiques. Les données précliniques indiquent que l’amine quaternaire ipratropium ne traverse pas le placenta ni la barrière hématoencéphalique.
Biotransformation
Après administration intraveineuse, environ 60 % de la dose sont métabolisés, probablement la majeure partie dans le foie par oxydation.
Les métabolites connus, qui sont formés par hydrolyse, deshydratation ou élimination du groupe hydroxy-méthyle dans le fragment acide tropique, montrent très peu ou pas d’affinité pour les récepteurs muscariniques et doivent être considérés comme inefficaces.
Élimination
La demi-vie de la phase d’élimination terminale est d’environ 1,6 heure.
L’ipratropium possède une clairance totale de 2,3 L/min et une clairance rénale de 0,9 L/min.
Dans une étude du bilan d’excrétion, l’excrétion rénale cumulée (6 jours) de la radioactivité liée au médicament (incluant le composé parent et tous ses métabolites) a représenté 72,1 % après administration intraveineuse, 9,3 % après administration orale et 3,2 % après inhalation. La radioactivité totale excrétée via les selles était de 6,3 % après administration intraveineuse, de 88,5 % après administration orale et de 69,4 % après inhalation. En ce qui concerne l’excrétion de la radioactivité liée au médicament après administration intraveineuse, l’excrétion principale se produit via les reins. La demi-vie d’élimination de la radioactivité liée au médicament (composé parent et métabolites) est de 3,6 heures.
5.3. Données de sécurité préclinique
Toxicité à dose unique
La toxicité aiguë par inhalation, administration orale et administration intraveineuse a été évaluée dans plusieurs espèces de rongeurs et de non-rongeurs.
En cas d’inhalation, la dose létale minimale chez le cochon d’Inde mâle est de 199 mg/kg.
Chez le rat, aucune mortalité n’a été observée jusqu’aux doses les plus élevées techniquement possibles (c’est-à-dire 0,05 mg/kg après 4 h d’administration ou 160 bouffées de bromure d’ipratropium, 0,02 mg/bouffée).
Les valeurs de DL 50 orale chez la souris, le rat et le lapin sont respectivement de 1 585, 1 925 et 1 920 mg/kg. La DL 50 intraveineuse chez la souris, le rat et le chien est, respectivement, de 13,6, 15,8 et environ 18,2 mg/kg. Les signes cliniques incluent une mydriase, une sécheresse de la muqueuse buccale, une dyspnée, des tremblements, des spasmes et/ou une tachycardie.
Toxicité à dose répétée
Des études de toxicité à dose répétée ont été menées chez des rats, des lapins, des chiens et des singes Rhésus.
Dans les études d’inhalation d’une durée allant jusqu’à 6 mois chez des rats, des chiens et des singes Rhésus, la NOAEL (dose sans effet indésirable observé) était de respectivement 0,38 mg/kg/jour, 0,18 mg/kg/jour et 0,8 mg/kg/jour. Une sécheresse de la muqueuse buccale et une tachycardie ont été notées chez les chiens. Aucune lésion histopathologique liée à la substance n’a été observée dans l’appareil bronchopulmonaire ni aucun autre organe. Chez le rat, la NOAEL après 18 mois d’administration orale était de 0,5 mg/kg/jour.
Des études de toxicité de l’inhalation à dose répétée, d’une durée allant jusqu’à 6 mois chez des rats et 3 mois chez des chiens, utilisant d’autres formulations (intranasale, avec propulseur HFA 134a et avec poudre de lactose) n’ont révélé aucune information supplémentaire sur le profil général de toxicité du bromure d’ipratropium.
L’administration intranasale pendant une durée allant jusqu’à 6 mois a révélé une NOEL (dose sans aucun effet) > 0,20 mg/kg/jour chez les chiens et a confirmé de précédentes études portant sur une administration intranasale pendant une durée allant jusqu’à 13 semaines. Des études de toxicité à dose répétée du bromure d’ipratropium ont montré que les profils toxicologiques de la formulation au HFA et de la préparation classique au CFC sont similaires.
Tolérance locale
Une solution aqueuse de bromure d’ipratropium (0,05 mg/kg) a été bien tolérée localement lors de l’administration à des rats par inhalation (administration unique sur 4 h). Dans les études de toxicité à dose répétée, le bromure d’ipratropium a été bien toléré localement.
Immunogénicité
Aucune anaphylaxie active ni réaction anaphylactique cutanée passive n’a été démontrée chez des cochons d’Inde.
Génotoxicité et carcinogénicité
Il n’a pas été observé de génotoxicité in vitro (test d’Ames), ni in vivo (test du micronoyau, test létal dominant chez la souris, essai cytogénétique sur des cellules de moelle osseuse de hamsters chinois).
Aucun effet tumorigène ni carcinogène n’a été démontré lors d’études à long terme chez des souris et des rats.
Toxicité de la reproduction et du développement
Des études visant à explorer l’influence possible du bromure d’ipratropium sur la fertilité, la toxicité embryonnaire et fœtale et le développement péri-/postnatal ont été conduites chez des souris, des rats et des lapins.
Des niveaux de dose orale élevés, c’est-à-dire de 1 000 mg/kg/jour chez le rat et de 125 mg/kg/jour chez le lapin, ont été associés à une toxicité maternelle dans les deux espèces et à une toxicité embryonnaire/fœtale chez le rat, où le poids des fœtus a été réduit. Il n’a pas été observé de malformations liées au traitement.
Les doses les plus élevées techniquement possibles pour l’inhalation au moyen de l’aérosol doseur, de 1,5 mg/kg/jour chez les rats et de 1,8 mg/kg/jour chez les lapins, n’ont été associées à aucun effet indésirable sur la reproduction.
Acide citrique anhydre, eau purifiée, éthanol anhydre.
Gaz de pressurisation : tétrafluoroéthane (HFA 134a).
3 ans en zones climatiques I et II
18 mois en zones climatiques III et IV
6.4. Précautions particulières de conservation
· à protéger des rayons du soleil et à ne pas exposer à une température supérieure à 25°C,
· ne pas percer ou brûler même après usage.
6.5. Nature et contenu de l'emballage extérieur
200 doses en flacon pressurisé (acier inoxydable) de 17 mL avec valve doseuse et embout buccal (polypropylène).
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
100-104 AVENUE DE FRANCE
75013 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 01/08/2022
ATROVENT20 microgrammes/dose, solution pour inhalation en flacon pressurisé
Bromure d’ipratropium anhydre
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
2. Quelles sont les informations à connaître avant d'utiliser ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé ?
3. Comment utiliser ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique anticholinergiques - code ATC : R03BB01
Qu’est-ce que ATROVENT 20 microgrammes/dose ?
ATROVENT 20 microgrammes/dose appartient à une famille de médicaments appelée les anticholinergiques.
Il agit en augmentant le diamètre des bronches.
Dans quels cas est-il utilisé ?
Votre médecin vous a prescrit ce médicament car vous avez besoin de recevoir cette substance active afin de traiter :
· les symptômes d’une crise d’asthme,
· ou des difficultés pour respirer provoquées par une maladie au long cours encombrant vos bronches et vos poumons (broncho-pneumopathie chronique obstructive).
Pour prendre ce médicament, vous utiliserez un flacon sous pression qui vous permettra de prendre ce médicament en utilisant votre respiration (également appelée voie inhalée).
Si vous avez une crise d’asthme aiguë ou des difficultés importantes pour respirer
Si vous avez une crise d’asthme aiguë ou une crise au cours de laquelle vous avez une difficulté importante pour respirer, ce médicament ne suffit pas à lui tout seul.
Vous devrez alors utiliser en complément un autre médicament par voie inhalée qui augmentera également le diamètre de vos bronches. Cet autre médicament contiendra une substance active appartenant à une famille de médicaments appelée les bêta2 mimétiques dont l’action est rapide et de courte durée. Un soulagement de la crise doit être observé rapidement après la prise de ces deux médicaments. En cas d’échec de cette association, consultez immédiatement votre médecin.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé?
N’utilisez jamais ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé :
· si vous êtes allergique à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6,
· si vous êtes allergique à une substance appartenant à la même famille (connue sous le nom d’atropine et ses dérivés),
· si l’utilisation de ce médicament a aggravé votre difficulté à respirer. Dans ce cas, vous ne devez en aucun cas renouveler la dose et consultez immédiatement votre médecin.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé.
Pour agir avec efficacité, ce médicament doit atteindre l’extrémité des petites bronches.
C’est pourquoi, l’efficacité du médicament peut être diminuée :
· si vos voies respiratoires (bronches, gorge ) sont encombrées,
· si vous avez une infection.
Si vous êtes dans l’une de ces situations, vous devez consulter votre médecin afin d’instaurer un traitement adapté.
Protection des yeux
La projection de ce médicament dans l’œil peut entraîner des troubles de la vue (flou, images colorées, sensation de voile devant l’œil) associés à une gêne ou à des douleurs.
Ces effets peuvent survenir en particulier :
si vous avez déjà eu dans le passé une maladie causée par une pression trop importante dans l’œil (appelée glaucome),
ou si vous êtes une personne susceptible de faire un glaucome.
Si vous êtes dans l’une de ces situations, vous devez :
prévenir votre médecin avant de manipuler ce médicament,
utiliser une protection (par exemple, porter des lunettes) afin d’éviter tout risque de projection de ce médicament dans l’œil,
et consulter immédiatement votre médecin si les troubles persistent.
Si vous avez reçu le médicament dans un œil, référez-vous aux rubriques 3 et 4 pour connaître les actions à entreprendre et les effets indésirables pouvant survenir.
Groupes particuliers de patients
Ce médicament doit être utilisé avec prudence :
chez les personnes de plus de 65 ans car ce médicament peut provoquer une difficulté voire une incapacité à uriner (et notamment chez les hommes qui ont déjà eu des troubles urinaires ou des problèmes de prostate),
chez les personnes atteintes d’une maladie appelée mucoviscidose car elles sont plus sensibles à certains troubles digestifs lorsqu’elles utilisent ce médicament.
Pendant le traitement
Vous devez arrêter de prendre ce médicament et consulter immédiatement votre médecin si vous développez une allergie. Les signes de l’allergie sont décrits à la rubrique 4.
La maladie peut s’aggraver et la dose qui vous soulage habituellement peut devenir insuffisante. Si les crises d’asthme et les difficultés à respirer deviennent plus fréquentes, dans ce cas, consultez rapidement votre médecin qui réévaluera votre traitement.
En cas d’apparition d’une oppression du thorax associée à une toux, une respiration sifflante ou des difficultés respiratoires survenant immédiatement après l’inhalation du produit (bronchospasme paradoxal pouvant mettre en jeu votre vie).
Enfants et adolescents
Sans objet.
Autres médicaments et ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Vous ne devez pas mélanger ce médicament avec d’autres médicaments ou solutions contenant un antiseptique (le chlorure de benzalkonium).
Vous devez indiquer à votre médecin si vous prenez l’un des médicaments suivants :
des médicaments pour traiter la dépression (les antidépresseurs imipraminiques),
des médicaments pour traiter une allergie (les antihistaminiques H1 atropiniques),
des médicaments pour traiter la maladie de Parkinson (les antiparkinsoniens anticholinergiques),
des médicaments pour traiter des contractions musculaires (les antispasmodiques atropiniques),
un médicament utilisé pour traiter certains troubles du rythme cardiaque (le disopyramide),
des médicaments utilisés pour traiter des troubles psychiatriques (les neuroleptiques phénothiaziniques),
un médicament utilisé pour traiter les troubles du comportement (la clozapine),
un anticholinestérasique (médicament utilisé dans le traitement de la maladie d’Alzheimer et dans certains cas de maladie de Parkinson),
un médicament à base de morphine (utilisé dans le traitement des douleurs importantes).
ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Il est préférable de ne pas utiliser ce médicament pendant la grossesse et l’allaitement. Si vous découvrez que vous êtes enceinte pendant le traitement ou si vous allaitez, consultez votre médecin, car lui seul peut juger du traitement le mieux adapté à votre cas.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé contient de l’éthanol
Ce médicament contient 8,415 mg d’alcool (éthanol) par dose.
La quantité par dose de ce médicament équivaut à moins de 1 ml de bière ou 1 ml de vin. La faible quantité d’alcool contenue dans ce médicament n’est pas susceptible d’entraîner d’effet notable.
3. COMMENT UTILISER ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin car celles-ci doivent être adaptées selon chaque patient. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Ne dépassez pas la dose quotidienne recommandée, que ce soit pendant la phase aiguë de traitement ou d’entretien.
Si le traitement n’apporte pas d’amélioration significative ou si votre état s’aggrave, demandez conseil à votre médecin qui réévaluera votre traitement. En cas d’aggravation de vos difficultés respiratoires, consultez immédiatement votre médecin.
La dose recommandée est de 1 à 2 bouffées à prendre :
soit uniquement quand vous avez une crise d’asthme aiguë ou des difficultés importantes pour respirer (en association avec un autre médicament). Cette dose peut éventuellement être répétée quelques minutes plus tard si nécessaire,
soit 2 à 4 fois par jour et à répartir régulièrement dans la journée si vous devez prendre ce médicament chaque jour et sur une longue durée (traitement de fond quotidien).
Dose maximale à ne pas dépasser
La dose quotidienne ne doit pas dépasser 16 bouffées par jour.
Si les troubles persistent, consultez immédiatement votre médecin.
Si vous avez l’impression que l’effet d’ATROVENT 20 microgrammes/dose est trop fort ou trop faible, consultez votre médecin ou votre pharmacien.
Voie d’administration
Ce médicament doit être administré par voie inhalée à l’aide d’un distributeur avec un embout qui se place à l’entrée de la bouche.
Ce médicament ne doit ni être avalé, ni être injecté.
Conseils concernant le flacon pressurisé
L’efficacité de ce médicament est en partie dépendante du bon usage de l’appareil utilisé.
Vous devez donc lire très attentivement le mode d’emploi. Au besoin, n’hésitez pas à demander à votre médecin de vous fournir des explications détaillées.
Protection des yeux
Il est recommandé de porter des lunettes de protection pour se protéger des risques de projection de ce médicament dans l’œil :
si vous (ou la personne qui administre ce médicament) avez déjà eu un glaucome,
ou si vous (ou la personne qui administre ce médicament) êtes une personne susceptible de faire un glaucome.
Si vous avez reçu le médicament dans les yeux, rincez abondamment à l’eau.
Mode d’administration
Si vous utilisez le flacon pour la 1ère fois, appuyez 2 fois sur l’embout buccal après avoir ôté le capuchon protecteur sans utiliser les 2 premières bouffées expulsées.
Si vous n’avez pas utilisé le flacon pendant 3 jours, réamorcez la valve en appuyant 1 fois sur l’embout buccal, sans utiliser la bouffée expulsée.
Contrôlez la technique d’inspiration devant une glace. Si une quantité importante de produit s’échappe par le nez ou par la bouche, les points suivants doivent être vérifiés :
· soit la pression sur la cartouche a été effectuée avant le début ou après la fin de l’inspiration,
· soit votre inspiration n’a pas été suffisamment profonde.
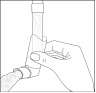
Pour utiliser ce médicament, veuillez suivre les étapes suivantes :
|
1. Ôtez le capuchon protecteur |
|
|
2. Videz vos poumons en expirant profondément. |
|
|
3. Présentez l’embout buccal à l’entrée de la bouche, le fond de la cartouche métallique dirigé vers le haut. |
|
|
4. Commencez à inspirer et pressez sur la cartouche métallique tout en continuant à inspirer lentement et profondément |
|
|
5. Retirez l’embout buccal et retenez votre respiration pendant au moins 10 secondes. L’embout buccal de l’appareil de propulsion doit, par mesure d’hygiène, être nettoyé après emploi. |
|
|
6. Replacez le capuchon protecteur après utilisation. |
|
|
7. Lorsque l’inhalateur n’a pas été utilisé pendant trois jours, la valve doit être actionnée une fois. |
|
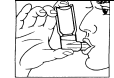
|
Le récipient n’étant pas transparent, il n’est pas possible de voir lorsqu’il est vide. L’inhalateur délivrera 200 bouffées (200 doses indiquées sur l’étiquette). Lorsque les 200 doses ont été utilisées (en général, après 3 semaines d’utilisation comme recommandé), la cartouche peut sembler contenir encore une petite quantité de liquide. L’inhalateur doit toutefois être remplacé afin d’être certain d’obtenir la bonne quantité délivrée de médicament à chaque utilisation. |
|
|
|
|
|
Nettoyez votre l’embout buccal de votre inhalateur au moins une fois par semaine. Il est important de maintenir l’embout buccal de l’inhalateur propre, afin d’éviter l’accumulation de médicament susceptible de bloquer le pulvérisateur. Pour le nettoyage, commencez par retirer le capuchon antipoussière, puis retirer la cartouche de l’embout buccal. Rincez l’embout buccal abondamment à l’eau chaude jusqu’à ce qu’aucune accumulation de médicament ni de poussière ne soit visible. |
|
|
Une fois le nettoyage terminé, secouez l’embout buccal et laissez-le sécher à l’air sans utiliser de système de chauffage. Lorsque l’embout buccal est sec, replacez la cartouche et le capuchon anti-poussière. |
|
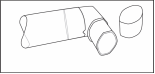
L’embout buccal en plastique est spécifiquement conçu pour être utilisé avec l’aérosol doseur ATROVENT 20 microgrammes/dose, afin de garantir l’administration de la quantité correcte de médicament. L’embout buccal ne doit jamais être utilisé avec un autre aérosol doseur et l’aérosol doseur ATROVENT 20 microgrammes/dose ne doit jamais être utilisé avec un embout buccal autre que celui fourni avec le produit. Le récipient est sous pression et ne doit en aucun cas être ouvert de force ni exposé à des températures supérieures à 50°C.
Durée du traitement
Conformez-vous à l’ordonnance de votre médecin.
Si vous avez utilisé plus de ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
La répétition abusive de la prise de ce médicament peut augmenter le risque que des effets indésirables surviennent (voir la rubrique 4 pour connaître les effets indésirables pouvant survenir avec ce médicament).
Si vous oubliez d’utiliser ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé:
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez d’utiliser ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé:
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables suivants nécessitent d’arrêter le traitement et de contacter ou de consulter un médecin :
une toux, une irritation localisée (nez, gorge, bouche) et plus rarement une augmentation de la difficulté à respirer. Dans ce cas, le médecin pourra décider de vous prescrire un autre traitement,
une allergie. Vous reconnaîtrez une allergie par des signes tels que urticaire, démangeaisons, rougeurs, boutons, difficultés à respirer, gonflement du visage, des lèvres, de la langue, de la gorge et du cou.
Les effets indésirables suivants peuvent survenir et disparaissent à l’arrêt du traitement :
des troubles du rythme cardiaque (battements trop rapides, trop lents ou irréguliers), une sensation anormale de perception des battements du cœur,
des troubles de la vue (problèmes d’accommodation visuelle),
des difficultés à uriner,
des vertiges,
des troubles digestifs tels qu’une constipation ou des vomissements.
La fréquence de survenue des effets indésirables est la suivante :
Fréquent : survient chez moins de 1 patient sur 10 mais plus de 1 patient sur 100,
Peu fréquent : survient chez moins de 1 patient sur 100 mais plus de 1 patient sur 1000,
Rare : survient chez moins de 1 patient sur 1000 mais plus de 1 patient sur 10000,
Non déterminée : la fréquence de survenue ne peut être estimée sur la base des données cliniques disponibles.
Les effets indésirables décrits ci-dessous ont été observés chez des patients prenant ce médicament et sont listés selon leur fréquence de survenue rapportée : fréquent, peu fréquent, rare ou avec une fréquence indéterminée.
Fréquent :
· Maux de tête,
· Sensations de vertige,
· Toux,
· Irritation localisée (nez, gorge, bouche),
· Sécheresse de la bouche,
· Nausées,
· Troubles de la motilité gastro-intestinale.
Peu fréquent :
· Réaction allergique (hypersensibilité),
· Réaction allergique sévère qui se manifeste par une difficulté respiratoire, une baisse importante de la pression artérielle (réaction anaphylactique),
· Glaucome par fermeture de l’angle,
· Augmentation de la pression intraoculaire,
· Dilatation de la pupille (mydriase),
· Douleur oculaire,
· Œdème de la cornée,
· Rougeur conjonctivale,
· Vision de halos lumineux autour des images lumineuses ou colorées,
· Vision trouble,
· Une sensation anormale de perception des battements du cœur (palpitations),
· Des troubles du rythme cardiaque (battements trop rapides, trop lents ou irréguliers),
· Oppression du thorax associée à une toux, une respiration sifflante ou des difficultés respiratoires survenant immédiatement après l’inhalation du produit (bronchospasme paradoxal),
· Spasme du larynx,
· Gonflement de la gorge (œdème pharyngé),
· Sécheresse de la gorge,
· Constipation,
· Vomissements,
· Diarrhées,
· Gonflement des lèvres ou de la langue (œdème buccal),
· Inflammation de la bouche (stomatite),
· Eruption cutanée (rash),
· Démangeaisons (prurit),
· Réactions allergiques sévères entraînant un œdème du visage ou de la gorge (œdème de Quincke),
· Des difficultés à uriner.
Rare :
· Des troubles de la vue (problèmes d’accommodation visuelle),
· Irrégularité du rythme cardiaque (fibrillation auriculaire),
· Augmentation du rythme du cœur (tachycardie),
· Urticaire.
Si vous avez reçu le médicament dans les yeux
vous pouvez ressentir les effets suivants : troubles de la vue (flou, images colorées, voile devant les yeux), dilatation des pupilles (mydriase), augmentation de la pression à l’intérieur de l’œil, douleur et rougeur de l’œil,
vous devez immédiatement rincer abondamment l’œil avec de l’eau. Si ces symptômes persistent, consultez d’urgence un médecin ou rendez-vous à l’hôpital le plus proche.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon, après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ne pas percer ou brûler, même après usage.
Conserver le flacon sous pression dans le conditionnement d’origine, à l’abri de la lumière à une température ne dépassant pas 25°C.
N’utilisez pas ce médicament si vous remarquez des signes visibles de détérioration.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ATROVENT 20 microgrammes/dose, solution pour inhalation en flacon pressurisé
· La substance active est :
Bromure d’ipratropium anhydre ................................................................... 20,00 microgrammes
Sous forme de bromure d’ipratropium monohydraté.
Pour une dose
· Les autres composants sont : l’acide citrique anhydre, l’eau purifiée et l’éthanol anhydre. Gaz de pressurisation : le tétrafluoroéthane (HFA 134a).
Ce médicament se présente sous forme d’une solution pour inhalation en flacon pressurisé.
Chaque flacon contient 200 doses.
Titulaire de l’autorisation de mise sur le marché
100-104 AVENUE DE FRANCE
75013 PARIS
Exploitant de l’autorisation de mise sur le marché
100-104 AVENUE DE FRANCE
75013 PARIS
BOEHRINGER INGELHEIM PHARMA GmbH & Co KG
BINGER STRASSE 173
55216 INGELHEIM AM RHEIN
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).