Dernière mise à jour le 01/06/2026
ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé
Indications thérapeutiques
ABIRATERONE BIOGARAN contient un médicament appelé acétate d’abiratérone. Il est utilisé chez les hommes adultes pour traiter le cancer de la prostate qui s’est disséminé dans d’autres parties du corps. ABIRATERONE BIOGARAN arrête la production de testostérone par votre corps, ce qui peut ralentir la croissance du cancer de la prostate.
Lorsque ABIRATERONE BIOGARAN est prescrit au stade précoce de la maladie répondant encore à un traitement hormonal, il est utilisé en association à un traitement qui diminue le taux de testostérone (suppression androgénique).
Lors de votre traitement par ce médicament, votre médecin vous prescrira également un autre médicament appelé prednisone ou prednisolone. Cela permettra de réduire vos risques de développer une pression artérielle élevée, d’accumuler une quantité excessive d’eau dans votre corps (rétention hydrique), ou de présenter des taux réduits d’un composant chimique appelé potassium dans votre sang.
Présentations
> plaquette(s) aluminium PVC polyéthylène PVDC de 60 comprimé(s)
Code CIP : 34009 302 413 0 1
Déclaration de commercialisation : 08/09/2022
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 871,44 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 872,46 €
- Taux de remboursement :100 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : BIOGARAN
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière annuelle
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- renouvellement non restreint
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 541 293 4
ANSM - Mis à jour le : 22/07/2022
ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé pelliculé contient 500 mg d’acétate d'abiratérone.
Excipient à effet notoire :
Chaque comprimé contient 64,6 mg de lactose (68 mg sous forme monohydratée).
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimés pelliculés violets, de forme ovale, gravés « 500 » sur une face, de dimensions 18,9 mm x 9,5 mm.
4.1. Indications thérapeutiques
ABIRATERONE BIOGARAN est indiqué en association avec la prednisone ou la prednisolone dans :
· le traitement du cancer métastatique de la prostate hormono-sensible (mHSPC) à haut risque nouvellement diagnostiqué chez les hommes adultes, en association avec un traitement par suppression androgénique (ADT) (voir rubrique 5.1)
· le traitement du cancer métastatique de la prostate résistant à la castration (mCRPC) chez les hommes adultes asymptomatiques ou peu symptomatiques, après échec d’un traitement par suppression androgénique et pour lesquels la chimiothérapie n’est pas encore cliniquement indiquée (voir rubrique 5.1)
· le traitement du cancer métastatique de la prostate résistant à la castration (mCRPC) chez les hommes adultes dont la maladie a progressé pendant ou après une chimiothérapie à base de docétaxel.
4.2. Posologie et mode d'administration
Ce médicament doit être prescrit par un professionnel de santé habilité.
Posologie
La dose recommandée est de 1 000 mg (deux comprimés de 500 mg) en une seule prise quotidienne et ne doit pas être administrée avec de la nourriture (voir ci-dessous « Mode d’administration »). La prise des comprimés avec la nourriture augmente l’exposition systémique à l’abiratérone (voir rubriques 4.5 et 5.2).
Dose de prednisone ou de prednisolone
Dans le mHSPC, ABIRATERONE BIOGARAN est utilisé en association avec 5 mg de prednisone ou de prednisolone par jour.
Dans le mCRPC, ABIRATERONE BIOGARAN est utilisé en association avec 10 mg de prednisone ou de prednisolone par jour.
La castration médicale par analogue de l’hormone de libération des gonadotrophines hypophysaires (LH-RH) doit être maintenue pendant la durée du traitement pour les patients n’ayant pas subi de castration chirurgicale.
Surveillance recommandée
Les taux de transaminases sériques doivent êtres dosés avant le début du traitement, toutes les deux semaines pendant les trois premiers mois de traitement et ensuite tous les mois.
La tension artérielle, le taux de potassium sérique et la rétention hydrique doivent être surveillés mensuellement. Cependant, les patients ayant un risque significatif d’insuffisance cardiaque congestive doivent être surveillés toutes les 2 semaines pendant les 3 premiers mois du traitement et ensuite tous les mois (voir rubrique 4.4).
Chez les patients ayant une hypokaliémie pré-existante ou ayant développé une hypokaliémie au cours du traitement par abiratérone, le maintien de la kaliémie à un taux ≥ 4,0 mM doit être envisagé. Chez les patients qui développent des toxicités de Grade ≥ 3 incluant hypertension artérielle, hypokaliémie, œdème et autres troubles d’origine non minéralocorticoïde, le traitement doit être interrompu et une prise en charge médicale appropriée instaurée. Le traitement par abiratérone ne doit pas être réintroduit tant que les symptômes de toxicité n’ont pas régressé au Grade 1 ou à l’état initial.
En cas d'oubli d'une dose quotidienne de ABIRATERONE BIOGARAN, ou de prednisone ou de prednisolone, il convient de reprendre le traitement le lendemain à la dose quotidienne habituelle.
Hépatotoxicité
Chez les patients développant une hépatotoxicité au cours du traitement (augmentation de l’alanine aminotransférase [ALAT] ou augmentation de l’aspartate aminotransférase [ASAT] de plus de 5 fois la limite supérieure de la normale [LSN]), le traitement doit être interrompu immédiatement (voir rubrique 4.4). Après le retour des tests fonctionnels hépatiques à leurs valeurs initiales, la reprise du traitement peut être effectuée à une dose réduite de 500 mg (un comprimé) une fois par jour. Chez les patients pour qui le traitement a été réintroduit, les taux de transaminases sériques doivent être surveillés au minimum toutes les 2 semaines pendant les trois premiers mois et ensuite tous les mois. Si l’hépatotoxicité réapparait à la dose réduite de 500 mg par jour, le traitement doit être arrêté.
Si les patients développent une hépatotoxicité sévère (ALAT ou ASAT 20 fois supérieurs à la LSN) à un moment quelconque au cours du traitement, celui-ci doit être arrêté et ne doit pas être réintroduit chez ces patients.
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère préexistante, Classe A de Child-Pugh.
En cas d’insuffisance hépatique modérée (Classe B de Child-Pugh), on observe une augmentation de l’exposition systémique à l’abiratérone d’environ quatre fois suite à l’administration de 1 000 mg en dose unique d’acétate d’abiratérone (voir rubrique 5.2). Il n’existe pas de données sur la sécurité et l’efficacité clinique suite à l’administration de doses répétées d’acétate d’abiratérone chez des patients ayant une insuffisance hépatique modérée ou sévère (Child-Pugh Classe B ou C). Aucun ajustement posologique ne peut être prévu. L’utilisation de abiratérone doit être évaluée avec précaution chez les patients atteints d’une insuffisance hépatique modérée, chez lesquels le bénéfice doit être nettement supérieur au risque potentiel (voir les rubriques 4.2 et 5.2). Abiratérone ne doit pas être utilisé chez les patients atteints d’une insuffisance hépatique sévère (voir les rubriques 4.3, 4.4 et 5.2).
Insuffisance rénale
Aucun ajustement posologique n’est nécessaire chez les patients insuffisants rénaux (voir rubrique 5.2). Cependant, il n’existe pas d’expérience clinique chez les patients présentant à la fois un cancer de la prostate et une insuffisance rénale sévère. La prudence est recommandée chez ces patients (voir rubrique 4.4).
Population pédiatrique
L’utilisation d’abiratérone dans la population pédiatrique n’est pas justifiée.
Mode d’administration
ABIRATERONE BIOGARAN est à usage oral. Les comprimés doivent être pris au moins une heure avant ou au moins deux heures après avoir mangé. Ceux-ci doivent être avalés en entier avec de l’eau.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Femmes enceintes ou susceptibles de l’être (voir rubrique 4.6).
· Insuffisance hépatique sévère [Classe C de Child-Pugh (voir les rubriques 4.2, 4.4 et 5.2)].
· L’association d’abiratérone et de prednisone/prednisolone avec du radium (Ra-223) est contre indiquée.
4.4. Mises en garde spéciales et précautions d'emploi
L’abiratérone peut entraîner une hypertension artérielle, une hypokaliémie et une rétention hydrique (voir rubrique 4.8) en raison de l'augmentation du taux de minéralocorticoïdes secondaire à l'inhibition du CYP17 (voir rubrique 5.1). L'administration concomitante d'un corticoïde réduit la stimulation de l'hormone adrénocorticotrope (ACTH), entraînant une baisse de l'incidence et de la gravité de ces effets indésirables. La prudence est recommandée lors du traitement des patients présentant des pathologies sous-jacentes pouvant être aggravées par une augmentation de la pression artérielle, par une hypokaliémie (par exemple, ceux traités par des glucosides cardiotoniques), ou par une rétention hydrique (par exemple, ceux présentant une insuffisance cardiaque, un angor sévère ou instable, un infarctus récent ou une arythmie ventriculaire et ceux avec une insuffisance rénale sévère).
L’abiratérone doit être utilisée avec prudence chez les patients présentant des antécédents de maladie cardiovasculaire. Les patients atteints d'hypertension artérielle non contrôlée, d'une maladie cardiaque cliniquement significative, telle qu’un infarctus du myocarde ou des événements thrombotiques artériels dans les 6 mois précédents, un angor sévère ou instable, une insuffisance cardiaque de classe III ou IV (étude 301) selon la New York Heart Association (NYHA) ou une insuffisance cardiaque de classe II à IV (études 3 011 et 302) ou avec une mesure de la fraction d’éjection cardiaque (FEVG) < 50 % ont été exclus des études de phase 3 menées avec l’abiratérone. Dans les études 3 011 et 302, les patients présentant une fibrillation auriculaire ou d’autres arythmies cardiaques nécessitant un traitement médical ont été exclus. La sécurité d’emploi chez les patients présentant une fraction d’éjection ventriculaire gauche (FEVG) < 50% ou atteints d'insuffisance cardiaque de classe III ou IV selon la NYHA (dans l’étude 301) ou d’insuffisance cardiaque de classe II à IV selon la NYHA (dans les études 3 011 et 302) n'a pas été établie (voir rubriques 4.8 et 5.1).
Avant de traiter les patients ayant un risque significatif d’insuffisance cardiaque congestive (par exemple antécédent d’insuffisance cardiaque, hypertension artérielle non contrôlée, ou troubles cardiaques comme une cardiopathie ischémique), il faut envisager la réalisation d’un bilan cardiaque (par exemple une échographie cardiaque). Avant le traitement par abiratérone, l’insuffisance cardiaque doit être traitée et la fonction cardiaque optimisée. Toute hypertension artérielle, hypokaliémie et rétention hydrique doit être corrigée et contrôlée. Pendant le traitement, la pression artérielle, la kaliémie, la rétention hydrique (prise de poids, œdèmes périphériques), et autres signes et symptômes d’insuffisance cardiaque congestive doivent être surveillés toutes les 2 semaines pendant 3 mois et ensuite tous les mois et les anomalies doivent être corrigées. Un allongement de l’intervalle QT a été observé chez des patients présentant une hypokaliémie au cours du traitement par ABIRATERONE BIOGARAN. Evaluez la fonction cardiaque, instaurez une prise en charge appropriée et envisagez l’arrêt de ce traitement en cas de détérioration cliniquement significative de la fonction cardiaque (voir rubrique 4.2).
Hépatotoxicité et insuffisance hépatique
Des élévations marquées du taux d'enzymes hépatiques entraînant l'arrêt du traitement ou une modification de la dose sont survenues lors des études cliniques contrôlées (voir rubrique 4.8). Les 5 taux de transaminases sériques doivent êtres dosés avant le début du traitement, toutes les deux semaines pendant les trois premiers mois de traitement puis tous les mois. En cas d'apparition de symptômes cliniques ou de signes révélant le développement d’une hépatotoxicité, les transaminases sériques doivent être dosées immédiatement. Si au cours du traitement les ALAT ou ASAT augmentent de plus de 5 fois par rapport à la LSN, le traitement doit être immédiatement interrompu et les fonctions hépatiques étroitement surveillées.
La reprise du traitement pourra se faire uniquement après le retour des tests fonctionnels hépatiques à leurs valeurs initiales et à doses réduites (voir rubrique 4.2).
Si les patients développent une hépatotoxicité sévère, (ALAT ou ASAT 20 fois supérieurs à la LSN) à un moment quelconque au cours du traitement, celui-ci devra être arrêté et ne sera pas réintroduit chez ces patients.
Les patients présentant une hépatite virale active ou symptomatique ont été exclus des essais cliniques ; il n’y a donc pas de données relatives à l’utilisation d’abiratérone dans cette population.
Il n’y a aucune donnée clinique de sécurité et d’efficacité concernant l’administration de doses multiples d’acétate d’abiratérone chez les patients atteints d’une insuffisance hépatique modérée ou sévère (Classe B ou C de Child-Pugh). L’utilisation d’abiratérone doit être évaluée avec précaution chez les patients atteints d’une insuffisance hépatique modérée, chez lesquels le bénéfice doit être nettement supérieur au risque potentiel (voir les rubriques 4.2 et 5.2).
L’abiratérone ne doit pas être utilisée chez les patients atteints d’une insuffisance hépatique sévère (voir les rubriques 4.2, 4.3 et 5.2).
De rares cas d’insuffisance hépatique aigüe et d’hépatite fulminante, dont certains d’issue fatale, ont été rapportés après commercialisation (voir rubrique 4.8).
Sevrage des corticoïdes et prise en charge des situations de stress
La prudence est recommandée et une surveillance de l'insuffisance corticosurrénale doit être mise en place en cas d’arrêt de l'administration de la prednisone ou de la prednisolone. En cas de poursuite de l'administration d’abiratérone après sevrage des corticoïdes, les patients doivent faire l'objet d'une surveillance afin de déceler les symptômes d'un surdosage en minéralocorticoïdes (voir les informations ci-dessus).
Chez les patients sous prednisone ou prednisolone sujets à un stress inhabituel, une augmentation de la dose de corticoïdes avant, pendant et après la période de stress peut être indiquée.
Densité osseuse
Une diminution de la densité osseuse peut survenir chez les hommes avec un cancer métastatique avancé de la prostate. L’utilisation d’abiratérone en association avec un glucocorticoïde peut augmenter cet effet.
Utilisation précédente de kétoconazole
Des taux de réponse plus faibles peuvent être attendus chez les patients qui ont été traités précédemment par du kétoconazole pour leur cancer de la prostate.
Hyperglycémie
L’utilisation de glucocorticoïdes pouvant augmenter l’hyperglycémie, la glycémie doit être fréquemment contrôlée chez les patients diabétiques.
Hypoglycémie
Des cas d’hypoglycémie ont été rapportés lors de l’administration de abiratérone associé à la prednisone/prednisolone à des patients diabétiques recevant de la pioglitazone ou du répaglinide (voir rubrique 4.5) ; par conséquent, la glycémie doit être surveillée chez les patients diabétiques.
Utilisation avec une chimiothérapie
La sécurité et l’efficacité de l’utilisation concomitante de abiratérone avec une chimiothérapie par agent cytotoxique n’a pas été établie (voir rubrique 5.1).
Intolérance aux excipients
Ce médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
Risques potentiels
Une anémie et un dysfonctionnement sexuel peuvent apparaitre chez les hommes souffrant d’un cancer métastatique de la prostate, y compris ceux traités par abiratérone.
Effets musculo-squelettiques
Des cas de myopathies et de rhabdomyolyse ont été rapportés chez des patients traités par abiratérone.
La plupart des cas se sont développés au cours des 6 premiers mois de traitement ; les patients se sont rétablis après l’arrêt de abiratérone. La prudence est recommandée chez les patients traités simultanément avec des médicaments connus pour être associés à une myopathie/rhabdomyolyse.
Interactions avec d’autres médicaments
En raison du risque de diminution de l’exposition à l’abiratérone, les inducteurs puissants du CYP3A4 doivent être évités au cours du traitement, à moins qu’il n’y ait pas d’alternative thérapeutique (voir rubrique 4.5).
Association d’abiratérone et de prednisone/prednisolone avec du radium (Ra-223)
Le traitement avec l’abiratérone et la prednisone/prednisolone en association avec du radium (Ra-223) est contre indiqué (voir rubrique 4.3). Les études cliniques ont montré une augmentation du risque de fractures et une tendance à l’augmentation de la mortalité chez les patients atteints d’un cancer de la prostate, asymptomatiques ou peu symptomatiques.
Il est recommandé de ne pas initier de traitement par du radium (Ra-223) moins de 5 jours après la dernière administration d’abiratérone en association avec la prednisone/prednisolone.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Effets de la nourriture sur l'acétate d'abiratérone
L'administration avec la nourriture augmente de façon significative l'absorption de l'acétate d'abiratérone. L'efficacité et la sécurité d’emploi lorsqu’il est administré avec de la nourriture n'ayant pas été établies, ce médicament ne doit pas être pris avec de la nourriture (voir rubriques 4.2 et 5.2).
Interactions avec d’autres médicaments
Effet potentiel d'autres médicaments sur l’exposition à l'abiratérone
Dans une étude pharmacocinétique clinique d’interaction chez des sujets sains pré-traités avec de la rifampicine, un inducteur puissant du CYP3A4, à la dose de 600 mg par jour pendant 6 jours, suivie d’une dose unique de 1000 mg d’acétate d’abiratérone, l’ASC∞ plasmatique moyenne de l’abiratérone était diminuée de 55%.
Les inducteurs puissants du CYP3A4 (comme par exemple la phénytoïne, la carbamazépine, la rifampicine, la rifabutine, la rifapentine, le phénobarbital, le millepertuis [Hypericum perforatum]) sont à éviter au cours du traitement, à moins qu’il n’y ait pas d’alternative thérapeutique.
Dans une autre étude pharmacocinétique clinique d’interaction chez des sujets sains, l'administration concomitante de kétoconazole, un inhibiteur puissant du CYP3A4, n’a pas eu d’effet cliniquement significatif sur la pharmacocinétique de l'abiratérone.
Effet potentiel sur l’exposition d'autres médicaments
L’abiratérone est un inhibiteur des enzymes hépatiques CYP2D6 et CYP2C8 métabolisant les médicaments. Dans une étude visant à déterminer les effets de l'acétate d'abiratérone (plus prednisone) sur une dose unique de dextrométhorphane comme substrat du CYP2D6, l'exposition systémique (ASC) du dextrométhorphane a été augmentée d’environ 2,9 fois. Une augmentation de l’ASC24 du dextrorphane, métabolite actif du dextrométhorphane, d'environ 33% a été observée.
La prudence est recommandée lors de l'administration avec des médicaments activés ou métabolisés par le CYP2D6, en particulier ceux ayant une marge thérapeutique étroite. Une diminution de la dose des médicaments à marge thérapeutique étroite métabolisés par le CYP2D6 doit être envisagée. Des exemples de médicaments métabolisés par le CYP2D6 incluent métoprolol, propanolol, désipramine, venlafaxine, halopéridol, rispéridone, propafénone, flécaïnide, codéine, oxycodone et tramadol (les trois derniers médicaments nécessitant le CYP2D6 pour la formation de leurs métabolites actifs analgésiques).
Dans une étude d'interaction médicamenteuse avec le CYP2C8 chez des sujets sains, l'ASC de la pioglitazone a été augmentée de 46% et les ASC de M-III et M-IV, les métabolites actifs de la pioglitazone, ont chacune diminué de 10% lorsque la pioglitazone était associée avec une dose unique de 1 000 mg d'acétate d'abiratérone. Les patients doivent être surveillés pour des signes de toxicité liés à un substrat du CYP2C8 à index thérapeutique étroit, s'ils sont utilisés de façon concomitante. Des exemples de médicaments métabolisés par le CYP2C8 incluent la pioglitazone et le répaglinide (voir rubrique 4.4).
In vitro, les métabolites principaux, le sulfate d'abiratérone et le sulfate de N-oxyde-abiratérone, inhibent l'absorption par le transporteur hépatique OATP1B1 ; en conséquence, cela peut augmenter les concentrations des médicaments éliminés par OATP1B1. Il n'existe pas de données cliniques disponibles pour confirmer l’interaction avec le transporteur.
Utilisation avec les médicaments allongeant l’intervalle QT
Le traitement par suppression androgénique étant susceptible d’allonger l’intervalle QT, la prudence est conseillée lorsque abiratérone est administré avec des médicaments connus pour allonger l’intervalle QT, ou des médicaments capables d’induire des torsades de pointes tels que les antiarythmiques de classe IA (par exemple quinidine, disopyramide) ou de classe III (par exemple amiodarone, sotalol, dofétilide, ibutilide), la méthadone, la moxifloxacine, les antipsychotiques, etc.
Utilisation avec la spironolactone
La spironolactone se lie aux récepteurs des androgènes et peut augmenter le taux d’antigène prostatique spécifique (PSA). L’utilisation avec abiratérone n’est pas recommandée (voir rubrique 5.1).
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Il n’y a pas de donnée chez l’Homme sur l’utilisation d’abiratérone lors de la grossesse et ce médicament ne doit pas être utilisé chez la femme en âge de procréer.
Contraception chez les hommes et les femmes
La présence de l'abiratérone ou de ses métabolites dans le sperme n’est pas connue. L’utilisation d’un préservatif est nécessaire en cas de rapport sexuel avec une femme enceinte. L’utilisation d’un préservatif associée à une autre méthode de contraception efficace est nécessaire en cas de rapport sexuel avec une femme en âge de procréer. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3).
Grossesse
L’abiratérone ne doit pas être utilisée chez la femme et il est contre-indiqué chez la femme enceinte ou susceptible de l’être (voir rubriques 4.3 et 5.3).
Allaitement
L’abiratérone ne doit pas être utilisée chez la femme.
Fertilité
L’acétate d’abiratérone a perturbé la fécondité des rats mâles et femelles, mais ces effets ont été entièrement réversibles (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
D’après une analyse des effets indésirables rapportés dans les différentes études de phase 3 menées avec abiratérone, les effets indésirables observés chez ≥ 10 % des patients étaient : œdème périphérique, hypokaliémie, hypertension artérielle, infection du tractus urinaire, augmentation de l’alanine aminotransférase et/ou augmentation de l’aspartate aminotransférase. Les autres effets indésirables importants incluent des affections cardiaques, une hépatotoxicité, des fractures et l’alvéolite allergique.
Les conséquences pharmacodynamiques du mécanisme d'action d’abiratérone peuvent entraîner une hypertension artérielle, une hypokaliémie et une rétention hydrique. Au cours des études de phase 3, des réactions indésirables minéralocorticoïdes attendues ont été observées plus fréquemment chez les patients traités par acétate d’abiratérone que chez les patients sous placebo : hypokaliémie 18% vs 8%, hypertension artérielle 22% vs 16%, rétention hydrique (œdème périphérique) 23% vs 17 %. Chez les patients traités par acétate d’abiratérone versus les patients traités par placebo, des cas d'hypokaliémie de grades 3 et 4 selon la CTCAE (version 4.0) ont été observés respectivement chez 6% versus 1%, des cas d’hypertension artérielle de grades 3 et 4 selon la CTCAE (version 4.0) ont été observés chez 7% versus 5%, et des cas de rétention hydrique (œdème périphérique) de grades 3 et 4 ont été observés respectivement chez 1% versus 1% des patients. Dans l'ensemble, les réactions minéralocorticoïdes ont été prises en charge médicalement avec succès. L'administration concomitante d'un corticoïde réduit l'incidence et la gravité de ces effets indésirables (voir rubrique 4.4).
Tableau récapitulatif des effets indésirables
Au cours d'études incluant des patients atteints de cancer de la prostate métastasique à un stade avancé traités par un analogue de la LH-RH ou traités précédemment par orchidectomie, l’abiratérone a été administré à une dose de 1 000 mg par jour en association avec la prednisone ou la prednisolone à faible dose (5 ou 10 mg par jour selon l’indication).
Les effets indésirables observés au cours des études cliniques et de l’expérience post-commercialisation sont énumérés ci-dessous par catégorie de fréquence. Les catégories sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Dans chaque catégorie de fréquence, les effets indésirables sont présentés dans l'ordre décroissant de gravité.
Tableau 1 : Effets indésirables observés au cours des essais cliniques et post-commercialisation
|
Classes de systèmes d’organes |
Effet indésirable et fréquence |
|
Infections et infestations |
Très fréquent : infection du tractus urinaire |
|
|
Fréquent : sepsis |
|
Troubles du système immunitaire |
Fréquence indéterminée : réactions anaphylactiques |
|
Affections endocriniennes |
Peu fréquent : insuffisance surrénalienne |
|
Troubles du métabolisme et de la nutrition |
Très fréquent : hypokaliémie |
|
Fréquent : hypertriglycéridémie |
|
|
Affections cardiaques |
Fréquent : insuffisance cardiaque*, angine de poitrine, fibrillation auriculaire, tachycardie |
|
Peu fréquent : autres arythmies |
|
|
Indéterminée : infarctus du myocarde, allongement de l’intervalle QT (voir rubriques 4.4 et 4.5) |
|
|
Affections vasculaires |
Très fréquent : hypertension artérielle |
|
Affections respiratoires, thoraciques et médiastinales |
Rare : alvéolite allergiquea |
|
Affections gastro-intestinales |
Très fréquent : diarrhée |
|
Fréquent : dyspepsie |
|
|
Affections hépatobiliaires |
Très fréquent : augmentation de l'alanine amino-transférase et/ou augmentation de l’aspartate amino-transféraseb |
|
Rare : hépatite fulminante, insuffisance hépatique aigüe |
|
|
Affections de la peau et du tissu sous- cutané |
Fréquent : rash |
|
Affections musculo-squelettiques et systémiques |
Peu fréquent : myopathie, rhabdomyolyse |
|
Affections du rein et des voies urinaires |
Fréquent : hématurie |
|
Troubles généraux et anomalies au site d'administration |
Très fréquent : œdème périphérique |
|
Lésions, intoxications et complications liées aux procédures |
Fréquent : fractures** |
* L'insuffisance cardiaque regroupe l'insuffisance cardiaque congestive, le dysfonctionnement ventriculaire gauche et la diminution de la fraction d'éjection.
** Les fractures incluent l’ostéoporose et toutes les fractures à l’exception des fractures pathologiques
a Notifications spontanées issues de l’expérience post-commercialisation
b L’augmentation de l’alanine aminotransférase et/ou de l’aspartate aminotransférase inclut l’augmentation du taux d’ALAT, l’augmentation du taux d’ASAT et les anomalies de la fonction hépatique.
Les effets indésirables de grade 3 selon la CTCAE (version 4.0) survenues chez les patients traités par acétate d’abiratérone ont été les suivants : hypokaliémie (5%), infection urinaire (2%), augmentation du taux d’alanine aminotransférase et/ou du taux d’aspartate aminotransférase (4%), hypertension artérielle (6%), fractures (2%), œdèmes périphériques, insuffisance cardiaque et fibrillation auriculaire (1% chacun). Une hypertriglycéridémie et une angine de poitrine de grade 3 selon la CTCAE (version 4.0) ont été observées chez moins de 1% des patients. Une infection du tractus urinaire, une augmentation du taux d’alanine aminotransférase et/ou une augmentation du taux d’aspartate aminotransférase, une hypokaliémie, une insuffisance cardiaque, une fibrillation auriculaire et des fractures de grade 4 selon la CTCAE (version 4.0) ont été observés chez moins de 1% des patients.
L’hypertension et l’hypokaliémie ont été observées à une fréquence plus élevée dans la population hormonosensible (étude 3 011). Une hypertension a été rapportée chez 36,7% des patients dans la population hormonosensible (étude 3 011), comparé à 11,8% et 20,2 % respectivement dans les études 301 et 302. Une hypokaliémie a été observée chez 20,4% des patients dans la population hormonosensible (étude 3 011) comparé à 19,2% et 14,9 % respectivement dans les études 301 et 302.
L’incidence et la gravité des évènements indésirables étaient plus élevées dans le sous-groupe de patients ayant un score de performance ECOG2 à l’inclusion et également chez les patients âgés (≥ 75 ans).
Description d'une sélection d'effets indésirables
Effets cardiovasculaires
Les patients atteints d'hypertension artérielle non contrôlée, d'une maladie cardiaque cliniquement significative, telle qu’un infarctus du myocarde ou un événement thrombotique artériel dans les 6 mois précédents, un angor sévère ou instable, une insuffisance cardiaque de classe III ou IV (étude 301) ou 10 une insuffisance cardiaque de classe II à IV (études 3 011 et 302) selon la NYHA ou avec une mesure de la fraction d’éjection cardiaque < 50% ont été exclus des trois études de phase 3. Tous les patients inclus (groupe traité par la substance active et groupe placebo) ont simultanément reçu un traitement de suppression androgénique, principalement par des analogues de la LH-RH, qui a été associé à des cas de diabète, d'infarctus du myocarde, d'accident vasculaire cérébral et de mort subite cardiaque. La fréquence des effets indésirables cardiovasculaires dans les études de phase 3 chez les patients sous acétate d’abiratérone versus les patients sous placebo a été : fibrillation auriculaire 2,6% vs2,0%, tachycardie 1,9% vs 1,0%, angine de poitrine 1,7% vs 0,8%, troubles cardiaques 0,7% vs 0,2%, et arythmie 0,7% vs 0,5%.
Hépatotoxicité
Des cas d'hépatotoxicité avec une élévation des taux d'ALAT, d'ASAT et de bilirubine totale ont été rapportés chez des patients traités par acétate d’abiratérone. Dans les études cliniques de phase 3, une hépatotoxicité de grades 3 et 4 (par exemple augmentation du taux d'ALAT ou d'ASAT de > 5 x LSN ou augmentation de la bilirubine de > 1,5 x LSN) a été rapportée chez 6% des patients traités par acétate d’abiratérone, généralement durant les trois premiers mois de traitement. Dans l’étude 3 011, une hépatotoxicité de grade 3 ou 4 a été observée chez 8,4 % des patients traités par abiratérone. Dix patients ayant reçu abiratérone ont arrêté le traitement en raison d’une hépatotoxicité ; deux présentaient une hépatotoxicité de grade 2, six une hépatotoxicité de grade 3 et deux une hépatotoxicité de grade 4. Aucun patient n’est décédé des suites d’une hépatotoxicité dans l’étude 3 011. Au cours des études cliniques de phase 3, les patients dont le taux initial d'ALAT ou d'ASAT était élevé se sont révélés plus susceptibles de présenter une augmentation des résultats des tests fonctionnels hépatiques que ceux commençant le traitement avec des valeurs normales. Lorsque des augmentations du taux d'ALAT ou d'ASAT > 5 x LSN ou de la bilirubine > 3 x LSN ont été observées, l’acétate d’abiratérone a été suspendu ou arrêté. Dans deux cas, une élévation importante des résultats des tests fonctionnels hépatiques est survenue (voir rubrique 4.4). Ces deux patients, avec une fonction hépatique initiale normale, ont présenté une élévation du taux d'ALAT ou d'ASAT de 15 à 40 x LSN et une élévation de la bilirubine de 2 à 6 x LSN. Après arrêt de l'administration du traitement, les valeurs des tests fonctionnels hépatiques de ces deux patients se sont normalisées et un des patients a été traité à nouveau, sans récurrence de ces augmentations. Dans l’étude 302, des élévations d’ALAT ou d’ASAT de grade 3 ou 4 ont été observées chez 35 (6,5%) des patients traités par acétate d’abiratérone. Les élévations du taux d’aminotransférase ont été normalisées chez tous les patients sauf 3 (2 avec de nouvelles métastases hépatiques multiples et 1 avec une augmentation des ASAT environ 3 semaines après la dernière administration d’acétate d’abiratérone). Dans les études cliniques de phase 3, les arrêts de traitement dus à l’augmentation des ALAT et des ASAT ou à des anomalies de la fonction hépatique ont été rapportés chez 1,1% des patients traités par acétate d’abiratérone et chez 0,6% des patients traités par placebo ; aucun décès lié à une toxicité hépatique n’a été rapporté.
Au cours des essais cliniques, le risque d'hépatotoxicité a été atténué par l'exclusion des patients présentant initialement une hépatite ou des anomalies significatives des tests de la fonction hépatique. Dans l’étude 3 011, les patients présentant initialement des taux d’ALAT et d’ASAT > 2,5 x LSN, un taux de bilirubine > 1,5 x LSN ou une hépatite virale active ou symptomatique ou une atteinte hépatique chronique, une ascite ou des troubles hémorragiques secondaires à un dysfonctionnement hépatique étaient exclus. Dans l’étude 301, les patients présentant un taux initial d'ALAT et d'ASAT ≥ 2,5 x LSN en l'absence de métastases hépatiques et > 5 x LSN en présence de métastases hépatiques ont été exclus.
Dans l’étude 302, les patients présentant des métastases hépatiques n’étaient pas éligibles et ceux ayant un taux initial d’ALAT et d’ASAT ≥ 2.5 x LSN ont été exclus. Les anomalies des tests fonctionnels hépatiques apparues chez les patients participant aux essais cliniques ont été prises en charge de manière active par l'interruption du traitement et la reprise éventuelle de celui-ci uniquement après retour des résultats des tests fonctionnels hépatiques à leur valeur initiale (voir rubrique 4.2). Les patients présentant une élévation du taux d'ALAT ou d'ASAT > 20 x LSN n'ont pas été retraités. La sécurité d'une reprise du traitement chez ces patients est inconnue. Le mécanisme de l'hépatotoxicité n'est pas connu.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
L’expérience de surdosage chez l’homme avec abiratérone est limitée.
Il n'y a pas d'antidote spécifique. En cas de surdosage, l'administration doit être suspendue et des mesures générales de prise en charge doivent être mises en place, incluant une surveillance de la survenue d’arythmies, d’une hypokaliémie et de signes et symptômes de rétention hydrique. La fonction hépatique doit également être évaluée.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d'action
L’acétate d’abiratérone est transformé in vivo en abiratérone, un inhibiteur de la biosynthèse des androgènes. Plus spécifiquement, l'abiratérone inhibe de manière sélective l'enzyme 17α-hydroxylase/C17,20-lyase (CYP17). Cette enzyme est exprimée et nécessaire lors de la biosynthèse des androgènes au niveau des testicules, des glandes surrénales et des tissus tumoraux prostatiques. Le CYP17 catalyse la conversion de la prégnénolone et de la progestérone en précurseurs de la testostérone, respectivement la DHEA et l'androstènedione, par 17α-hydroxylation et rupture de la liaison C17,20. L'inhibition du CYP17 entraîne également une augmentation de la production de minéralocorticoïdes par les glandes surrénales (voir rubrique 4.4).
Le cancer de la prostate sensible aux androgènes répond aux traitements qui diminuent les taux d’androgènes. Les traitements suppresseurs des androgènes, tels que les analogues de la LH-RH ou l'orchidectomie, réduisent la production d'androgènes dans les testicules mais n'affectent pas leur production par les glandes surrénales ni dans la tumeur. Administré en même temps que des analogues de la LH-RH (ou que l'orchidectomie), le traitement par abiratérone abaisse le taux de testostérone sérique à un niveau indétectable (par les méthodes de dosage commercialisées).
Effets pharmacodynamiques
L’abiratérone diminue les taux sériques de testostérone et des autres androgènes à des niveaux inférieurs à ceux atteints par l’utilisation des analogues de la LH-RH seuls ou par l’orchidectomie. Ceci est dû à l’inhibition sélective de l’enzyme CYP17 nécessaire à la biosynthèse des androgènes. Le PSA sert de biomarqueur chez les patients atteints de cancer de la prostate. Lors d'une étude clinique de phase 3 chez des patients en échec de chimiothérapie contenant des taxanes, 38% des patients traités par acétate d’abiratérone ont vu leur PSA baisser d'au moins 50% par rapport à la valeur initiale, contre 10% pour le groupe placebo.
Efficacité et sécurité clinique
L'efficacité a été établie au cours de trois études cliniques (études 3 011, 302 et 301) de phase 3, multicentriques, randomisées, contrôlées versus placebo, chez des patients atteints de mHSPC et de mCRPC. L’étude 3 011 incluait des patients atteints de mHSPC nouvellement diagnostiqué (au cours des 3 mois précédant la randomisation) et présentant des facteurs pronostiques de haut risque. Le pronostic à haut risque était défini par la présence d’au moins 2 des 3 facteurs de risque suivants : (1) score de Gleason ≥ 8 ; (2) présence d’au moins 3 lésions à la scintigraphie osseuse ; (3) présence de métastase viscérale mesurable (hors atteinte ganglionnaire). Dans le bras actif, abiratérone était administré à la dose de 1 000 mg par jour en association avec une faible dose de 5 mg de prednisone une fois par jour en complément d’un ADT (analogue de la LH-RH ou orchidectomie), qui était considéré comme le traitement de référence. Les patients du groupe contrôle ont reçu un ADT et des placebos à la place d’abiratérone et de la prednisone. Les patients inclus dans l’étude 302 n’avaient pas eu de chimiothérapie antérieure par docétaxel ; alors que les patients inclus dans l’étude 301 avaient déjà reçu au préalable du docétaxel. Les patients étaient traités par un analogue de la LH-RH ou avaient précédemment été traités par orchidectomie. Dans le groupe recevant la substance active, abiratérone a été administré à une dose de 1 000 mg par jour en association avec la prednisone ou la prednisolone à une dose faible de 5 mg, deux fois par jour. Les patients du groupe témoin ont reçu un placebo et de la prednisone ou de la prednisolone à une dose faible de 5 mg, deux fois par jour.
Les modifications du PSA sérique pris isolément ne sont pas toujours prédictives du bénéfice clinique. Ainsi, dans toutes les études, il était recommandé que les patients poursuivent leurs traitements jusqu’à ce que les critères d’arrêt soient remplis pour chaque étude comme spécifié ci-après. Dans toutes les études, l’utilisation de spironolactone n’était pas autorisée car la spironolactone se lie aux récepteurs des androgènes et peut augmenter le PSA.
Étude 3 011 (patients atteints d’un mHSPC à haut risque nouvellement diagnostiqué)
Dans l’étude 3 011 (n = 1 199), l’âge médian des patients inclus était de 67 ans. Par origine ethnique, le nombre de patients traités par abiratérone était de 832 patients caucasiens (69,4%), 246 patients asiatiques (20,5%), 25 patients noirs ou afro-américains (2,1%), 80 patients d’origine autre (6,7%), 13 patients d’origine inconnue/non rapportée (1,1%) et 3 patients originaires d’Alaska ou amérindiens (0,3%). Chez 97 % des patients, le score de performance ECOG était de 0 ou 1. Les patients présentant des métastases cérébrales connues, une hypertension non contrôlée, une pathologie cardiaque significative ou une insuffisance cardiaque de classe NYHA II à IV étaient exclus. Les patients précédemment traités pour leur cancer métastatique de la prostate par un traitement pharmacologique, une radiothérapie ou une chirurgie étaient exclus, excepté ceux ayant reçu moins de 3 mois de traitement par ADT, ou une séance de radiothérapie palliative ou ayant subi une intervention chirurgicale visant à traiter les symptômes résultant de la maladie métastatique. Les co-critères principaux d’évaluation de l’efficacité étaient la survie globale (OS) et la survie sans progression radiologique (rPFS). Le score médian d’évaluation de la douleur à l’inclusion, mesuré à l’aide du questionnaire Brief Pain Inventory Short Form (BPI-SF), était de 2,0 dans le groupe de traitement comme dans le groupe placebo. Outre les co-critères principaux d’évaluation, les bénéfices du traitement ont également été évalués d’après le délai de survenue de complications osseuses (SRE), le délai jusqu’au traitement suivant pour le cancer de la prostate, le délai jusqu’à instauration d’une chimiothérapie, le délai jusqu’à progression de la douleur et le délai jusqu’à progression du PSA. Le traitement a été poursuivi jusqu’à progression de la maladie, retrait du consentement, survenue d’une toxicité inacceptable ou décès du patient.
La survie sans progression radiologique était définie comme le délai entre la randomisation et la survenue d’une progression radiologique ou le décès du patient, toutes causes confondues. La progression radiologique incluait la progression visible à l’imagerie par scintigraphie osseuse (selon les critères modifiés du PCWG2) ou la progression des lésions des tissus mous visible au scanner ou à l’IRM (selon les critères RECIST 1.1).
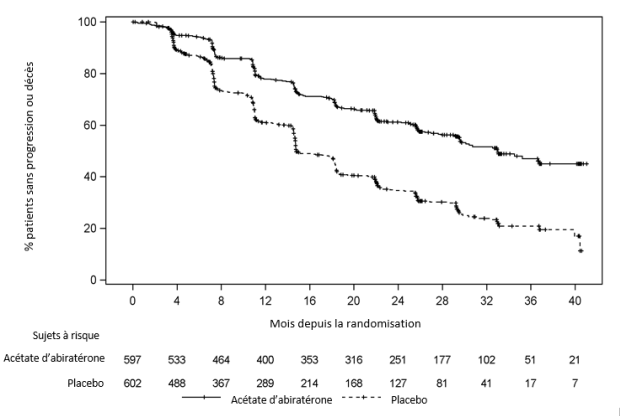
Une différence significative en termes de rPFS a été observée entre les groupes de traitement (voir Tableau 2 et Figure 1).
Tableau 2 : Analyse stratifiée de la survie sans progression radiologique ; Population en Intention de Traiter (étude PCR 3 011)
|
|
AA+P |
Placebo |
|
Sujets randomisés |
597 |
602 |
|
Evènements |
239 (40,0%) |
354 (58,8%) |
|
Censurés |
358 (60,0%) |
248 (41,2%) |
|
Délai de survenue de l’évènement (mois) Médiane (IC à 95%) |
33,02 (29,57; NE) |
14,78 (14,69; 18,27) |
|
Intervalle |
(0,0+; 41,0+) |
(0,0+; 40,6+) |
|
pa |
(0,0+; 41,0+) |
(0,0+; 40,6+) |
|
Hazard Ratio (IC à 95%)b |
< 0,0001 0,466 (0,394; 0,550) |
|
Note: += observation censurée, NE = non évaluable. La progression radiologique et le décès sont pris en compte dans la définition de l’évènement rPFS. AA+P = sujets ayant reçu de l’acétate d’abiratérone et de la prednisone.
a La valeur de p est dérivée d’un test du log-rank ajusté sur le statut du score de performance ECOG (0/1 ou 2) et l’atteinte viscérale (présence ou absence).
b Le Hazard Ratio est calculé à partir d’un modèle à risques proportionnels stratifié. Un Hazard Ratio < 1 est en faveur de AA+P.
Figure 1 : Courbe de Kaplan-Meier de la survie sans progression radiologique ; Population en Intention de Traiter (étude PCR3 011)
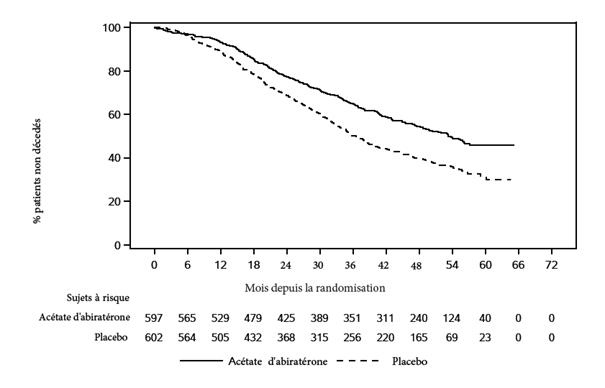
Une amélioration statistiquement significative de l’OS a été observée en faveur du traitement AA+P plus ADT, avec une réduction de 34 % du risque de décès comparé au groupe placebo plus ADT (HR = 0,66 ; IC à 95 % : 0,56 à 0,78 ; p< 0,0001) (voir Tableau 3 et Figure 2).
Tableau 3 : Survie Globale des patients traités par abiratérone ou placebo dans l’étude PCR3011 (Analyse en Intention de Traiter)
|
Survie Globale |
Abiratérone et Prednisone |
Placebo |
|
|
(N=597) |
(N=602) |
|
Décès (%) |
275 (46%) |
343 (57%) |
|
Survie médiane (mois) |
53,3 |
36,5 |
|
(IC à 95%) Hazard ratio (IC à 95%)1 |
(48,2; NE) 0,66 (0,56 ; 0,78) |
(33,5 ; 40,0) |
NE=non évaluable
1 Le Hazard Ratio est dérivé d’un modèle à risques proportionnels stratifié. Un Hazard Ratio < 1 est en faveur d’abiratérone associé à la prednisone.
Figure 2 : Courbe de Kaplan-Meier de la Survie Globale ; Population en Intention de Traiter dans l’étude PCR3 011
Les analyses en sous-groupes sont toutes favorables au traitement par abiratérone. Au sein des différents sous-groupes préspécifiés, l’effet du traitement par AA+P sur la rPFS et l’OS a été favorable et cohérent avec la population générale de l’étude, excepté pour le sous-groupe de score ECOG 2 pour lequel aucune tendance en termes de bénéfice n’a été observée. Cependant la faible taille d’échantillon (n=40) ne permet pas de tirer de conclusion valide de ce résultat.
Outre les améliorations observées au niveau de la survie globale et de la rPFS, le bénéfice d’un traitement par abiratérone vs placebo a été démontré sur tous les critères secondaires d’évaluation définis de façon prospective.
Étude 302 (patients n’ayant pas eu de chimiothérapie antérieure)
Cette étude a inclus des patients n’ayant pas eu de chimiothérapie antérieure qui étaient asymptomatiques ou peu symptomatiques et pour lesquels la chimiothérapie n’était pas encore cliniquement indiquée. Un score ne dépassant pas 0-1 sur l’échelle BPI-SF (Brief Pain Inventory-Short Form) au cours des dernières 24 heures était considéré comme asymptomatique, et un score de 2-3 était considéré comme peu symptomatique.
Dans l’étude 302, (n=1 088) l’âge médian des patients inclus était de 71 ans pour les patients traités par abiratérone plus prednisone ou prednisolone et de 70 ans pour les patients traités par placebo plus prednisone ou prednisolone. Le nombre de patients traités par abiratérone par groupe ethnique était de 520 sujets de race blanche (95,4%), 15 sujets de race noire (2,8%), 4 sujets asiatiques (0,7%) et 6 sujets d’autres groupes ethniques (1,1%). Dans chacun des deux bras, le score de performance à l’échelle de l’Eastern Cooperative Oncology Group (ECOG) était de 0 pour 76% des patients et de 1 pour 24% des patients. Cinquante pourcents des patients n’avaient que des métastases osseuses, 31% des patients avaient des métastases osseuses et des tissus mous ou des ganglions lymphatiques et 19% des patients avaient seulement des métastases des tissus mous ou des ganglions lymphatiques. Les patients présentant des métastases viscérales étaient exclus. Les co-critères primaires d’efficacité étaient la survie globale et la survie sans progression radiologique (rPFS). En plus de ces co-critères primaires, le bénéfice a également été évalué par le délai jusqu’à l’utilisation des opiacés pour les douleurs cancéreuses, le délai jusqu’à l’instauration d’une chimiothérapie, le délai jusqu’à la 15 détérioration du score de performance à l’échelle ECOG ≥ 1 point et le délai jusqu’à l’augmentation du PSA sur la base des critères PCWG2 (Prostate Cancer Working Group-2). Les traitements à l’étude ont été arrêtés à l’apparition d’une progression clinique sans équivoque. Les traitements pouvaient aussi être arrêtés au moment de la confirmation de la progression radiologique, laissée à la discrétion de l’investigateur.
La survie sans progression radiologique (rPFS) a été évaluée à l’aide d’études d’imagerie séquentielle comme défini par les critères PCWG2 (pour les lésions osseuses) et par les critères RECIST (Response Evaluation Criteria In Solid Tumors) modifiés (pour les lésions des tissus mous). L’analyse de la rPFS était basée sur une évaluation centralisée de la progression radiologique.
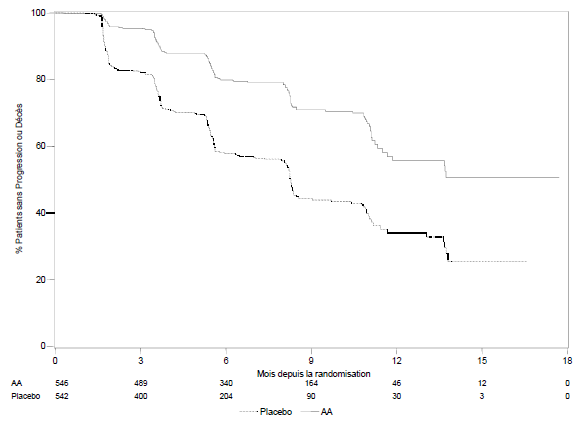
Lors de l’analyse planifiée de la rPFS il y avait 401 événements ; soit 150 (28%) patients traités par abiratérone et 251 (46%) patients traités par placebo ayant progressé radiologiquement ou étant décédés. Une différence significative de rPFS entre les groupes de traitement a été observée (voir Tableau 4 et Figure 3)
Tableau 4 : Étude 302 : Survie sans progression radiologique des patients traités par abiratérone ou par placebo en association avec la prednisone ou la prednisolone et des analogues de la LH-RH ou une orchidectomie préalable
NE= Non évaluable
* La valeur de p est dérivée d’un test du log-rank ajusté sur le statut du score de performance ECOG (0 ou 1)
** Un Hazard Ratio < 1 est en faveur d’abiratérone
Figure 3 : Courbe de Kaplan Meier de survie sans progression radiologique pour les patients traités par abiratérone ou placebo en association avec la prednisone ou la prednisolone, et des analogues de la LH-RH ou une orchidectomie préalable
AA = Abiratérone
Cependant, des données ont continué à être collectées dans le cadre de la seconde analyse intermédiaire de la survie globale (OS). L’analyse radiologique de la rPFS par l’investigateur, réalisée dans le cadre de l’analyse de suivi de sensibilité, est présentée dans le tableau 5 et la figure 4.
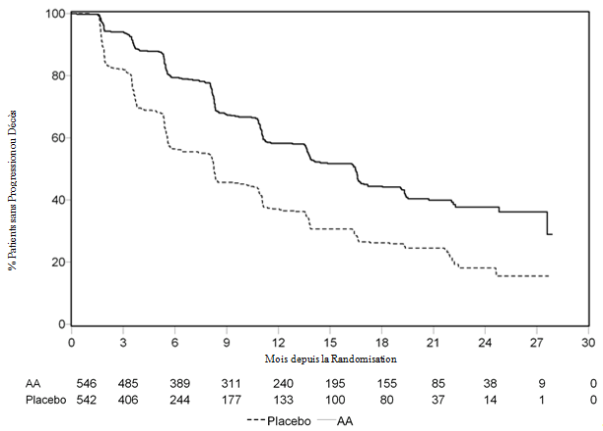
Six cent sept (607) sujets ont eu une progression radiologique ou sont décédés : 271 (50%) dans le groupe acétate d’abiratérone et 336 (62%) dans le groupe placebo. Le traitement par acétate d’abiratérone a diminué le risque de progression radiologique ou de décès de 47% par rapport au placebo (HR = 0,530 ; IC à 95% : [0,451 - 0,623], p < 0,0001). La rPFS médiane était de 16,5 mois dans le groupe acétate d’abiratérone et de 8,3 mois dans le groupe placebo.
Tableau 5 : Étude 302 : Survie sans progression radiologique des patients traités par abiratérone ou par placebo en association avec la prednisone ou la prednisolone et des analogues de la LH-RH ou une orchidectomie préalable (à la seconde analyse intermédiaire de l’OS, évaluation par l’investigateur)
|
|
Abiratérone (N=546) |
Placebo (N=542) |
|
Survie sans progression radiologique (rPFS) |
|
|
|
Progression ou décès (%) |
271 (50%) |
336 (62%) |
|
rPFS médiane en mois |
16,5 |
8,3 |
|
(IC à 95%) |
(13,80 - 16,79) |
(8,05 – 9,43) |
|
p* |
< 0,0001 |
|
|
Hazard Ratio ** |
0,530 |
|
|
(IC à 95%) |
(0,451 - 0,623) |
|
* La valeur de p est dérivée d’un test du log-rank ajusté sur le statut du score de performance ECOG (0 ou 1)
** Un Hazard Ratio < 1 est en faveur d’abiratérone
Figure 4 : Courbe de Kaplan Meier de survie sans progression radiologique pour les patients traités par abiratérone ou placebo en association avec la prednisone ou la prednisolone et des analogues de la LH-RH ou une orchidectomie préalable (à la seconde analyse intermédiaire, évaluation par l’investigateur)
AA= Abiratérone
Une analyse intermédiaire (AI) planifiée de l’OS a été menée après l’observation de 333 décès. La levée d’aveugle s’est basée sur l’amplitude du bénéfice clinique observé et un traitement par abiratérone a été proposé aux patients du groupe placebo. La survie globale était plus longue pour le groupe traité par abiratérone que pour celui traité par placebo avec une réduction de 25% du risque de décès (HR = 0,752 ; IC à 95% : [0,606 - 0,934], p=0,0097), mais les données de survie globale n’étaient pas matures et les résultats intermédiaires n’atteignaient pas le seuil d’arrêt statistiquement significatif prédéfini (voir Tableau 6). La survie a continué à être suivie après cette AI.
L’analyse finale planifiée de la survie globale a été menée après l’observation de 741 décès (durée médiane de suivi de 49 mois). Soixante-cinq pourcent des patients traités par abiratérone sont décédés (354 sur 546), comparé à 71% des patients traités par placebo (387 sur 542). Un bénéfice statistiquement significatif en termes de survie globale a été démontré dans le groupe traité par abiratérone avec une réduction du risque de décès de 19,4% (HR=0,806 ; IC à 95% : [0,697 - 0,931], p=0,0033) et une amélioration dans la médiane de survie globale de 4,4 mois (abiratérone : 34,7 mois, placebo : 30,3 mois) (voir Tableau 6 et Figure 5). Cette amélioration a été démontrée alors que 44 % des patients du groupe placebo avaient reçu un traitement ultérieur par abiratérone.
Tableau 6 : Étude 302 : Survie globale des patients traités par abiratérone ou par placebo en association avec la prednisone ou la prednisolone et des analogues de la LH-RH ou une orchidectomie préalable
|
|
Abiratérone (N=546) |
Placebo (N=542) |
|
Analyse intermédiaire de la survie |
|
|
|
Décès (%) |
147 (27%) |
186 (34%) |
|
Survie médiane (mois) |
Non atteint |
27,2 |
|
(IC à 95%) |
(NE – NE) |
(25,95 – NE) |
|
p* |
0,0097 |
|
|
Hazard Ratio** |
0,752 |
|
|
(IC à 95%) |
(0,606 - 0,934) |
|
|
Analyse finale de la survie |
|
|
|
Décès |
354 (65%) |
387 (71%) |
|
Survie globale médiane en mois (IC à 95%) |
34,7 (32,7 - 36,8) |
30,3 (28,7 - 33,3) |
|
p* |
0,0033 |
|
|
Hazard Ratio** (IC à 95%) |
0,806 (0,697 - 0,931) |
|
NE= Non Évaluable
* La valeur de p est dérivée d’un test du log-rank ajusté sur le statut du score de performance ECOG (0 ou 1)
** Un Hazard Ratio < 1 est en faveur d’abiratérone
Figure 5 : Courbe de Kaplan Meier de survie pour les patients traités par abiratérone ou placebo en association avec la prednisone ou la prednisolone et des analogues de la LH-RH ou une orchidectomie préalable, analyse finale
AA=Abiratérone
Outre l’observation de l’amélioration de la survie globale et de la rPFS, l’ensemble des critères secondaires était en faveur d’abiratérone comme suit :
Le délai médian jusqu’à progression du PSA, basé sur les critères PCWG2, était de 11,1 mois pour les patients recevant abiratérone et de 5,6 mois pour les patients recevant le placebo (HR = 0,488 ; IC à 95% : [0,420 - 0,568], p < 0,0001). Le délai jusqu’à progression du PSA était approximativement doublé avec le traitement par abiratérone (HR = 0,488). La proportion de patients avec une réponse confirmée sur le PSA était plus grande dans le groupe abiratérone que dans le groupe placebo (62% vs 24% ; p < 0,0001). Une augmentation significative du nombre de réponses tumorales complètes et partielles a été observée chez les patients atteints de lésions des tissus mous mesurables et traités par abiratérone.
Délai jusqu’à l’utilisation des opiacés pour les douleurs cancéreuses : lors de l’analyse finale, le délai médian jusqu’à l’utilisation des opiacés pour les douleurs liées au cancer de la prostate était de 33,4 mois pour les patients recevant abiratérone et de 23,4 mois pour les patients recevant le placebo (HR = 0,721 ; IC à 95% : [0,614 - 0,846], p < 0,0001).
Délai jusqu’à l’instauration d’une chimiothérapie par agent cytotoxique : le délai médian était de 25,2 mois pour les patients recevant abiratérone et de 16,8 mois pour les patients recevant le placebo (HR = 0,580 ; IC à 95% : [0,487 - 0,691], p < 0,0001)
Délai jusqu’à la détérioration du score de performance à l’échelle ECOG ≥ 1 point : le délai médian était de 12,3 mois pour les patients recevant abiratérone et de 10,9 mois pour les patients recevant le placebo (HR = 0,821 ; IC à 95% : [0,714 - 0,943], p = 0,0053).
Les critères suivants d’évaluation de cette étude démontrent un avantage statistiquement significatif en faveur du traitement par abiratérone :
La réponse objective : Une réponse objective a été définie comme la proportion de patients avec une maladie mesurable parvenant à une réponse complète ou partielle selon les critères RECIST (la taille initiale des ganglions lymphatiques devait être ≥ 2 cm pour être considérée comme une lésion-cible). La proportion des patients avec une maladie mesurable initiale ayant une réponse objective était de 36% pour le groupe abiratérone et de 16% dans le groupe placebo (p < 0,0001).
Douleur : Le traitement par abiratérone a réduit significativement le risque de progression du score moyen de la douleur de 18% par rapport au traitement par placebo (p = 0,0490). Le délai médian jusqu’à la progression était de 26,7 mois dans le groupe abiratérone et de 18,4 mois dans le groupe placebo.
Délai jusqu’à la dégradation du FACT-P (score total) : le traitement par abiratérone a réduit le risque de dégradation du FACT-P (score total) de 22% par rapport au placebo (p = 0,0028). Le délai médian jusqu’à la dégradation du FACT-P (score total) était de 12,7 mois dans le groupe abiratérone et de 8,3 mois dans le groupe placebo.
Étude 301 (patients ayant eu une chimiothérapie antérieure)
Les patients inclus dans l’étude 301 avaient déjà été traités par docétaxel. Il n’était pas nécessaire que les patients présentent une progression de la maladie sous docétaxel, étant donné que le traitement avait pu être arrêté suite à la toxicité de la chimiothérapie. Les patients ont été maintenus sous traitement à l’étude jusqu’à l’observation d’une progression du PSA (confirmée par une augmentation de 25% par rapport à l’état initial/nadir) ainsi que jusqu’à progression radiologique telle que définie au protocole et progression symptomatique ou clinique. Les patients ayant antérieurement reçu un traitement par le kétoconazole pour un cancer de la prostate ont été exclus de cette étude. Le critère primaire d’efficacité était la survie globale.
L’âge médian des patients inclus était de 69 ans (intervalle [39 – 95]). Le nombre de patients traités par abiratérone par groupe ethnique comportait 737 (93,2%) sujets de race blanche, 28 (3,5%) sujets de race noire, 11 (1,4%) sujets asiatiques et 14 (1,8%) sujets d’autres groupes ethniques. Onze pourcent 20 des patients inclus présentaient un score de performance de 2 à l’échelle ECOG, 70% présentaient des signes radiologiques de progression de la maladie, avec ou sans progression du PSA, 70% avaient déjà reçu une chimiothérapie par un agent cytotoxique et 30% en avaient reçu deux. 11% des patients traités par abiratérone présentaient des métastases hépatiques.
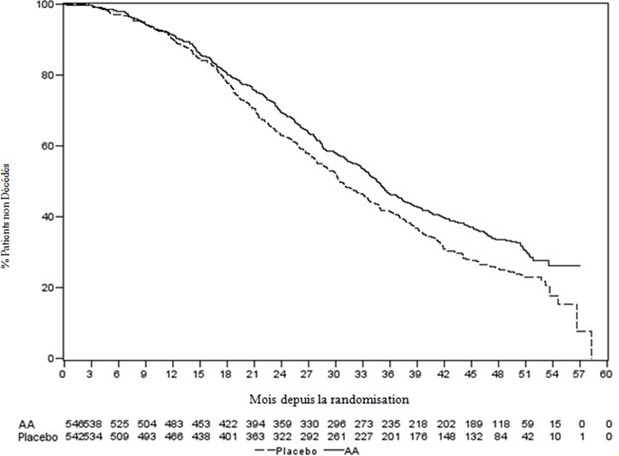
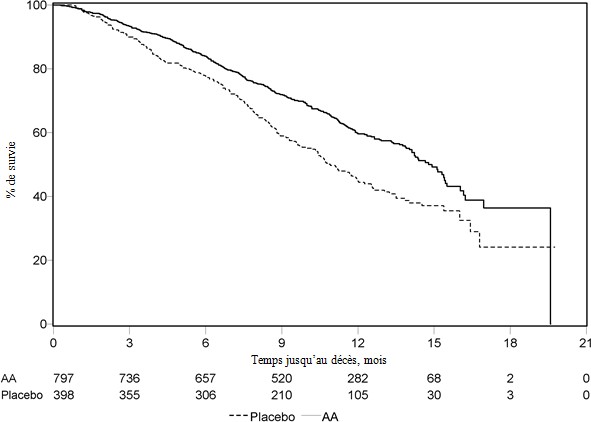
Une analyse programmée, réalisée après observation de 552 décès, a montré que 42% (333 sur 797) des patients traités par abiratérone contre 55% (219 sur 398) des patients sous placebo sont décédés. Une amélioration statistiquement significative de la médiane de survie globale a été observée chez les patients traités par abiratérone (voir Tableau 7).
Tableau 7 : Survie globale des patients traités par abiratérone ou par placebo en association avec la prednisone ou la prednisolone et des analogues de la LH-RH ou une orchidectomie préalable
|
|
Abiratérone (N=797) |
Placebo (N=398) |
|
Analyse primaire de survie Décès (%) |
333 (42%) |
219 (55%) |
|
Survie médiane (mois) |
14,8 |
10,9 |
|
(IC à 95%) |
(14,1 - 15,4) |
(10,2 - 12,0) |
|
pa |
< 0,0001 |
|
|
Hazard Ratio (IC à 95%)b |
0,646 (0,543 - 0,768) |
|
|
Analyse de survie mise à jour |
|
|
|
Décès (%) |
501 (63%) |
274 (69%) |
|
Survie médiane (mois) |
15,8 |
11,2 |
|
(IC à 95%) |
(14,8 - 17,0) |
(10,4 - 13,1) |
|
Hazard Ratio (IC à 95%)b |
0,740 (0,638 - 0,859) |
|
a La valeur de p est dérivée d’un test du log-rank ajusté sur le statut du score de performance ECOG (0-1 vs 2), le score de douleur (absent vs présent), le nombre de cures de chimiothérapies antérieures (1 vs. 2), et le type de progression de la pathologie (PSA uniquement vs. radiologique)
b Le Hazard Ratio est dérivé d’un modèle à risques proportionnels stratifié. Un Hazard Ratio < 1 est en faveur d’abiratérone.
À chaque évaluation après les premiers mois de traitement, la proportion de patients toujours en vie était plus importante dans le groupe abiratérone comparé au groupe contrôle (voir Figure 6).
Figure 6 : Courbes de survie de Kaplan Meier pour les patients traités par abiratérone ou placebo en association avec la prednisone ou la prednisolone, et des analogues de la LH-RH ou une orchidectomie préalable
AA= Abiratérone
Les analyses de survie par sous-groupe révèlent un bénéfice de survie constant pour le traitement par abiratérone (voir Figure 7).
Figure 7 : Survie globale par sous-groupe : Hazard Ratio et intervalle de confiance à 95%
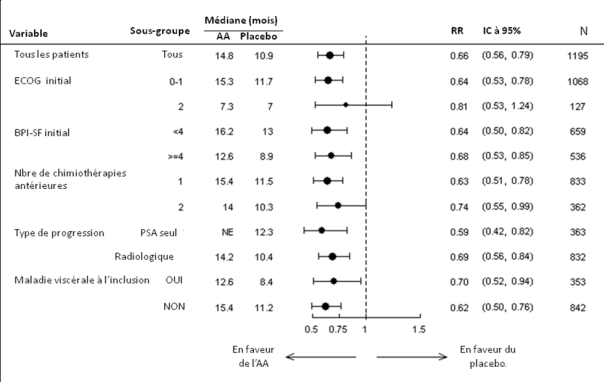
AA= Abiratérone, BPI=Brief Pain Inventory, IC=intervalle de confiance, ECOG=indice de performance de l'Eastern Cooperative Oncology Group, HR=Hazard Ratio, NE=non évaluable
Outre l'observation de l'amélioration de la survie globale, l'ensemble des critères secondaires étaient en faveur d’abiratérone et étaient statistiquement significatifs, après ajustements en analyse multivariée, comme suit :
Les patients traités par abiratérone présentaient un taux de réponse sur le PSA total (défini comme une baisse ≥ 50% par rapport à la valeur initiale) significativement plus élevé que ceux sous placebo, 38% vs 10%, p < 0,0001.
Le temps médian jusqu’à progression du PSA était de 10,2 mois pour les patients traités par abiratérone et de 6,6 mois pour les patients sous placebo (RR= 0,580 ; IC à 95% : [0,462 - 0,728], p < 0,0001).
La survie médiane sans progression radiologique était de 5,6 mois pour les patients traités par abiratérone et de 3,6 mois pour les patients sous placebo (HR = 0,673 ; IC à 95% : [0,585 - 0,776], p < 0,0001).
Douleur
La proportion de patients ayant ressenti un soulagement de la douleur était, d'un point de vue statistique, significativement plus élevée pour le groupe traité par abiratérone que pour le groupe placebo (44% vs 27%, p = 0,0002). Un patient répondant au soulagement de la douleur était défini comme un patient ayant ressenti une diminution d’au moins 30% par rapport à la valeur initiale du score de la pire douleur sur l'échelle BPI-SF, au cours des dernières 24 heures sans augmentation du score d’utilisation des antalgiques, observée lors de deux évaluations consécutives, à quatre semaines d'intervalle. Le soulagement de la douleur a été évalué uniquement chez les patients présentant un score initial de douleur ≥ 4 et avec au moins un score de soulagement de la douleur évalué en cours de traitement (N=512).
Une plus faible proportion de patients traités par abiratérone a ressenti une augmentation de la douleur comparativement aux patients sous placebo à 6 mois (22% vs 28%), 12 mois (30% vs 38%) et 18 mois (35% vs 46%). L'augmentation de la douleur a été définie comme une augmentation ≥ 30%, par rapport à la valeur initiale, du score de la pire douleur sur l'échelle BPI-SF au cours des 24 heures précédentes, sans baisse du score d'utilisation des analgésiques observée lors de deux visites consécutives ou une augmentation ≥ 30% du score d'utilisation des analgésiques observée lors de deux visites consécutives. Le temps avant progression de la douleur au 25e percentile était de 7,4 mois pour le groupe traité par abiratérone contre 4,7 mois pour le groupe placebo.
Complications osseuses
Une plus faible proportion de patients dans le groupe abiratérone a présenté des complications osseuses par rapport au groupe placebo à 6 mois (18% vs 28%), 12 mois (30% vs 40%) et 18 mois (35% vs 40%). Le temps avant la survenue d’une complication osseuse au 25e percentile a été deux fois plus élevé dans le groupe abiratérone que celui du groupe contrôle : 9,9 mois versus 4,9 mois. Une complication osseuse est définie comme une fracture spontanée, une compression médullaire, une irradiation palliative des os ou une intervention chirurgicale sur des os.
Population pédiatrique
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec abiratérone dans tous les sous-groupes de la population pédiatrique dans le cancer avancé de la prostate. Voir rubrique 4.2 pour des informations concernant l’usage pédiatrique.
5.2. Propriétés pharmacocinétiques
Absorption
Après administration orale d’acétate d’abiratérone à jeun, la concentration plasmatique maximale d'abiratérone est atteinte après environ 2 heures.
Comparé à une administration à jeun, l'administration d’acétate d’abiratérone avec la nourriture entraîne une augmentation de l’exposition systémique moyenne à l'abiratérone jusqu’à 10 fois (pour l'ASC) et jusqu’à 17 fois (pour la Cmax), en fonction de la teneur en graisses des aliments. En raison de la variabilité normale du contenu et de la composition des repas, la prise d'abiratérone avec les repas peut entraîner des degrés d’exposition très variables. Ainsi, l'abiratérone ne doit pas être pris avec de la nourriture. Il doit être pris au moins une heure avant ou au moins deux heures après le repas. Les comprimés doivent être avalés en entier, avec de l'eau (voir rubrique 4.2).
Distribution
Dans le plasma humain, la fixation protéique de la 14C-abiratérone est de 99,8%. Le volume de distribution apparent est d'environ 5 630 L, ce qui suggère une large distribution de l'abiratérone vers les tissus périphériques.
Biotransformation
Après administration orale de gélules de 14C-acétate d'abiratérone, l'acétate d'abiratérone est hydrolysé en abiratérone, elle-même éliminée par plusieurs mécanismes dont la sulfatation, l'hydroxylation et l'oxydation, principalement au niveau du foie. La majorité de la radioactivité circulante (environ 92%) se trouve sous forme de métabolites de l’abiratérone. Sur 15 métabolites détectables, deux métabolites principaux, le sulfate d'abiratérone et le sulfate de N-oxyde-abiratérone, représentent chacun environ 43% de la radioactivité totale.
Élimination
D'après les données recueillies chez les sujets sains, la demi-vie plasmatique moyenne de l'abiratérone est d'environ 15 heures. Après administration orale de 1 000 mg de 14C-acétate d'abiratérone, environ 88% de la dose radioactive est retrouvée dans les selles et environ 5% dans l'urine. Les principaux 24 composés présents dans les selles sont l'acétate d'abiratérone et l'abiratérone sous forme inchangée (respectivement environ 55% et 22% de la dose administrée).
Insuffisance hépatique
La pharmacocinétique de l'acétate d’abiratérone a été étudiée chez des sujets atteints d'insuffisance hépatique préexistante légère ou modérée (respectivement Classes A et B de Child-Pugh) et chez des sujets contrôles sains. L'exposition systémique à l'abiratérone après administration d'une dose unique de 1 000 mg par voie orale a augmenté d'environ 11% et 260% respectivement chez les sujets atteints d'insuffisance hépatique préexistante légère et modérée. La demi-vie moyenne de l'abiratérone est prolongée jusqu'à environ 18 heures chez les sujets atteints d’insuffisance hépatique légère et jusqu’à environ 19 heures chez les sujets atteints d’insuffisance hépatique modérée.
Dans une autre étude, la pharmacocinétique de l’abiratérone a été étudiée chez des sujets atteints d'insuffisance hépatique sévère (Classe C de Child-Pugh) préexistante (n=8) et chez 8 sujets contrôles sains ayant une fonction hépatique normale. L'ASC de l’abiratérone a augmenté d'environ 600% et la fraction libre de médicament a augmenté d'environ 80% chez les sujets atteints d'insuffisance hépatique sévère comparés aux sujets ayant une fonction hépatique normale.
Aucun ajustement de la dose n'est nécessaire chez les patients atteints d'insuffisance hépatique préexistante légère.
L’utilisation d’acétate d’abiratérone doit être évaluée avec précaution chez les patients atteints d'insuffisance hépatique modérée chez lesquels le bénéfice doit être nettement supérieur au risque potentiel (voir les rubriques 4.2 et 4.4). L’acétate d’abiratérone ne doit pas être utilisé chez les patients atteints d’une insuffisance hépatique sévère (voir rubriques 4.2, 4.3 et 4.4).
Pour les patients qui développent une hépatotoxicité en cours de traitement, un arrêt du traitement et une adaptation de la dose peuvent être nécessaires (voir rubriques 4.2 et 4.4).
Insuffisance rénale
La pharmacocinétique de l’acétate d’abiratérone a été comparée chez des patients atteints d’insuffisance rénale terminale sous hémodialyse stable versus des patients contrôles appariés ayant une fonction rénale normale. L’exposition systémique à l’abiratérone après administration d’une dose unique de 1 000 mg par voie orale n’a pas augmenté chez les sujets atteints d’insuffisance rénale terminale sous dialyse. L’administration chez des patients présentant une insuffisance rénale, incluant une insuffisance rénale sévère ne nécessite pas de réduction de la dose (voir rubrique 4.2). Cependant, il n’existe pas d’expérience clinique chez les patients présentant à la fois un cancer de la prostate et une insuffisance rénale sévère. La prudence est recommandée chez ces patients.
5.3. Données de sécurité préclinique
Dans les études de fécondité chez le rat mâle et femelle, l’acétate d’abiratérone a réduit la fécondité ; ceci était complètement réversible en 4 à 16 semaines après l’arrêt de l’acétate d’abiratérone.
Dans une étude de toxicité chez le rat, l’acétate d’abiratérone a affecté la grossesse, incluant une diminution de la survie et du poids du fœtus. Des effets sur les organes génitaux externes ont été observés bien que l’acétate d’abiratérone n’ait pas été tératogène.
Dans ces études de fécondité et de toxicité réalisées chez le rat, tous les effets ont été rapportés à l’activité pharmacologique de l’acétate d’abiratérone.
Outre les modifications des organes génitaux observées lors de toutes les études de toxicité chez l'animal, les données non cliniques issues des études classiques de sécurité pharmacologique, de toxicité à doses répétées, de génotoxicité et de potentiel carcinogène n'ont pas révélé de risque particulier pour l'homme. Dans une étude de 6 mois chez la souris transgénique (Tg.rasH2), l’acétate d’abiratérone n’était pas carcinogène. Dans une étude de carcinogénicité de 24 mois chez le rat, l’acétate d’abiratérone a augmenté l’incidence des néoplasmes des cellules interstitielles au niveau des testicules. Ce résultat est considéré comme lié à l’action pharmacologique de l’abiratérone et spécifique au rat. L’acétate d’abiratérone n’était pas carcinogène chez la rate.
La substance active, l’abiratérone, présente un risque environnemental pour l’environnement aquatique, notamment pour les poissons.
Croscarmellose sodique
Laurilsulfate de sodium
Povidone (E1201)
Cellulose microcristalline (E460)
Lactose monohydraté
Silice colloïdale anhydre 5E551)
Stéarate de magnésium (E470b)
Pelliculage
Alcool polyvinylique (E1203)
Dioxyde de titane (E171)
Macrogol (E1521)
Talc (E553b)
Oxyde de fer rouge (E172)
Oxyde de fer noir (E172).
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Plaquettes (Aluminium/OPA/Aluminium/PVC) ou (Aluminium/PVC/PE/PVDC) contenant 56 ou 60 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Ce médicament peut induire un risque pour l’environnement aquatique (voir rubrique 5.3).
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 412 6 4 : 56 comprimés pelliculés sous plaquettes (Aluminium/OPA/Aluminium/PVC).
· 34009 302 412 7 1 : 60 comprimés pelliculés sous plaquettes (Aluminium/OPA/Aluminium/PVC).
· 34009 302 412 9 5 : 56 comprimés pelliculés sous plaquettes (Aluminium/PVC/PE/PVDC).
· 34009 302 413 0 1 : 60 comprimés pelliculés sous plaquettes (Aluminium/PVC/PE/PVDC).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Prescription initiale hospitalière annuelle, réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie.
Renouvellement non restreint.
ANSM - Mis à jour le : 22/07/2022
ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé
Acétate d’abiratérone
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé ?
3. Comment prendre ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
ABIRATERONE BIOGARAN contient un médicament appelé acétate d’abiratérone. Il est utilisé chez les hommes adultes pour traiter le cancer de la prostate qui s’est disséminé dans d’autres parties du corps. ABIRATERONE BIOGARAN arrête la production de testostérone par votre corps, ce qui peut ralentir la croissance du cancer de la prostate.
Lorsque ABIRATERONE BIOGARAN est prescrit au stade précoce de la maladie répondant encore à un traitement hormonal, il est utilisé en association à un traitement qui diminue le taux de testostérone (suppression androgénique).
Lors de votre traitement par ce médicament, votre médecin vous prescrira également un autre médicament appelé prednisone ou prednisolone. Cela permettra de réduire vos risques de développer une pression artérielle élevée, d’accumuler une quantité excessive d’eau dans votre corps (rétention hydrique), ou de présenter des taux réduits d’un composant chimique appelé potassium dans votre sang.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé ?
Ne prenez jamais ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé :
· si vous êtes allergique à l’acétate d’abiratérone ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
· si vous êtes une femme, surtout si vous êtes enceinte. ABIRATERONE BIOGARAN ne doit être utilisé que chez les patients de sexe masculin.
· si vous avez une lésion sévère du foie.
· en association avec du radium (Ra-223) (utilisé pour traiter le cancer de la prostate).
Ne prenez pas ce médicament si l’un des cas ci-dessus vous concerne. En cas de doute, parlez-en à votre médecin ou à votre pharmacien avant de prendre ce médicament.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé.
· si vous avez des troubles du foie
· si on vous a dit que vous aviez une pression artérielle élevée ou une insuffisance cardiaque ou un faible taux sanguin de potassium (un faible taux sanguin de potassium peut augmenter le risque de troubles du rythme cardiaque)
· si vous avez eu d’autres problèmes cardiaques ou vasculaires
· si vous avez un rythme cardiaque rapide ou irrégulier
· si vous êtes essoufflé
· si vous avez pris du poids rapidement
· si vous avez un gonflement des pieds, des chevilles ou des jambes
· si vous avez pris comme traitement du cancer de la prostate, un médicament appelé kétoconazole
· au sujet de la nécessité de prendre Abiratérone avec de la prednisone ou de la prednisolone
· au sujet des effets possibles sur vos os
· si vous avez une glycémie (taux de sucre dans le sang) élevée.
Informez votre médecin si on vous a dit que vous aviez une maladie cardiaque ou vasculaire, y compris des troubles du rythme cardiaque (arythmie), ou si vous recevez des médicaments pour traiter ces maladies.
Informez votre médecin si vous présentez un jaunissement de la peau ou des yeux, des urines plus foncées, des nausées ou des vomissements sévères, car il pourrait s’agir de signes ou symptômes révélateurs de problèmes hépatiques. Une défaillance des fonctions du foie (appelée insuffisance hépatique aigüe), pouvant être fatale, peut survenir dans de rares cas.
Une diminution des globules rouges, une diminution du désir sexuel (libido), une faiblesse musculaire et/ou des douleurs musculaires peuvent apparaître.
ABIRATERONE BIOGARAN ne doit pas être administré en association avec du radium (Ra-223) en raison d’une possible augmentation du risque de fractures ou décès.
Si vous prévoyez d’utiliser du radium (Ra-223) après un traitement avec Abiratérone et prednisone/prednisolone, vous devez attendre 5 jours avant de commencer le traitement avec du radium (Ra-223).
Si vous n’êtes pas sûr(e) que l’un de ces cas vous concerne, parlez-en à votre médecin ou à votre pharmacien avant de prendre ce médicament.
Surveillance des paramètres sanguins
ABIRATERONE BIOGARAN peut affecter votre foie, bien qu’il soit possible que vous ne présentiez pas de symptômes. Lors de votre traitement par ce médicament, votre médecin contrôlera périodiquement votre bilan sanguin afin de rechercher tout effet sur votre foie.
Enfants et adolescents
Ce médicament ne doit pas être utilisé chez les enfants et les adolescents. Si ABIRATERONE BIOGARAN est accidentellement ingéré par un enfant ou un adolescent, allez immédiatement à l’hôpital et prenez la notice avec vous pour la montrer au médecin urgentiste.
Autres médicaments et ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé
Demandez conseil à votre médecin ou pharmacien avant de prendre tout médicament.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Ceci est important parce que ABIRATERONE BIOGARAN peut augmenter les effets d’un certain nombre de médicaments, incluant les médicaments pour le cœur, les tranquillisants, certains médicaments contre le diabète, les médicaments à base de plantes (par exemple le millepertuis) et autres. Votre médecin peut vouloir changer la dose de ces médicaments. De plus, certains médicaments peuvent augmenter ou diminuer les effets de ABIRATERONE BIOGARAN. Ceci peut entrainer des effets indésirables ou faire que ABIRATERONE BIOGARAN ne marche pas aussi bien qu’il le devrait.
Le traitement par suppression androgénique peut augmenter le risque de troubles du rythme cardiaque. Informez votre médecin si vous prenez des médicaments
· utilisés pour traiter les troubles du rythme cardiaque (par exemple quinidine, procaïnamide, amiodarone et sotalol) ;
· connus pour augmenter le risque de troubles du rythme cardiaque [par exemple la méthadone (utilisé comme antidouleur et lors de cures de désintoxication d’une addiction aux opiacés), la moxifloxacine (un antibiotique), les antipsychotiques (utilisés pour les maladies mentales graves)].
Informez votre médecin si vous prenez l’un des médicaments mentionnés ci-dessus.
ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé avec des aliments
· Ce médicament ne doit pas être pris avec de la nourriture (voir rubrique 3 : « Prise du médicament »).
· La prise de ABIRATERONE BIOGARAN avec de la nourriture peut provoquer des effets indésirables.
ABIRATERONE BIOGARAN ne doit pas être utilisé chez la femme.
· Ce médicament peut causer des malformations chez l’enfant à naître s’il est pris par des femmes enceintes.
· Si vous avez des rapports sexuels avec une femme susceptible de tomber enceinte, utilisez un préservatif et une autre méthode de contraception efficace.
· Si vous avez des relations sexuelles avec une femme enceinte, utilisez un préservatif pour protéger l’enfant à naître.
Conduite de véhicules et utilisation de machines
Il est peu probable que ce médicament ait un effet sur votre capacité à conduire un véhicule et à utiliser certains outils ou machines.
ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé contient du lactose et du sodium
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé ?
Combien en prendre
La dose recommandée est de 1 000 mg (deux comprimés) une fois par jour.
Prise du médicament
· Prenez ce médicament par la bouche.
· Ne prenez pas ABIRATERONE BIOGARAN avec de la nourriture.
· Prenez ABIRATERONE BIOGARAN au moins une heure avant ou au moins deux heures après avoir mangé (Voir rubrique 2 : « ABIRATERONE BIOGARAN avec des aliments »).
· Avalez les comprimés en entier avec de l’eau.
· Ne cassez pas les comprimés.
· ABIRATERONE BIOGARAN est pris avec un médicament appelé prednisone ou prednisolone. Prendre la prednisone ou la prednisolone en suivant exactement les indications de votre médecin.
· Vous devez prendre la prednisone ou la prednisolone tous les jours pendant votre traitement par ABIRATERONE BIOGARAN.
· La dose de prednisone ou de prednisolone que vous prenez peut devoir être modifiée en cas de survenue d’une urgence médicale. Votre médecin vous préviendra si vous devez changer la dose de prednisone ou de prednisolone que vous prenez. N’arrêtez pas de prendre la prednisone ou la prednisolone à moins que votre médecin ne vous le demande.
Votre médecin peut également vous prescrire d’autres médicaments pendant votre traitement par ABIRATERONE BIOGARAN et la prednisone ou la prednisolone.
Si vous avez pris plus de ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé que vous n’auriez dû
Si vous avez pris plus que vous n’auriez dû, prévenez immédiatement votre médecin ou allez immédiatement à l’hôpital.
Si vous oubliez de prendre ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé
· Si vous oubliez de prendre ABIRATERONE BIOGARAN ou la prednisone ou la prednisolone, prenez votre dose habituelle le jour suivant.
· Si vous oubliez de prendre ABIRATERONE BIOGARAN ou la prednisone ou la prednisolone durant plus d’un jour, contactez votre médecin sans délai.
Si vous arrêtez de prendre ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé
N’arrêtez pas de prendre ABIRATERONE BIOGARAN ou la prednisone ou la prednisolone à moins que votre médecin ne vous l’ait dit.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Arrêtez de prendre ABIRATERONE BIOGARAN et consultez immédiatement votre médecin si vous ressentez l’un des effets suivants :
· Faiblesse musculaire, contractions musculaires ou forts battements du cœur (palpitations). Ces derniers peuvent être le signe d’un faible taux de potassium dans votre sang.
Les autres effets indésirables incluent :
Très fréquents (peuvent affecter plus d’1 personne sur 10) :
Rétention d’eau dans vos jambes ou vos pieds, faible taux sanguin de potassium, augmentation des résultats des tests de la fonction hépatique, pression artérielle élevée, infection des voies urinaires, diarrhée.
Fréquents (peuvent affecter jusqu’à 1 personne sur 10) :
Taux élevés de graisses dans votre sang, douleur thoracique, rythme cardiaque irrégulier (fibrillation auriculaire), insuffisance cardiaque, rythme cardiaque rapide, infections sévères appelées sepsis, fractures osseuses, indigestion, sang dans les urines, éruption cutanée.
Peu fréquents (peuvent affecter jusqu’à 1 personne sur 100) :
Troubles des glandes surrénales (en lien avec un déséquilibre des quantités en sel et en eau), rythme cardiaque anormal (arythmie), faiblesse musculaire et/ou douleur musculaire.
Rares (peuvent affecter jusqu’à 1 personne sur 1 000) :
Irritation du poumon (également appelée alvéolite allergique).
Défaillance des fonctions du foie (également appelée insuffisance hépatique aigüe).
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
Crise cardiaque, modifications de l’ECG - électrocardiogramme (allongement de l’intervalle QT), et réactions allergiques sévères avec difficulté à avaler ou à respirer, gonflement du visage, des lèvres, de la langue ou de la gorge, ou éruption cutanée avec démangeaisons.
Une perte osseuse peut apparaitre chez les hommes traités pour le cancer de la prostate. Abiratérone en association avec la prednisone ou la prednisolone peut augmenter la perte osseuse.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et la plaquette. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé
· La substance active est l’acétate d’abiratérone. Chaque comprimé pelliculé contient 500 mg d’acétate d’abiratérone.
· Les autres composants sont :
Noyau : Croscarmellose sodique, laurilsulfate de sodium, povidone (E1201), cellulose microcristalline (E460), lactose monohydraté, silice colloïdale anhydre (E551), stéarate de magnésium (E470b) (voir la rubrique 2 « ABIRATERONE BIOGARAN contient du lactose et du sodium »).
Pelliculage : Alcool polyvinylique (E1203), dioxyde de titane (E171), macrogol (E1521), talc (E553b), oxyde de fer rouge (E172), oxyde de fer noir (E172).
Qu’est-ce que ABIRATERONE BIOGARAN 500 mg, comprimé pelliculé et contenu de l’emballage extérieur
ABIRATERONE BIOGARAN se présente sous la forme de comprimés pelliculés violets, de forme ovale, comportant la mention « 500 » sur une face.
Les comprimés pelliculés sont disponibles sous plaquettes (Aluminium-OPA/Aluminium/PVC) ou en (Aluminium/PVC/PE/PVDC) contenant 56 ou 60 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
Exploitant de l’autorisation de mise sur le marché
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
AHARNON STREET
LIMASSOL INDUSTRIAL ESTATE
3056 LIMASSOL
CHYPRE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).