Dernière mise à jour le 29/06/2026
VERSATIS 700 mg, emplâtre médicamenteux
Indications thérapeutiques
Classe pharmacothérapeutique : anesthésique local, amides - code ATC : N01BB02
On vous a prescrit VERSATIS 700 mg, emplâtre médicamenteux pour traiter une affection douloureuse appelée « douleur neuropathique post-zostérienne ». Celle-ci se manifeste généralement par des symptômes locaux tels que des brûlures, des élancements, des coups de poignard ou des fourmillements.
Présentations
> 4 sachet(s) papier polyéthylène aluminium copolymère d'éthylène et d'acide méthacrylique de 5 emplâtre(s)
Code CIP : 382 854-4 ou 34009 382 854 4 4
Déclaration de commercialisation : 02/07/2018
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 38,14 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 39,16 €
- Taux de remboursement :65%
> 6 sachet(s) papier polyéthylène aluminium copolymère d'éthylène et d'acide méthacrylique de 5 emplâtre(s)
Code CIP : 382 856-7 ou 34009 382 856 7 3
Déclaration de commercialisation : 01/02/2008
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 57,01 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 58,03 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 20/01/2016 | Renouvellement d'inscription (CT) | Le service médical rendu par VERSATIS reste important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| IV (Mineur) | Avis du 06/10/2010 | Nouvel examen suite au dépôt de nouvelles données | Au vu des nouvelles données cliniques disponibles, VERSATIS apporte une amélioration du service médical rendu mineure (de niveau IV) dans la prise en charge des douleurs post-zostériennes de l'adulte. |
| V (Inexistant) | Avis du 19/06/2008 | Inscription (CT) | VERSATIS n'apporte pas d'amélioration du service médical rendu (ASMR V) par rapport à la stratégie thérapeutique actuelle. |
ANSM - Mis à jour le : 31/01/2024
VERSATIS 700 mg, emplâtre médicamenteux
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque emplâtre médicamenteux (10 cm x 14 cm) contient 700 mg (5% m/m) de lidocaïne.
Excipients à effet notoire :
Parahydroxybenzoate de méthyle 14 mg,
Parahydroxybenzoate de propyle 7 mg,
Propylène glycol 700 mg.
Pour la liste complète des excipients, voir rubrique 6.1.
Emplâtre blanc d'hydrogel avec une base adhésive, collé à un support non-tissé de téréphtalate de polyéthylène embossé "Lidocaïne 5%" et recouvert d’un film protecteur de téréphtalate de polyéthylène.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Adultes et sujets âgés
Appliquer les emplâtres sur la zone douloureuse une fois par jour, pendant une période maximale de 12 heures par 24 heures.
N’utilisez que le nombre d’emplâtres nécessaires à l’efficacité du traitement. Si nécessaire, les emplâtres peuvent être découpés à la taille requise avec des ciseaux avant d’enlever le film protecteur. Au total, utilisez 3 emplâtres au maximum en même temps.
L’emplâtre doit être appliqué tel quel, sur une peau sèche et non irritée (après cicatrisation des vésicules de zona).
L’emplâtre ne doit pas être appliqué plus de 12 heures. Il est nécessaire de respecter un intervalle de 12 heures avant l’application de l’emplâtre suivant. L’emplâtre peut être appliqué indifféremment pendant la journée ou la nuit.
L’emplâtre doit être appliqué sur la peau immédiatement après l’ouverture du sachet et après avoir enlevé le film protecteur. Les poils de la zone concernée doivent être coupés avec des ciseaux (ne pas raser).
L’efficacité du traitement sera évaluée au bout de 2 à 4 semaines. Le traitement sera interrompu en cas d’inefficacité au terme de cette période (durant l’application ou durant la période sans emplâtre) car les risques potentiels liés au traitement peuvent l’emporter sur les bénéfices attendus (voir rubriques 4.4 et 5.1).
L’utilisation de VERSATIS au long cours dans les essais cliniques montre que le nombre d’emplâtres utilisés diminue dans le temps. C’est pourquoi le traitement sera réévalué périodiquement pour réduire éventuellement le nombre d’emplâtres nécessaires ou bien pour allonger la période de 12 heures sans emplâtre.
Insuffisance rénale
Un ajustement de la posologie n’est pas nécessaire chez les patients présentant une insuffisance rénale légère ou modérée. VERSATIS devra être utilisé avec précaution chez les patients présentant une insuffisance rénale sévère (voir rubrique 4.4).
Insuffisance hépatique
Un ajustement de la posologie n’est pas nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée. VERSATIS devra être utilisé avec précaution chez les patients présentant une insuffisance rénale sévère (voir rubrique 4.4).
Population pédiatrique
La tolérance et l’efficacité de VERSATIS chez les enfants au-dessous de 18 ans n’ont pas été évaluées. Aucune donnée n’est disponible.
Hypersensibilité connue à la substance active ou aux excipients mentionnés à la rubrique 6.1.
L’emplâtre est également contre-indiqué chez les patients présentant une hypersensibilité connue aux autres anesthésiques locaux de type amide comme par exemple la bupivacaïne, l’étidocaïne, la mépivacaïne et la prilocaïne.
L’emplâtre ne doit pas être appliqué sur une peau inflammatoire ou lésée, telle que des lésions actives de zona, des dermatites ou des plaies.
4.4. Mises en garde spéciales et précautions d'emploi
L’emplâtre ne doit pas être appliqué sur des muqueuses. Eviter le contact de l’emplâtre avec l’œil.
L’emplâtre contient du propylène glycol (E1520) qui peut causer une irritation de la peau. Il contient également du parahydroxybenzoate de méthyle (E218) et du parahydroxybenzoate de propyle (E216) qui peuvent provoquer des réactions allergiques (parfois retardées).
L’emplâtre doit être utilisé avec prudence chez les patients présentant une insuffisance cardiaque sévère, une insuffisance rénale sévère ou une insuffisance hépatique sévère. Un des métabolites de la lidocaïne, la 2,6 xylidine s’est révélé génotoxique et carcinogène chez le rat (voir rubrique 5.3). Des métabolites secondaires se sont révélés mutagènes. La signification clinique de ces données est inconnue.
En conséquence, un traitement au long cours avec VERSATIS n’est justifié que dans le cas où un bénéfice thérapeutique est observé pour le patient (voir rubrique 4.2).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude spécifique d'interaction n'a été réalisée. Aucune interaction médicamenteuse n’a été observée au cours des études cliniques.
Les concentrations plasmatiques maximales de lidocaïne observées au cours des études cliniques avec l’emplâtre étant faibles (voir rubrique 5.2), une interaction pharmacocinétique cliniquement significative est peu probable.
Bien que l'absorption de la lidocaïne par la peau soit faible, VERSATIS doit être utilisé avec prudence chez les patients recevant des anti-arythmiques de la classe I (par exemple tocaïnide, méxilétine) ou d'autres anesthésiques locaux. Le risque d’addition des effets systémiques ne peut être exclu.
4.6. Fertilité, grossesse et allaitement
Grossesse
La lidocaïne traverse la barrière placentaire. Néanmoins, en clinique, il n’y a pas de donnée pertinente concernant l'utilisation de la lidocaïne chez les femmes enceintes.
Les études chez l’animal ne retrouvent pas de tératogénie potentielle en relation avec la lidocaïne (voir rubrique 5.3).
Chez la femme, le risque potentiel n’est pas connu. Par conséquent, VERSATIS ne doit pas être utilisé pendant la grossesse sauf en cas de nécessité.
La lidocaïne est excrétée dans le lait maternel. Néanmoins, il n'y a aucune étude chez les femmes allaitantes. Compte tenu d’un rapide métabolisme de la lidocaïne essentiellement hépatique, de très faibles taux de lidocaïne sont susceptibles d’être retrouvés dans le lait maternel.
Fertilité
Aucune donnée clinique sur la fertilité n’est disponible. Les études chez l’animal n’ont pas montré d’effet sur la fertilité des femelles.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables sont classés par fréquence et par ordre de sévérité décroissante.
Des effets indésirables ont été observés chez environ 16% des patients, essentiellement à type de réactions locales dues à la forme pharmaceutique du médicament.
Les effets indésirables les plus fréquemment rapportés sont des réactions locales au niveau du site d’application (brûlures, dermatites, érythèmes, prurit, rash, irritation de la peau et vésicules ).
Dans le tableau ci-dessous, les effets indésirables rapportés au cours des études cliniques chez des patients souffrant de douleurs neuropathiques post-zostériennes et traités par l’emplâtre sont classés par système de classe d’organe et par fréquence: très fréquent (³ 1/10) ; fréquent (³ 1/100 ; < 1/10), peu fréquent (³ 1/1000 ; < 1/100), rare (³ 1/10 000 ; < 1/1000), très rare (< 1/10 000) ; inconnu (la fréquence ne peut pas être estimée à partir des données disponibles).
|
Système de classe d’organe / fréquence |
Effets indésirables |
|
Affections de la peau et des tissus sous cutanés |
|
|
Peu fréquentes |
Lésions cutanées |
|
Blessures, empoisonnement et complications procédurales |
|
|
Peu fréquentes |
Blessures |
|
Réactions au niveau du site d’application et troubles de l’état général |
|
|
Très fréquentes |
Réactions au niveau du site d’application |
Autres réactions observées chez des patients traités par l’emplâtre dans des conditions normales d’utilisation :
|
Système de classe d’organe / fréquence |
Effets indésirables |
|
Blessures, empoisonnement et complications procédurales |
|
|
Très rares |
Blessures |
|
Troubles du système immunitaire |
|
|
Très rare |
Réaction anaphylactique, hypersensibilité |
Tous les effets indésirables étaient essentiellement d'intensité légère à modérée, et moins de 5% d’entre eux ont entraîné l’arrêt du traitement.
La survenue d’effets indésirables systémiques est peu probable compte tenu des faibles concentrations circulantes de lidocaïne (voir la rubrique 5.2). Les effets indésirables rapportés sont semblables à ceux observés avec les autres agents anesthésiques locaux de type amide (voir la rubrique 4.9).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Des interactions médicamenteuses connues lors de l’utilisation de lidocaïne par voie systémique avec un béta-bloquant ou des inhibiteurs du CYP3A4 (par ex. dérivés imidazole, macrolides) ou des agents antiarythmiques pourraient être observées en cas de surdosage.
En cas de surdosage, les emplâtres devront être retirés et des mesures cliniquement adaptées devront être prises. Il n’y a pas d’antidote connu à la lidocaïne.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Anesthésique local, amides - Code ATC : N01BB02
Mécanisme d’action
VERSATIS présente un double mode d’action ; une action pharmacologique lors de la diffusion de la lidocaïne et une protection mécanique de l’emplâtre qui protège la zone hyper sensible.
La lidocaïne contenue dans l’emplâtre diffuse de façon continue dans la peau, produisant un effet antalgique local.
Le mécanisme d’action serait lié à une stabilisation des membranes neuronales entraînant une diminution d’activité des canaux sodiques aboutissant ainsi à une diminution de la douleur.
Efficacité clinique
La prise en charge de la douleur post-zostérienne (DPZ) est difficile. VERSATIS a prouvé son efficacité dans le traitement symptomatique de l’allodynie liée aux douleurs neuropathiques post-zostériennes (voir rubrique 4.2).
L'efficacité de VERSATIS a été démontrée au cours d’études dans la douleur neuropathique post-zostérienne.
Deux principales études contrôlées ont été réalisées pour évaluer l'efficacité de la lidocaïne 700 mg.
Dans la première étude, les patients ont été sélectionnés parmi une population considérée comme répondeuse au produit. Il s’agissait d’un essai croisé randomisé au cours duquel les patients ont été traités avec la lidocaïne 700 mg, suivis de 14 jours de placebo ou inversement. Le critère principal était le délai de sortie de l’essai pour aggravation de la douleur ; les patients sortaient de l’essai si le score de soulagement de la douleur diminuait d’au moins 2 points sur une échelle d’évaluation en 6 points. 32 patients ont participé, dont 30 ont terminé l’étude. Le délai de sortie moyen pour le groupe placebo est de 4 jours et de 14 jours pour le groupe recevant le traitement actif (p < 0.001). Aucun des patients recevant le traitement actif n’est sorti prématurément de l’étude au cours des 2 semaines de traitement.
Dans la deuxième étude, 265 patients souffrant de douleurs post-zostériennes ont été sélectionnés et ont reçu la lidocaïne 700 mg pendant 8 semaines. Au cours de cet essai en ouvert, non contrôlé, environ 50% de patients ont présenté un soulagement de leur douleur d’au moins 4 points sur une échelle de 6 points (allant de aggravation à soulagement complet).
A l’issue de cette période, 71 patients ont été randomisés pour recevoir soit du placebo, soit lidocaïne 700 mg pendant 2 à 14 jours. Le critère principal était défini comme une absence d’efficacité au cours de 2 jours consécutifs lorsque le soulagement était de 2 points inférieurs à celui d’une réponse normale obtenue sur une échelle de 6 points (échelle allant de aggravation à soulagement complet) et entrainant une interruption de traitement. Neuf patients sur 36 (groupe ’traitement actif’) et 16 patients sur 35 (groupe ’placebo’) sont sortis prématurément de l’essai pour absence de bénéfice lié au traitement.
Des analyses à posteriori de cette étude ont montré que la réponse initiale est indépendante de la durée des douleurs neuropathiques post-zostériennes pré-existantes. Cependant, dans la deuxième phase de l’étude après randomisation du traitement actif versus placebo, les patients qui souffraient de douleurs neuropathiques post-zostériennes depuis plus de 12 mois maintenaient un bénéfice avec le traitement actif, alors que ceux qui étaient sous placebo sortaient plus rapidement de l’étude par manque d’efficacité.
Dans une étude contrôlée, en ouvert portant sur 98 patients atteints de douleurs post-zostériennes, les résultats obtenus avec VERSATIS montre une efficacité comparable avec ceux à la prégabaline et un profil de tolérance favorable.
5.2. Propriétés pharmacocinétiques
Aux doses maximales recommandées d’emplâtre médicamenteux de lidocaïne 700 mg (3 emplâtres appliqués simultanément pendant 12 heures), 3 ± 2 % de la dose totale de lidocaïne est retrouvé au niveau plasmatique après administrations unique ou réitérée.
Une analyse en cinétique de population des études cliniques d’efficacité chez des patients souffrant de douleurs neuropathiques post-zostériennes ont révélé une concentration maximale moyenne de lidocaïne de 45 ng/ml après application simultanée de 3 emplâtres, 12 heures par jour, jusqu’à un an. Cette concentration est similaire à celle observée dans des études pharmacocinétiques chez des patients souffrant de douleurs post-zostériennes (52 ng/ml) et chez des volontaires sains (85 ng/ml et 125 ng/ml).
Aucune tendance à l’accumulation n’est observée pour la lidocaïne et ses métabolites MEGX, GX et 2,6 xylidine, l’état d’équilibre pour ces concentrations étant atteint dans les 4 premiers jours.
En augmentant le nombre d’emplâtres de 1 à 3 appliqués simultanément, le passage systémique augmente proportionnellement moins que le nombre d’emplâtres utilisés.
Distribution
Après une administration intraveineuse de lidocaïne à des volontaires sains, le volume de distribution observé est de 1.3 ± 0.4 l/kg (n = 15). Le volume de distribution de la lidocaïne n'est pas lié à l’âge. Le volume de distribution est diminué chez les patients présentant une insuffisance cardiaque congestive et augmenté chez les patients présentant une affection hépatique.
Lors de l'application de l’emplâtre, approximativement 70 % de lidocaïne est lié aux protéines plasmatiques. La lidocaïne passe les barrières placentaires et hémato-encéphalique vraisemblablement par diffusion passive.
Biotransformation
La lidocaïne est transformée rapidement par le foie en un certain nombre de métabolites. La lidocaïne est métabolisée par N-déalkylation en monoethylglycinexylidide (MEGX) et en glycinexylidide (GX), moins actifs que la lidocaïne et présentes à de faibles concentrations. Celles-ci sont hydrolysées en 2,6-xylidine, elle-même transformée en 4-hydroxy-2,6-xylidine conjuguée.
La 2,6-xylidine, a une activité pharmacologique mal connue mais présente un potentiel carcinogène chez le rat (voir rubrique 5.3).
Une analyse pharmacocinétique de population a montré une concentration maximale moyenne en 2,6-xylidine de 9 ng/ml après applications quotidiennes répétées de l’emplâtre jusqu’à un an. Ceci a été confirmé par une étude pharmacocinétique de phase I. Aucune donnée n’est disponible concernant le métabolisme de la lidocaïne dans la peau.
Élimination
La lidocaïne et ses métabolites sont excrétés par les reins. Plus de 85% de la dose administrée est retrouvée dans l'urine sous forme de métabolites ou inchangée. Moins de 10% de la dose de lidocaïne excrétée est inchangée.
Le principal métabolite excrété est un conjugué de 4-hydroxy-2,6-xylidine, représentant 70 à 80% de la dose totale excrétée dans l’urine.
Chez l’Homme, la 2,6-xylidine est excrétée dans l'urine à une concentration inférieure à 1% de la dose appliquée. La demi-vie d'élimination de la lidocaïne après application de l’emplâtre chez des volontaires sains est de 7,6 heures.
L'élimination de la lidocaïne et de ses métabolites peut être retardée en cas d'insuffisance cardiaque, rénale ou hépatique.
5.3. Données de sécurité préclinique
Le chlorhydrate de lidocaïne n'a montré aucune génotoxicité in vitro ou in vivo. Son produit d'hydrolyse et son métabolite, 2,6-xylidine, ont montré une activité génotoxique dans plusieurs études en particulier après activation métabolique.
La carcinogénicité n’a pas été étudiée avec la lidocaïne. De telles études réalisées avec le métabolite 2,6-xylidine mélangé dans la nourriture de rats mâles et femelles ont montré une cytotoxicité et une hyperplasie de l'épithélium olfactif nasal, et une apparition de carcinomes et d’adénomes de la cavité nasale. Des modifications tumorales ont également été retrouvées dans le foie et dans les tissus sous-cutanés. Le risque chez l’Homme étant inconnu, un traitement à long terme à fortes doses de lidocaïne devra être évité.
La lidocaïne n'a aucun effet sur la reproduction, la fertilité, le développement embryo-foetal ou la tératogénicité chez le rat femelle à des concentrations plasmatiques jusqu’à plus de 50 fois supérieures à celles observés chez l’Homme.
Les études chez l’animal sont incomplètes en ce qui concerne la fertilité chez le mâle, la parturition ou le développement postnatal.
Glycérol, sorbitol liquide, carmellose sodique, propylène glycol (E1520), urée, kaolin lourd, acide tartrique, gélatine, alcool polyvinylique, glycinate d’aluminium, édétate disodique, parahydroxybenzoate de méthyle (E218), parahydroxybenzoate de propyle (E216), acide polyacrylique, polyacrylate de sodium, eau purifiée.
Tissu de support :
Polyéthylène téréphtalate (PET)
Film protecteur :
Polyéthylène téréphtalate (PET)
Durée de conservation après première ouverture : 14 jours.
6.4. Précautions particulières de conservation
Ne pas mettre au réfrigérateur. Ne pas le congeler.
Après 1ère ouverture : conserver le sachet soigneusement fermé à l’abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
Sachet refermable composé de papier/polyéthylène/aluminium/copolymère acide méta-acrylique contenant 5 emplâtres.
Chaque boîte contient 5, 10, 20, 25 ou 30 emplâtres. Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Après son utilisation, l’emplâtre contient encore de la substance active. L’emplâtre usagé doit être plié en 2 par la moitié, côté adhésif vers l'intérieur afin que la couche auto-adhésive ne soit pas à l’extérieur, puis jeté.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
TOUR PACIFIC
11-13 COURS VALMY
92800 PUTEAUX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 382 853 8 3 : emplâtre médicamenteux en sachet (Papier/PE/Aluminium/Copolymère acide méta acrylique). Boîte de 10.
· 34009 382 854 4 4 : emplâtre médicamenteux en sachet (Papier/PE/Aluminium/Copolymère acide méta acrylique). Boîte de 20.
· 34009 382 855 0 5 : emplâtre médicamenteux en sachet (Papier/PE/Aluminium/Copolymère acide méta acrylique). Boîte de 25.
· 34009 382 856 7 3 : emplâtre médicamenteux en sachet (Papier/PE/Aluminium/Copolymère acide méta acrylique). Boîte de 30.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
ANSM - Mis à jour le : 31/01/2024
VERSATIS 700 mg, emplâtre médicamenteux
Lidocaïne
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que VERSATIS 700 mg, emplâtre médicamenteux et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser VERSATIS 700 mg, emplâtre médicamenteux ?
3. Comment utiliser VVERSATIS 700 mg, emplâtre médicamenteux ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver VERSATIS 700 mg, emplâtre médicamenteux ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE VERSATIS 700 mg, emplâtre médicamenteux ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : anesthésique local, amides - code ATC : N01BB02
On vous a prescrit VERSATIS 700 mg, emplâtre médicamenteux pour traiter une affection douloureuse appelée « douleur neuropathique post-zostérienne ». Celle-ci se manifeste généralement par des symptômes locaux tels que des brûlures, des élancements, des coups de poignard ou des fourmillements.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER VERSATIS 700 mg, emplâtre médicamenteux ?
N’utilisez jamais VERSATIS 700 mg, emplâtre médicamenteux :
· si vous êtes allergique (hypersensible) à la lidocaïne ou à l’un des autres composants contenus dans ce médicament (listés en rubrique 6).
· si vous avez eu une allergie à d'autres produits semblables à la lidocaïne, tel que la bupivacaïne, l’étidocaïne, la mépivacaïne et la prilocaïne.
· si vous avez des lésions cutanées ou des blessures ouvertes.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser VERSATIS.
Si vous avez une insuffisance hépatique ou rénale sévère ou en cas de problème cardiaque grave, prévenez votre médecin.
VERSATIS doit être appliqué uniquement après cicatrisation des lésions cutanées. Il ne doit pas être appliqué sur ou autour de l’œil ou de la bouche.
VERSATIS n’a pas été étudié chez des patients de moins de 18 ans ; il n’est donc pas recommandé de l’utiliser dans cette population.
La lidocaïne est transformée dans votre foie en plusieurs composés. Il a été montré qu’à de très fortes doses et après un traitement très long, un de ces composés, la 2,6 xylidine a provoqué des tumeurs chez le rat. La signification clinique de ces données est inconnue.
Enfants et adolescents
VERSATIS n’a pas été étudié chez les enfants de moins de 18 ans. Par conséquent, l’utilisation n’est pas recommandée dans cette population.
Autres médicaments et VERSATIS 700 mg, emplâtre médicamenteux
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
VERSATIS 700 mg, emplâtre médicamenteux avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
VERSATIS ne doit pas être utilisé pendant la grossesse excepté en cas de nécessité clairement avérée.
Il n’existe aucune donnée clinique chez la femme allaitante. En utilisant VERSATIS, de très faibles taux de lidocaïne peuvent être retrouvés dans le sang circulant. Un effet sur les nourrissons en cours d’allaitement est peu probable.
Conduite de véhicules et utilisation de machines
Un effet de VERSATIS sur l’aptitude à conduire et à utiliser des machines est peu probable. Par conséquent, vous pouvez conduire ou utiliser des machines tout en étant traité par VERSATIS.
VERSATIS 700 mg, emplâtre médicamenteux contient du propylène glycol, du parahydroxybenzoate de méthyle et du parahydroxybenzoate de propyle.
Les emplâtres contiennent du propylène glycol (E1520) qui peut entrainer une irritation de la peau qui peuvent entrainer des réactions allergiques. Il contient également du parahydroxybenzoate de méthyle (E218) et du parahydroxybenzoate de propyle (E216) qui peuvent entrainer des réactions allergiques, y compris après l’arrêt du traitement.
3. COMMENT UTILISER VERSATIS 700 mg, emplâtre médicamenteux ?
La dose recommandée est de 1 à 3 emplâtres VERSATIS à appliquer sur les zones douloureuses de votre peau. VERSATIS peut être découpé à la taille requise pour s’adapter à la zone douloureuse. N’utilisez pas plus de 3 emplâtres en même temps.
Les emplâtres doivent être enlevés après 12 heures d’utilisation, de sorte que vous ayez une période de 12 heures sans emplâtre. Vous pouvez choisir d’appliquer VERSATIS indifféremment pendant la journée ou la nuit.
Habituellement, vous sentirez un soulagement de votre douleur le premier jour d’utilisation de VERSATIS, mais le soulagement complet peut prendre jusqu'à 2 à 4 semaines. Passé ce délai, si vous continuez à souffrir, parlez-en à votre médecin car les risques potentiels liés au traitement peuvent l’emporter sur les bénéfices attendus (voir rubrique 2 QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER VERSATIS, paragraphe « Avertissements et précautions »)
A intervalles réguliers, votre médecin jugera avec vous l’intérêt de continuer VERSATIS.
Précautions à prendre avant d’appliquer VERSATIS 700 mg, emplâtre médicamenteux
· En cas de présence de poils sur la zone douloureuse, coupez-les à l’aide de ciseaux. Ne les rasez pas.
· La peau doit être propre et sèche avant l’application de l’emplâtre afin qu’il puisse adhérer correctement à la peau.
· Les crèmes et lotions pourront être utilisées sur la peau durant la période sans emplâtre.
· Si vous avez pris un bain ou une douche récemment, vous devrez attendre que votre peau se refroidisse avant l’application de l’emplâtre.
Application cutanée :
|
|
Mode d’utilisation |
|
|
Etape 1 : ouvrir le sachet et prendre un ou plusieurs emplâtres · Déchirez ou découpez le sachet en suivant la ligne pointillée. · Si vous utilisez des ciseaux, faites attention à ne pas endommager les emplâtres et le système de fermeture. · Sortez un ou plusieurs emplâtres selon la taille de la zone douloureuse sur votre peau. |
|
|
Etape 2 : refermez le sachet · Refermez le sachet correctement après utilisation en exerçant une pression sur le système de fermeture. · Les emplâtres contiennent de l’eau et pourraient sécher si le sachet n'était pas fermé correctement. |
|
|
Etape 3 : découpez l’emplâtre, si nécessaire · Si nécessaire, découpez l’emplâtre à la taille requise en fonction de la zone douloureuse avant de retirer le film protecteur. |
|
|
Etape 4 : retirez le film protecteur · Retirez le film protecteur de l’emplâtre. · Essayez de ne pas toucher la partie collante de l’emplâtre. |
|
|
Etape 5 : appliquez l’emplâtre et pressez le fermement sur votre peau · N’appliquez pas plus de 3 emplâtres sur la zone douloureuse de votre peau. · Pressez l’emplâtre sur votre peau au moins 10 secondes afin d’être sûr que l’emplâtre adhère fermement. · Assurez-vous que la totalité de l’emplâtre colle à la peau ainsi que les côtés. |
|
|
Ne gardez l’emplâtre que pour une durée maximale de 12 heures Il est important que l’emplâtre ne soit pas en contact plus 12 heures avec la peau. Par exemple, si les douleurs s’intensifient la nuit, vous appliquerez l’emplâtre à 7 heures le soir et vous le retirerez à 7 heures le matin. Si les douleurs sont plus importantes le jour, vous appliquerez l’emplâtre à 7 heures le matin et vous le retirerez à 7 heures le soir. |


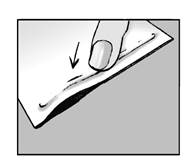

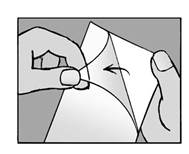
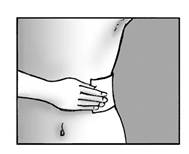
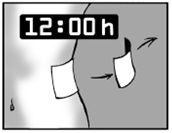
Se baigner, se doucher, nager :
Lorsque vous utilisez l’emplâtre, évitez au possible le contact avec l’eau.
Vous devrez prendre votre bain, votre douche ou nager uniquement lorsque vous ne portez pas l’emplâtre. Si vous venez de prendre votre bain ou votre douche, attendre que votre peau se refroidisse avant d’appliquer l’emplâtre.
Si l’emplâtre se décolle :
Dans de rares cas l’emplâtre peut se décoller, ou devenir incollable. Si c’est le cas, essayez de le coller à nouveau sur la même zone. S’il ne colle toujours pas, retirez le et placez un nouvel emplâtre sur la même zone douloureuse.
Comment enlever VERSATIS 700 mg, emplâtre médicamenteux :
Enlevez lentement l’emplâtre usagé. S'il ne se retire pas facilement, vous pouvez l’imbiber d’eau chaude pendant quelques minutes avant de le retirer.
Si vous avez oublié d'enlever l’emplâtre après 12 heures :
Dès que vous vous en apercevez, enlevez l’emplâtre usagé. L’application suivante devra avoir lieu au moins 12 heures après le retrait de l’emplâtre précédent.
Si vous avez utilisé plus d’emplâtre que vous n’auriez dû :
Si vous utilisez plus d’emplâtres que nécessaire ou si vous les appliquez trop longtemps, cela risque d’augmenter le risque de survenue d’effets indésirables.
Si vous oubliez d’utiliser VERSATIS 700 mg, emplâtre médicamenteux :
Après 12 heures sans emplâtre, si vous avez oublié d’appliquer un nouvel emplâtre, vous devez coller un nouvel emplâtre dès que possible.
Si vous arrêtez d’utiliser VERSATIS 700 mg, emplâtre médicamenteux :
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Effets indésirables ou symptômes importants et conduite à tenir si vous êtes concerné :
Si une irritation ou une sensation de brûlure apparaissent lorsque vous utilisez l’emplâtre, celui-ci devra être enlevé. Un nouvel emplâtre pourra être de nouveau appliqué lorsque l’irritation aura disparu.
Autres effets indésirables pouvant apparaitre :
Très fréquents (peut concerner plus de 1 personne sur 10) :
Réactions locales au niveau du site d’application : rougeur, éruption, démangeaisons, sensation de brûlure, dermatite, et petites boursouflures.
Peu fréquents (peut concerner jusqu’à 1 personne sur 100) :
Blessures et plaies cutanées.
Très rares (peut concerner jusqu’à 1 personne sur 10 000) :
Plaies ouvertes, réactions allergiques sévères et allergie.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER VERSATIS 700 mg, emplâtre médicamenteux ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le sachet et la boîte après la mention EXP. La date de péremption fait référence au dernier jour de ce mois.
Ne pas mettre au réfrigérateur, ne pas congeler.
Après première ouverture, conserver le sachet soigneusement fermé à l’abri de la lumière.
Durée de conservation après première ouverture du sachet : 14 jours.
Ne pas utiliser ce médicament si le sachet est endommagé, ceux-ci risqueraient de sécher et de ne plus coller.
Comment jeter VERSATIS 700 mg, emplâtre médicamenteux
Les emplâtres usagés contiennent encore de la substance active qui pourrait être nocive pour les autres. Pliez-les en deux, les faces collantes entre elles et jetez-les de telle sorte que ni les enfants ni les animaux de compagnie n’y aient accès.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient VERSATIS 700 mg, emplâtre médicamenteux
· La substance active est la lidocaïne.
Chaque emplâtre (de dimension 10 cm x 14 cm) contient 700 mg (5% m/m) de lidocaïne.
· Les autres composants (excipients) sont :
Couche auto-adhésive : Glycérol, sorbitol liquide, carmellose sodique, propylène glycol (E1520), urée, kaolin lourd, acide tartrique, gélatine, alcool polyvinylique, glycinate d’aluminium, édétate disodique, parahydroxybenzoate de méthyle (E218), parahydroxybenzoate de propyle (E216), acide polyacrylique, polyacrylate de sodium, eau purifiée.
Tissu de support et film protecteur : polyéthylène téréphtalate (PET).
Qu’est-ce que VERSATIS 700 mg, emplâtre médicamenteux et contenu de l’emballage extérieur
L’emplâtre médicamenteux mesure 14 cm sur 10 cm. Il est constitué de tissu blanc sur marqué avec "Lidocaïne 5%". Les emplâtres sont réunis par 5 dans un sachet refermable.
Chaque boîte contient 5, 10, 20, 25 ou 30 emplâtres regroupés dans 1, 2, 4, 5, ou 6 sachets, respectivement. Toutes les présentations peuvent ne pas être toutes commercialisées.
Titulaire de l’autorisation de mise sur le marché
TOUR PACIFIC
11-13 COURS VALMY
92800 PUTEAUX
Exploitant de l’autorisation de mise sur le marché
TOUR PACIFIC
11-13 COURS VALMY
92800 PUTEAUX
ZIEGLERSTRASSE 6
52078 AACHEN
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).