Dernière mise à jour le 03/08/2026
DRAFORA, solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique : sans objet.
DRAFORA, solution injectable est un médicament homéopathique traditionnellement utilisé chez l’adulte en cas d’arthrose et d’arthropathies dégénératives.
Composition en substances actives
-
Solution ( Composition pour une ampoule de 1 mL )
- > arnica montana pour préparations homéopathiques 0,167 g (15 DH)
- > betula alba, folium pour préparations homéopathiques 0,167 g (4 DH)
- > cartilago pour préparations homéopathiques 0,167 g (8 DH)
- > equisetum arvense, décocté pour préparations homéopathiques 0,167 g (15 DH)
- > formica rufa pour préparations homéopathiques 0,167 g (10 DH)
- > mandragora officinarum, décocté pour préparations homéopathiques 0,167 g (4 DH)
Présentations
> 8 ampoule(s) en verre de 1 ml
Code CIP : 34009 302 431 2 1
Déclaration de commercialisation : 03/01/2023
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 02/07/2022
DRAFORA, solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Arnica montana 15DH........................................................................................................... 0,167 g
Betula alba, folium 4DH........................................................................................................ 0,167 g
Cartilago 8DH....................................................................................................................... 0,167 g
Equisetum arvense, décocté 15DH........................................................................................ 0,167 g
Formica rufa 10DH............................................................................................................... 0,167 g
Mandragora officinarum, décocté 4DH.................................................................................. 0,167 g
Pour une ampoule de 1 mL.
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Une injection sous-cutanée d’une fois par jour à une fois par semaine pendant 4 semaines. Le traitement pourra être renouvelé si nécessaire.
Mode d’administration
Voie injectable sous-cutanée.
Tenir le corps de l’ampoule à la verticale et saisir la tête entre le pouce et l’index au niveau du point rouge. Une pression par effet levier permet de casser l’ampoule au niveau de l’étranglement. Le contenu de l’ampoule est prélevé en une fois au moyen d’une seringue stérile munie d’une aiguille stérile. L’air est chassé de la seringue.
L'injection doit être réalisée dans les tissus sous-cutanés, de préférence dans l’abdomen, dans la partie supérieure de la cuisse ou dans la partie supérieure du bras. L'aiguille doit être introduite complètement dans l'épaisseur du pli cutané formé par le pouce et l'index. Il convient de veiller à ne pas pénétrer un vaisseau sanguin. Le produit doit être injecté lentement et à vitesse constante dans des conditions d’aseptie adéquates. Garder le pli de peau maintenu entre le pouce et l'index pendant l'injection.
DRAFORA, solution injectable ne doit pas être administré par voie intraveineuse (IV) ou voie intramusculaire (IM).
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Enfants et adolescents de moins de 18 ans.
4.4. Mises en garde spéciales et précautions d'emploi
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose administrée, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée.
4.6. Fertilité, grossesse et allaitement
En l’absence de données expérimentales et cliniques, et par mesure de précaution, l’utilisation de ce médicament est à éviter pendant la grossesse et l’allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Sans objet.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Médicament homéopathique.
En l’absence de données scientifiques, l’indication de ce médicament repose sur l’usage homéopathique traditionnel de ses composants.
5.2. Propriétés pharmacocinétiques
Sans objet.
5.3. Données de sécurité préclinique
Véhicules utilisés pour la dilution finale : chlorure de sodium, eau pour préparations injectables.
3 ans.
A utiliser immédiatement après ouverture de l’ampoule.
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
1 mL de solution en ampoule (verre incolore de type I). Boîte de 8 ampoules.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Pour les instructions de manipulation et d’injection des ampoules, se référer à la notice.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
9 RUE EUGENE JUNG
68330 HUNINGUE
[Tel, fax, e-Mail : à compléter ultérieurement par le titulaire]
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· CIP
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation: JJ mois AAAA
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
JJ mois AAAA
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 02/07/2022
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que DRAFORA, solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser DRAFORA, solution injectable ?
3. Comment utiliser DRAFORA, solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver DRAFORA, solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE DRAFORA, solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : sans objet.
DRAFORA, solution injectable est un médicament homéopathique traditionnellement utilisé chez l’adulte en cas d’arthrose et d’arthropathies dégénératives.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER DRAFORA, solution injectable ?
N’utilisez jamais DRAFORA, solution injectable :
· si vous êtes allergique aux substances actives ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· Enfants et adolescents de moins de 18 ans.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien ou infirmier/ère avant d’utiliser DRAFORA, solution injectable.
Enfants et adolescents
Sans objet.
Autres médicaments et DRAFORA, solution injectable
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
DRAFORA, solution injectable avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
En l’absence de données expérimentales et cliniques, et par mesures de précaution, l’utilisation de ce médicament est à éviter pendant la grossesse et l’allaitement.
Conduite de véhicules et utilisation de machines
Sans objet.
DRAFORA, solution injectable contient du chlorure de sodium et de l’eau pour préparations injectables.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose administrée, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER DRAFORA, solution injectable ?
Médicament réservé à l’adulte.
Une injection sous-cutanée d’une fois par jour à une fois par semaine pendant 4 semaines. Le traitement pourra être renouvelé si nécessaire.
Mode d’administration
Voie injectable sous-cutanée.
DRAFORA, solution injectable est administré par injection dans le tissu sous-cutané (injection sous la peau). Il est recommandé de varier la localisation des injections. Réaliser l’injection immédiatement après ouverture de l’ampoule.
Tenir le corps de l’ampoule à la verticale et saisir la tête entre le pouce et l’index au niveau du point rouge. Une pression par effet levier permet de casser l’ampoule au niveau de l’étranglement. Le contenu de l’ampoule est prélevé en une fois au moyen d’une seringue stérile munie d’une aiguille stérile. L’air est chassé de la seringue.
L'injection doit être réalisée dans les tissus sous-cutanés, de préférence dans l’abdomen, dans la partie supérieure de la cuisse ou dans la partie supérieure du bras. L'aiguille doit être introduite complètement dans l'épaisseur du pli cutané formé par le pouce et l'index. Il convient de veiller à ne pas pénétrer un vaisseau sanguin. Le produit doit être injecté lentement et à vitesse constante dans des conditions d’aseptie adéquates. Garder le pli de peau maintenu entre le pouce et l'index pendant l'injection.
DRAFORA, solution injectable ne doit pas être administré par voie intraveineuse (IV) ou voie intramusculaire (IM).
|
Vous pouvez réaliser vous-même les injections sous-cutanées, si vous vous sentez capable de les faire. Dans ce cas, assurez-vous d’avoir bien compris comment préparer et administrer le médicament. Suivez exactement les instructions précises données par votre médecin. Un mode d’emploi détaillé est disponible à la fin de cette notice. Si vous ne vous sentez pas en mesure de réaliser vous-même les injections, demandez à ce qu’elles soient réalisées par votre infirmier/ère. Pour toutes questions, contactez votre médecin ou votre pharmacien. |
Utilisation chez les enfants et les adolescents
Sans objet.
Si vous avez utilisé plus de DRAFORA, solution injectable que vous n’auriez dû
Sans objet.
Si vous oubliez d’utiliser DRAFORA, solution injectable
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez d’utiliser DRAFORA, solution injectable
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, votre pharmacien ou votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER DRAFORA, solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boite et l’ampoule. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation avant ouverture de l’ampoule.
A utiliser immédiatement après ouverture de l’ampoule.
Ne pas conserver l’ampoule après ouverture.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient DRAFORA, solution injectable
· Les substances actives sont :
.... Arnica montana 15DH ..................................................................................................... 0,167 g
.... Betula alba, folium 4DH .................................................................................................. 0,167 g
.... Cartilago 8DH.................................................................................................................. 0,167 g
.... Equisetum arvense, décocté 15DH .................................................................................. 0,167 g
.... Formica rufa 10 DH ......................................................................................................... 0,167 g
.... Mandragora officinarum, décocté 4 DH ............................................................................ 0,167 g
Pour une ampoule de 1 mL
· Les autres composants sont :
Chlorure de sodium, eau pour préparations injectables, utilisés pour la dilution finale.
Qu’est-ce que DRAFORA, solution injectable et contenu de l’emballage extérieur
L’emballage contient 8 ampoules de 1 mL de DRAFORA, solution injectable.
Titulaire de l’autorisation de mise sur le marché
9 RUE EUGENE JUNG
68330 HUNINGUE
Exploitant de l’autorisation de mise sur le marché
LABORATOIRES WELEDA
9 RUE EUGENE JUNG
68330 HUNINGUE
MÖHLERSTRASSE 3-5
D-73525 SCHWÄBISCH GMÜND
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
MM/AAAA
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
MODE D’EMPLOI DE DRAFORA, solution injectable
Votre médecin ou votre infirmier/ère vous aura montré comment utiliser les ampoules de DRAFORA, solution injectable et comment pratiquer une injection sous-cutanée (sous la peau). Cependant, lisez attentivement les informations ci-dessous avant d’utiliser une ampoule. Si vous n'êtes pas sûr de vous pour effectuer l'injection ou si vous avez des questions, demandez de l'aide à votre médecin ou votre infirmier/ère.
Ce dont j'ai besoin :
Pour effectuer l'injection, vous aurez besoin du matériel suivant :
1. Une ampoule de DRAFORA, solution injectable.
2. Un coton imbibé d'alcool ou équivalent afin de désinfecter la zone d’injection.
3. Une seringue stérile de 2 mL sous blister.
4. Une aiguille stérile (par exemple de taille 25G), sous blister, pour injecter la solution en sous-cutanée.
5. Un récipient collecteur pour seringues et aiguilles (à se procurer en pharmacie).
Si vous pratiquez vous-même l’injection, les seringues et aiguilles vous seront habituellement prescrites par votre médecin. Si elle est pratiquée par votre infirmier/ère, celui/celle-ci utilisera son propre matériel.
Le site d’injection :
· Le site d’injection est l’endroit de votre corps où vous allez effectuer l’injection. DRAFORA, solution injectable doit être administré par voie sous-cutanée. Il convient de pincer la peau pour faire un pli afin d’accéder au tissu sous-cutané au-delà de la zone graisseuse.
· Pour chaque injection, choisissez un site différent de la précédente pour éviter un endolorissement ou une irritation cutanée.
· Evitez les injections sur des sites sensibles ou présentant une irritation cutanée.
Pour commencer :
Lorsque vous êtes prêt à effectuer l'injection, suivez soigneusement les étapes ci-dessous :
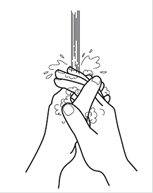
· Lavez-vous soigneusement les mains.
· Préparez votre matériel d’injection :
o une ampoule de DRAFORA
o une seringue de 2 mL sous blister
o une aiguille pour injection sous-cutanée (par exemple de taille 25G) sous blister
o un coton alcoolisé.
· Utilisez des nouvelles aiguilles et des nouvelles seringues jetables pour chaque nouvelle injection. N'utilisez qu'une seule fois les seringues et les aiguilles. Ne partagez jamais les aiguilles et les seringues.
· Inspectez visuellement l'ampoule. La solution est limpide, incolore. NE L’UTILISEZ PAS si elle est cassée, si le liquide semble trouble, s’il contient des particules et n’est pas incolore. Si tel est le cas, rapportez la boîte complète à la pharmacie.
Comment injecter DRAFORA, solution injectable :
|
|
Etape 1 : Lavez-vous soigneusement les mains. |
|
|
|
|
|
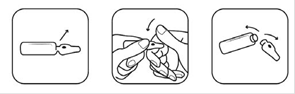
Etape 2. Cassez l’ampoule La solution injectable DRAFORA est contenue dans une ampoule autocassable. Le point coloré à la partie supérieure indique la zone de rupture du col de l’ampoule. Tenir l’ampoule par sa partie cylindrique entre le pouce et l’index de la main gauche (si vous êtes droitier(e)). Tapoter l’ampoule avec le dos de l’index droit afin de faire éventuellement descendre le liquide présent dans la partie supérieure. Tenir fermement l’ampoule, le point coloré face à vous. Saisir la tête de l’ampoule entre le pouce et l’index (le pouce sur le point coloré) puis exercer une pression vers l’arrière. Une fois l’ampoule ouverte, la placer à la verticale sur une surface propre et plate. |
|
|
|
|
|
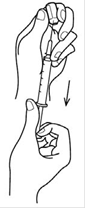
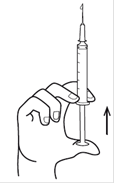
Etape 3 : Prélevez le produit Décapuchonner une seringue à usage unique stérile munie d'une aiguille stérile. Le cas échéant, éliminer l'air présent dans la seringue en poussant le piston à fond. Introduire l'aiguille dans l'ampoule et aspirer le liquide. Une fois le liquide aspiré en totalité dans la seringue, vérifier l'absence d'air dans la seringue. Si nécessaire, expulser l'air en tenant la seringue en position verticale pointe vers le haut. |
|
|
|
|
|
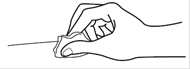
Etape 4 : Nettoyez la zone d’injection avec le coton imbibé d’alcool, tout en gardant la seringue dans une main. |
|
|
Etape 5. Tenir le corps en plastique de la seringue verticalement d’une main, et de l’autre tapoter le corps (sans toucher l’aiguille) de façon à faire remonter à la surface l’air éventuellement contenu dans la seringue. |
|
|
|
|
|
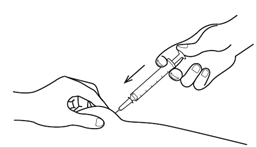
Etape 6. Procédez à l’injection Pincer franchement la peau (sans comprimer) de façon à avoir un pli d’au moins 2 cm d’épaisseur entre le pouce et l’index (plus en présence de tissu graisseux). Planter l’aiguille tout droit d’un coup sec avec toute la main, sans appuyer avec le pouce. L’aiguille doit faire un angle d’environ 60 à 90° avec la peau à l’endroit de la piqûre, et environ 45° avec sa surface (il n’y a pas de risque d’atteindre un organe interne lorsque l’injection est pratiquée de cette manière). Une fois que l’aiguille a presque totalement pénétré dans la peau, injecter lentement tout le contenu de la seringue en appuyant sur le piston avec le pouce. Relâcher lentement le pli cutané et retirez délicatement l’aiguille. Remettez le capuchon sur l’aiguille. |
|
|
|
|
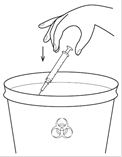
|
Etape 7. Eliminez la seringue et l’aiguille usagée dans un récipient collecteur rigide fermé, conformément à la réglementation en vigueur. |