Dernière mise à jour le 29/06/2026
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie contient une substance de synthèse, appelée fondaparinux sodique. Il empêche le facteur de coagulation Xa (« dix-A ») d’agir dans le sang, et ainsi prévient la formation de caillots non souhaités (thromboses) dans les vaisseaux sanguins.
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie est indiqué pour traiter les patients adultes ayant un caillot dans les vaisseaux sanguins de leurs jambes (thrombose veineuse profonde) et/ou poumons (embolie pulmonaire).
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 22/08/2022
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Fondaparinux sodique........................................................................................................... 10 mg
Pour une seringue pré-remplie de 0,8 mL.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable en seringue pré-remplie.
La solution est limpide et incolore à légèrement jaune.
pH : 5,7-7,5.
Osmolalité : 255-315 mOsm/kg d’eau.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
La posologie recommandée de fondaparinux est de 7,5 mg (pour les patients dont le poids est compris entre 50 et 100 kg) une fois par jour, administrée par injection sous-cutanée. Pour les patients dont le poids est inférieur à 50 kg, la posologie recommandée est de 5 mg. Pour les patients dont le poids est supérieur à 100 kg, la posologie recommandée est de 10 mg.
Le traitement sera poursuivi pendant au moins 5 jours et jusqu’à ce que la posologie adéquate du traitement anticoagulant oral instauré en relais soit atteinte (International Normalized Ratio compris entre 2 et 3). Un traitement anticoagulant concomitant par voie orale doit être initié dès que possible et généralement dans les 72 heures. La durée moyenne d’administration dans les études cliniques était de 7 jours et l’expérience clinique au-delà de 10 jours est limitée.
Populations particulières
Sujets âgés
Aucune adaptation posologique n’est nécessaire. Chez les patients de 75 ans et plus, le fondaparinux doit être utilisé avec précaution, du fait de la dégradation de la fonction rénale liée à l’âge (voir rubrique 4.4).
Insuffisance rénale
Le fondaparinux doit être utilisé avec précaution chez les patients ayant une insuffisance rénale modérée (voir rubrique 4.4).
Il n’y a pas d’expérience dans le sous-groupe des patients de poids élevé (> 100 kg) et ayant une insuffisance rénale modérée (clairance de la créatinine comprise entre 30 et 50 ml/min). Dans ce sous-groupe, après une posologie initiale de 10 mg une fois par jour, une diminution de la posologie quotidienne à 7,5 mg peut être envisagée sur la base des données de modélisation pharmacocinétique (voir rubrique 4.4).
Le fondaparinux ne doit pas être utilisé chez les patients ayant une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) (voir rubrique 4.3).
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients atteints d'une insuffisance hépatique légère à modérée. Chez les patients ayant une insuffisance hépatique sévère, le fondaparinux doit être utilisé avec précaution : ce groupe de patients n’ayant pas été étudié (voir rubriques 4.4 et 5.2).
Population pédiatrique
L’utilisation du fondaparinux n’est pas recommandée chez l’enfant de moins de 17 ans, étant donné l’insuffisance des données de tolérance et d’efficacité (voir rubriques 5.1 et 5.2).
Mode d’administration
Le fondaparinux doit être injecté par voie sous-cutanée profonde, le patient étant en position allongée. Les sites d'injection doivent être alternés entre la ceinture abdominale antérolatérale et postérolatérale, alternativement du côté droit et du côté gauche. Pour éviter toute perte de médicament lors de l'utilisation de la seringue pré-remplie, ne pas purger la bulle d'air de la seringue avant d'effectuer l'injection. L'aiguille doit être introduite perpendiculairement sur toute sa longueur dans l'épaisseur d'un pli cutané réalisé entre le pouce et l'index ; ce pli cutané doit être maintenu pendant toute la durée de l'injection.
Pour des instructions supplémentaires sur l'utilisation, la manipulation et l'élimination, voir rubrique 6.6.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1
· Saignement évolutif cliniquement significatif
· Endocardite bactérienne aiguë
· Insuffisance rénale sévère (clairance de la créatinine < 30 ml/min).
4.4. Mises en garde spéciales et précautions d'emploi
Voie sous cutanée uniquement. Le fondaparinux ne doit pas être injecté par voie intramusculaire.
L’expérience du traitement des patients hémodynamiquement instables par fondaparinux est limitée, et il n’y a pas d’expérience chez les patients nécessitant une thrombolyse, une embolectomie, ou la mise en place d’un filtre cave.
Hémorragie
Le fondaparinux doit être utilisé avec précaution en cas de risque hémorragique accru, notamment troubles acquis ou congénitaux de la coagulation (par exemple, numération plaquettaire < 50 000/mm3), maladie ulcéreuse gastro-intestinale en poussée, hémorragie intracrânienne récente ou dans les suites récentes d'une intervention chirurgicale cérébrale, rachidienne ou ophtalmique, et dans les populations particulières mentionnées ci-dessous.
Comme pour les autres anticoagulants, le fondaparinux doit être utilisé avec précaution chez les patients qui ont bénéficié d’une intervention chirurgicale récente (< 3 jours) et seulement lorsqu’une hémostase chirurgicale a été établie.
Les traitements susceptibles d’accroître le risque hémorragique ne doivent pas être administrés en association avec le fondaparinux. Ces traitements comprennent : désirudine, agents fibrinolytiques, antagonistes du récepteur GP IIb/IIIa, héparine, héparinoïdes ou Héparines de Bas Poids Moléculaire (HBPM). Lors du traitement d’évènements thromboemboliques veineux, un traitement concomitant par antivitamine K sera administré selon les modalités définies à la rubrique 4.5. Les autres médicaments antiagrégants plaquettaires (acide acétylsalicylique, dipyridamole, sulfinpyrazone, ticlopidine ou clopidogrel) et les AINS doivent être utilisés avec précaution. Si l'association ne peut être évitée, une surveillance particulière s'impose.
Rachianesthésie/anesthésie péridurale
Chez les patients recevant du fondaparinux à titre curatif pour le traitement d’évènements thromboemboliques veineux, à la différence du traitement préventif, les anesthésies péridurales ou les rachianesthésies ne doivent pas être utilisées lors d’actes chirurgicaux.
Sujets âgés
Les sujets âgés présentent un risque accru de saignement. Une dégradation de la fonction rénale apparaissant généralement avec l’âge, les patients âgés peuvent présenter une réduction de l’élimination et un accroissement des concentrations plasmatiques de fondaparinux (voir rubrique 5.2). Chez les patients de moins de 65 ans, de 65 à 75 ans et de plus de 75 ans, traités aux doses recommandées pour des TVP ou des EP, l’incidence des hémorragies était respectivement de 3,0 %, 4,5 % et 6,5 %. Les incidences correspondantes pour les patients recevant enoxaparine aux doses recommandées pour le traitement d’une TVP étaient respectivement de 2,5 %, 3,6 % et 8,3 %, alors que les incidences chez les patients recevant une Héparine non fractionnée aux doses recommandées pour le traitement d’une EP étaient respectivement 5,5 %, 6,6 % et 7,4 %. Chez les patients âgés, le fondaparinux doit être utilisé avec précaution (voir rubrique 4.2).
Sujets de faible poids
L’expérience clinique est limitée chez les patients présentant un poids corporel < 50 kg. Le fondaparinux doit être utilisé avec prudence à une dose quotidienne de 5 mg dans cette population (voir rubriques 4.2 et 5.2).
Insuffisance rénale
Le risque de saignement augmente avec la dégradation de la fonction rénale. Le fondaparinux est essentiellement excrété par le rein. L’incidence des hémorragies chez les patients traités aux doses recommandées pour une TVP ou une EP, et ayant une fonction rénale normale, une insuffisance rénale légère, modérée ou sévère, était respectivement de 3,0 % (34/1132), 4,4 % (32/733), 6,6 % (21/318), et 14,5 % (8/55). Les incidences correspondantes pour les patients traités par enoxaparine aux doses recommandées pour le traitement d’une TVP étaient respectivement de 2,3 % (13/559), 4,6 % (17/368), 9,7 % (14/145) et 11,1 % (2/18), alors que les incidences chez les patients traités par une Héparine non fractionnée aux doses recommandées pour le traitement d’une EP étaient de respectivement 6,9 % (36/523), 3,1 % (11/352), 11,1 % (18/162) et 10,7 % (3/28).
Le fondaparinux est contre-indiqué chez l’insuffisant rénal sévère (clairance de la créatinine < 30 ml/min) et doit être utilisé avec précaution chez l’insuffisant rénal modéré (clairance de la créatinine entre 30 et 50 ml/min). La durée de traitement ne doit pas être supérieure à celle évaluée dans les études cliniques (en moyenne 7 jours) (voir rubriques 4.2, 4.3 et 5.2).
Il n’y a pas d’expérience dans le sous-groupe des patients ayant à la fois un poids élevé (> 100 kg) et une insuffisance rénale modérée (clairance de la créatinine comprise entre 30 et 50 ml/min). Le fondaparinux doit être utilisé avec précaution chez ces patients. Après une posologie initiale de 10 mg une fois par jour, une diminution de la posologie quotidienne à 7,5 mg peut être envisagée sur la base des données de modélisation pharmacocinétique (voir rubrique 4.2).
Insuffisance hépatique sévère
L’utilisation du fondaparinux doit être envisagée avec précaution en raison d'un risque hémorragique accru dû au déficit en facteurs de coagulation chez l’insuffisant hépatique sévère (voir rubrique 4.2).
Patients présentant une thrombopénie induite par l'héparine
Le fondaparinux doit être utilisé avec prudence chez les patients ayant des antécédents de TIH (Thrombocytopénie Induite par l’Héparine). L’efficacité et la tolérance du fondaparinux n’ont pas été étudiées de façon formelle chez les patients ayant une TIH de type II. Le fondaparinux ne se lie pas au facteur IV plaquettaire et il n’existe habituellement pas de réaction croisée avec le sérum des patients ayant une thrombocytopénie induite par l’héparine (TIH) de type II. Toutefois, de rares déclarations spontanées de TIH chez les patients traités par fondaparinux ont été rapportées.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est à dire qu’il est dit essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Dans les études cliniques réalisées avec du fondaparinux, les anticoagulants oraux (warfarine) n’ont pas modifié les paramètres pharmacocinétiques du fondaparinux ; à la dose de 10 mg, utilisée dans les études d’interaction, le fondaparinux n’a pas modifié l’effet de la warfarine sur l’INR.
Les antiagrégants plaquettaires (acide acétylsalicylique), les AINS (piroxicam) et la digoxine n'ont pas modifié les paramètres pharmacocinétiques du fondaparinux. A la dose de 10 mg utilisée dans les études d’interaction, le fondaparinux n'a pas modifié le temps de saignement sous traitement par acide acétylsalicylique ou piroxicam, ni la pharmacocinétique de la digoxine à l'état d'équilibre.
4.6. Fertilité, grossesse et allaitement
Grossesse
Aucune donnée clinique concernant des grossesses exposées n’est actuellement disponible. Les études conduites chez l'animal ne sont pas suffisantes pour exclure un effet sur la gestation, le développement embryonnaire ou fœtal, la mise bas ou le développement post-natal, du fait d’une exposition limitée. Le fondaparinux ne doit pas être utilisé chez la femme enceinte à moins d’une nécessité absolue.
Chez le rat, le fondaparinux est excrété dans le lait mais il n'existe pas de données concernant un éventuel passage du fondaparinux dans le lait maternel. L’allaitement n’est pas recommandé pendant le traitement par fondaparinux. L'absorption orale par l'enfant est cependant peu probable.
Fertilité
Aucune donnée de l'effet du fondaparinux sur la fertilité chez l'homme n'est disponible. Les études chez l’animal n’ont pas montré d’effet sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés.
Les effets indésirables graves les plus fréquemment rapportés avec le fondaparinux sont des complications à type de saignement (dans diverses localisations incluant de rares cas de saignements intracrâniens/intracérébraux ou rétropéritonéaux). Le fondaparinux doit être utilisé avec précaution chez les patients ayant un risque accru d’hémorragie (voir rubrique 4.4.).
La tolérance du fondaparinux a été évaluée chez 2517 patients traités pour des évènements thromboemboliques veineux pendant en moyenne 7 jours. Les effets indésirables les plus fréquents étaient les complications hémorragiques (voir rubrique 4.4).
Les effets indésirables rapportés par les investigateurs comme au moins possiblement liés au fondaparinux sont présentés au sein de chaque classe organe par ordre décroissant de fréquence de survenue (très fréquent : ³ 1/10 ; fréquent : ³ 1/100 à < 1/10 ; peu fréquent : ³ 1/1.000 à < 1/100 ; rare : ³ 1/10.000 à < 1/1.000 ; très rare : < 1/10.000) et de sévérité.
|
Classe-organe MedDRA |
Effets indésirables chez des patients traités pour des évènements thromboemboliques veineux (ETV)1 |
|
Affections hématologiques et du système lymphatique |
Fréquent : saignement (gastro-intestinal, hématurie, hématome, épistaxis, hémoptysie, hémorragie utérovaginale, hémarthrose, oculaire, purpura, ecchymose) Peu fréquent : anémie, thrombocytopénie. Rare : autres saignements (hépatique, rétropéritonéal, intracrânien/intracérébral) thrombocytopénie |
|
Affections du système immunitaire |
Rare : réaction allergique (incluant de très rares cas d’angiœdème, de réaction anaphylactoïde/anaphylactique) |
|
Troubles du métabolisme et de la nutrition |
Rare : augmentation de l’azote non-protéique (ANP)2 |
|
Affections du système nerveux |
Peu fréquent : céphalée Rare : vertige |
|
Affections gastro-intestinales |
Peu fréquent : nausées, vomissements Rare : douleur abdominale |
|
Affections hépatobiliaires |
Peu fréquent : anomalie de la fonction hépatique, augmentation des enzymes hépatiques |
|
Affections de la peau et du tissu sous-cutané |
Rare : éruption érythémateuse, prurit |
|
Troubles généraux et anomalies au site d'administration |
Peu fréquent : douleur, œdème Rare : réaction au site d’injection |
1 les effets indésirables rapportés isolément n’ont pas été pris en compte sauf s’ils étaient médicalement pertinents.
2 ANP signifie azote non-protéique comme l’urée, l’acide urique, l’acide aminé, etc.
Au cours de la surveillance post-marketing de rares cas de gastrite, de constipation, de diarrhée et de bilirubinémie ont été rapportés.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
L'administration de doses de fondaparinux supérieures à celles recommandées peut conduire à une augmentation du risque de saignement. Il n’existe pas d’antidote connu au fondaparinux.
Un surdosage associé à des complications hémorragiques doit conduire à l'arrêt du traitement et à la recherche de l’origine du saignement. L'instauration d'un traitement approprié tel que l’hémostase chirurgicale, la transfusion de sang ou de plasma frais, ou la plasmaphérèse, doit être envisagée.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : agent antithrombotique, code ATC : B01AX05
Effets pharmacodynamiques
Le fondaparinux est un inhibiteur synthétique et sélectif du Facteur X activé (Xa). L’activité antithrombotique du fondaparinux est le résultat de l’inhibition sélective du Facteur Xa par l’antithrombine III (antithrombine). En se liant sélectivement à l’antithrombine, le fondaparinux potentialise (environ 300 fois) l'inhibition naturelle du Facteur Xa par l’antithrombine. L'inhibition du Facteur Xa interrompt la cascade de la coagulation, en inhibant aussi bien la formation de la thrombine que le développement du thrombus. Le fondaparinux n’inactive pas la thrombine (Facteur II activé) et n’a pas d’effet sur les plaquettes.
Aux doses utilisées pour le traitement, le fondaparinux ne modifie pas, de façon cliniquement pertinente, les tests de coagulation de routine tels que le temps de céphaline activé (TCA), le temps de coagulation activé (ACT) ou le taux de prothrombine (TP) /International Normalised Ratio (INR) dans le plasma, ni le temps de saignement ou l'activité fibrinolytique. Toutefois, de rares déclarations spontanées d'élévation du TCA ont été enregistrées.
A doses plus élevées, le TCA peut être modifié de façon modérée. A la dose de 10 mg utilisée dans les études d’interaction, le fondaparinux n’a pas modifié, de façon significative, l’effet de la warfarine sur l’INR.
Il n’existe habituellement pas de réaction croisée entre le fondaparinux et le sérum des patients ayant une thrombopénie induite par l’héparine (TIH). De rares cas de TIH ont toutefois été rapportés spontanément chez des patients traités par fondaparinux.
Etudes cliniques
Le programme de développement clinique du fondaparinux dans le traitement des événements thromboemboliques veineux a été conçu pour démontrer l’efficacité du fondaparinux dans le traitement des thromboses veineuses profondes (TVP) et des embolies pulmonaires (EP). Plus de 4874 patients ont été étudiés dans des essais cliniques contrôlés de phase II et III.
Traitement des thromboses veineuses profondes
Dans un essai clinique, randomisé, en double-aveugle, chez des patients ayant un diagnostic confirmé de thrombose veineuse profonde aiguë symptomatique, le fondaparinux administré en une injection sous-cutanée par jour de 5 mg (poids inférieur à 50 kg), 7,5 mg (poids compris entre 50 et 100 kg) ou 10 mg (poids supérieur à 100 kg), a été comparé à l’enoxaparine 1 mg/kg administré en injection sous-cutanée deux fois par jour. Un total de 2192 patients a été traité dans les deux groupes les patients ont été traités au moins 5 jours, et jusqu’à 26 jours (en moyenne 7 jours). Les 2 groupes de patients ont reçu un traitement par anti-vitamine K, généralement initié dans les 72 heures après la première administration des produits étudiés et continué pendant 90 ± 7 jours, avec des adaptations régulières de posologie pour atteindre un INR de 2-3. Le critère principal d’efficacité était un critère combiné associant récidive confirmée d’évènements thromboemboliques veineux (ETEV) symptomatique non fatal et ETEV fatal rapportés dans les 97 jours. Il a été démontré que le fondaparinux est non-inférieur à l’énoxaparine (taux d’ETEV de 3,9 % et 4,1 % respectivement).
Pendant la phase initiale du traitement, des saignements majeurs ont été observés chez 1,1 % des patients traités par le fondaparinux et 1,2 % de ceux traités par l’énoxaparine.
Traitement des embolies pulmonaires
Un essai clinique randomisé, en ouvert, a été mené chez des patients ayant une embolie pulmonaire aiguë symptomatique. Le diagnostic a été confirmé par des tests objectifs (scanner pulmonaire, angiographie pulmonaire ou tomodensitométrie hélicoïdale). Les patients nécessitant une thrombolyse, une embolectomie, ou la mise en place d’un filtre cave ont été exclus. Les patients randomisés pouvaient avoir été traités dans un premier temps par une Héparine non fractionnée pendant la phase de sélection, mais les patients traités pendant plus de 24 heures avec des anticoagulants à dose thérapeutique ou ayant une hypertension non contrôlée, étaient exclus. Le fondaparinux administré en une injection sous-cutanée par jour de 5 mg (poids inférieur à 50 kg), 7,5 mg (poids compris entre 50 et 100 kg) ou 10 mg (poids supérieur à 100 kg), a été comparé à l’héparine non fractionnée, administrée en bolus IV (5000 UI) suivi d’une perfusion IV continue ajustée pour maintenir le TCA entre 1,5 et 2,5 fois la valeur témoin. Un total de 2184 patients ont été traités; dans les deux groupes, les patients ont été traités au moins 5 jours et jusqu’à 22 jours (en moyenne 7 jours). Les 2 groupes de patients ont reçu un traitement par anti-vitamine K, généralement initié dans les 72 heures après la première administration du produit étudié et continué pendant 90 ± 7 jours, avec des adaptations régulières de la posologie pour atteindre un INR de 2-3. Le critère principal d’efficacité était un critère combiné associant récidive confirmée d’ETEV symptomatique non fatal et ETEV fatal rapportés dans les 97 jours. Il a été démontré que le fondaparinux est non-inférieur à l’héparine non fractionnée (taux d’ETEV de 3,8 % et 5,0 % respectivement).
Pendant la phase initiale du traitement, des saignements majeurs ont été observés chez 1,3 % des patients traités par le fondaparinux et 1,1 % de ceux traités par l’héparine non fractionnée.
Etude pharmacocinétique pilote de recherche de dose du fondaparinux chez les enfants atteints de thrombose veineuse profonde
Une étude clinique a été réalisée en ouvert chez 24 enfants (10 âgés de 1 à 5 ans, avec un poids compris entre 8 et 20 kg; 7 âgés de 6 à 12 ans avec un poids compris entre 17 et 47 kg et 7 âgés de 13 à 18 ans avec un poids compris entre 47 et 130 kg) présentant une thrombose veineuse diagnostiquée à l’inclusion et traités par du fondaparinux. La plupart des enfants étaient d’origine hispanique (67%) et de sexe masculin (58%). Le fondaparinux a été administré à une dose initiale de 0,1 mg/kg en sous-cutanée une fois par jour ; la dose a été adaptée pour atteindre des concentrations maximales de fondaparinux sodique de 0,5 à 1 mg/L après 4 heures. La durée médiane de traitement dans cette étude était de 3,5 jours.
La plupart des enfants (88%) ont atteint les concentrations cibles de fondaparinux 4 heures après la première dose de fondaparinux. Deux patients ont présenté des saignements pendant l'étude. L’un a présenté une encéphalopathie hypertensive accompagnée d’une hémorragie intracrânienne au 5ème jour du traitement, conduisant à l'arrêt du fondaparinux. Une hémorragie gastro-intestinale mineure a été signalée chez un autre patient au 5ème jour du traitement, entrainant l'arrêt temporaire du fondaparinux. Aucune conclusion sur l'efficacité clinique ne peut être tirée de cette étude non contrôlée.
5.2. Propriétés pharmacocinétiques
Les paramètres pharmacocinétiques du fondaparinux sodique ont été déterminés à partir des concentrations plasmatiques de fondaparinux quantifiées par l’activité anti-Xa. Seul le fondaparinux peut être utilisé pour étalonner le test anti-Xa (les standards internationaux des héparines, ou des HBPM ne sont pas appropriés dans ce cas). Par conséquent, la concentration en fondaparinux est exprimée en milligrammes (mg).
Après administration sous-cutanée, le fondaparinux est entièrement et rapidement absorbé (biodisponibilité absolue 100 %). Après une injection unique sous-cutanée de 2,5 mg de fondaparinux chez le volontaire sain jeune, la concentration plasmatique maximale (Cmax moyenne = 0,34 mg/l) est obtenue 2 heures après l’administration. Les valeurs des concentrations plasmatiques correspondant à la moitié de la Cmax moyenne sont atteintes 25 minutes après l’administration.
Chez le volontaire sain âgé, la pharmacocinétique du fondaparinux administré par voie sous-cutanée est linéaire entre 2 et 8 mg. A une injection par jour, l'état d’équilibre des concentrations plasmatiques est obtenu en 3 à 4 jours, avec une Cmax et une ASC augmentées d’un facteur de 1,3.
Chez les patients ayant bénéficié d’une prothèse de hanche et recevant une dose quotidienne de 2,5 mg de fondaparinux, les paramètres pharmacocinétiques du fondaparinux [moyennes (coef. variation)] estimés à l’état d’équilibre sont : Cmax (mg/l) : 0,39 (31 %), Tmax (h) : 2,8 (18 %) et Cmin (mg/l) : 0,14 (56 %). En raison de l’âge plus élevé des patients ayant eu une fracture de hanche, les concentrations plasmatiques du fondaparinux à l’état d’équilibre sont : Cmax (mg/l) : 0,50 (32 %) et Cmin (mg/l) : 0,19 (58 %).
Dans le traitement des thromboses veineuses profondes et des embolies pulmonaires, chez les patients traités par le fondaparinux 5 mg (poids inférieur à 50 kg), 7,5 mg (poids compris entre 50 et 100 kg) ou 10 mg (poids supérieur à 100 kg) une fois par jour, l’ajustement de la posologie selon le poids du patient permet une exposition au produit similaire quelle que soit la catégorie de poids. Chez les patients ayant un ETEV et recevant quotidiennement le fondaparinux à la posologie proposée, les paramètres pharmacocinétiques du fondaparinux [moyennes (coef. variation)] estimés à l’état d’équilibre sont : Cmax (mg/l) : 1,41 (23 %), Tmax (h) : 2,4 (8 %) et Cmin (mg/l) : 0,52 (45 %). Les 5ème et 95ème percentiles associés sont respectivement 0,97 et 1,92 pour Cmax (mg/l), et 0,24 et 0,95 pour Cmin (mg/l).
Distribution
Le volume de distribution du fondaparinux est faible (7 à 11 litres). In vitro, le fondaparinux se lie fortement et spécifiquement à l’ATIII ; cette liaison est dépendante des doses et des concentrations plasmatiques (de 98,6 % à 97,0 % pour des concentrations de 0,5 à 2 mg/l).
Le fondaparinux ne se lie pas significativement aux autres protéines plasmatiques, y compris au facteur plaquettaire IV (FP4). Le fondaparinux ne se liant pas significativement aux protéines plasmatiques à l'exception de l'antithrombine, aucune interaction avec d'autres médicaments par déplacement de la liaison protéique n'est attendue.
Biotransformation
Il n'existe aucun élément en faveur d'un métabolisme du fondaparinux, et en particulier de formation de métabolites actifs, bien que celui-ci n’ait pas été complètement évalué.
Le fondaparinux n'inhibe pas les cytochromes P450 (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ou CYP3A4) in vitro. Aucune interaction du fondaparinux avec d'autres médicaments n'est donc attendue in vivo par inhibition du métabolisme lié au CYP.
Élimination
La demi-vie (t½) d’élimination est d’environ 17 heures chez les sujets sains jeunes, et d’environ 21 heures chez les sujets sains âgés. 64 à 77 % du fondaparinux est excrété par le rein sous forme inchangée.
Populations particulières
Population pédiatrique
Des données limitées sont disponibles dans la population pédiatrique (voir rubrique 5.1).
Sujets âgés
Compte tenu de la possible altération de la fonction rénale liée à l’âge, la capacité à éliminer le fondaparinux peut être réduite chez les sujets âgés. Chez le sujet de plus de 75 ans, ayant subi une intervention de chirurgie orthopédique et recevant une dose quotidienne de 2,5 mg de fondaparinux, la clairance plasmatique estimée était 1,2 à 1,4 fois inférieure à celle des sujets de moins de 65 ans. Un résultat similaire est observé chez les patients traités pour thrombose veineuse profonde ou embolie pulmonaire.
Insuffisance rénale
Comparée aux patients avec une fonction rénale normale (clairance de la créatinine > 80 ml/min), ayant subi une intervention de chirurgie orthopédique et recevant une dose quotidienne de 2,5 mg de fondaparinux, la clairance plasmatique est 1,2 à 1,4 fois inférieure chez les patients ayant une insuffisance rénale légère (clairance de la créatinine entre 50 et 80 ml/min), et en moyenne 2 fois inférieure chez les patients ayant une insuffisance rénale modérée (clairance de la créatinine entre 30 et 50 ml/min). En cas d’insuffisance rénale sévère (clairance de la créatinine < 30 ml/min), la clairance plasmatique est environ 5 fois plus faible qu’en cas de fonction rénale normale. La demi-vie terminale d’élimination est respectivement de 29 h et de 72 h chez les patients insuffisants rénaux modérés et sévères. Un résultat similaire est observé chez les patients traités pour thrombose veineuse profonde ou embolie pulmonaire.
Poids corporel
La clairance plasmatique du fondaparinux augmente avec le poids (9 % par 10 kg de poids).
Sexe
Après ajustement au poids corporel, aucune différence liée au sexe n’a été mise en évidence.
Origine ethnique
Les différences de pharmacocinétique liées à l’ethnie n’ont pas été étudiées prospectivement. Cependant, des études réalisées chez des sujets sains asiatiques (japonais) n’ont pas mis en évidence de profil pharmacocinétique particulier en comparaison aux sujets sains de type caucasien. Aucune différence dans les clairances plasmatiques n’a été observée entre les patients d'origine ethnique noire et caucasienne après chirurgie orthopédique.
Insuffisance hépatique
Lors de l’administration d’une dose unique en sous-cutanée de fondaparinux chez des sujets ayant une insuffisance hépatique modérée (Child-Pugh B), la Cmax et l’ASC des concentrations totales (i.e. liées et non liées) ont diminué respectivement de 22% et 39%, par rapport aux sujets ayant une fonction hépatique normale. Cette baisse des concentrations plasmatiques de fondaparinux a été attribuée à une réduction de la liaison à l’ATIII secondaire à une diminution des concentrations plasmatiques en ATIII chez les sujets ayant une insuffisance hépatique, avec pour conséquence une augmentation de la clairance rénale du fondaparinux. Par conséquent, les concentrations de fondaparinux non liées ne devraient pas être changées chez les patients ayant une insuffisance hépatique légère à modérée. Aucun ajustement posologique n'est nécessaire d’après les résultats des études pharmacocinétiques.
La pharmacocinétique du fondaparinux n'a pas été étudiée chez des patients ayant une insuffisance hépatique sévère (voir rubriques 4.2 et 4.4).
5.3. Données de sécurité préclinique
Les données non cliniques issues des études classiques de pharmacologie générale, et de génotoxicité n'ont pas révélé de risque particulier pour l'homme. Les études de toxicité par administration réitérée et les études sur la reproduction n’ont pas révélé de risques particuliers, mais n’ont pas permis d’établir de façon adéquate des marges de sécurité, du fait d’une exposition limitée des espèces animales.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
2 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Seringue en verre transparent de type I (1 ml) munie d’une aiguille et d’un capuchon d’élastomère de chlorobutyl pour le piston. L’ensemble est conditionné dans une barquette contenue dans une boîte.
La seringue se présente avec un piston violet et un système de sécurité automatique.
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie est disponible en boîte de 2, 7, 10, 20 et 30 seringues pré-remplies.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
L'injection sous-cutanée est réalisée de la même façon qu'avec une seringue classique.
Les solutions parentérales doivent être examinées visuellement avant l’administration afin de vérifier l’absence de particules ou de décoloration.
Les instructions concernant l’auto-administration sont présentées dans la notice.
Le système de sécurité des seringues pré-remplies de FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml a été conçu avec un système de sécurité, destiné à éviter les piqûres accidentelles après injection.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Usage unique.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
9 AVENUE EDOUARD BELIN
92500 RUEIL-MALMAISON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 649 4 8 : solution injectable en seringue pré-remplie (verre) de 0,8 ml ; boîte de 2.
· 34009 300 649 5 5 : solution injectable en seringue pré-remplie (verre) de 0,8 ml ; boîte de 7.
· 34009 300 649 6 2 : solution injectable en seringue pré-remplie (verre) de 0,8 ml ; boîte de 10.
· 34009 550 236 1 6 : solution injectable en seringue pré-remplie (verre) de 0,8 ml ; boîte de 20.
· 34009 550 236 2 3 : solution injectable en seringue pré-remplie (verre) de 0,8 ml ; boîte de 30.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 22/08/2022
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie
Fondaparinux sodique
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ?
3. Comment utiliser FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ET DANS QUELS CAS EST-IL UTILISE ?
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie contient une substance de synthèse, appelée fondaparinux sodique. Il empêche le facteur de coagulation Xa (« dix-A ») d’agir dans le sang, et ainsi prévient la formation de caillots non souhaités (thromboses) dans les vaisseaux sanguins.
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie est indiqué pour traiter les patients adultes ayant un caillot dans les vaisseaux sanguins de leurs jambes (thrombose veineuse profonde) et/ou poumons (embolie pulmonaire).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ?
N'utilisez jamais FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie :
· si vous êtes allergique au fondaparinux sodique ou à l'un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6) ;
· si vous saignez de manière excessive ;
· si vous souffrez d'une infection cardiaque bactérienne ;
· si vous souffrez d'une affection rénale sévère.
Parlez-en à votre médecin si vous pensez être concerné. Si c’est le cas, vous ne devez pas utiliser FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie :
· si vous avez déjà présenté, pendant un traitement par héparine ou par des médicaments apparentés à l’héparine, des complications ayant entraîné une diminution du nombre de plaquettes dans le sang (thrombopénie induite par l'héparine)
· si vous avez un risque de saignement incontrôlé (hémorragie) incluant :
o ulcère de l'estomac ;
o troubles hémorragiques ;
o hémorragie cérébrale récente (hémorragie intracrânienne) ;
o opération récente du cerveau, de la colonne vertébrale ou des yeux ;
· si vous souffrez d'une maladie sévère du foie ;
· si vous souffrez d'une insuffisance rénale ;
· si vous êtes âgé de 75 ans ou plus.
Parlez-en à votre médecin si cela vous concerne.
Enfants et adolescents
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie n’a pas été étudié chez les enfants et chez les adolescents de moins de 17 ans.
Autres médicaments et FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Si vous prenez, avez récemment pris ou pourriez prendre un autre médicament, y compris un médicament obtenu sans ordonnance, parlez-en à votre médecin ou à votre pharmacien. Certains médicaments peuvent modifier l’action de FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ou FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie peut modifier leur action.
Sans objet.
Grossesse et allaitement
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ne doit pas être prescrit à une femme enceinte sauf en cas de réelle nécessité.
L’allaitement n’est pas recommandé pendant le traitement avec FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie. Si vous êtes enceinte, ou que vous allaitez, si vous pensez être enceinte ou envisagez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Sans objet.
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie contient du sodium.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Posologie
|
Votre poids |
Dose usuelle |
|
Moins de 50 kg |
5 mg par jour |
|
Entre 50 et 100 kg |
7,5 mg par jour |
|
Plus de 100 kg |
10 mg par jour. La dose peut être réduite à 7,5 mg par jour si vous souffrez d’une insuffisance rénale modérée. |
L’injection doit être réalisée à peu près à la même heure tous les jours.
Mode d’administration
· FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie est administré par injection sous la peau (sous-cutanée), dans un pli cutané réalisé dans la partie inférieure de l’abdomen. Les seringues sont préremplies avec la dose exacte qui vous est nécessaire. Il existe des seringues différentes pour les dosages 2,5 mg, 5 mg, 7,5 mg et 10 mg. Voir pages suivantes pour le mode d’emploi détaillé.
· Ne pas injecter FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie dans un muscle.
Durée du traitement
FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie vous protège contre une maladie grave, vous devez donc continuer votre traitement aussi longtemps que votre médecin vous l’a indiqué.
Si vous avez utilisé plus de FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie que vous n’auriez dû :
Demandez conseil à votre médecin ou votre pharmacien dès que possible en raison du risque accru d'hémorragie.
Si vous oubliez d’utiliser FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie :
· Prenez la dose prescrite dès que vous constatez l’oubli. Ne pratiquez pas une double injection pour compenser celle que vous avez oubliée.
· En cas de doute, contactez votre médecin ou votre pharmacien.
Si vous arrêtez d’utiliser FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie :
Si vous interrompez votre traitement avant la fin de la durée prescrite par votre médecin, le caillot peut ne pas être traité correctement et un nouveau caillot de sang peut se former dans les veines de vos jambes ou poumons. Contactez votre médecin ou votre pharmacien avant d'interrompre votre traitement.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Situations que vous devez surveiller
Réactions allergiques sévères (anaphylaxie) : Elles surviennent très rarement (jusqu’à 1 personne sur 10 000) chez les patients prenant FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie. Les signes comprennent :
· gonflement, parfois du visage ou de la bouche (angiœdème), entraînant une difficulté à avaler ou à respirer ;
· collapsus
Contactez immédiatement un médecin si vous ressentez ces symptômes. Arrêtez de prendre FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie.
Les autres effets indésirables sont :
Effets indésirables fréquents
Ils peuvent affecter plus de 1 personne sur 100 traitées par FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie :
· saignement (par exemple du foyer opératoire, d’un ulcère de l’estomac préexistant, du nez, ecchymoses).
Effets indésirables peu fréquents
Ils peuvent affecter jusqu’à 1 personne sur 100 traitées par FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie.
· gonflement (œdème) ;
· maux de tête ;
· douleur ;
· se sentir ou être nauséeux (nausées ou vomissements) ;
· nombre bas de globules rouges (anémie) ;
· nombre bas de plaquettes (cellules nécessaires à la formation de caillots sanguins) ;
· augmentation des enzymes du bilan hépatique.
Effets indésirables rares
Ils peuvent affecter jusqu’à 1 personne sur 1 000 traitées par FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie.
· réaction allergique (incluant démangeaisons, gonflement, éruption) ;
· saignements internes cérébraux, hépatiques ou abdominaux ;
· éruption ;
· vertiges ;
· douleur et gonflement au point d’injection ;
· nombre élevé de plaquettes (cellules nécessaires à la formation de caillots sanguins) ;
· augmentation de la quantité d’azote non-protéique dans le sang ;
· douleur abdominale ;
· démangeaisons ;
· indigestion ;
· diarrhée ou constipation ;
· augmentation de la bilirubine (une substance produite par le foie) dans le sang.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette de la seringue pré-remplie et la boîte après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne pas utiliser ce médicament :
· si vous constatez la présence de particules dans la solution, ou si la solution est d’une couleur anormale ;
· si vous constatez que la seringue est endommagée ;
· si vous avez ouvert la seringue sans l’utiliser tout de suite.
Elimination des seringues
Ne jetez aucun médicament ni seringue au tout-à-l’égout ni avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Fondaparinux sodique..................................................................................................... 10 mg
Pour une seringue pré-remplie de 0,8 mL.
· Les autres composants sont : le chlorure de sodium, l’eau pour préparations injectables et l’acide chlorhydrique et/ou l’hydroxyde de sodium pour l’ajustement du pH.
Ce médicament est une solution injectable limpide et incolore à légèrement jaune, contenue dans une seringue pré-remplie à usage unique avec un piston violet et un système de sécurité automatique.
Il est disponible en boîtes de 2, 7, 10, 20 et 30 seringues pré-remplies.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
9 AVENUE EDOUARD BELIN
92500 RUEIL-MALMAISON
Exploitant de l’autorisation de mise sur le marché
9 AVENUE EDOUARD BELIN
92500 RUEIL-MALMAISON
DR. REDDY´S LABORATORIES (UK) LTD.
6 RIVERVIEW ROAD
BEVERLEY
EAST YORKSHIRE HU17 0LD
ROYAUME-UNI
OU
betapharm Arzneimittel GmbH
Kobelweg 95
86156 Augsburg
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
INSTRUCTIONS ETAPE PAR ETAPE D’UTILISATION DU FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie
|
Instructions pour l’auto-injection |
||
|
Les différentes parties de la seringue de sécurité de FONDAPARINUX SODIQUE REDDY PHARMA 10 mg/0,8 ml, solution injectable en seringue pré-remplie sont les suivantes : |
||
|
1. Capuchon de protection de l’aiguille 2. Piston 3. Ailettes appui-doigts 4. Manchon de sécurité |
|
|
|
Seringue AVANT UTILISATION
|
Seringue APRÈS UTILISATION
|
|
|
1. Lavez-vous les mains soigneusement avec de l'eau et du savon. Essuyez-les avec une serviette. 2. Asseyez-vous ou allongez-vous dans une position confortable. Choisissez un endroit dans la partie inférieure du ventre (abdomen) à au moins 5 cm sous le nombril (Figure A). Changez (alternez) de côté pour chaque injection, tantôt du côté droit tantôt du côté gauche de la partie inférieure de l’abdomen. Si vous avez des questions, contactez votre infirmier/ère ou votre médecin. |
|
|
|
3. Nettoyez la zone de l’injection avec un coton imprégné d'alcool. |
||
|
4. Retirez le capuchon de protection de l’aiguille en le tirant (Figure B). Jetez le capuchon de protection de l’aiguille. Pour éviter toute infection, ne touchez pas l’aiguille et ne la mettez pas en contact avec une surface quelle qu’elle soit avant l’injection. La présence d’une petite bulle d’air dans la seringue est normale. Pour s’assurer de ne pas perdre de médicament, n’essayez pas de retirer les bulles d’air de la seringue avant l’injection. |
|
|
|
5. Pincez doucement la peau que vous avez nettoyée pour faire un pli. Maintenez ce pli entre le pouce et l’index de la même main pendant toute la durée de l’injection (Figure C). |
|
|
|
6. Tenez fermement la seringue dans votre autre main à l’aide des ailettes appui-doigts. Insérez l’aiguille dans toute sa longueur perpendiculairement (à un angle de 90°) dans le pli de peau (Figure D). |
|
|
|
7. Injectez tout le médicament contenu dans la seringue en poussant le piston aussi loin que possible. (Figure E). |
|
|
|
8. Retirez la seringue du site d’injection en gardant votre doigt sur le piston. |
|
|
|
9. Orientez l’aiguille loin de vous-même et des autres et activez le manchon de sécurité en poussant fermement sur le piston. Le manchon de sécurité recouvrira automatiquement l’aiguille et vous entendrez un « clic » qui confirmera l’activation de la protection. |
|
|
|
Respectez les instructions de votre infirmier/ère ou de votre médecin concernant l’élimination des seringues et aiguilles utilisées. Il peut exister des lois sur l’élimination des seringues et des aiguilles utilisées et des récipients de stockage. REMARQUES : · Le manchon de sécurité ne peut être activé qu’une fois la seringue vidée. · L’activation du manchon de sécurité doit être effectuée uniquement une fois l’aiguille retirée de la peau du patient. · Ne pas remettre le capuchon de protection de l’aiguille après l’injection. · Le manchon de sécurité ne doit pas être stérilisé. · L’activation du manchon de sécurité peut provoquer des éclaboussures minimes de liquide. Pour une sécurité optimale, activez le système en l’orientant vers le bas, loin de vous-même et des autres. |
||
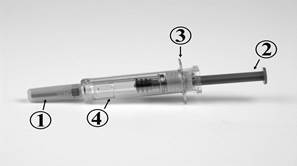
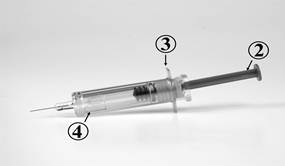
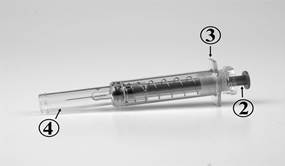
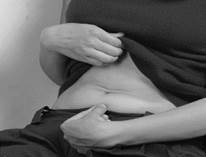
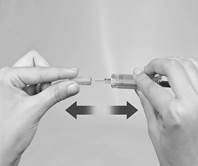 Figure B.
Figure B. Figure C.
Figure C.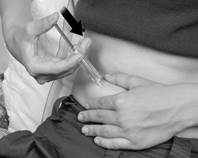 Figure D.
Figure D.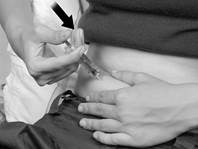 Figure E.
Figure E.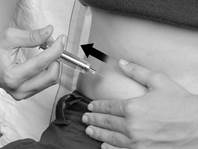 Figure F.
Figure F.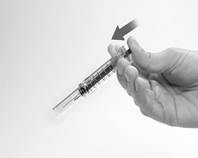 Figure G.
Figure G.