Dernière mise à jour le 01/06/2026
ZOPHREN 8 mg, lyophilisat oral
Indications thérapeutiques
Classe pharmacothérapeutique – code ATC : ANTAGONISTE DE LA SEROTONINE - A04AA01.
(A : appareil digestif et métabolisme)
Ce médicament est indiqué dans la prévention et le traitement des nausées et des vomissements causés par la chimiothérapie ou la radiothérapie.
Ce médicament est réservé à l’adulte et à l’enfant à partir de 6 mois.
Présentations
> plaquette(s) PVC-Aluminium de 2 lyophilisat(s)
Code CIP : 343 948-1 ou 34009 343 948 1 2
Déclaration de commercialisation : 04/01/1999
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 4,71 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 5,73 €
- Taux de remboursement :65%
> plaquette(s) PVC-Aluminium de 4 lyophilisat(s)
Code CIP : 343 949-8 ou 34009 343 949 8 0
Déclaration de commercialisation : 04/01/1999
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 9,32 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 10,34 €
- Taux de remboursement :65%
> plaquette(s) PVC-Aluminium de 10 lyophilisat(s)
Code CIP : 343 951-2 ou 34009 343 951 2 3
Déclaration de commercialisation : 04/01/1999
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 27/06/2018 | Renouvellement d'inscription (CT) | Le service médical rendu par ZOPHREN reste important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l\'aide)
ANSM - Mis à jour le : 14/10/2025
ZOPHREN 8 mg, lyophilisat oral
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Ondansétron.................................................................................................................... 8,0000 mg
Pour un lyophilisat.
Excipients à effet notoire : chaque lyophilisat contient 1,250 mg d’aspartam (E951), 0,110 mg de parahydroxybenzoate de méthyle sodique, 0,014 mg de parahydroxybenzoate de propyle sodique peut contenir jusqu’à 0,063 mg d’éthanol à 96 % et une quantité d’alcool benzylique n’excédant pas 0,250 mg.
Pour la liste complète des excipients, voir rubrique 6.1.
Lyophilisat oral.
4.1. Indications thérapeutiques
Prévention des nausées et vomissements aigus induits par la chimiothérapie cytotoxique moyennement émétisante chez l'adulte.
Prévention et traitement des nausées et vomissements retardés induits par la chimiothérapie cytotoxique moyennement à hautement émétisante chez l'adulte et l'enfant à partir de 6 mois.
Prévention et traitement des nausées et vomissements induits par la radiothérapie hautement émétisante chez l'adulte.
4.2. Posologie et mode d'administration
Voie orale.
Le lyophilisat peut être absorbé avec ou sans eau.
Posologie
Adultes
Nausées et vomissements induits par la chimiothérapie cytotoxique et la radiothérapie
La dose initiale habituelle est de 8 mg administrée 2 heures avant la chimiothérapie moyennement émétisante ou la radiothérapie.
Pour la prévention et le traitement des nausées et vomissements retardés, la dose est de 8 mg administrée toutes les 12 heures par voie orale sur une durée moyenne de 2 à 3 jours pouvant aller jusqu'à 5 jours.
Dans certaines circonstances (utilisation de substances cytotoxiques très émétisantes et/ou prescrites à très fortes doses, facteurs liés au patient tels que sujets jeunes, de sexe féminin, ayant l'expérience de phénomènes émétiques lors de précédents traitements cytotoxiques...), une association à une corticothérapie et/ou une dose plus élevée d’ondansétron par voie orale ou injectable pourront être utilisées d'emblée.
Population pédiatrique
Chez l’enfant de moins de 6 ans, la forme comprimé n’étant pas adaptée en raison du risque de fausse route, les formes lyophilisat (dissout dans un verre d’eau) et sirop sont donc recommandées.
Nausées et vomissements induits par la chimiothérapie cytotoxique chez les enfants à partir de 6 mois et les adolescents
La dose peut être calculée à partir de la surface corporelle (Body Surface Area BSA) ou du poids – cf ci-après. La dose journalière calculée à partir du poids est supérieure à celle calculée à partir de la surface corporelle (voir rubriques 4.4 et 5.1).
Il n’y a pas de données issues d’essais cliniques contrôlés sur l’utilisation de Zophren dans la prévention des nausées et vomissements retardés ou prolongés induits par les traitements cytotoxiques. Il n’y a pas de données issues d’essais cliniques contrôlés sur l’utilisation de Zophren pour les nausées et vomissements induits par la radiothérapie chez l’enfant.
Dose calculée à partir de la surface corporelle : Initiation par voie IV puis relais par voie orale :
Zophren doit être administré immédiatement avant la chimiothérapie en une dose intraveineuse unique de 5 mg/m², n’excédant pas 8 mg. Un relais par voie orale peut débuter douze heures plus tard et pourra être poursuivi pendant 5 jours (Tableau 1).
La dose totale sur 24 heures (répartie en plusieurs prises) ne doit pas excéder la dose adulte de
32 mg.
Tableau 1 : Dose calculée à partir de la surface corporelle pour les chimiothérapies – Enfants âgés de plus de 6 mois et adolescents
|
Surface Corporelle |
Jour 1(a,b) |
Jours 2-6(b) |
|
< 0,6 m² |
5 mg/m² IV puis 2 mg forme sirop après 12 h |
2 mg forme sirop toutes les 12 h |
|
≥ 0,6 m² à ≤ 1,2 m² |
5 mg/m² IV puis 4 mg forme sirop, compriméc ou lyophilisat oral après 12 h |
4 mg forme sirop, compriméc ou lyophilisat oral toutes les 12 h |
|
> 1,2 m² |
5 mg/m² IV ou 8 mg IV puis 8 mg forme sirop, compriméc ou lyophilisat oral après 12 h |
8 mg forme sirop, compriméc ou lyophilisat oral toutes les 12 h |
a La dose intraveineuse ne doit pas excéder 8 mg.
b La dose totale sur 24 heures ne doit pas excéder la dose adulte de 32 mg.
c La forme comprimé n’est pas adaptée chez l’enfant de moins de 6 ans.
Dose calculée à partir du poids : Initiation par voie IV puis relais par voie orale :
Zophren doit être administré immédiatement avant la chimiothérapie en une dose intraveineuse unique de 0,15 mg/kg, n’excédant pas 8 mg. Deux doses intraveineuses supplémentaires pourront ensuite être administrées à intervalles de 4 heures.
Un relais par voie orale peut débuter douze heures plus tard et pourra être poursuivi jusqu’à 5 jours (Tableau 2).
La dose totale sur 24 heures (répartie en plusieurs prises) ne doit pas dépasser la dose adulte de
32 mg.
Tableau 2 : Dose calculée à partir du poids pour les chimiothérapies – Enfants âgés de plus de 6 mois et adolescents
|
Poids |
Jour 1(a,b) |
Jours 2-6(b) |
|
≤ 10 kg |
Jusqu’à 3 doses de 0,15 mg/kg IV toutes les 4 h |
2 mg forme sirop, toutes les 12 h |
|
> 10 kg |
Jusqu’à 3 doses de 0,15 mg/kg IV toutes les 4 h |
4 mg forme sirop, compriméc ou lyophilisat oral toutes les 12 h |
a La dose intraveineuse ne doit pas excéder 8 mg.
b La dose totale sur 24 heures ne doit pas excéder la dose adulte de 32 mg.
c La forme comprimé n’est pas adaptée chez l’enfant de moins de 6 ans.
Nausées et vomissements post-opératoires chez les enfants et les adolescents
Aucune étude n’a été conduite sur l’administration des formes orales de Zophren dans la prévention ou le traitement des nausées et vomissements post-opératoires. Dans cette indication, la forme injectable est recommandée.
Patients âgés
Chez les patients âgés de 65 ans et plus, les posologies recommandées sont les mêmes que celles recommandées chez l’adulte.
Insuffisants hépatiques
Il est recommandé de ne pas dépasser une dose totale journalière de 8 mg chez ces patients.
Insuffisants rénaux
Il n’est pas nécessaire de modifier la posologie journalière, la fréquence d’administration ou la voie d’administration chez ces patients.
Patients métaboliseurs lents
Le métabolisme de la spartéine et de la débrisoquine au niveau du cytochrome P450 n'est pas modifié. Aucune adaptation posologique n'est donc nécessaire chez ce type de patients.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Utilisation concomitante d’apomorphine (voir rubrique 4.5).
En raison de la présence d’aspartam, ce médicament est contre-indiqué en cas de phénylcétonurie.
4.4. Mises en garde spéciales et précautions d'emploi
Mises en garde spéciales
Un bilan cardiovasculaire doit être effectué en cas de survenue de douleurs thoraciques et de syncope, ou de troubles du rythme cardiaque.
Prendre en compte le risque éventuel d’hypersensibilité croisée avec les autres antagonistes des récepteurs 5-HT3.
Les événements respiratoires, pouvant constituer des signes précurseurs de réactions d’hypersensibilité, doivent être traités de façon symptomatique et les cliniciens doivent y porter une attention particulière.
L'ondansétron prolonge l'intervalle QT de façon dose-dépendante (voir rubrique 5.1). De plus, des cas de Torsade de Pointes ont été rapportés après commercialisation chez des patients traités par ondansétron. L'utilisation de l'ondansétron n’est pas recommandée chez les patients présentant un syndrome du QT long congénital. L'ondansétron doit être administré avec prudence chez les patients susceptibles de développer ou ayant un allongement de l'intervalle QTc, y compris les patients présentant des anomalies électrolytiques, une insuffisance cardiaque congestive, des bradyarythmies ou les patients prenant d'autres médicaments susceptibles d'entraîner un allongement de l'intervalle QT ou des anomalies électrolytiques.
Des cas d’ischémie myocardique ont été rapportés chez des patients traités par ondansétron. Chez certains patients, principalement lors de l’administration intraveineuse, les symptômes apparaissent immédiatement après l’administration d’ondansétron. Les patients doivent être alertés des signes et des symptômes d’ischémie myocardique.
L'hypokaliémie et l'hypomagnésémie doivent être corrigées avant l'administration d'ondansétron.
Des cas de syndrome sérotoninergique (avec troubles de la conscience, dysautonomie et troubles neuromusculaires) ont été rapportés après commercialisation suite à l'utilisation concomitante d'ondansétron et de médicaments sérotoninergiques (y compris les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS), les inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSNa) et les agonistes-antagonistes morphiniques comme la buprénorphine).
Les manifestations cutanées évoquant une nécrolyse épidermique toxique imposent l’arrêt immédiat et définitif du traitement.
Population pédiatrique
Les patients pédiatriques recevant de l’ondansétron avec des agents de chimiothérapie hépatotoxiques doivent être étroitement surveillés par rapport au risque d’anomalie fonctionnelle hépatique.
Nausées et vomissements induits par les traitements cytotoxiques
En calculant la dose en mg/kg et lors de l’administration de 3 doses à intervalles de 4 heures, la dose totale journalière sera plus élevée que lors de l’administration d’une dose de 5 mg/m² suivie d’une dose orale. L’efficacité comparative de ces deux schémas thérapeutiques n’a pas été étudiée dans les essais cliniques. Une comparaison inter-essais indique une efficacité similaire pour les deux options (voir rubrique 5.1).
Précautions d'emploi
En cas d'insuffisance hépatique modérée ou sévère, les paramètres pharmacocinétiques de l'ondansétron sont significativement modifiés : réduction de la clairance plasmatique totale, augmentation de la demi-vie plasmatique (voir rubrique 4.2).
L'ondansétron pouvant favoriser un syndrome occlusif, il convient de surveiller attentivement le transit des patients en cours de traitement (voir rubrique 4.8).
Ce médicament contient du « Parahydroxybenzoate » et peut provoquer des réactions allergiques (éventuellement retardées).
Ce médicament contient de l’alcool benzylique (n’excédant pas 0,250 mg par lyophilisat). L’alcool benzylique peut provoquer des réactions allergiques. Il n’est pas recommandé d’utiliser ce produit pendant plus d’une semaine chez les jeunes enfants (moins de 3 ans).
Ce médicament contient de faibles quantités d’éthanol (alcool), inférieures à 100 mg par lyophilisat oral.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par lyophilisat oral, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L'ondansétron doit être utilisé avec prudence en cas d’administration concomitante avec les médicaments entraînant un allongement de l'intervalle QT et/ou des anomalies électrolytiques (voir rubrique 4.4).
L'utilisation de l'ondansétron avec les médicaments prolongeant l’intervalle QT peut entraîner un allongement supplémentaire de l'intervalle QT. L'utilisation concomitante d'ondansétron et de médicaments cardiotoxiques (par exemple les anthracyclines comme la doxorubicine, la daunorubicine ou le trastuzumab), d’antibiotiques (comme l'érythromycine), de kétoconazole, d’antiarythmiques (tel que l’amiodarone) et de bêta-bloquants (tels que l'aténolol ou le timolol) peuvent augmenter le risque d'arythmie (voir rubrique 4.4).
Des cas de syndrome sérotoninergique (incluant troubles de la conscience, dysautonomie et troubles neuromusculaires) ont été rapportés après commercialisation à la suite de l'utilisation concomitante d'ondansétron et de médicaments sérotoninergiques (y compris les ISRS, les IRSNa et les agonistes-antagonistes morphiniques comme la buprénorphine). Si l’utilisation d’ondansétron avec des médicaments sérotoninergiques est cliniquement justifiée, une surveillance appropriée du patient est recommandée (voir rubrique 4.4).
+ Apomorphine
Sur la base de cas rapportés d’hypotension profonde et de perte de conscience lorsque l’ondansétron est administré avec du chlorhydrate d’apomorphine, l’utilisation concomitante avec l’apomorphine est contre-indiquée.
+ Tramadol
Des données issues de petites études indiquent que l’ondansétron réduirait l’effet analgésique du tramadol.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Les femmes en âge de procréer doivent envisager d’utiliser un moyen de contraception.
Grossesse
Sur la base de l’expérience acquise en matière d’études épidémiologiques chez l’homme, l’ondansétron est présumé provoquer des malformations orofaciales au cours du premier trimestre de la grossesse.
Dans le cadre d’une étude de cohorte comprenant 1,8 million de grossesses, l’utilisation d’ondansétron pendant le premier trimestre a été associée à un risque accru de fentes labiales (3 cas supplémentaires pour 10 000 femmes traitées ; risque relatif ajusté, 1,24, (IC 95 % 1,03-1,48)).
Les études épidémiologiques disponibles sur les malformations cardiaques révèlent des résultats contradictoires.
Les études sur des animaux n’indiquent aucun effet nocif direct ou indirect en ce qui concerne la toxicité pour la reproduction.
L’ondansétron ne doit pas être utilisé au cours du premier trimestre de la grossesse.
Allaitement
En cas d'allaitement ou de désir d'allaitement et compte tenu du passage de l'ondansétron dans le lait maternel, l'utilisation de ce produit est déconseillée.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Selon les résultats des tests psychomoteurs, l’ondansétron n’altère pas la performance et n’entraîne pas de sédation et aucun effet n’est attendu compte-tenu de la pharmacologie de l’ondansétron. Toutefois, l’attention doit être attirée sur le fait que certains effets indésirables sont susceptibles d’altérer la capacité de conduite.
Les effets indésirables sont listés ci-dessous par classe-organe et par fréquence. Les fréquences sont définies en : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Les évènements très fréquents, fréquents et peu fréquents ont généralement été déterminés à partir des données des études cliniques. L’incidence correspondante dans le bras placebo a été prise en compte. Les évènements rares, très rares et de fréquence indéterminée ont généralement été déterminés à partir des données spontanées post-commercialisation.
Les fréquences ci-dessous ont été estimées sur la base des recommandations posologiques standard d’ondansétron.
Tableau 3 Effets indésirables observés chez les patients traités par Zophren
|
Affections du système immunitaire |
|
|
Rare |
Réactions allergiques immédiates, quelquefois sévères incluant des réactions anaphylactiques |
|
Affections du système nerveux |
|
|
Très fréquent |
Céphalées |
|
Peu fréquent |
Syndromes extra-pyramidaux, tels que des crises oculogyres, des dystonies, des dyskinésies, ainsi que des convulsions ont été signalés (observés sans preuve définitive de séquelles cliniques persistantes) |
|
Rare |
Vertiges principalement au cours des injections IV rapides |
|
Affections oculaires |
|
|
Rare |
Troubles visuels transitoires (par exemple vision trouble) principalement au cours des injections IV |
|
Très rare |
Cécité transitoire principalement lors de l’administration d’ondansétron par voie injectable |
|
La majorité des cas de cécité qui ont été signalés se sont résolus dans les 20 minutes. La plupart des patients avaient reçu une chimiothérapie comprenant du cisplatine. Quelques cas de cécité transitoire ont été déclarés comme étant d’origine corticale. |
|
|
Affections cardiaques |
|
|
Peu fréquent |
Troubles du rythme, douleurs thoraciques avec ou sans décalage du segment ST, bradycardie |
|
Rare |
Allongement de l’intervalle QT (incluant des torsades de pointes). |
|
Fréquence indéterminée |
Ischémie myocardique |
|
Affections vasculaires |
|
|
Fréquent |
Bouffées de chaleur ou flush |
|
Peu fréquent |
Hypotension |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Peu fréquent |
Hoquets |
|
Affections gastro-intestinales |
|
|
Fréquent |
Constipation, pouvant, dans de rares cas, se compliquer d'iléus ou d'occlusion intestinale, en particulier chez des patients présentant des facteurs de risque associés : ralentisseurs du transit, antécédents de chirurgie digestive |
|
Affections hépatobiliaires |
|
|
Peu fréquent |
Anomalies biologiques hépatiques |
|
Ces évènements ont été observés fréquemment chez les patients recevant une chimiothérapie avec du cisplatine. |
|
|
Troubles de la peau et des tissus sous-cutanés |
|
|
Très rare |
Manifestations cutanées de type nécrolyse épidermique toxique (syndrome de Stevens Johnson et syndrome de Lyell) |
Population pédiatrique
Le profil d’effets indésirables chez les enfants et les adolescents était comparable à celui observé chez l’adulte.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
Signes et symptômes
Peu d’informations concernant le surdosage d’ondansétron sont disponibles. Un surdosage en ondansétron peut entraîner les effets indésirables déjà mentionnés dans la rubrique 4.8. Les manifestations qui ont été signalées incluent des troubles visuels, une constipation sévère, une hypotension et un épisode vaso-vagal avec un bloc auriculo-ventriculaire transitoire du second degré.
L'ondansétron prolonge l'intervalle QT de façon dose-dépendante. Un contrôle de l'ECG est recommandé en cas de surdosage.
Population pédiatrique
Des cas compatibles avec un syndrome sérotoninergique ont été rapportés dans la population pédiatrique suite à un surdosage accidentel en ondansétron par voie orale (ingestion estimée supérieure à 4 mg/kg) chez des nourrissons et des enfants âgés de 12 mois à 2 ans.
Traitement
Il n’existe pas d’antidote spécifique de l’ondansétron. Par conséquent, en cas de surdosage, seule une thérapeutique symptomatique appropriée sera instaurée.
Toute prise en charge complémentaire doit être définie en fonction de l’état clinique ou des recommandations des centres antipoison, lorsqu'elles sont disponibles.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : ANTAGONISTE DE LA SEROTONINE, code ATC : A04AA01.(A : appareil digestif et métabolisme)
L'ondansétron est un antagoniste des récepteurs 5-HT3 à la sérotonine, impliquée dans les phénomènes de réflexe émétique.
L'administration d'ondansétron ne modifie pas les taux sériques de prolactine.
Etudes cliniques
Allongement de l'intervalle QT
Une étude croisée randomisée en double aveugle, contrôlée versus contrôle positif (moxifloxacine) et versus placebo, a évalué l'effet de l'ondansétron sur l'intervalle QTc chez 58 adultes sains, de sexe féminin et masculin. Des doses d'ondansétron de 8 mg et 32 mg, administrées par voie intraveineuse sur 15 minutes ont été utilisées. A la plus forte dose testée de 32 mg, la différence moyenne maximale du QTcF [limite supérieure de l'IC à 90 %] observée par rapport au placebo était de 19,6 [21,5] msec après ajustement par rapport aux valeurs initiales. A la plus faible dose testée de 8 mg, la différence moyenne maximale du QTcF [limite supérieure de l'IC à 90 %] observée par rapport au placebo était de 5,8 [7,8] msec après ajustement par rapport aux valeurs initiales. Dans cette étude, aucune mesure de QTcF supérieure à 480 msec, ni d’allongement de l’intervalle QTcF supérieur à 60 msec n'ont été observés. Les mesures électrocardiographiques des intervalles PR ou QRS n’ont pas été modifiées de manière significative au cours de l’étude.
Population pédiatrique
Nausées et vomissements induits par les traitements cytotoxiques
L’efficacité de l’ondansétron dans le contrôle des épisodes émétiques et des nausées induits par une chimiothérapie anticancéreuse a été évaluée dans un essai randomisé en double-aveugle chez 415 patients âgés de 1 à 18 ans (S3AB3006). Les jours de chimiothérapie, les patients recevaient soit 5 mg/m² d’ondansétron intraveineux + 4 mg d’ondansétron oral après 8-12 h ou 0,45 mg/kg d’ondansétron intraveineux + placebo oral après 8-12 h.
Après chimiothérapie, les deux groupes ont reçu 4 mg d’ondansétron sirop deux fois par jour pendant 3 jours. Le taux de contrôle complet des épisodes émétiques était au minimum de 49 % (5 mg/m² intraveineux + 4 mg d’ondansétron oral) et 41 % (0,45 mg/kg intraveineux + placebo oral).
Après chimiothérapie, les deux groupes ont reçu 4 mg d’ondansétron sirop deux fois par jour pendant 3 jours.
Aucune différence dans l’incidence globale ou dans la nature des effets indésirables n’a été observée entre les deux groupes de traitement.
Un essai randomisé en double-aveugle avec contrôle placebo (S3AB4003) chez 438 patients âgés de 1 à 17 ans a démontré un contrôle complet des épisodes émétiques chez :
· 73 % des patients lorsque l’ondansétron était administré par voie intraveineuse à une dose de 5 mg/m2 associé à 2-4 mg de dexamethasone par voie orale,
· 71 % des patients lorsque l’ondansétron était administré en sirop à une dose de 8 mg + 2-4 mg de dexamethasone par voie orale les jours de chimiothérapie.
Après chimiothérapie, les deux groupes ont reçu 4 mg d’ondansétron sirop deux fois par jour pendant 2 jours.
Aucune différence dans l’incidence globale ou dans la nature des effets indésirables n’a été observée entre les deux groupes de traitement.
L’efficacité de l’ondansétron chez 75 enfants âgés de 6 à 48 mois a été étudiée dans un essai ouvert, non-comparatif, à un seul bras (S3A40320). Tous les enfants ont reçu trois doses d’ondansétron de 0,15 mg/kg par voie IV, administrées 30 minutes avant le début de la chimiothérapie puis quatre et huit heures après la première dose. Le contrôle complet des épisodes émétiques a été atteint chez
56 % des patients.
Une autre étude en ouvert, non-comparative, à un seul bras (S3A239) a étudié l’efficacité d’une dose d’ondansétron par voie IV de 0,15 mg/kg suivie de deux doses d’ondansétron par voie orale de 4 mg chez des enfants âgés de moins de 12 ans et 8 mg chez les enfants âgés de plus de 12 ans (nombre total des enfants inclus n = 28). Le contrôle complet des épisodes émétiques a été atteint chez 42 % des patients.
Nausées et vomissements post-opératoires
L’efficacité de l’ondansétron pris en dose unique dans la prévention des nausées et vomissements post-opératoires a été évaluée dans un essai randomisé en double-aveugle versus placebo chez 670 patients âgés de 1 à 24 mois (âge après conception ≥ 44 semaines, poids ≥ 3 kg). Les sujets inclus devaient subir une intervention chirurgicale programmée, sous anesthésie générale et avaient un statut ASA ≤ III. Une dose unique d’ondansétron de 0,1 mg/kg a été administrée dans les cinq minutes suivant l’induction de l’anesthésie. La proportion de sujets ayant eu au moins un épisode émétique pendant les 24 heures d’évaluation (ITT) était supérieure pour les patients ayant reçu le placebo que pour ceux ayant reçu de l’ondansétron (28 % vs. 11 %, p < 0,0001).
Quatre études menées en double-aveugle versus placebo ont été effectuées chez 1469 patients masculins et féminins (âgés de 2 à 12 ans) subissant une anesthésie générale. Les patients ont été randomisés afin de recevoir une dose IV unique d’ondansétron (0,1 mg/kg pour les patients pédiatriques pesant 40 kg ou moins, 4 mg pour les patients pédiatriques pesant plus de 40 kg ; nombre de patients = 735) ou le placebo (nombre de patients = 734). Le traitement à l’étude a été administré sur une durée d’au moins 30 secondes, immédiatement avant ou après l’induction de l’anesthésie. L’ondansétron a été significativement plus efficace que le placebo dans la prévention des nausées et vomissements. Les résultats de ces études sont résumés dans le tableau 4.
Tableau 4 : Prévention et traitement des nausées et vomissements post-opératoires chez des patients pédiatriques – Réponse au traitement sur 24 heures
|
Etude |
Critère d’évaluation |
Ondansétron % |
Placebo % |
Valeur p |
|
S3A380 |
CR |
68 |
39 |
≤ 0,001 |
|
S3GT09 |
CR |
61 |
35 |
≤ 0,001 |
|
S3A381 |
CR |
53 |
17 |
≤ 0,001 |
|
S3GT11 |
Pas de nausées |
64 |
51 |
0,004 |
|
S3GT11 |
Pas de vomissements |
60 |
47 |
0.004 |
CR = pas d’épisodes émétiques, de traitement de secours ou sortie d’étude.
5.2. Propriétés pharmacocinétiques
Après administration orale, l’absorption d’ondansétron est rapide : des concentrations plasmatiques maximales voisines de 30 ng/ml sont atteintes 1,5 heures environ après une dose de 8 mg.
La biodisponibilité absolue est approximativement de 60 %. La biodisponibilité moyenne chez les sujets masculins sains après administration d’un comprimé unique de 8 mg est approximativement de 55 à 60 %. La demi-vie d’élimination terminale est d’environ 3 heures. Le volume de distribution à l’état d’équilibre est d’environ 140 l. La liaison aux protéines plasmatiques est de 70 à 76 %. L’ondansétron est métabolisé principalement par voie hépatique par de multiples voies enzymatiques. La majorité des métabolites excrétés (> 40 % de la dose administrée) sont des glucuronides et des conjugués de sulfate de 8-hydroxyondansétron, qui est pharmacologiquement actif mais ne peut pas être détecté (< 2 ng/ml) dans la circulation systémique après administration orale ou IV d’ondansétron. Les métabolites d’ondansétron sont principalement excrétés dans les urines. Moins de 5 % de la dose résorbée sont excrétés sous forme inchangée dans les urines. La pharmacocinétique demeure inchangée en cas d’administration réitérée.
La demi-vie d’élimination peut être prolongée jusqu’à 5 heures chez le sujet âgé.
Populations particulières de patients
Sexe
Des différences liées au sexe ont été montrées dans le devenir de l’ondansétron ; les sujets féminins ont une vitesse et intensité d’absorption accrues après administration d’une dose orale ainsi qu’une réduction de la clairance systémique et du volume de distribution (ajusté selon le poids).
Enfants et adolescents (âgés de 1 mois à 17 ans)
Chez les patients pédiatriques âgés de 1 à 4 mois (n = 19) ayant subi une chirurgie, la clairance normalisée par le poids était d’environ 30 % plus lente que chez les patients âgés de 5 à 24 mois (n = 22) mais comparable à celle des patients âgés de 3 à 12 ans.
La demi-vie dans la population pédiatrique âgée de 1 à 4 mois était en moyenne de 6,7 heures et de 2,9 heures chez les patients âgés de 5 à 24 mois et de 3 à 12 ans. Les différences des paramètres pharmacocinétiques dans la population de patients âgés de 1 à 4 mois peuvent être expliquées en partie par le pourcentage supérieur d’eau corporelle totale chez les nouveau-nés et les nourrissons et un volume de distribution plus élevé pour les principes actifs hydrosolubles tels que l’ondansétron.
Chez les patients pédiatriques âgés de 3 à 12 ans ayant subi une chirurgie programmée avec anesthésie générale, les valeurs absolues pour la clairance et le volume de distribution de l’ondansétron étaient réduits en comparaison des valeurs des patients adultes. Ces deux paramètres augmentaient de manière linéaire avec le poids et à partir de l’âge de 12 ans, les valeurs approchaient celles des jeunes adultes. Lorsque la clairance et le volume de distribution étaient normalisés par le poids corporel, les valeurs de ces paramètres étaient similaires entre les différents groupes d’âge. L’utilisation de doses en fonction du poids permet de compenser les modifications liées à l’âge et est efficace pour la normalisation de l’imprégnation systémique chez les patients pédiatriques.
Une analyse de pharmacocinétique de population a été effectuée sur 428 patients (patients souffrant de cancer, patients ayant subi une chirurgie et volontaires sains) âgés de 1 mois à 44 ans après administration intraveineuse d’ondansétron. En se basant sur cette analyse, l’imprégnation systémique en ondansétron (ASC) après administration orale ou intraveineuse chez les enfants et les adolescents était comparable à celle des adultes, à l’exception des nourrissons âgés de 1 à 4 mois. Le volume de distribution était lié à l’âge et était plus faible chez les adultes que chez les nourrissons et les enfants. La clairance était liée au poids mais pas à l’âge à l’exception des nourrissons âgés de 1 à 4 mois. Il est difficile de conclure s’il y avait une réduction supplémentaire de la clairance liée à l’âge chez les nourrissons de 1 à 4 mois ou tout simplement une variabilité inhérente au faible nombre de patients étudiés dans cette tranche d’âge. Etant donné que les patients âgés de moins de 6 mois recevront seulement une dose unique en cas de nausées et vomissements post-opératoires, il est peu probable qu’une clairance diminuée soit cliniquement significative.
Personnes âgées
Des études de phase I menées chez les sujets âgés sains ont montré une légère diminution de la clairance et une augmentation de la demi-vie de l’ondansétron liées à l'âge. Cependant, en raison de la grande variabilité interindividuelle, il a été constaté des chevauchements importants entre les paramètres pharmacocinétiques des sujets âgés de moins de 65 ans et ceux des sujets âgés de 65 ans et plus. De manière générale, aucune différence n'a été observée en termes de sécurité ou d'efficacité chez les sujets âgés de moins de 65 ans et les sujets âgés de 65 ans et plus atteints de cancer et inclus dans des essais cliniques portant sur la prévention et le traitement des nausées et vomissements aigus induits par la chimiothérapie cytotoxique pour soutenir une posologie différente chez les personnes âgées de 65 ans et plus.
Cependant, en se basant sur des modélisations plus récentes des concentrations plasmatiques et des réponses en fonction de l’exposition à l'ondansétron, un effet plus important sur l’allongement de l’intervalle QTcF est attendu chez les patients ≥ 75 ans comparativement aux patients de moins de 65 ans. Les informations concernant les posologies de l’ondansétron administré par voie intraveineuse chez les patients âgés de 65 à 74 ans et chez les patients ≥ 75 ans sont détaillées dans la rubrique 4.2.
5.3. Données de sécurité préclinique
Une étude sur des canaux ioniques cardiaques humains clonés a montré que l’ondansétron pouvait, potentiellement affecter la repolarisation cardiaque par l’intermédiaire d’un blocage des canaux potassiques hERG à des concentrations susceptibles d’être atteintes en thérapeutique, par voie parentérale en particulier.
In vivo, un allongement de l’intervalle QT a été observé chez des chats anesthésiés après administration intraveineuse, mais à des doses plus de cent fois supérieures aux doses pharmacologiquement efficaces. Des effets similaires n’ont pas été observés chez des singes cynomolgus. Des modifications transitoires de l’ECG ont été rapportées en clinique (voir rubriques 4.4 et 5.1).
Aucun effet carcinogène n’a été observé au cours des études réalisées sur 2 ans chez le rat et la souris avec des doses orales d’ondansétron allant jusqu’à respectivement 10 mg/kg/j et 30 mg/kg/j (environ 4 et 6 fois la dose orale maximale recommandée de 24 mg par jour basée sur la surface corporelle chez l’homme).
L’ondansétron n’était pas mutagène dans les tests standards de mutagénicité.
Gélatine, mannitol, aspartam, parahydroxybenzoate de méthyle sodique, parahydroxybenzoate de propyle sodique, arôme fraise (éthanol à 96 %, propylèneglycol, alcool benzylique, sodium).
Sans objet.
3 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30°C.
6.5. Nature et contenu de l'emballage extérieur
2, 4, 6 ou 10 lyophilisats sous plaquette (PVC/aluminium)
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Ne pas pousser le lyophilisat à l'extérieur de l'alvéole sans en avoir préalablement retiré la pellicule protectrice.
Retirer le feuillet de protection de la plaquette et pousser doucement le lyophilisat à l'extérieur de l'alvéole.
Le lyophilisat peut être, soit placé sur le bout de la langue où il se dissoudra en quelques secondes, soit administré après dissolution dans un peu d'eau.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
SANDOZ
92300 LEVALLOIS-PERRET
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 343 948 1 2 : 2 lyophilisats sous plaquette (PVC/aluminium)
· 34009 343 949 8 0 : 4 lyophilisats sous plaquette (PVC/aluminium)
· 34009 343 950 6 2 : 6 lyophilisats sous plaquette (PVC/aluminium)
· 34009 343 951 2 3 : 10 lyophilisats sous plaquette (PVC/aluminium)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I
ANSM - Mis à jour le : 14/10/2025
ZOPHREN 8 mg, lyophilisat oral
Ondansétron
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou à votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ZOPHREN 8 mg, lyophilisat oral et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre ZOPHREN 8 mg, lyophilisat oral ?
3. Comment prendre ZOPHREN 8 mg, lyophilisat oral ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ZOPHREN 8 mg, lyophilisat oral ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ZOPHREN 8 mg, lyophilisat oral ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique – code ATC : ANTAGONISTE DE LA SEROTONINE - A04AA01.
(A : appareil digestif et métabolisme)
Ce médicament est indiqué dans la prévention et le traitement des nausées et des vomissements causés par la chimiothérapie ou la radiothérapie.
Ce médicament est réservé à l’adulte et à l’enfant à partir de 6 mois.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE ZOPHREN 8 mg, lyophilisat oral ?
Ne prenez jamais ZOPHREN 8 mg, lyophilisat oral :
· si vous êtes allergique à l’ondansétron ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· en cas de phénylcétonurie (maladie héréditaire dépistée à la naissance) en raison de la présence d’aspartam.
· si vous prenez de l’apomorphine (utilisée dans le traitement de la maladie de Parkinson).
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre ZOPHREN 8 mg, lyophilisat oral.
Faites attention avec ZOPHREN 8 mg, lyophilisat oral :
· si vous avez déjà eu des problèmes cardiaques ou si vous souffrez de problèmes cardiaques,
o signaler à votre médecin que vous prenez du Zophren avant toute autre prescription (d’autres médicaments peuvent agir sur le cœur),
o prévenir immédiatement un médecin en cas de douleurs thoraciques, de syncopes ou de troubles du rythme cardiaque,
· si votre analyse de sang présente des anomalies concernant le taux de potassium, de sodium et de magnésium (déséquilibre électrolytique),
· si vous souffrez de symptômes respiratoires ; ils peuvent être les signes précoces d’une allergie (hypersensibilité),
· si vous avez des antécédents d’allergie à un autre médicament de la même classe. Vous devez prendre en compte le risque d’allergie avec ce médicament,
· si vous souffrez d’insuffisance hépatique modérée ou sévère (défaillance des fonctions du foie),
· si vous souffrez de constipation sévère et prolongée.
Enfants
Sans objet.
Autres médicaments et ZOPHREN 8 mg, lyophilisat oral
Ne prenez jamais Zophren :
· si vous prenez de l’apomorphine (utilisée dans le traitement de la maladie de Parkinson).
Prévenez votre médecin :
· si vous prenez d'autres médicaments pouvant entraîner des anomalies de l'électrocardiogramme (ECG) et/ou un déséquilibre électrolytique.
· si vous prenez des médicaments anticancéreux (en particulier les anthracyclines ou le trastuzumab).
· si vous prenez des médicaments pour traiter les infections (comme l'érythromycine).
· si vous prenez des médicaments utilisés pour la maladie de Cushing (kétoconazole).
· si vous prenez des médicaments anti-arythmiques (comme l’amiodarone) ou bêta bloquants (comme l'aténolol ou le timolol) pour traiter des troubles du rythme cardiaque.
· si vous prenez des médicaments pour traiter la dépression ou l’anxiété appelés inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) ou inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSNa).
· si vous prenez du tramadol, un médicament utilisé pour traiter les fortes douleurs.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance.
ZOPHREN 8 mg, lyophilisat oral avec des aliments et boissons
Sans objet.
Grossesse et allaitement
Il est conseillé de ne pas utiliser Zophren pendant le premier trimestre de la grossesse. En effet, Zophren peut augmenter légèrement le risque que l’enfant naisse avec un bec de lièvre et/ou une fente palatine (orifices ou fentes dans la lèvre supérieure et/ou le palais). Si vous êtes déjà enceinte ou que vous pensez l’être, ou si vous prévoyez une grossesse, demandez conseil à votre médecin ou à votre pharmacien avant de prendre Zophren. Si vous êtes une femme en âge de procréer, il est conseillé d’utiliser un moyen de contraception efficace.
En cas d’allaitement ou de désir d’allaitement, l’utilisation de ce médicament est déconseillée, compte tenu de son passage dans le lait maternel.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Zophren peut entraîner des effets indésirables pouvant diminuer votre capacité de conduite.
Ne conduisez pas si des effets indésirables diminuant votre capacité de conduite apparaissent ou persistent.
ZOPHREN 8 mg, lyophilisat oral contient de l’aspartam (E951), du parahydroxybenzoate de méthyle sodique et du parahydroxybenzoate de propyle sodique, de l’alcool benzylique et de l’éthanol à 96 %.
Ce médicament contient 1,250 mg d’aspartam par lyophilisat.
L’aspartam contient une source de phénylalanine. Peut être dangereux pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de phénylalanine ne pouvant être éliminée correctement.
Ce médicament contient du « Parahydroxybenzoate » et peut provoquer des réactions allergiques (éventuellement retardées).
Ce médicament contient 0,00005 mg d’alcool benzylique par lyophilisat oral de 8 mg. L’alcool benzylique peut provoquer des réactions allergiques.
L’alcool benzylique est associé à un risque d’effets secondaires graves y compris des problèmes respiratoires (appelés « syndrome de suffocation ») chez les jeunes enfants.
Ne pas utiliser pendant plus d’une semaine chez les jeunes enfants (moins de 3 ans), sauf avis contraire de votre médecin ou de votre pharmacien.
Demandez conseil à votre médecin ou à votre pharmacien si vous êtes enceinte ou si vous allaitez. De grandes quantités d’alcool benzylique peuvent s’accumuler dans votre corps et entraîner des effets secondaires (appelés «acidose métabolique»).
Demandez conseil à votre médecin ou votre pharmacien si vous souffrez d’une maladie du foie ou du rein. De grandes quantités d’alcool benzylique peuvent s’accumuler dans votre corps et entraîner des effets secondaires (appelés «acidose métabolique»).
Ce médicament contient moins de 0,06 mg d’alcool (éthanol) par lyophilisat oral de 8 mg équivalent à 0,23% w/w. La quantité dans un lyophilisat oral de 8 mg de ce médicament équivaut à moins de 1 ml de bière et 1 ml de vin.
La faible quantité d’alcool contenue dans ce médicament n’est pas susceptible d’entraîner d’effet notable.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par lyophilisat oral, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE ZOPHREN 8 mg, lyophilisat oral ?
Posologie
Respectez toujours la posologie indiquée par votre médecin. En cas de doute, consultez votre médecin ou votre pharmacien.
La dose habituellement recommandée est de :
· Chez l’adulte : 1 à 2 lyophilisats par jour.
· Chez l’enfant : se conformer à l’ordonnance du médecin.
Chez l’enfant de moins de 6 ans, dissoudre le lyophilisat dans un verre d’eau, en raison du risque de fausse route.
Mode d'administration
Voie orale.
Ne pas pousser le lyophilisat à l’extérieur de l’alvéole sans en avoir préalablement retiré la pellicule protectrice.
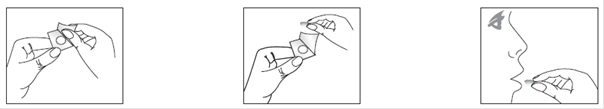
|
1) Retirer le feuillet de protection de la plaquette. |
2) Poussez doucement le lyophilisat à l’extérieur de l’alvéole. |
3) Le lyophilisat peut être, soit placé sur le bout de la langue où il se dissoudra en quelques secondes, soit administré après dissolution dans un peu d'eau. |
Durée du traitement
1 à 5 jours.
Si vous avez pris plus de ZOPHREN 8 mg, lyophilisat oral que vous n’auriez dû
Si vous ou votre enfant avez pris plus de ZOPHREN 8 mg, lyophilisat oral que vous n’auriez dû, contactez votre médecin ou allez directement à l’hôpital. Emportez la boite du médicament avec vous.
Si vous oubliez de prendre ZOPHREN 8 mg, lyophilisat oral
Ne prenez pas de dose double pour compenser la dose que vous avez oubliée de prendre.
Si vous arrêtez de prendre ZOPHREN 8 mg, lyophilisat oral
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Certains effets indésirables peuvent être graves. Arrêtez de prendre le médicament et contactez immédiatement votre médecin si vous remarquez :
Une réaction allergique sévère pouvant être immédiate : les signes peuvent inclure :
· Une éruption cutanée avec démangeaisons (urticaire),
· Un gonflement du visage ou de la gorge (angioedème). Vous pourrez alors avoir du mal à respirer,
· Un malaise.
Une ischémie myocardique : les signes peuvent inclure :
· Une douleur soudaine dans la poitrine,
· Une oppression thoracique.
Autres effets indésirables possibles :
· maux de tête, bouffées de chaleur, hoquets,
· anomalies biologiques hépatiques,
· constipation pouvant dans de rares cas se compliquer d’occlusion intestinale en particulier chez certains patients avec des facteurs de risque associés (prise de médicaments ralentisseurs du transit, antécédent de chirurgie digestive),
· baisse de la pression artérielle, douleurs au niveau du thorax, troubles du rythme cardiaque (pouvant entraîner une perte soudaine de connaissance) et bradycardie (ralentissement du rythme cardiaque),
· mouvements anormaux des yeux, raideur anormale des muscles et tremblements, convulsions,
· réactions cutanées rares et graves, caractérisées par des bulles et un détachement de la peau, imposant l’arrêt immédiat du traitement,
· troubles visuels transitoires, voire de très rares cas de cécité transitoire ont été décrits,
· vertiges, principalement au cours des injections rapides.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ZOPHREN 8 mg, lyophilisat oral ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 30°C.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ZOPHREN 8 mg, lyophilisat oral
· La substance active est :
Ondansétron....................................................................................................................... 8,00 mg
Pour un lyophilisat oral.
· Les autres composants sont :
gélatine, mannitol, aspartam, parahydroxybenzoate de méthyle sodique, parahydroxybenzoate de propyle sodique, arôme fraise.
Qu’est-ce que ZOPHREN 8 mg, lyophilisat oral et contenu de l’emballage extérieur
Ce médicament se présente sous forme de lyophilisat oral.
Boîte de 2, 4, 6 ou 10.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
SANDOZ
9 PLACE MARIE-JEANNE BASSOT
92300 LEVALLOIS-PERRET
Exploitant de l’autorisation de mise sur le marché
SANDOZ
9 PLACE MARIE-JEANNE BASSOT
92300 LEVALLOIS-PERRET
LEK PHARMACEUTICALS D.D.
VEROVSKOVA ULICA 57
1526 LJUBLJANA
SLOVENIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).