Dernière mise à jour le 03/08/2026
AMBRISENTAN EG 10 mg, comprimé pelliculé
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : Anti-hypertenseurs, autres anti-hypertenseurs - code ATC : C02KX02
AMBRISENTAN EG contient la substance active « ambrisentan ». Il appartient à une classe de médicaments appelés « autres anti-hypertenseurs » (utilisés pour traiter une pression artérielle élevée).
Il est utilisé pour traiter l’hypertension artérielle pulmonaire (HTAP) chez l’adulte. L’HTAP est définie par une élévation de la pression artérielle dans les vaisseaux sanguins (les artères pulmonaires) qui transportent le sang entre le cœur et les poumons. Chez les patients atteints d’HTAP, ces artères se rétrécissent, et le cœur doit fournir un effort supplémentaire pour pomper le sang. Ceci provoque une fatigue, des vertiges et des essoufflements.
AMBRISENTAN EG élargit les artères pulmonaires et facilite ainsi le pompage du sang par le cœur. Ceci fait diminuer la pression sanguine et réduit les symptômes.
AMBRISENTAN EG peut aussi être utilisé en association avec d’autres médicaments pour traiter l’HTAP.
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 04/12/2020
AMBRISENTAN EG 10 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Ambrisentan........................................................................................................................... 10 mg
Pour un comprimé pelliculé.
Excipients à effet notoire :
Chaque comprimé contient environ 95 mg de lactose (sous forme monohydratée), 0,21 mg de lécithine (de soja), 0,405 mg de rouge Allura AC (E129) et 0,67 mg de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé biconvexe, de forme oblongue, rose, comportant la mention « 10 » gravée sur une face, l’autre face étant lisse, dont la longueur nominale est d’environ 11,1 mm et dont la largeur nominale est d’environ 5,6 mm.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Le traitement doit être instauré par un médecin expérimenté dans le traitement de l'hypertension artérielle pulmonaire.
Posologie
L’ambrisentan en monothérapie
AMBRISENTAN EG doit être débuté par voie orale à la dose de 5 mg une fois par jour et peut être augmenté à 10 mg par jour en fonction de la réponse clinique et de la tolérance.
L’ambrisentan en association avec le tadalafil
Utilisé en association avec le tadalafil, la dose préconisée d’AMBRISENTAN EG est de 10 mg une fois par jour.
Dans l’étude AMBITION, les patients recevaient 5 mg par jour d’ambrisentan durant les 8 premières semaines et la dose quotidienne était ensuite augmentée à 10 mg en fonction de la tolérance (voir rubrique 5.1). Associé au tadalafil à la dose de 20 mg, le traitement par ambrisentan était débuté à la dose de 5 mg. En fonction de la tolérance, la dose de tadalafil était ensuite augmentée à 40 mg après 4 semaines et celle de l'ambrisentan était augmentée à 10 mg après 8 semaines. Plus de 90 % des patients ont suivi ce schéma posologique. Les doses pouvaient aussi être baissées en fonction de la tolérance.
Des données limitées suggèrent que l'interruption brutale de l’ambrisentan n'est pas associée à un effet rebond avec aggravation de l’hypertension artérielle pulmonaire.
En cas de co-administration avec la ciclosporine A, la dose d'ambrisentan doit être limitée à 5 mg une fois par jour et le patient devra faire l'objet d'une étroite surveillance (voir rubriques 4.5 et 5.2).
Populations particulières
Sujets âgés
Aucune adaptation posologique n'est nécessaire chez les patients de plus de 65 ans (voir rubrique 5.2).
Patients insuffisants rénaux
Aucune adaptation posologique n'est nécessaire chez les patients insuffisants rénaux (voir rubrique 5.2). Les données d'utilisation de l’ambrisentan étant limitées chez les patients atteints d'insuffisance rénale sévère (clairance de la créatinine < 30 mL/min), le traitement doit être instauré avec précaution dans cette population et une attention particulière doit être portée en cas d’augmentation de la dose à 10 mg d’ambrisentan.
Patients insuffisants hépatiques
L’ambrisentan n'a pas été étudié chez les sujets présentant une insuffisance hépatique (avec ou sans cirrhose). Comme les principales voies métaboliques de l'ambrisentan sont la glucurono-conjugaison et l'oxydation avec élimination par voie biliaire, l'insuffisance hépatique pourrait augmenter l'exposition systémique (Cmax et ASC) à l’ambrisentan. Par conséquent, un traitement par ambrisentan ne doit pas être instauré chez les patients présentant une insuffisance hépatique sévère ou une augmentation des aminotransférases hépatiques cliniquement significative (plus de 3 fois la limite supérieure de la normale [> 3 x LSN] ; voir rubriques 4.3 et 4.4).
Population pédiatrique
La sécurité et l'efficacité de l’ambrisentan chez les enfants et les adolescents âgés de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Il est recommandé d’avaler le comprimé entier, au cours ou en dehors des repas. Il est recommandé de ne pas couper, écraser ou mâcher le comprimé.
Grossesse (voir rubrique 4.6).
Femmes en âge de procréer n’utilisant pas de méthode de contraception fiable (voir rubriques 4.4 et 4.6).
Allaitement (voir rubrique 4.6).
Insuffisance hépatique sévère (avec ou sans cirrhose) (voir rubrique 4.2).
Taux initial des aminotransférases hépatiques (aspartate aminotransférase [ASAT] et/ou alanine aminotransférase [ALAT] > 3 × LSN (voir rubriques 4.2 et 4.4).
Fibrose pulmonaire idiopathique avec ou sans hypertension pulmonaire associée (voir rubrique 5.1).
4.4. Mises en garde spéciales et précautions d'emploi
Le rapport bénéfice/risque de l’ambrisentan n'a pas été établi chez les patients atteints d'hypertension artérielle pulmonaire (HTAP) en classe fonctionnelle I (classification OMS).
L'efficacité de l’ambrisentan en monothérapie n'a pas été établie chez les patients atteints d'hypertension artérielle pulmonaire en classe fonctionnelle IV (classification OMS). En cas de détérioration de l'état clinique, un traitement recommandé pour le stade sévère de la maladie (par ex., époprosténol) doit être envisagé.
Fonction hépatique
Des anomalies de la fonction hépatique ont été associées à l'HTAP. Des cas compatibles avec une hépatite auto-immune, incluant une possible exacerbation d’hépatite auto-immune sous-jacente, des cas d’atteinte hépatique et d’élévations des enzymes hépatiques potentiellement liés au traitement ont été observés avec l’ambrisentan (voir rubriques 4.8 et 5.1). Les aminotransférases hépatiques sériques (ALAT et ASAT) doivent par conséquent être dosées avant l’instauration du traitement par ambrisentan et le traitement ne doit pas être instauré chez les patients dont les taux sériques d'ASAT et/ou ALAT sont supérieurs à 3 fois la limite supérieure de la normale (3 × LSN) (voir rubrique 4.3).
Les patients doivent faire l'objet d'une surveillance afin de déceler tout signe d’atteinte hépatique et un suivi mensuel des taux d'ALAT et ASAT est recommandé. Si un patient présente une augmentation cliniquement significative, prolongée et inexpliquée des taux d'ALAT et/ou d'ASAT, ou si cette augmentation s'accompagne de signes ou de symptômes d'atteinte hépatique (par ex., ictère), la prise d’ambrisentan doit être interrompue.
Chez les patients ne présentant pas de symptôme clinique d'atteinte hépatique ou d'ictère, la réintroduction de l’ambrisentan après normalisation des taux sériques d'enzymes hépatiques peut être envisagée. L'avis d'un médecin spécialiste hépatologue est recommandé.
Taux d'hémoglobine
Des diminutions du taux d'hémoglobine et de l'hématocrite ont été associées aux antagonistes des récepteurs de l'endothéline (ARE), y compris à l’ambrisentan. Dans la majorité des cas, la diminution a été observée au cours des 4 premières semaines de traitement et le taux d'hémoglobine s'est ensuite généralement stabilisé. Au cours de l'étude d'extension en ouvert des études cliniques pivots de phase III, les diminutions moyennes des taux d'hémoglobine observées sur le long terme par rapport aux taux à l'inclusion (de 0,9 jusqu'à 1,2 g/dL) ont persisté jusqu'à 4 ans de traitement par ambrisentan. Depuis la mise sur le marché, des cas d'anémie nécessitant une transfusion sanguine ont été rapportés (voir rubrique 4.8).
L'instauration du traitement par ambrisentan n'est pas recommandée chez des patients présentant une anémie cliniquement significative. Un contrôle des taux d'hémoglobine et/ou de l'hématocrite est recommandé pendant la prise d’ambrisentan, par exemple 1 mois et 3 mois après le début du traitement et ensuite périodiquement selon la pratique clinique. Si une diminution cliniquement significative de l'hémoglobine ou de l'hématocrite est observée, alors que d'autres causes ont été exclues, une diminution de la dose ou une interruption du traitement devra être envisagée. L’incidence des anémies était plus élevée lorsque l’ambrisentan était associé au tadalafil (15 % de la fréquence des évènements indésirables), comparativement au traitement en monothérapie par ambrisentan ou tadalafil (7 % et 11 % respectivement).
Rétention hydrique
Des œdèmes périphériques ont été observés avec les antagonistes des récepteurs de l'endothéline (ARE), y compris avec l’ambrisentan. La plupart des cas d'œdèmes périphériques rapportés au cours des études cliniques avec l’ambrisentan ont été d’intensité légère à modérée ; cependant, ils peuvent être plus fréquents et d’intensité plus importante chez les patients d’âge ≥ 65 ans. Les œdèmes périphériques ont été plus fréquemment rapportés avec la dose de 10 mg d’ambrisentan dans les études cliniques à court terme (voir rubrique 4.8).
Des cas de rétention hydrique survenant dans les semaines suivant la mise en route d'un traitement par l’ambrisentan ont été rapportés depuis sa commercialisation. Certains cas ont nécessité un traitement diurétique ou une hospitalisation pour traitement de la surcharge hydrique ou d'une décompensation cardiaque. En cas de rétention hydrique préexistante, le traitement par ambrisentan ne sera instauré qu'après traitement approprié de la surcharge hydrique.
Si une rétention hydrique, avec ou sans prise de poids associée, apparait au cours du traitement par ambrisentan, les investigations appropriées devront être menées afin de distinguer si elle est liée à la prise d’ambrisentan ou à une décompensation cardiaque sous-jacente, et décider en conséquence de la nécessité d'un traitement spécifique ou de l'arrêt du traitement par ambrisentan. L’incidence des œdèmes périphériques était plus élevée lorsque l’ambrisentan était associé au tadalafil (45 % de la fréquence des évènements indésirables), comparativement aux traitements en monothérapie par ambrisentan ou tadalafil (38 % et 28 % respectivement). Les œdèmes périphériques sont survenus en majorité au cours du premier mois de traitement.
Femmes en âge de procréer
Chez la femme en âge de procréer, une grossesse doit être exclue avant la mise en route du traitement par AMBRISENTAN EG et doit ensuite être évitée par une méthode de contraception fiable. En cas de doute sur le choix du mode de contraception adapté à la patiente, la consultation d'un médecin spécialiste gynécologue doit être envisagée. Des tests de grossesse mensuels sont recommandés pendant le traitement par ambrisentan (voir rubriques 4.3 et 4.6).
Maladie veino-occlusive pulmonaire
Des cas d'œdème pulmonaire ont été rapportés avec des médicaments vasodilatateurs, tels que les antagonistes des récepteurs de l'endothéline (ARE), lorsqu'ils sont utilisés chez des patients ayant une maladie veino-occlusive pulmonaire. Par conséquent, si des patients atteints d'hypertension artérielle pulmonaire développent un œdème pulmonaire aigu sous traitement par ambrisentan, la possibilité d'une maladie pulmonaire veino-occlusive devra être évoquée.
Utilisation concomitante avec d'autres médicaments
Les patients traités par ambrisentan doivent faire l'objet d'une étroite surveillance lorsqu'ils débutent un traitement par rifampicine (voir rubriques 4.5 et 5.2).
Excipients
AMBRISENTAN EG, comprimé pelliculé contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
AMBRISENTAN EG, comprimé pelliculé contient l’agent colorant azoïque rouge Allura AC (E129), qui peut provoquer des réactions allergiques.
AMBRISENTAN EG, comprimé pelliculé contient de la lécithine de soja. Si un patient est hypersensible à l’arachide ou au soja, AMBRISENTAN EG ne doit pas être utilisé (voir rubrique 4.3).
AMBRISENTAN EG, comprimé pelliculé contient moins de 1 mmol (23 mg) de sodium par comprimé c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Dans des études réalisées in vitro et in vivo chez l'animal, l'ambrisentan n'a provoqué ni inhibition ni induction des enzymes impliquées dans les phases I ou II du métabolisme des médicaments aux concentrations utilisées en thérapeutique, ce qui suggère un faible potentiel d'interactions sur le profil pharmacocinétique des médicaments métabolisés par ces voies.
L'induction potentielle de l'activité du CYP3A4 par l'ambrisentan a été étudiée chez des volontaires sains ; les résultats suggèrent une absence d'effet inducteur de l'ambrisentan sur l'isoenzyme CYP3A4.
Ciclosporine A
A l'état d'équilibre, la co-administration d'ambrisentan et de ciclosporine A a doublé l'exposition à l'ambrisentan chez des volontaires sains. Ce phénomène peut être dû à l'inhibition par la ciclosporine A des transporteurs et des enzymes métaboliques impliqués dans la pharmacocinétique de l'ambrisentan. Par conséquent, en cas de co-administration avec la ciclosporine A, la dose d'ambrisentan doit être limitée à 5 mg une fois par jour (voir rubrique 4.2). L'administration de doses multiples d'ambrisentan n'a pas eu d'effet sur l'exposition à la ciclosporine A et aucun ajustement posologique de la ciclosporine A n'est justifié.
Rifampicine
L’administration concomitante de rifampicine (qui est un inhibiteur du transporteur polypeptide des anions organiques [OATP], un puissant inducteur des cytochromes CYP3A et 2C19, et un inducteur de la glycoprotéine P (P-gp) et des uridine-diphospho-glucuronosyltransférases [UGT]), a été associée à une augmentation transitoire (environ 2 fois) de l'exposition à l'ambrisentan suite à l’instauration du traitement chez des volontaires sains. Toutefois, à J8, l’administration à l’état d’équilibre de la rifampicine n'a pas entraîné d’effet cliniquement significatif sur l'exposition à l'ambrisentan. Les patients traités par ambrisentan doivent faire l'objet d'une étroite surveillance lorsqu'ils débutent un traitement par rifampicine (voir rubriques 4.4 et 5.2).
Inhibiteurs de la phosphodiestérase
L'administration concomitante d'ambrisentan et d'un inhibiteur de la phosphodiestérase, que ce soit le sildénafil ou le tadalafil (tous deux substrats du CYP3A4) chez des volontaires sains, n'a pas entraîné de modifications significatives de la pharmacocinétique de l'inhibiteur de la phosphodiestérase ni de celle de l'ambrisentan (voir rubrique 5.2).
Autres traitements ciblés de l’HTAP
L'efficacité et la sécurité de l’ambrisentan lors de l’administration concomitante avec d'autres traitements de l'hypertension artérielle pulmonaire (par ex., prostanoïdes et stimulateurs de la guanylate cyclase soluble) n'ont pas été spécifiquement étudiées dans des essais cliniques contrôlés menés chez des patients atteints d'HTAP (voir rubrique 5.1). Au vu des données disponibles concernant la métabolisation, aucune interaction médicamenteuse n’est attendue avec les stimulateurs de la guanylate cyclase soluble ou les prostanoïdes (voir rubrique 5.2). Cependant, aucune étude spécifique d’interactions médicamenteuses n’a été réalisée avec ces médicaments. Par conséquent, la prudence est recommandée en cas d'administration concomitante.
Contraceptifs oraux
Dans une étude clinique menée chez des volontaires sains, l’administration à l’état d’équilibre pharmacocinétique de 10 mg d'ambrisentan une fois par jour n'a pas entraîné de modifications significatives de la pharmacocinétique d'une dose unique d'un contraceptif oral associant l'éthinylestradiol et la noréthindrone (voir rubrique 5.2). Selon cette étude pharmacocinétique, l'ambrisentan ne devrait pas avoir d'effet significatif sur l'exposition aux œstrogènes ou à la progestérone des contraceptifs oraux.
Warfarine
L'ambrisentan n'a eu aucun effet sur la pharmacocinétique à l'état d'équilibre et sur l'activité anticoagulante de la warfarine chez le volontaire sain (voir rubrique 5.2). Par ailleurs, la warfarine n'a eu aucun effet cliniquement significatif sur la pharmacocinétique de l'ambrisentan. De plus, chez les patients atteints d'hypertension artérielle pulmonaire, il n'a pas été observé de retentissement sur la dose hebdomadaire utilisée des anticoagulants de type warfarine nécessaire, sur le temps de prothrombine (TP) et sur le rapport normalisé international (INR).
Kétoconazole
L'administration de kétoconazole (puissant inhibiteur du CYP3A4) à l'état d'équilibre pharmacocinétique n'a pas entraîné d'augmentation cliniquement significative de l'exposition à l'ambrisentan (voir rubrique 5.2).
Effet de l'ambrisentan sur les transporteurs de xénobiotiques
In vitro, l'ambrisentan n'a eu aucun effet inhibiteur sur les transporteurs humains à des concentrations cliniquement significatives, incluant la glycoprotéine P (P-gp), la BCRP (breast cancer resistance protein), la MRP2 (multi-drug resistance protein isoform-2), la BSEP (bile salt export pump), les OATP1B1 et OATP1B3 (organic anion transporting polypeptides) et le NTCP (sodium-dependent taurocholate co-transporting polypeptide).
L’ambrisentan est un substrat de la glycoprotéine P (P-gp).
Des études in vitro effectuées sur des hépatocytes de rat ont également montré que l'ambrisentan n’était pas un inducteur des protéines P-gp, BSEP ou MRP2.
L’administration à l’état d’équilibre de l'ambrisentan chez des volontaires sains n'a pas eu d'effets cliniquement significatifs sur la pharmacocinétique de la digoxine administrée en dose unique, substrat de la P-gp (voir rubrique 5.2).
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Chez les femmes en âge de procréer, le traitement par ambrisentan ne doit être instauré qu’après obtention d'un test de grossesse négatif et à condition qu’une méthode de contraception fiable soit utilisée. Des tests mensuels de grossesse sont recommandés au cours du traitement par ambrisentan.
Grossesse
L’ambrisentan est contre-indiqué durant la grossesse (voir rubrique 4.3). Les études réalisées chez l'animal ont montré que l'ambrisentan était tératogène. Il n'y a pas de données chez l'Homme.
Les femmes traitées par ambrisentan doivent être informées du risque pour le fœtus et un traitement alternatif devra être instauré en cas de grossesse (voir rubriques 4.3, 4.4 et 5.3).
Allaitement
Il n'existe pas de données sur le passage de l'ambrisentan dans le lait maternel humain. L'excrétion de l'ambrisentan dans le lait n'a pas été étudiée chez l'animal. L’allaitement est par conséquent contre-indiqué pendant le traitement par ambrisentan (voir rubrique 4.3).
Fertilité masculine
Le développement d'une atrophie tubulaire testiculaire lié à l'administration chronique des antagonistes des récepteurs de l'endothéline (ARE) a été observé chez les animaux mâles, y compris avec l’ambrisentan (voir rubrique 5.3). Bien qu'il n’ait pas été clairement mis en évidence d'effet délétère sur le nombre de spermatozoïdes lors d'une administration au long cours de l’ambrisentan dans l'étude ARIES-E, l'administration chronique d'ambrisentan a été associée à des modifications des marqueurs de la spermatogénèse. Une diminution de la concentration plasmatique de l'inhibine B et une augmentation de la concentration plasmatique de la FSH ont été observées. L'effet de l'ambrisentan sur la fertilité chez l'Homme n'est pas connu mais l'éventualité d'une altération de la spermatogenèse ne peut être exclue. L'administration chronique d'ambrisentan n'a pas été associée à une modification de la testostérone plasmatique dans les études cliniques.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
La tolérance de l’ambrisentan a été étudiée chez plus de 1 200 patients atteints d'hypertension artérielle pulmonaire en monothérapie et/ou en association lors d’essais cliniques (voir rubrique 5.1). Les effets indésirables observés dans les essais cliniques contrôlés versus placebo de 12 semaines sont répertoriés ci-dessous par classes de systèmes d’organes et par fréquence de survenue.
Les informations provenant des études à long terme non contrôlées contre placebo (ARIES-E et AMBITION [en association avec le tadalafil]) figurent également ci-dessous. Il n'a pas été identifié de nouvel effet indésirable avec le traitement à long terme ou avec l’ambrisentan en association avec le tadalafil. Le profil de tolérance observé dans les études non contrôlées sur une durée plus longue (en moyenne 79 semaines) est similaire à celui observé dans les études court terme. Les effets indésirables recueillis depuis la mise sur le marché sont également présentés.
Les effets indésirables les plus fréquemment observés avec l'ambrisentan ont été : œdème périphérique, rétention hydrique et céphalées (incluant céphalées sinusales et migraine). L’utilisation de la dose la plus forte (10 mg) a été associée à une incidence plus élevée de ces effets indésirables, et une tendance à une majoration de la sévérité des œdèmes périphériques a été observée chez les sujets ≥ 65 ans dans les études cliniques à court-terme (voir rubrique 4.4).
Liste tabulée des effets indésirables
La convention suivante a été utilisée pour la classification des fréquences : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Pour les effets indésirables dose-dépendants, les fréquences de survenue correspondent à la dose la plus élevée d’ambrisentan. Les fréquences ne tiennent pas compte d'autres facteurs incluant des variations de durée d'étude, les pathologies associées et les caractéristiques des patients à l'inclusion. Les fréquences de survenue des effets indésirables reposent sur l'expérience des essais cliniques et peuvent ne pas refléter les fréquences des événements indésirables survenant dans la pratique clinique. Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
|
|
Ambrisentan (ARIES-C et depuis la commercialisation) |
Ambrisentan (AMBITION et ARIES-E) |
Association avec le tadalafil (AMBITION) |
|
Affections hématologiques et du système lymphatique |
|||
|
Anémie (diminution de l’hémoglobine, diminution de l'hématocrite) |
Fréquent1 |
Très fréquent |
Très fréquent |
|
Affections du système immunitaire |
|||
|
Réactions d'hypersensibilité (par ex., angiœdème, éruption cutanée, prurit) |
Peu fréquent |
Fréquent |
Fréquent |
|
Affections du système nerveux |
|||
|
Céphalées (y compris céphalées sinusales, migraine) |
Très fréquent2 |
Très fréquent |
Très fréquent |
|
Sensations vertigineuses |
Fréquent3 |
Très fréquent |
Très fréquent |
|
Affections oculaires |
|||
|
Vision floue, troubles visuels |
Fréquence indéterminée4 |
Fréquent |
Fréquent |
|
Affections de l’oreille et du labyrinthe |
|||
|
Acouphènes |
NR |
NR |
Fréquent |
|
Perte soudaine de l’audition |
NR |
NR |
Peu fréquent |
|
Affections cardiaques |
|||
|
Insuffisance cardiaque |
Fréquent5 |
Fréquent |
Fréquent |
|
Palpitations |
Fréquent |
Très fréquent |
Très fréquent |
|
Affections vasculaires |
|||
|
Hypotension |
Fréquent3 |
Fréquent |
Fréquent |
|
Bouffées vasomotrices |
Fréquent |
Fréquent |
Très fréquent |
|
Syncope |
Peu fréquent3 |
Fréquent |
Fréquent |
|
Affections respiratoires, thoraciques et médiastinales |
|||
|
Epistaxis |
Fréquent3 |
Fréquent |
Fréquent |
|
Dyspnée |
Fréquent3,6 |
Très fréquent |
Très fréquent |
|
Congestion des voies respiratoires hautes (par ex., nez, sinus), sinusite, rhinopharyngite, rhinite |
Fréquent7 |
||
|
Rhinopharyngite |
Très fréquent |
Très fréquent |
|
|
Sinusite, rhinite |
Fréquent |
Fréquent |
|
|
Congestion nasale |
Très fréquent |
Très fréquent |
|
|
Affections gastro-intestinales |
|||
|
Nausées, vomissements, diarrhée |
Fréquent3 |
||
|
Nausées |
Très fréquent |
Très fréquent |
|
|
Vomissements |
Fréquent |
Très fréquent |
|
|
Diarrhée |
Très fréquent |
Très fréquent |
|
|
Douleurs abdominales |
Fréquent |
Fréquent |
Fréquent |
|
Constipation |
Fréquent |
Fréquent |
Fréquent |
|
Affections hépatobiliaires |
|||
|
Atteinte hépatique (voir rubrique 4.4) |
Peu fréquent3, 8 |
NR |
NR |
|
Hépatite auto-immune (voir rubrique 4.4) |
Peu fréquent3,8 |
NR |
NR |
|
Elévation des transaminases hépatiques |
Fréquent3 |
NR |
NR |
|
Affections de la peau et du tissu sous-cutané |
|||
|
Eruption cutanée |
NR |
Fréquent9 |
Très fréquent9 |
|
Troubles généraux et anomalies au site d’administration |
|||
|
Œdème périphérique, rétention hydrique |
Très fréquent |
Très fréquent |
Très fréquent |
|
Douleur/gêne thoracique |
Fréquent |
Fréquent |
Très fréquent |
|
Asthénie |
Fréquent3 |
Fréquent |
Fréquent |
|
Fatigue |
Fréquent3 |
Très fréquent |
Très fréquent |
NR – non rapporté
1 Voir paragraphe « Description de certains effets indésirables ».
2 La fréquence des céphalées apparait plus élevée avec 10 mg d’ambrisentan.
3 Données issues de la pharmacovigilance depuis la mise sur le marché et fréquences de survenue estimées à partir des fréquences observées dans les essais cliniques contrôlés contre placebo.
4 Données issues de la pharmacovigilance depuis la mise sur le marché.
5 La plupart des cas d'insuffisance cardiaque rapportés étaient associés à une rétention hydrique. Données issues de la pharmacovigilance depuis la mise sur le marché et fréquences de survenue estimées par une modélisation statistique à partir des données issues des essais cliniques contrôlés contre placebo.
6 Des cas d'aggravation de dyspnée d'étiologie indéterminée ont été rapportés peu de temps après le début du traitement par ambrisentan.
7 L'incidence des congestions nasales s’est révélée dose-dépendante pendant le traitement par ambrisentan.
8 Des cas d'hépatite auto-immune, incluant des cas d'exacerbation d'hépatite auto-immune, et d'atteinte hépatique ont été rapportés au cours du traitement par ambrisentan.
9 Eruption cutanée incluant des éruptions érythémateuses, des éruptions généralisées, des éruptions papuleuses et des éruptions prurigineuses.
Description de certains effets indésirables
Diminution de l'hémoglobine
Depuis la mise sur le marché, des cas d’anémie nécessitant une transfusion sanguine ont été rapportés (voir rubrique 4.4). La fréquence de diminution de l'hémoglobine (anémie) apparaît plus élevée avec 10 mg d’ambrisentan. Durant les 12 semaines d’études cliniques de phase III contrôlées versus placebo, les taux moyens d'hémoglobine ont diminué chez les patients des groupes ambrisentan et ces diminutions ont été détectées dès la quatrième semaine (diminution de 0,83 g/dL). Les variations par rapport à la valeur à l’inclusion ont semblé se stabiliser au cours des huit semaines suivantes. Au total, 17 patients (6,5 %) des groupes traités par ambrisentan ont présenté une diminution du taux d'hémoglobine d'au moins 15 % par rapport à l’inclusion et le taux était inférieur à la limite inférieure de la normale.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Il n'y a pas de données rapportées chez des patients atteints d'hypertension artérielle pulmonaire traités avec plus de 10 mg par jour d’ambrisentan. Chez des volontaires sains, l’administration d'une dose unique de 50 et 100 mg (5 à 10 fois la dose maximale recommandée) a été associée à des céphalées, des bouffées vasomotrices, des sensations vertigineuses, des nausées et des congestions nasales.
Du fait de son mécanisme d'action, un surdosage par ambrisentan peut potentiellement entraîner une hypotension (voir rubrique 5.3). L'hypotension induite peut être sévère et nécessiter un traitement de réanimation. Aucun antidote n'est disponible.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Anti-hypertenseurs, autres anti-hypertenseurs, code ATC : C02KX02.
Mécanisme d’action
L’ambrisentan est un antagoniste sélectif des récepteurs de type A de l'endothéline (ETA), actif par voie orale appartenant à la catégorie des acides propioniques. L'endothéline joue un rôle important dans la physiopathologie de l'hypertension artérielle pulmonaire.
· L’ambrisentan est un antagoniste ETA puissant (Ki 0,016 nM) et hautement sélectif (environ 4 000 fois plus sélectif pour ETA par rapport à ETB).
· L’ambrisentan bloque les récepteurs de sous-type ETA, principalement localisés sur les cellules des muscles lisses vasculaires et sur les cardiomyocytes, ce qui empêche l'activation des médiateurs de l'endothéline, messagers secondaires dans le processus de vasoconstriction et de prolifération des cellules musculaires lisses.
· La sélectivité d'ambrisentan pour le récepteur ETA plus que pour le récepteur ETB permet de respecter la production médiée par le récepteur ETB de vasodilatateurs comme le monoxyde d'azote et la prostacycline.
Efficacité et sécurité clinique
Deux essais pivots de phase III, multicentriques, randomisés, en double aveugle, contrôlés contre placebo ont été réalisés (ARIES-1 et 2). L'essai ARIES-1, incluant 201 patients, a comparé l’ambrisentan 5 mg et l’ambrisentan 10 mg au placebo. L'essai ARIES-2, incluant 192 patients, a comparé l’ambrisentan 2,5 mg et l’ambrisentan 5 mg au placebo. Dans ces deux études, l’ambrisentan a été ajouté au traitement pré-existant, qui pouvait inclure une association de digoxine, d'anticoagulants, de diurétiques, d'oxygène et de vasodilatateurs (inhibiteurs des canaux calciques et inhibiteurs ACE). Les patients inclus présentaient une HTAP idiopathique ou une HTAP associée à une collagénose systémique. La majorité des patients était en classe fonctionnelle II (38,4 %) ou III (55,0 %) selon la classification OMS. Les patients atteints d'insuffisance hépatique préexistante (cirrhose ou taux d’aminotransférases cliniquement significatif) et les patients utilisant d'autres thérapies de l'hypertension artérielle pulmonaire (par exemple, prostanoïdes) ont été exclus. Les paramètres hémodynamiques n'ont pas été évalués durant ces études.
Le critère principal défini pour ces études de phase III était l'amélioration de la capacité à l'exercice mesurée par le changement par rapport à la valeur à l’inclusion des résultats au test de marche de 6 minutes à 12 semaines. Dans les deux études, le traitement par ambrisentan a entraîné une amélioration significative du test de marche à 6 minutes pour chacune des doses d’ambrisentan étudiées.
Dans les études ARIES-1 et ARIES- 2, l'amélioration de la distance moyenne parcourue durant le test de marche pendant 6 minutes à la semaine 12 était, après ajustement de l'effet placebo, respectivement de 30,6 m (IC à 95 % : 2,9 à 58,3 ; p = 0,008) et de 59,4 m (IC à 95 % : 29,6 à 89,3 ; p < 0,001) dans le groupe ambrisentan 5 mg. Pour l’étude ARIES-1, l'amélioration de la distance moyenne parcourue durant le test de marche pendant 6 minutes à la semaine 12 était de 51,4 m (IC à 95 % : 26,6 à 76,2 ; p < 0,001) dans le groupe ambrisentan 10 mg.
Une analyse combinée des études de phase III (ARIES-C) conduite selon des modalités pré-spécifiées dans le protocole a retrouvé une amélioration sur la moyenne des différences observées par rapport au placebo des tests de marche pendant 6 minutes : 44,6 m (IC à 95 % : 24,3 à 64,9 ; p < 0,001) dans le groupe ambrisentan 5 mg et 52,5 m (IC à 95 % : 28,8 à 76,2 ; p < 0,001) dans le groupe ambrisentan 10 mg.
Dans l’étude ARIES-2 (groupe recevant des doses combinées), l’ambrisentan a significativement retardé l'aggravation clinique de l’HTAP par rapport au placebo (p < 0,001), le rapport de risque a démontré une réduction de 80 % (IC à 95 % : 47 % à 92 %). Cette évaluation englobait : le décès, la transplantation, l'hospitalisation pour HTAP, la septostomie auriculaire, l’ajout d'autres agents thérapeutiques pour le traitement de l’HTAP. Une amélioration statistiquement significative dans le domaine concernant les fonctions physiques évalué par le questionnaire d'échelle de qualité de vie SF-36 Health Survey, a été observée pour le groupe recevant les doses combinées d’ambrisentan (3,41 ± 6,96) par rapport au placebo (-0,20 ± 8,14, p = 0,005). Le traitement par ambrisentan a amélioré de façon statistiquement significative la dyspnée évaluée par l'Index de Dyspnée de Borg (IDB) à la semaine 12 après ajustement de l’effet placebo de -1,1 (IC à 95 % : -1,8 à -0,4 ; p = 0,019 ; groupe recevant la dose combinée).
Données à long terme
Les patients inclus dans les études ARIES-1 et 2 étaient éligibles pour entrer dans l’étude d'extension à long terme en ouvert ARIES-E (n = 383). L'exposition moyenne calculée sur l'ensemble des données des 2 études était d'environ 145 ± 80 semaines, et l'exposition maximale d'environ 295 semaines. Les critères d'évaluation principaux de cette étude étaient l'incidence et la sévérité des événements indésirables associés à une administration au long cours de l'ambrisentan, incluant les anomalies des tests sanguins de la fonction hépatique. La tolérance observée à long terme dans cette étude était globalement superposable à celle observée dans les études contrôlées comparativement au placebo de 12 semaines.
Le taux de survie des patients sous ambrisentan (tous groupes de doses d’ambrisentan combinés) à 1, 2 et 3 ans était respectivement de 93 %, 85 % et 79 %.
Dans une étude conduite en ouvert (AMB222), l’ambrisentan a été administré chez 36 patients afin d'évaluer l'incidence de l'augmentation des taux sériques d'aminotransférases chez les patients qui avaient précédemment arrêté d’autres traitements avec un antagoniste des récepteurs de l'endothéline en raison d'anomalies des taux d'aminotransférases. Pendant une durée moyenne de 53 semaines de traitement par ambrisentan, aucun des patients inclus n'a présenté un taux d'ALAT sérique confirmé > 3 × LSN nécessitant un arrêt définitif du traitement. Cinquante pour cent des patients étaient passés de 5 mg à 10 mg d’ambrisentan pendant cette période.
L'incidence cumulée des anomalies des taux d'aminotransférases sériques > 3 × LSN dans l'ensemble des études cliniques de phase II et III (y compris les extensions en ouvert) a été de 17 sur 483 patients sur une durée d'exposition moyenne de 79,5 semaines. Soit un taux d'évènements de 2,3 pour 100 patients-années exposés à l’ambrisentan. Au cours de l'étude d'extension à long terme en ouvert ARIES-E, le risque à 2 ans de développer des élévations des aminotransférases sériques > 3 × LSN chez les patients traités par ambrisentan était de 3,9 %.
Autres informations cliniques
Dans l'étude de phase II (AMB220), une amélioration des paramètres hémodynamiques a été observée après 12 semaines de traitement chez les patients (n = 29) présentant une HTAP. Le traitement par ambrisentan a entraîné une augmentation de l'index cardiaque moyen, une diminution de la pression artérielle pulmonaire moyenne et une diminution de la résistance vasculaire pulmonaire moyenne.
Une diminution de la pression artérielle systolique et diastolique a été rapportée sous traitement par ambrisentan. Dans des études cliniques contrôlées contre placebo d'une durée de 12 semaines, la réduction moyenne des pressions artérielles systolique et diastolique entre les valeurs à l'inclusion et celles observées à la fin du traitement ont été, respectivement, de 3 mm Hg et 4,2 mm Hg. Au cours de l'étude d'extension en ouvert ARIES E, les diminutions moyennes des pressions artérielles systolique et diastolique ont persisté jusqu'à 4 ans de traitement par ambrisentan.
Aucun effet cliniquement significatif relatif à la pharmacocinétique de l’ambrisentan ou du sildénafil n’a été observé pendant une étude d’interaction médicamenteuse effectuée chez des volontaires sains, et l’association a été bien tolérée. Dans les études ARIES-E et AMB222, 22 patients (5,7 %) et 17 patients (47 %), respectivement, ont reçu un traitement concomitant d’ambrisentan et de sildénafil. Aucun problème d'intolérance supplémentaire n'a été détecté chez ces patients.
Efficacité clinique en association avec le tadalafil
Une étude clinique de phase III conduite en fonction de la survenue d’événements, multicentrique en double aveugle utilisant un comparateur actif a été menée afin d'évaluer l’efficacité de l’association thérapeutique en première intention d’ambrisentan et de tadalafil (bithérapie) comparativement à un traitement en monothérapie par l’ambrisentan ou le tadalafil chez 500 patients naïfs de traitement pour l’HTAP, randomisés selon le ratio 2/1/1 (AMB112565/AMBITION). Aucun des patients n’avait reçu de placebo seul. L'analyse principale reposait sur la comparaison des données en bithérapie avec celles regroupées des deux en monothérapie. Des comparaisons entre le groupe en bithérapie et chacun des deux groupes en monothérapie ont également été effectuées. Les patients ayant une anémie, une rétention hydrique ou des maladies rares de la rétine cliniquement significatives selon le jugement de l’investigateur, étaient exclus de l’étude. Les patients ayant des valeurs d’ALAT et d’ASAT >2 × LSN à l’inclusion ont également été exclus.
A l’inclusion, 96 % des patients étaient naïfs de tout traitement spécifique de l’HTAP et le délai médian entre le diagnostic et l’inclusion dans l’étude était de 22 jours. Les patients débutaient leur traitement avec 5 mg d’ambrisentan et 20 mg de tadalafil puis les doses étaient augmentées à 40 mg de tadalafil à la 4ème semaine et à 10 mg d’ambrisentan à la 8ème semaine sauf en cas d'intolérance. La durée médiane de traitement en double aveugle pour la bithérapie était supérieure à 1,5 an.
Le critère principal était le délai de survenue du premier échec clinique défini par :
· la survenue du décès, ou
· une hospitalisation pour aggravation de l’HTAP,
· une progression de la maladie,
· une réponse clinique à long terme jugée non satisfaisante.
La moyenne d’âge des patients était de 54 ans (ET 15 ; 18– 75 ans). A l’inclusion, les patients étaient en classe fonctionnelle II (31 %) et III (69 %) (classification de l’OMS). Par ordre de fréquence, l'étiologie de l'HTAP dans la population de l'étude était : HTAP idiopathique ou familiale (56 %), HTAP associée à une connectivite (37 %), HTAP associée à la prise de médicaments ou une exposition à des toxines (3 %), HTAP associée à une malformation cardiaque congénitale opérée (2 %) et HTAP associée à une infection par le VIH (2 %). Les patients en classe fonctionnelle II et III (classification de l’OMS) avaient une distance moyenne de marche de 6 minutes de 353 mètres à l’inclusion.
Résultats sur le critère principal combiné
Le traitement par bithérapie a réduit le risque de 50 % (rapport de risque [HR] : 0,502 [IC à 95 % : 0,348 ; 0,724], p = 0,0002) du critère composite de survenue du premier échec clinique jusqu'à la dernière visite d'évaluation par rapport aux monothérapies groupées (figure 1 et tableau 1 ci-dessous). L'effet du traitement observé était principalement en relation avec une réduction de 63 % des hospitalisations dans le groupe traité par la bithérapie. Cet effet a été observé précocement et s’est maintenu dans le temps. L’efficacité de la bithérapie sur le critère principal a été retrouvée lors de la comparaison avec chacune des monothérapies et dans les sous-groupes d'âge, d'origine ethnique, de région géographique, d'étiologie (HTAP idiopathique ou familiale et HTAP associée à une connectivite). L'effet était significatif pour les patients en classe fonctionnelle II et pour les patients en classe fonctionnelle III.
Figure 1
IC à 95 %
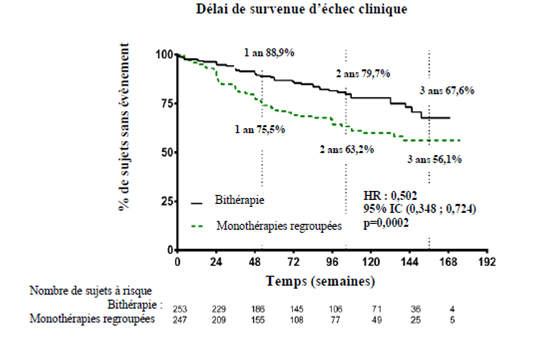
Tableau 1
|
|
Ambrisentan + tadalafil (N = 253) |
Monothérapies regroupées (N = 247) |
Ambrisentan en monothérapie (N = 126) |
Tadalafil en monothérapie (N = 121) |
|
Délai de survenue du premier échec clinique (adjudiqué) |
||||
|
Echec clinique, nbre (%) |
46 (18 %) |
77 (31 %) |
43 (34 %) |
34 (28 %) |
|
Rapport de risque (IC à 95 %) |
|
0,502 (0,348 ; 0,724) |
0,477 (0,314 ; 0,723) |
0,528 (0,338 ; 0,827) |
|
Valeur de p, test du log-rank |
|
0,0002 |
0,0004 |
0,0045 |
|
Evènements du critère principal composite (délai de survenue du premier échec clinique) (adjudiqué) |
||||
|
Décès (toutes causes confondues) |
9 (4 %) |
8 (3 %) |
2 (2 %) |
6 (5 %) |
|
Hospitalisation pour aggravation de l’HTAP |
10 (4 %) |
30 (12 %) |
18 (14 %) |
12 (10 %) |
|
Progression de la maladie |
10 (4 %) |
16 (6 %) |
12 (10 %) |
4 (3 %) |
|
Réponse clinique insatisfaisante à long terme |
17 (7 %) |
23 (9 %) |
11 (9 %) |
12 (10 %) |
|
Délai de survenue de la première hospitalisation pour aggravation de l’HTAP (adjudiqué) |
||||
|
Première hospitalisation, nbre (%) |
19 (8 %) |
44 (18 %) |
27 (21 %) |
17 (14 %) |
|
Rapport de risque (IC à 95 %) |
|
0,372 |
0,323 |
0,442 |
|
Valeur de p, test du log-rank |
|
0,0002 |
< 0,0001 |
0,0124 |
Critères secondaires
Les critères secondaires ont été examinés.
Tableau 2
|
Critères secondaires (changement entre la valeur initiale et la semaine 24) |
Ambrisentan + tadalafil |
Monothérapies regroupées |
Différence [Intervalle de confiance] |
Valeur de p |
|
NT-proBNP (% de réduction |
-67,2 |
-50,4 |
Différence en % : -33,8 ; [IC à 95 % : -44,8 ; -20,7] |
p < 0,0001 |
|
% de sujets ayant une réponse clinique satisfaisante à la semaine 24 |
39 |
29 |
Odds ratio 1,56 ; [IC à 95 % : 1,05 ; 2,32] |
p = 0,026 |
|
Distance de marche pendant 6 minutes (mètres, changement médian) |
49,0 |
23,8 |
22,75 m ; [IC à 95 % : 12,00 ; 33,50] |
p < 0,0001 |
Fibrose pulmonaire idiopathique
Une étude a été conduite chez 492 patients (ambrisentan N = 329, placebo N = 163) atteints de fibrose pulmonaire idiopathique, parmi lesquels 11 % présentaient une hypertension pulmonaire associée (groupe 3 de la classification OMS) ; cette étude a été prématurément arrêtée lorsqu'il a été mis en évidence que l'objectif sur le critère de jugement principal d'efficacité ne pourrait pas être atteint (étude ARTEMIS-IPF). Quatre-vingt-dix patients traités par ambrisentan (27 %) ont présenté des événements correspondant à une progression de la fibrose pulmonaire idiopathique (incluant des hospitalisations pour aggravations respiratoires) ou un décès comparativement à 28 patients (17 %) dans le groupe placebo. L'ambrisentan est par conséquent contre-indiqué chez les patients ayant une fibrose pulmonaire idiopathique avec ou sans hypertension pulmonaire associée (voir rubrique 4.3).
5.2. Propriétés pharmacocinétiques
L’ambrisentan est rapidement absorbé chez l’Homme. Après administration orale, les concentrations plasmatiques maximales (Cmax) de l’ambrisentan sont obtenues généralement en 1,5 heure environ après la prise, à jeun ou non. La Cmax et l’aire sous la courbe de la concentration plasmatique en fonction du temps (ASC) augmentent proportionnellement à la dose, dans l'intervalle posologique administré. L’état d'équilibre est généralement obtenu après 4 jours de doses répétées.
Une étude de l'influence de l'alimentation portant sur l’administration d’ambrisentan chez des volontaires sains à jeun et après un repas riche en graisses a indiqué que la Cmax était diminuée de 12 % tandis que l’ASC demeurait identique. Cette diminution du pic de concentration n’est pas cliniquement significative et, par conséquent, l’ambrisentan peut être pris avec ou sans aliments.
Distribution
L’ambrisentan est fortement lié aux protéines plasmatiques. In vitro, l’ambrisentan était lié, en moyenne, à 98,8 % aux protéines plasmatiques indépendamment de la concentration dans l'intervalle de 0,2 à 20 microgrammes/mL.
L’ambrisentan est lié principalement à l’albumine (96,5 %) et, dans une moindre mesure, à la glycoprotéine acide alpha1.
L’ambrisentan est faiblement distribué dans les érythrocytes, avec un rapport moyen sang/plasma de 0,57 chez l’homme et de 0,61 chez la femme.
Biotransformation
L’ambrisentan n’est pas un ARE du groupe des sulfonamides (groupe des acides propioniques).
L’ambrisentan est glucurono-conjugué par plusieurs isoenzymes UGT (UGT1A9S, UGT2B7S et UGT1A3S) sous forme de glucurono-conjugué d’ambrisentan (13 %). L’ambrisentan subit également un métabolisme oxydatif, principalement par le CYP3A4 et, dans une moindre mesure, par le CYP3A5 et le CYP2C19 pour former le 4-hydroxyméthyl ambrisentan (21 %), ensuite glucurono-conjugué en glucurono-conjugué de 4-hydroxyméthyl ambrisentan (5 %). L’affinité de liaison du 4-hydroxyméthyl ambrisentan au récepteur de l'endothéline humaine est 65 fois moindre que celle de l’ambrisentan. Par conséquent, aux concentrations observées dans le plasma (environ 4 % par rapport à l’ambrisentan parent), le 4-hydroxyméthyl ambrisentan ne devrait pas contribuer à l’activité pharmacologique de l’ambrisentan.
Des données in vitro indiquent que l’ambrisentan à une concentration de 300 μM entraîne moins de 50 % d’inhibition des isoenzymes UGT1A1, UGT1A6, UGT1A9, UGT2B7 (jusqu’à 30 %) ou des isoenzymes du cytochrome P450 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 et 3A4 (jusqu’à 25 %). In vitro, l’ambrisentan n’a pas d’effet inhibiteur sur les transporteurs humains à des concentrations cliniquement significatives, y compris P-gp, BCRP, MRP2, BSEP, OATP1B1, OATP1B3 et NTCP. En outre, les modèles in vitro sur hépatocytes de rats n’ont pas mis en évidence d’effet inducteur d’ambrisentan sur l’expression des protéines MRP2, P-gp ou BSEP. L’analyse globale des données in vitro suggère qu’ambrisentan, à des concentrations cliniquement significatives, (Cmax plasmatique jusqu'à 3,2 μM) ne devrait pas avoir d’effet sur UGT1A1, UGT1A6, UGT1A9, UGT2B7 ou les isoenzymes du cytochrome P450 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4 ou les protéines de transport BSEP, BCRP, P-gp, MRP2, OATP1B1/3 ou NTCP.
Les effets de l’ambrisentan administré à l'état d’équilibre (10 mg, une fois par jour) sur les propriétés pharmacocinétiques et pharmacodynamiques d’une seule dose de warfarine (25 mg), mesurés par le taux de prothrombine (TP) et le rapport normalisé international (INR), ont été étudiés chez 20 volontaires sains. L’ambrisentan n’a eu aucun effet cliniquement significatif sur la pharmacocinétique ou la pharmacodynamique de la warfarine. De même, la co-administration avec la warfarine n’a pas modifié les propriétés pharmacocinétiques de l’ambrisentan (voir rubrique 4.5).
L’effet de l'administration de sildénafil pendant 7 jours (20 mg, trois fois par jour) sur la pharmacocinétique d’une seule dose d’ambrisentan, et les effets de l’administration d’ambrisentan pendant 7 jours (10 mg, une fois par jour) sur la pharmacocinétique d’une seule dose de sildénafil ont été étudiés chez 19 volontaires sains. À l’exception d’une augmentation de 13 % de la Cmax de sildénafil après co-administration avec l’ambrisentan, il n'y a eu aucun changement dans les paramètres pharmacocinétiques du sildénafil, du N-desméthyl-sildénafil et de l’ambrisentan. Cette faible augmentation de la Cmax du sildénafil n’est pas considérée comme cliniquement significative (voir rubrique 4.5).
Les effets de l'ambrisentan, administré aux doses permettant d'atteindre l'état d'équilibre pharmacocinétique (10 mg, une fois par jour), sur les propriétés pharmacocinétiques d’une seule dose de tadalafil et les effets de l’administration aux doses permettant d'obtenir l'état d'équilibre pharmacocinétique de tadalafil (40 mg, une fois par jour), sur les propriétés pharmacocinétiques d’une seule dose d'ambrisentan, ont été étudiés chez 23 volontaires sains. L’ambrisentan n’a eu aucun effet cliniquement significatif sur la pharmacocinétique du tadalafil. De même, l'administration concomitante de tadalafil n’a pas eu d'effets sur les paramètres pharmacocinétiques de l’ambrisentan (voir rubrique 4.5).
Les effets des doses répétées de kétoconazole (400 mg, une fois par jour) sur la pharmacocinétique d’une seule dose de 10 mg d’ambrisentan ont été étudiés chez 16 volontaires sains. Les expositions systémiques de l'ambrisentan mesurées par l’aire sous la courbe des concentrations plasmatiques (ASC(0-inf)) et la Cmax ont été augmentées respectivement de 35 % et de 20 %. Cette modification de l'exposition systémique ne devrait pas avoir de retentissement cliniquement significatif et, par conséquent, l’ambrisentan peut être administré de façon concomitante avec le kétoconazole.
Les effets de doses répétées de ciclosporine A (100– 150 mg deux fois par jour) sur la pharmacocinétique à l'état d'équilibre de l'ambrisentan (5 mg une fois par jour), et les effets de doses répétées d'ambrisentan (5 mg une fois par jour) sur la pharmacocinétique à l'état d'équilibre de la ciclosporine A (100– 150 mg deux fois par jour) ont été étudiés chez des volontaires sains. La Cmax et l'ASC(0-τ) de l'ambrisentan ont augmenté (respectivement 48 % et 121 %) en présence de doses multiples de ciclosporine A. Sur la base de ces modifications, la dose d'ambrisentan doit être limitée à 5 mg une fois par jour en cas de co-administration avec la ciclosporine A (voir rubrique 4.2). Toutefois, l'administration de doses multiples d'ambrisentan n'a pas eu d'effet cliniquement significatif sur l'exposition à la ciclosporine A et aucun ajustement posologique de la ciclosporine A n'est justifié.
Les effets de l'administration en dose unique ou en doses répétées de rifampicine (600 mg une fois par jour) sur les paramètres pharmacocinétiques de l'ambrisentan (10 mg une fois par jour) à l'état d'équilibre ont été étudiés chez des volontaires sains. Après l'initiation du traitement par rifampicine, une augmentation transitoire de l'ASC(0– τ) (121 % et 116 % après l'administration de la première et de la seconde dose de rifampicine) a été observée, probablement due à une inhibition de l'OATP par la rifampicine. Toutefois, il n'a pas été observé de modification cliniquement significative de l'exposition systémique de l'ambrisentan au 8ème jour suivant l'administration de rifampicine en doses répétées. Les patients traités par ambrisentan doivent faire l'objet d'une étroite surveillance lorsqu'ils débutent un traitement par rifampicine (voir rubriques 4.4 et 4.5).
Les effets de doses répétées d’ambrisentan (10 mg) sur la pharmacocinétique d’une seule dose de digoxine ont été étudiés chez 15 volontaires sains. L'administration de doses répétées d'ambrisentan augmente légèrement l'ASC0-last et les concentrations résiduelles de la digoxine, la Cmax de la digoxine est augmentée de 29 %. L'augmentation de l’exposition à la digoxine en présence de doses multiples d'ambrisentan n'a pas été considérée cliniquement significative, et aucun ajustement de dose de la digoxine n’est nécessaire (voir rubrique 4.5).
Les effets de l'administration de l’ambrisentan pendant 12 jours (10 mg, une fois par jour) sur les paramètres pharmacocinétiques d’une dose unique d'un contraceptif oral associant de l'éthinylestradiol (35 μg) et de la noréthindrone (1 mg) ont été étudiés chez des femmes volontaires saines. La Cmax et l'ASC(0-∞) ont été légèrement diminuées pour l'éthinylestradiol (respectivement 8 % et 4 %) et légèrement augmentées pour la noréthindrone (respectivement 13 % et 14 %). Ces modifications d'expositions systémiques de l'éthinylestradiol ou de la noréthindrone sont considérées comme faibles et ne devraient pas avoir de retentissement cliniquement significatif (voir rubrique 4.5).
Elimination
L’ambrisentan et ses métabolites sont éliminés principalement dans la bile après le métabolisme hépatique et/ou extra-hépatique. Environ 22 % de la dose administrée sont retrouvés dans l’urine après administration orale, dont 3,3 % d’ambrisentan inchangé. La demi-vie d’élimination plasmatique chez l’Homme est comprise entre 13,6 et 16,5 heures.
Populations particulières
D’après les résultats d’une analyse pharmacocinétique de population réalisée sur des volontaires sains et des patients atteints d’hypertension artérielle pulmonaire, les propriétés pharmacocinétiques de l’ambrisentan n’ont pas été significativement influencées par le sexe ou l’âge (voir rubrique 4.2).
Insuffisance rénale
L'élimination rénale ou la clairance rénale (excrétion) de l’ambrisentan ne sont pas significatifs. Dans une analyse pharmacocinétique de population, la clairance de la créatinine était une covariable affectant la pharmacocinétique de l’ambrisentan de façon statistiquement significative. L’intensité de la diminution de la clairance est modeste (20 à 40 %) chez les patients souffrant d’insuffisance rénale modérée et cette diminution est, par conséquent, peu susceptible d’avoir une pertinence clinique quelconque. Toutefois, la prudence s’impose chez les patients présentant une insuffisance rénale sévère (voir rubrique 4.2).
Insuffisance hépatique
Les principales voies métaboliques de l’ambrisentan sont la glucurono-conjugaison et l’oxydation, avec élimination dans la bile ; par conséquent, l’insuffisance hépatique devrait augmenter l'exposition systémique (Cmax et ASC) de l’ambrisentan. Une analyse pharmacocinétique de population a montré que la clairance orale de l'ambrisentan diminuait en fonction de l'augmentation de la bilirubine. Toutefois, l’effet de la bilirubine apparait mineur : pour une élévation du taux de bilirubine de 4,5 mg/dL (par rapport à la valeur normale de 0,6 mg/dL), la clairance de l’ambrisentan serait environ 30 % moindre. La pharmacocinétique de l’ambrisentan n'a pas été étudiée chez les sujets présentant une insuffisance hépatique avec ou sans cirrhose. Par conséquent, un traitement par ambrisentan ne doit pas être instauré chez les patients souffrant d'insuffisance hépatique sévère ou présentant une augmentation des aminotransférases hépatiques cliniquement significative (plus de 3 fois la limite supérieure de la normale (> 3 x LSN) (voir rubriques 4.3 et 4.4).
5.3. Données de sécurité préclinique
L’ambrisentan n'a pas montré un effet inhibiteur du transport des acides biliaires ou une hépatotoxicité.
Chez le rongeur, lors de l’administration chronique d’ambrisentan à des niveaux d’exposition inférieurs aux doses thérapeutiques utilisées chez l’Homme, une inflammation et des modifications de l’épithélium de la cavité nasale ont été observées. Et, dans une moindre mesure, chez le chien, des réponses inflammatoires légères ont été observées lors de l’administration chronique d’ambrisentan à des doses élevées répétées correspondant à une exposition 20 fois plus importante que celle observée chez les patients.
Une hyperplasie osseuse des cornets ethmoïdaux a été observée dans la cavité nasale de rats traités par l'ambrisentan, à des niveaux d'exposition 3 fois supérieurs à l'ASC en clinique humaine. L’hyperplasie de l’os nasal n'a pas été observée avec l’ambrisentan chez les souris ou les chiens. Chez le rat, une hyperplasie osseuse des cornets ethmoïdaux est reconnue comme une conséquence de l'inflammation nasale, compte tenu de l'expérience acquise avec d'autres composés.
Utilisé en concentrations élevées sur des cellules de mammifères in vitro, l’ambrisentan s'est révélé clastogène. Les tests sur bactéries n'ont pas retrouvé d'effet mutagène et il n'a pas été observé de génotoxicité chez les rongeurs dans deux études menées in vivo.
Aucun potentiel carcinogène n'a été mis en évidence dans des études par voie orale de 2 ans menées chez le rat et la souris. Une légère augmentation de l'incidence des fibroadénomes mammaires, tumeurs bénignes, a été observée chez le rat mâle, uniquement à la dose la plus élevée. L'exposition systémique avec l’ambrisentan chez le rat mâle à cette dose, mesurée par l'aire sous la courbe (ASC) à l'état d'équilibre, était 6 fois celle obtenue à la dose clinique de 10 mg/jour.
Une atrophie tubulaire testiculaire, occasionnellement associée à une aspermie, a été observée dans des études de toxicité et de fertilité effectuées avec des doses orales répétées proches des doses utilisées en thérapeutique (sans marge de sécurité) sur des souris et des rats mâles. Les modifications au niveau testiculaire n'ont pas entièrement régressé au cours du suivi mené après arrêt du traitement. Cependant aucune modification n’a été observée au cours des études chez les chiens, jusqu'à la 39ème semaine à une exposition de 35 fois la dose utilisée chez l'Homme sur la base de l’ASC. Chez les rats mâles, aucun effet de l’ambrisentan sur la mobilité des spermatozoïdes n’a été mis en évidence, quelle que soit la dose testée (jusqu’à 300 mg/kg/jour). Une légère réduction (< 10 %) du pourcentage de spermatozoïdes morphologiquement normaux a été observée à la dose de 300 mg/kg/jour, mais pas à celle de 100 mg/kg/jour (> 9 fois l'imprégnation clinique obtenue à une dose de 10 mg/jour). L'effet de l'ambrisentan sur la fertilité masculine humaine n'est pas connu.
L’ambrisentan s'est révélé tératogène chez le rat et le lapin. Des anomalies de la mâchoire inférieure, de la langue et/ou du palais ont été observées à toutes les doses testées. De plus, l’étude chez le rat a montré une augmentation de l’incidence des défauts du septum interventriculaire et des troncs vasculaires, des anomalies de la thyroïde et du thymus, une ossification du basi-sphénoïde et la survenue de cas de position de l’artère ombilicale du côté gauche de la vessie au lieu du côté droit. La tératogénicité est associée à la classe des antagonistes des récepteurs aux endothélines.
L’administration d’ambrisentan à des rats femelles à partir de la fin de la gestation et pendant la lactation a provoqué des anomalies du comportement maternel, une réduction de la survie des portées et de la capacité reproductive de la descendance (avec l'observation de petits testicules à l'autopsie) lors d’une exposition de 3 fois l'ASC de la dose maximale recommandée chez l'Homme.
Chez des rats juvéniles, auxquels l’ambrisentan a été administré par voie orale une fois par jour aux jours 7 à 26, 36 ou 62 après la naissance, une diminution du poids du cerveau (-3 % à -8 %) sans modification morphologique ni neuro-comportementale a été constatée après observation de sons respiratoires, d’apnée et d’hypoxie. Ces effets sont apparus suite à des expositions correspondant à environ 1,8 à 7 fois l’exposition pédiatrique humaine de 10 mg (de 9 à 15 ans), sur la base de l’ASC. La pertinence clinique de ces résultats pour la population pédiatrique n’est pas totalement élucidée.
Cellulose microcristalline (E460), lactose monohydraté, croscarmellose sodique (E468), stéarate de magnésium (E572).
Pelliculage
Poly (alcool vinylique) (E1203), dioxyde de titane (E171), macrogol/polyéthylène glycol (E1521), talc (E553b), rouge Allura AC (E129), lécithine (de soja) (E322).
Pour les plaquettes blanches en PVC/PVDC/Aluminium
30 mois
Pour les plaquettes transparentes en PVC/PE/PVDC/Aluminium
30 mois
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
Plaquettes blanches en PVC/PVDC/Aluminium et plaquettes transparentes en PVC/PE/PVDC.
Boîtes de 10 ou 30 comprimés pelliculés sous plaquettes ou de 10 × 1 ou 30 × 1 comprimé pelliculé sous plaquettes prédécoupées pour délivrance à l’unité.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
EG LABO - LABORATOIREES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 959 8 7 : 10 comprimés sous plaquettes (PVC/PVDC/Aluminium).
· 34009 301 959 9 4 : 30 comprimés sous plaquettes (PVC/PVDC/Aluminium).
· 34009 301 960 0 7 : 10X1 comprimés sous plaquettes prédécoupées unitaires (PVC/PVDC/Aluminium).
· 34009 301 960 1 4 : 30X1 comprimés sous plaquettes prédécoupées unitaires (PVC/PVDC/Aluminium).
· 34009 301 960 2 1 : 10 comprimés sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 301 960 4 5 : 30 comprimés sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 301 960 5 2 : 10X1 comprimés sous plaquettes prédécoupées unitaires (PVC/PE/PVDC/Aluminium).
· 34009 301 960 6 9 : 30X1 comprimés sous plaquettes prédécoupées unitaires (PVC/PE/PVDC/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière réservée aux spécialistes et/ou aux services spécialisés en pneumologie, en cardiologie ou en médecine interne.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 04/12/2020
AMBRISENTAN EG 10 mg, comprimé pelliculé
Ambrisentan
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que AMBRISENTAN EG 10 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre AMBRISENTAN EG 10 mg, comprimé pelliculé ?
3. Comment prendre AMBRISENTAN EG 10 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver AMBRISENTAN EG 10 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE AMBRISENTAN EG 10 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Anti-hypertenseurs, autres anti-hypertenseurs - code ATC : C02KX02
AMBRISENTAN EG contient la substance active « ambrisentan ». Il appartient à une classe de médicaments appelés « autres anti-hypertenseurs » (utilisés pour traiter une pression artérielle élevée).
Il est utilisé pour traiter l’hypertension artérielle pulmonaire (HTAP) chez l’adulte. L’HTAP est définie par une élévation de la pression artérielle dans les vaisseaux sanguins (les artères pulmonaires) qui transportent le sang entre le cœur et les poumons. Chez les patients atteints d’HTAP, ces artères se rétrécissent, et le cœur doit fournir un effort supplémentaire pour pomper le sang. Ceci provoque une fatigue, des vertiges et des essoufflements.
AMBRISENTAN EG élargit les artères pulmonaires et facilite ainsi le pompage du sang par le cœur. Ceci fait diminuer la pression sanguine et réduit les symptômes.
AMBRISENTAN EG peut aussi être utilisé en association avec d’autres médicaments pour traiter l’HTAP.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE AMBRISENTAN EG 10 mg, comprimé pelliculé ?
Ne prenez jamais AMBRISENTAN EG 10 mg, comprimé pelliculé :
· si vous êtes allergique à l’ambrisentan, à l’arachide, au soja ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si vous êtes enceinte, si vous prévoyez d’être enceinte ou si vous pourriez le devenir parce que vous n’utilisez pas une méthode de contraception fiable. Veuillez lire les informations indiquées au paragraphe « Grossesse » ;
· si vous allaitez. Lisez les informations du paragraphe « Allaitement » ;
· si vous souffrez d’une maladie du foie. Parlez-en à votre médecin ; il décidera si vous pouvez prendre ce médicament ;
· si vous présentez une fibrose pulmonaire idiopathique (maladie du tissu pulmonaire dont la cause n'est pas connue).
Avertissements et précautions
Contactez votre médecin avant de prendre ce médicament si vous avez :
· des problèmes au niveau du foie ;
· une anémie (diminution du nombre de globule rouges dans le sang) ;
· un gonflement au niveau des mains, des chevilles ou des pieds causé par une accumulation de liquide (œdème périphérique) ;
· une maladie des poumons caractérisée par l’obstruction des veines pulmonaires (maladie pulmonaire veino-occlusive).
→ Votre médecin décidera si vous pouvez prendre AMBRISENTAN EG.
Vous devrez effectuer régulièrement des analyses de sang
Avant un traitement par AMBRISENTAN EG, puis à intervalles réguliers lorsque vous prendrez ce médicament, votre médecin vous prescrira des analyses de sang afin de vérifier :
· si vous souffrez d’anémie ;
· si votre foie fonctionne normalement.
→ Il est important de faire régulièrement ces analyses de sang pendant toute la durée du traitement par AMBRISENTAN EG.
Signes pouvant indiquer que votre foie ne fonctionne pas correctement :
· perte d’appétit ;
· envie de vomir (nausées) ;
· vomissements ;
· fièvre ;
· douleur d'estomac (maux de ventre) ;
· coloration jaune de la peau ou du blanc des yeux (jaunisse) ;
· urines foncées ;
· démangeaisons de la peau.
Si vous présentez l’un de ces signes :
→ Informez-en immédiatement votre médecin.
Enfants et adolescents
AMBRISENTAN EG n’est pas recommandé chez les enfants et les adolescents âgés de moins de 18 ans, sa sécurité et son efficacité n’étant pas connues dans ce groupe d'âge.
Autres médicaments et AMBRISENTAN EG 10 mg, comprimé pelliculé
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Votre médecin peut être amené à modifier votre dose d’AMBRISENTAN EG si vous commencez un traitement par ciclosporine A (un médicament utilisé après une transplantation d'organe ou dans le traitement du psoriasis).
Si vous prenez de la rifampicine (un antibiotique utilisé dans le traitement d'infections graves), vous serez attentivement suivi par votre médecin lors de l’instauration de votre traitement par AMBRISENTAN EG.
Si vous prenez d’autres médicaments utilisés dans le traitement de l'hypertension artérielle pulmonaire (par exemple, iloprost, époprosténol, sildénafil), une surveillance par votre médecin pourra être nécessaire.
→ Informez votre médecin ou votre pharmacien si vous prenez l’un de ces médicaments.
AMBRISENTAN EG 10 mg, comprimé pelliculé avec des aliments
Sans objet.
Grossesse, allaitement et fertilité
Grossesse
AMBRISENTAN EG peut présenter des risques pour le bébé conçu avant, pendant ou peu après le traitement.
→ Si vous êtes en âge d’être enceinte, vous devez utiliser une méthode de contraception fiable pendant le traitement par AMBRISENTAN EG. Parlez-en à votre médecin.
→ Ne prenez pas AMBRISENTAN EG si vous êtes enceinte ou si vous envisagez une grossesse.
→ Si vous découvrez que vous êtes enceinte ou si vous pensez être enceinte pendant votre traitement par AMBRISENTAN EG, consultez immédiatement votre médecin.
Si vous êtes une femme en âge d'être enceinte, votre médecin vous demandera de réaliser un test de grossesse avant de prendre AMBRISENTAN EG, puis à intervalles réguliers tout au long de votre traitement par ce médicament.
Allaitement
Le passage d’AMBRISENTAN EG dans le lait maternel n'est pas connu.
→ Vous ne devez pas allaiter pendant que vous prenez AMBRISENTAN EG. Parlez-en à votre médecin.
Fertilité
Si vous êtes un homme et que vous prenez AMBRISENTAN EG, il est possible que ce médicament diminue le nombre de vos spermatozoïdes. Si vous avez des questions ou des inquiétudes à ce sujet, parlez-en à votre médecin.
Conduite de véhicules et utilisation de machines
AMBRISENTAN EG peut provoquer des effets indésirables tels qu'une baisse de la pression artérielle, des sensations de vertige ou de la fatigue (voir rubrique 4), qui peuvent altérer votre aptitude à conduire des véhicules ou à utiliser des machines. Les symptômes de votre maladie peuvent également compromettre votre aptitude à conduire des véhicules ou à utiliser des machines.
→ Ne conduisez pas et n’utilisez pas de machines si vous ne vous sentez pas bien.
AMBRISENTAN EG 10 mg, comprimé pelliculé contient du lactose, de la lécithine (de soja), du rouge Allura AC (E129) et du sodium
AMBRISENTAN EG, comprimé pelliculé contient du lactose. Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
AMBRISENTAN EG, comprimé pelliculé contient de la lécithine de soja. Si vous êtes allergique à l’arachide ou au soja, ne pas utiliser ce médicament (voir rubrique 2 « Ne prenez jamais AMBRISENTAN EG 10 mg, comprimé pelliculé »).
AMBRISENTAN EG, comprimé pelliculé contient un agent colorant appelé rouge Allura AC (E129), qui peut provoquer des réactions allergiques (voir rubrique 4).
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE AMBRISENTAN EG 10 mg, comprimé pelliculé ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Quelle dose d’AMBRISENTAN EG 10 mg, comprimé pelliculé faut-il prendre
La dose habituelle d’AMBRISENTAN EG est de un comprimé pelliculé de 5 mg, une fois par jour. Votre médecin peut décider d'augmenter votre dose à 10 mg, une fois par jour.
Si vous prenez de la ciclosporine A, ne prenez pas plus d'un comprimé pelliculé de 5 mg d’AMBRISENTAN EG une fois par jour.
Comment prendre AMBRISENTAN EG 10 mg, comprimé pelliculé
Il est préférable de prendre votre comprimé chaque jour à la même heure. Avalez le comprimé entier, avec un grand verre d’eau, sans le couper, le croquer ni le mâcher. Vous pouvez prendre AMBRISENTAN EG pendant ou en dehors des repas.
Si vous avez pris plus d’AMBRISENTAN EG 10 mg, comprimé pelliculé que vous n’auriez dû
Si vous avez pris trop de comprimés, vous serez davantage susceptible d’avoir des effets indésirables tels que des maux de tête, des bouffées de chaleur, des sensations de vertige, des nausées (envie de vomir), ou une pression artérielle basse pouvant entraîner des étourdissements :
→ Demandez conseil à votre médecin ou à votre pharmacien si vous avez pris plus de comprimés que ce qui vous a été prescrit.
Si vous oubliez de prendre AMBRISENTAN EG 10 mg, comprimé pelliculé
Si vous avez oublié de prendre une dose d’AMBRISENTAN EG, prenez un comprimé dès que vous vous en apercevez puis continuez normalement le traitement.
→ Ne prenez pas deux doses en même temps pour compenser la dose que vous avez oublié de prendre.
N’arrêtez pas de prendre AMBRISENTAN EG 10 mg, comprimé pelliculé sans l’avis de votre médecin
AMBRISENTAN EG est un traitement que vous devrez continuer à prendre pour contrôler votre HTAP.
→ N’arrêtez pas de prendre AMBRISENTAN EG sans en avoir discuté avec votre médecin et obtenu son accord.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Situations nécessitant votre vigilance, ainsi que celle de votre médecin :
Réactions allergiques
Il s’agit d’un effet indésirable fréquent pouvant concerner jusqu’à une personne sur 10. Vous pouvez présenter une éruption cutanée, des démangeaisons ou un gonflement (habituellement au niveau du visage, des lèvres, de la langue ou de la gorge), susceptibles d'entraîner des difficultés pour respirer ou avaler.
Gonflement (œdème), plus particulièrement au niveau des chevilles et des pieds
Il s’agit d’un effet indésirable très fréquent qui peut concerner plus d’une personne sur 10.
Insuffisance cardiaque
Elle est due au fait que le cœur ne pompe pas suffisamment de sang, ce qui entraîne un essoufflement, une intense fatigue et un gonflement au niveau des chevilles et des jambes. Il s’agit d’un effet indésirable fréquent, pouvant concerner jusqu’à une personne sur 10.
Anémie (diminution du nombre de globules rouges dans le sang)
Il s’agit d’un trouble sanguin pouvant entraîner une fatigue, une faiblesse, un essoufflement et généralement une sensation de malaise, et nécessitant parfois une transfusion sanguine. C’est un effet indésirable très fréquent qui peut concerner plus d’une personne sur 10.
Hypotension (pression artérielle basse)
Ceci peut entraîner des étourdissements. Il s’agit d’un effet indésirable fréquent qui peut concerner jusqu’à une personne sur 10.
→ Informez immédiatement votre médecin si vous observez ces effets indésirables ou s'ils surviennent soudainement après la prise d’AMBRISENTAN EG.
Il est important que vous effectuiez régulièrement des analyses de sang afin de rechercher une éventuelle anémie et de vérifier que votre foie fonctionne correctement. Assurez-vous également d'avoir pris connaissance des informations mentionnées à la rubrique 2, aux paragraphes « Vous devrez effectuer régulièrement des analyses de sang » et « Signes pouvant indiquer que votre foie ne fonctionne pas correctement ».
Les autres effets indésirables incluent :
Effets indésirables très fréquents :
· maux de tête ;
· sensations vertigineuses ;
· palpitations (accélération ou irrégularité des battements cardiaques) ;
· aggravation de l'essoufflement peu de temps après le début du traitement par AMBRISENTAN EG ;
· écoulement ou sensation de nez bouché, congestion ou douleur dans les sinus ;
· nausées ;
· diarrhée ;
· sensation de fatigue.
En association avec le tadalafil (autre médicament de l’HTAP)
Peuvent également survenir en plus de ce qui est décrit ci-dessus :
· bouffées de chaleur (rougeurs de la peau) ;
· vomissements ;
· éruption cutanée ;
· douleur ou gêne dans la poitrine.
Effets indésirables fréquents :
· vision floue ou autres altérations de la vision ;
· évanouissements ;
· résultats anormaux des analyses de sang destinées à contrôler la fonction hépatique ;
· écoulement du nez ;
· constipation ;
· douleur d'estomac (maux de ventre) ;
· douleur ou gêne dans la poitrine ;
· bouffées de chaleur (rougeurs de la peau) ;
· vomissements ;
· sensation de faiblesse générale ;
· saignement de nez ;
· éruption cutanée.
En association avec le tadalafil
Peuvent également survenir en plus de ce qui est décrit ci-dessus à l’exception des résultats anormaux des analyses de sang destinées à contrôler la fonction hépatique :
· bourdonnements d'oreilles (acouphènes) seulement lorsqu’AMBRISENTAN EG est associé avec le tadalafil.
Effets indésirables peu fréquents :
· atteinte du foie ;
· inflammation du foie provoquée par le propre système de défense de l’organisme (hépatite auto-immune).
En association avec le tadalafil
· perte soudaine de l’audition.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER AMBRISENTAN EG 10 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et la plaquette après EXP.
La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation concernant la température ; à conserver dans l’emballage d’origine, à l’abri de la lumière.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient AMBRISENTAN EG 10 mg, comprimé pelliculé
· La substance active est :
Ambrisentan..................................................................................................................... 10 mg
Pour un comprimé pelliculé.
· Les autres composants sont : cellulose microcristalline (E460), lactose monohydraté, croscarmellose sodique (E468), stéarate de magnésium (E572), ply (alcool vinylique), dioxyde de titane (E171), macrogol/polyéthylène glycol (E1521), talc (E553b), rouge Allura AC (E129) et lécithine (de soja) (E322).
Qu’est-ce que AMBRISENTAN EG 10 mg, comprimé pelliculé et contenu de l’emballage extérieur
AMBRISENTAN EG 10 mg, comprimé pelliculé se présente sous forme d’un comprimé pelliculé biconvexe, de forme oblongue, rose, comportant la mention « 10 » gravée sur une face, l’autre face étant lisse, dont la longueur nominale est d’environ 11,1 mm et dont la largeur nominale est d’environ 5,6 mm.
AMBRISENTAN EG 10 mg, comprimé pelliculé est disponible en boîtes de 10 ou 30 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
EG LABO - LABORATOIREES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
Exploitant de l’autorisation de mise sur le marché
EG LABO - LABORATOIREES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
18TH KM MARATHONOS AVE,
153 51 PALLINI ATTIKI
GRECE
ou
DELORBIS PHARMACEUTICALS LTD,
L7 ATHINON STR., ERGATES INDUSTRIAL AREA,
2643 ERGATES, LEFKOSIA,
CHYPRE
ou
STADA Arzneimittel AG
Stadastrasse 2-18
61118 Bad Vilbel
allemagne
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).