Dernière mise à jour le 03/08/2026
COLISTIMETHATE SODIQUE AMDIPHARM 1 MUI, poudre et solvant pour inhalation par nébuliseur
Indications thérapeutiques
Classe pharmacothérapeutique: Autres antibactériens – polymyxines, code ATC: J01XB01
Ce médicament appartient à une famille d’un antibiotique de la famille des polymixines. Il contient du colistiméthate sodique qui est un précurseur de la colistine.
L’administration de ce médicament par inhalation doit être réservée au traitement des infections pulmonaires chroniques chez les patients atteints de mucoviscidose.
COLISTIMETHATE SODIQUE AMDIPHARM est utilisé quand ces infections sont causées par une bactérie spécifique appelée Pseudomonas aeruginosa.
Présentations
> 1 flacon(s) de poudre en verre - 1 ampoule(s) de solvant en verre de 3 ml
Code CIP : 365 058-9 ou 34009 365 058 9 6
Déclaration de commercialisation : 02/01/2007
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 11,33 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 12,35 €
- Taux de remboursement :65%
> 5 flacon(s) de poudre en verre - 5 ampoule(s) de solvant en verre de 3 ml
Code CIP : 34009 301 888 2 8
Déclaration de commercialisation : 01/07/2021
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 56,03 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 57,05 €
- Taux de remboursement :65 %
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 04/12/2019 | Inscription (CT) | Le service médical rendu par la spécialité COLIMYCINE 1 MUI, poudre et solvant pour inhalation par nébuliseur est important dans l’indication de l’AMM. |
| Important | Avis du 06/12/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par les spécialités COLIMYCINE (forme injectable et inhalée) reste important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 04/12/2019 | Inscription (CT) | La spécialité COLIMYCINE 1 MUI, poudre et solvant pour inhalation par nébuliseur n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres présentations déjà inscrites. |
Autres informations
- Titulaire de l'autorisation : AMDIPHARM Ltd
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière semestrielle
- renouvellement non restreint
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 523 184 1
ANSM - Mis à jour le : 18/03/2026
COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Colistiméthate sodique................................................................................................ 1 000 000 U.I.
Quantité correspondant en colistiméthate sodique à................................................................. 80 mg
Pour un flacon de poudre.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour inhalation par nébuliseur.
4.1. Indications thérapeutiques
COLISTIMETHATE SODIQUE AMDIPHARM est indiqué chez l’adulte et l’enfant dans la prise en charge des infections pulmonaires chroniques dues à Pseudomonas aeruginosa chez les patients atteints de mucoviscidose (voir rubrique 5.1).
Il convient de tenir compte des recommandations officielles concernant l'utilisation appropriée des antibactériens.
4.2. Posologie et mode d'administration
Il est recommandé que le colistiméthate sodique (CMS) soit administré sous une surveillance médicale qui requiert une expérience appropriée pour son utilisation.
Posologie
La posologie peut être adaptée en fonction de la sévérité de l’infection et de la réponse clinique.
Posologies recommandées :
Administration par inhalation
Adultes, adolescents et enfants de ≥ 2 ans
1-2 M U.I. deux à trois fois par jour (maximum 6 M U.I. / jour)
Enfants < 2 ans
0,5-1 M U.I. deux fois par jour (max 2 M U.I. / jour)
Les recommandations cliniques en rapport avec les schémas thérapeutiques, y compris sur la durée de traitement, la périodicité et l’administration concomitante à d'autres antibiotiques doivent être respectées.
Sujet âgé
Aucune adaptation posologique n’est jugée nécessaire.
Insuffisance rénale
Aucune adaptation posologique n’est jugée nécessaire, toutefois la prudence est recommandée chez les insuffisants rénaux (voir rubriques 4.4 et 5.2).
Insuffisance hépatique
Aucune adaptation posologique n’est jugée nécessaire.
Mode d'administration
Pour une utilisation par inhalation
La solution pour nébulisation est préparée extemporanément, en solubilisant la poudre de colistiméthate sodique à 1 M U.I. dans son solvant : 3 ml d'une solution isotonique de chlorure de sodium.
Une légère formation de mousse peut être observée.
COLISTIMETHATE SODIQUE AMDIPHARM est destinée à être administré par voie inhalée par nébulisation à l'aide d'un nébuliseur et d'un compresseur (système de nébulisation pneumatique) adaptés.
La solution est versée dans le nébuliseur et administrée à l'aide du nébuliseur adapté (PARI LC STAR), couplé au compresseur pneumatique (PARI TURBO BOY N). Le compresseur possède les caractéristiques suivantes: débit de service de 5 l/min avec buse de 0.48 mm, pression statique de 3.2 bar et pression de service de 1.6 bar (avec buse de 0.48 mm).
Tout appareil ayant des caractéristiques similaires peut être utilisé à condition que la granulométrie de la solution nébulisée de colistiméthate sodique ait été validée.
Pour l'utilisation et l'entretien du matériel, suivre les instructions du fabricant.
Dans une solution aqueuse, le colistiméthate sodique est hydrolysé en colistine, la substance active. Pour les précautions particulières concernant le dispositif et la manipulation des solutions reconstituées, voir rubrique 6.6.
Si d'autres traitements sont en cours, ils doivent être pris dans l'ordre recommandé par le médecin.
Tableau de conversion des posologies :
Dans l'Union Européenne (UE), la dose de colistiméthate sodique (CMS) doit être prescrite et administrée seulement en unités internationales (UI). L'étiquetage du produit indique le nombre d’UI par flacon.
Des confusions et des erreurs médicamenteuses ont eu lieu en raison des différentes expressions de la dose en termes d’activité. Aux Etats-Unis, et dans d’autres parties du monde, la dose exprimée est en milligrammes d’activité de colistine base (mg ACB).
Le tableau de conversion suivant est mentionné pour information et les valeurs doivent être seulement considérées comme indicatives et approximatives.
Tableau de conversion en CMS
|
Activité |
≈ masse de CMS (mg)* |
|
|
UI |
≈ mg ACB |
|
|
12 500 |
0.4 |
1 |
|
150 000 |
5 |
12 |
|
1 000 000 |
34 |
80 |
|
4 500 000 |
150 |
360 |
|
9 000 000 |
300 |
720 |
*Activité indicative de la substance active = 12 500 UI/mg
· Hypersensibilité à la substance active, à la colistine base, aux autres antibiotiques de la famille des polymyxines ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Ce médicament ne doit généralement pas être utilisé en cas :
· d'intolérance antérieure aux aérosols de colistine, notamment en cas de bronchospasme résistant à l'administration préalable de béta2 mimétique,
· d'association avec les aminosides (voie parentérale) (voir rubrique 4.5).
Néphrotoxicité
La colistine est éliminée exclusivement par voie rénale et est néphrotoxique. Elle n'est pas dialysable par hémodialyse.
Bien que les concentrations plasmatiques de colistine obtenues après administration inhalée soient faibles chez les sujets aux fonctions rénales normales, ce médicament est à utiliser avec précaution chez les patients en insuffisance rénale dialysés et non dialysés, et chez les très jeunes enfants au regard de l’immaturité de la fonction rénale, en évaluant le bénéfice/risque du traitement pour chaque patient.
Chez ces patients, il convient d'utiliser la dose efficace la plus faible, de surveiller l'apparition d'éventuelles paresthésies péribuccales et de contrôler les fonctions rénales chez les patients non dialysés.
En cas de besoin, il peut être utile de mesurer la concentration plasmatique de colistine afin de mieux adapter la posologie (voir rubrique 5.2).
En cas de traitement prolongé ou de traitement concomitant par des médicaments néphrotoxiques, les mêmes surveillances sont à mettre en œuvre (voir rubrique 4.5).
Bronchospasme
L'inhalation de ce médicament est susceptible de provoquer un bronchospasme ; des cas ont été rapportés avec la colistine inhalée ; le retentissement est d'autant plus important que le VEMS de base est abaissé. La première dose de ce médicament doit être administrée sous surveillance après désencombrement bronchique, avec utilisation d'un bronchodilatateur avant la nébulisation si cela fait partie du traitement habituel du patient.
Le volume expiratoire maximum-seconde (VEMS) sera mesuré avant et dans les 30 minutes après la nébulisation. En cas d'apparition d'un bronchospasme induit par le traitement chez un patient ne recevant pas de bronchodilatateur, répéter le test à un autre moment en utilisant un bronchodilatateur. L'apparition d'un bronchospasme malgré l'administration préalable d'un bronchodilatateur peut être le signe d'une réaction allergique.
Si une réaction allergique est suspectée, le traitement par ce médicament doit être interrompu. Tout bronchospasme doit faire l'objet d'un traitement médical approprié.
Hémoptysie
L'inhalation de solutions nébulisées est susceptible d'induire une toux réflexe. L'utilisation de ce médicament inhalé chez les patients ayant présenté une hémoptysie sévère récente ne doit être envisagée que si les bénéfices du traitement sont plus importants que les risques de déclencher une nouvelle hémorragie.
Hypersensibilité
En cas de réaction allergique, le traitement par colistiméthate sodique doit être interrompu et des mesures adaptées doivent être mises en place.
Myasthénie
Le colistiméthate sodique est connu pour réduire la libération présynaptique de l'acétylcholine à la jonction neuro-musculaire et doit être utilisé chez les patients atteints de myasthénie avec la plus grande prudence et seulement si cette prescription est absolument nécessaire.
Porphyrie
Le colistiméthate sodique doit être utilisé avec une extrême prudence chez les patients présentant une porphyrie.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L'utilisation concomitante de colistiméthate sodique par voie intraveineuse avec d'autres médicaments potentiellement néphrotoxiques ou neurotoxiques doit être initiée avec beaucoup de prudence.
Il faut être prudent lors de l’utilisation concomitante avec d’autres formulations à base de colistiméthate sodique étant donné que l’expérience est limitée et qu’une toxicité cumulative est possible.
Aucune étude d'interaction in vivo n'a été réalisée. Le mécanisme de transformation du colistiméthate sodique en substance active, la colistine, n’est pas caractérisé. Le mécanisme de la clairance de la colistine, y compris au niveau rénal, est également inconnu. Le colistiméthate sodique ou la colistine n'ont induit aucune activité des enzymes P 450 (CYP) testées (CYP1A2, 2B6, 2C8, 2C9, 2C19 et 3A4/5) dans les études in vitro menées sur des hépatocytes humains.
Les interactions médicamenteuses potentielles doivent être prises en compte lorsque COLISTIMETHATE SODIQUE AMDIPHARM est co-administré avec des médicaments connus pour inhiber ou activer le métabolisme enzymatique ou des médicaments connus pour être des substrats pour les transporteurs rénaux.
En raison des effets de la colistine sur la libération d’acétylcholine, les myorelaxants non dépolarisants doivent être utilisés avec prudence chez les patients recevant du colistiméthate sodique car leurs effets pourraient être prolongés (voir rubrique 4.4).
Un traitement concomitant de colistiméthate sodique avec les macrolides tels que l'azithromycine et la clarithromycine, ou les fluoroquinolones comme la norfloxacine et la ciprofloxacine, doit être envisagé avec prudence chez les patients présentant une myasthénie (voir rubrique 4.4).
Médicaments néphrotoxiques
L’utilisation conjointe de médicaments ayant une toxicité rénale propre augmente le risque de néphrotoxicité. Si une telle association est nécessaire, il faut renforcer la surveillance biologique rénale.
Les médicaments concernés sont représentés notamment par les produits de contraste iodés, les aminosides, les organoplatines, le méthotrexate à fortes doses, certains antiviraux (tels les « ciclovirs », le foscarnet), la pentamidine, la ciclosporine ou le tacrolimus.
Association faisant l’objet de précautions d'emploi
+ Curares
Potentialisation des curares lorsque l'antibiotique est administré par voie parentérale avant, pendant ou après l'agent curarisant. Surveiller le degré de curarisation en fin d'anesthésie.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données animales ne mettent pas en évidence d'effet malformatif du colistiméthate sodique lorsqu'elle est administrée par voie générale (IV ou IM).
Bien que les données cliniques soient insuffisantes, compte tenu du bénéfice thérapeutique et du très faible passage systémique, l'utilisation du colistiméthate sodique par voie inhalée est envisageable au cours de la grossesse.
Allaitement
Compte tenu de l'immaturité digestive du nouveau-né et du risque de néphrotoxicité, la prescription de ce médicament par voie orale ou parentérale n'est pas recommandée en cas d'allaitement. Après administration par voie inhalée, les concentrations sanguines de colistine chez l'adulte traité sont 100 fois moindres que celles mesurées après administration parentérale de colistine.
Fertilité
Sans objet.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Après administration I.V. de colistiméthate sodique, une neurotoxicité, effet dose-dépendant, caractérisée par des étourdissements, une confusion ou des troubles visuels, a été rapportée. L'administration par voie inhalée génère en principe un faible passage systémique du produit. Si ces effets surviennent, il convient de ne pas conduire de véhicule ou d'utiliser des machines.
Les fréquences sont définies comme suit :
· Très fréquent: supérieur à 10%,
· Fréquent: 1% à 10%,
· Occasionnel: 0,1% à 1%,
· Rare: 0,01% à 0,1%,
· Très rare: inférieur à 0,01%.
Système respiratoire
· Fréquents : baisse du VEMS de plus de 10%, associée ou non à des signes cliniques.
Bronchospasme, dyspnée et oppression thoracique observés surtout chez les patients ayant un VEMS de base abaissée. Augmentation de la toux et augmentation des expectorations comme au cours de toute exposition à un aérosol.
· Occasionnels : pharyngites, altération de la voix.
· Rares : hémoptysie.
Réactions d'hypersensibilité
· Rares : de type respiratoire, bronchospasme ; de type cutané: rash; voire réaction anaphylactique plus sévère.
Effets systémiques
En cas d'insuffisance rénale ou d'administration à doses très élevées, la survenue des effets indésirables systémiques ne peut être exclue :
· insuffisance rénale,
· troubles neuropsychiques: paresthésies péribuccales et des extrémités, désorientation temporospatiale, syndrome confusionnel. En cas d'anesthésie récente ou association avec des agents curarisants, risque de bloc neuromusculaire et notamment de paralysie respiratoire.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Symptômes
Le surdosage peut provoquer des apnées, une faiblesse musculaire et une insuffisance rénale. Il n'existe pas d'antidote spécifique.
Prise en charge
La prise en charge du surdosage repose sur le traitement symptomatique et des mesures prévues pour augmenter l'élimination du colistiméthate sodique tel que la diurèse osmotique à l'aide de mannitol. La colistine n'est pas dialysable.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique: Autres antibactériens – polymyxines, code ATC: J01XB01
La colistine est un antibiotique polypeptidique de la famille des polymyxines, du groupe des polymyxines E.
SPECTRE D'ACTIVITE ANTI-BACTERIENNE
Compte-tenu des concentrations bronchiques obtenues après inhalation, le spectre d’activité antibactérienne correspondant à cette voie d’administration et pour un traitement à visée locale est mentionné ci-après.
La prévalence de la résistance acquise peut varier en fonction de la géographie et du temps pour certaines espèces. Il est donc utile de disposer d’informations sur la prévalence de la résistance locale, surtout pour le traitement d’infections sévères. Ces données ne peuvent apporter qu’une orientation sur les probabilités de la sensibilité d’une souche bactérienne à cet antibiotique.
Lorsque la variabilité de la prévalence de la résistance en France est connue pour une espèce bactérienne, elle est indiquée dans le tableau ci-dessous.
|
Catégories |
Fréquence de résistance acquise en France (> 10%) (valeurs extrêmes) |
|
ESPÈCES SENSIBLES |
|
|
Aérobies à Gram négatif |
|
|
Alcaligenes xylosoxidans |
|
|
Haemophilus influenzae |
|
|
Pseudomonas aeruginosa* |
|
|
Stenotrophomonas maltophilia |
|
|
ESPÈCES RÉSISTANTES |
|
|
Aérobies à Gram positif |
|
|
Cocci et bacilles |
|
|
Aérobies à Gram négatif |
|
|
Burkholderia cepacia |
|
|
Anaérobies |
|
|
Cocci et bacilles |
* Efficacité clinique démontrée pour les souches sensibles dans les indications cliniques approuvées.
5.2. Propriétés pharmacocinétiques
Biotransformation
Dans l'organisme, la colistiméthate sodique s'hydrolyse pour une part en libérant de la colistine; par ailleurs, colistiméthate et colistine sont l'un et l'autre métabolisés en produits inactifs.
Elimination
La voie principale d'élimination du colistiméthate et des différents métabolites formés est urinaire tandis que l'élimination de la colistine paraît essentiellement de nature métabolique.
Cependant, les concentrations plasmatiques sont suffisamment faibles pour éviter une accumulation importante chez l'insuffisant rénal et justifier une réduction de la posologie par nébulisation.
5.3. Données de sécurité préclinique
Aucun essai pré-clinique n'a été effectué avec la colistine administrée par voie inhalée.
Solvant : chlorure de sodium, eau pour préparations injectables.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
3 ans.
L’hydrolyse du colistiméthate sodique est significativement augmentée lorsqu’il est reconstitué et dilué en-dessous de sa concentration micellaire critique d’environ 80 000 UI par ml.
Les solutions en-dessous de cette concentration doivent être utilisées immédiatement.
La stabilité chimique et physique en cours d’utilisation de la solution reconstituée dans le flacon d’origine, avec une concentration ≥ 80 000 UI/ml, a été démontrée pendant 24 heures à 2-8°C.
D’un point de vue microbiologique, sauf si la méthode d’ouverture / reconstitution / dilution exclut le risque de contamination microbienne, le produit doit être utilisé immédiatement.
Si la solution n’est pas utilisée immédiatement, les durées et conditions de conservation sont de la responsabilité de l’utilisateur.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas + 25°C.
Pour les conditions de conservation du médicament après reconstitution, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre en flacon (verre incolore de type I) de 10 ml avec bouchon (chlorobutyl) gris.
3 ml de solvant en ampoule (verre incolore de type I).
Boîte de 1, 5, 15, 30, 60 flacons de poudre +1, 5, 15, 30, 60 ampoules de solvant.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Reconstituer le contenu du flacon avec de l’eau pour préparations injectables afin de produire une solution hypotonique ou avec un mélange 50 :50 d’eau pour préparations injectables et de chlorure de sodium à 0,9 % pour produire une solution isotonique, ou avec l’ampoule de solvant contenant 3 ml de solution de chlorure de sodium à 0,9 % pour produire une solution hypertonique.
Le volume de reconstitution doit être selon les instructions pour l’utilisation du nébuliseur, et n’est normalement pas plus de 4 ml.
Au cours de la reconstitution, mélanger doucement pour éviter de faire mousser.
La solution reconstituée est une préparation aqueuse stérile, apyrogène, pour usage unique. Cette solution ne contenant pas de conservateur, l'intégralité du contenu d'un flacon doit être utilisée immédiatement après ouverture. Les flacons ouverts ne doivent jamais être conservés pour être réutilisés et toute solution restante doit être éliminée.
Veuillez-vous conformer aux instructions du fabricant sur l’utilisation appropriée du nébuliseur à utiliser avec COLISTIMETHATE SODIQUE AMDIPHARM, poudre et solvant pour solution pour inhalation par nébuliseur.
Ne pas dissoudre COLISTIMETHATE SODIQUE AMDIPHARM avant le moment de l’utilisation (voir étape 5).
|
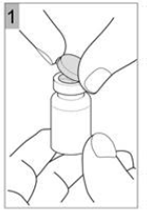
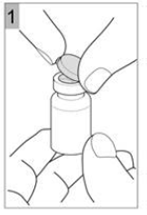
Etape 1 Prenez un flacon de COLISTIMETHATE SODIQUE AMDIPHARM et tapotez doucement le flacon en verre de telle sorte que la poudre se dépose au fond. Cela permet de vous assurer d'obtenir la dose appropriée de médicament. Ouvrir le flacon en soulevant le capuchon en plastique du dessus (Figure 1). |
|
|
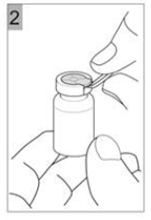
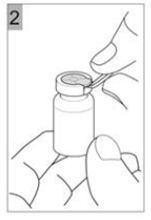
Etape 2 Tirer le capuchon en plastique avec la bague métallique (Figure 2). |
|
|
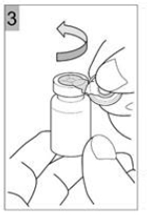
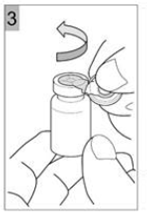
Etape 3 Déchirer le capuchon en plastique avec un mouvement de torsion dans le sens de la flèche. Une bonne exécution vous permettra de déchirer l'ouverture de l'anneau de métal. Déchirer l'anneau métallique. Retirer l'anneau métallique du flacon (Figure 3). Jetez la bague et le capuchon. |
|
|
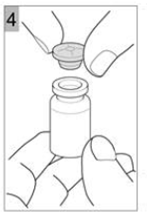
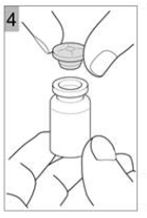
Etape 4 Retirez délicatement le bouchon (Figure 4). |
|
|
Etape 5 Ajouter 3 mL de solution isotonique stérile de chlorure de sodium dans le flacon. Afin d’éviter de faire mousser, agiter doucement le flacon jusqu’à ce que la poudre soit dissoute. N’utilisez pas COLISTIMETHATE SODIQUE AMDIPHARM si vous remarquez des particules visibles dans la solution après dissolution. |
|
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
AMDIPHARM LIMITED
NORTHWOOD CRESCENT
NORTHWOOD
DUBLIN 9 D09 V504
IRLANDE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 365 058 9 6 : poudre en flacon (verre) + 3 ml de solvant en ampoule (verre), boite de 1.
· 34009 301 888 2 8 : poudre en flacon (verre) + 3 ml de solvant en ampoule (verre), boite de 5.
· 34009 380 467 3 1 : poudre en flacon (verre) + 3 ml de solvant en ampoule (verre), boite de 15.
· 34009 380 469 6 0 : poudre en flacon (verre) + 3 ml de solvant en ampoule (verre), boite de 30.
· 34009 380 470 4 2 : poudre en flacon (verre) + 3 ml de solvant en ampoule (verre), boite de 60.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I.
Médicament soumis à prescription initiale hospitalière de 6 mois. Renouvellement non restreint.
ANSM - Mis à jour le : 18/03/2026
COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur
Colistiméthate sodique
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur ?
3. Comment prendre COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique: Autres antibactériens – polymyxines, code ATC: J01XB01
Ce médicament appartient à une famille d’un antibiotique de la famille des polymixines. Il contient du colistiméthate sodique qui est un précurseur de la colistine.
L’administration de ce médicament par inhalation doit être réservée au traitement des infections pulmonaires chroniques chez les patients atteints de mucoviscidose.
COLISTIMETHATE SODIQUE AMDIPHARM est utilisé quand ces infections sont causées par une bactérie spécifique appelée Pseudomonas aeruginosa.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur ?
N’utilisez jamais COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur :
· si vous êtes allergique au colistiméthate sodique, à la colistine, à d’autres polymyxines ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur :
· si vous avez ou avez eu des problèmes au niveau des reins
· si vous êtes atteint de myasthénie (fatigue musculaire)
· si vous êtes atteint de porphyrie
· si vous souffrez d’asthme.
Prévenez immédiatement votre médecin ou un autre professionnel de santé si une réaction allergique sévère se produit pendant le traitement par COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant car le traitement par COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant devra être arrêté.
· en cas de prise d'autres médicaments qui peuvent avoir un impact sur le fonctionnement de vos reins (antibiotiques de type aminosides),
· si vous avez eu récemment d'importants crachats de sang en toussant car les médicaments inhalés peuvent être à l'origine de cette toux; votre médecin jugera de l'intérêt à continuer ce traitement.
L'utilisation de médicaments inhalés comme celui-ci peut provoquer un bronchospasme (gêne respiratoire). Votre médecin surveillera la première administration de ce médicament et contrôlera votre fonction pulmonaire avant et après.
Chez les prématurés et les nouveau-nés, une attention particulière doit être prise lors de l'utilisation de COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant car les reins ne sont pas encore totalement développés.
Enfants
Sans objet.
Autres médicaments et COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Ne jamais mélanger ou diluer ce médicament avec un autre médicament dans le nébuliseur.
· Médicaments qui peuvent affecter le fonctionnement de vos reins. Prendre ces médicaments en même temps que COLISTIMETHATE SODIQUE AMDIPHARM peut augmenter le risque de défaillance de vos reins.
· Médicaments qui peuvent affecter votre système nerveux. Prendre ces médicaments en même temps que COLISTIMETHATE SODIQUE AMDIPHARM peut augmenter le risque d'effets indésirables sur votre système nerveux.
· Médicaments appelés relaxants musculaires, souvent utilisés au cours d’une anesthésie générale. COLISTIMETHATE SODIQUE AMDIPHARM peut augmenter les effets de ces médicaments. Si vous devez recourir à une anesthésie générale, informez l’anesthésiste que vous prenez COLISTIMETHATE SODIQUE AMDIPHARM.
Si vous souffrez de myasthénie et que vous prenez également d'autres antibiotiques appelés macrolides (tels que l’azithromycine, la clarithromycine ou l’érythromycine) ou des antibiotiques appelés fluoroquinolones (comme l’ofloxacine, la norfloxacine et la ciprofloxacine), prendre COLISTIMETHATE SODIQUE AMDIPHARM augmente encore plus le risque de faiblesse musculaire et de difficultés respiratoires.
Recevoir COLISTIMETHATE SODIQUE AMDIPHARM par perfusion en même temps que colistiméthate sodique par inhalation peut augmenter votre risque d'effets indésirables.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur avec des aliments ou boissons
Sans objet.
Grossesse et allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Grossesse
Ce médicament ne sera utilisé pendant la grossesse que sur les conseils de votre médecin. Si vous découvrez que vous êtes enceinte pendant le traitement, consultez rapidement votre médecin: lui seul pourra adapter le traitement à votre état.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Allaitement
Compte tenu de l'immaturité digestive du nouveau-né et d'un risque d'atteinte rénale, la prescription de ce médicament par voie orale ou injectable n'est pas recommandée en cas d'allaitement. Cependant, après administration par voie inhalée, les concentrations sanguines de colistine chez l'adulte traité sont très faibles.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Sans objet.
COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur contient :
Sans objet.
3. COMMENT UTILISER COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur ?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Posologie
Conformez-vous à la posologie prescrite par votre médecin.
La dose recommandée est de :
|
|
Dose recommandée |
Dose maximale journalière |
|
Adultes et adolescents (âgés de 12 à 17 ans) Enfants (âgés de 2 à 11 ans) |
1 - 2 millions d’unités 2 à 3 fois par jour |
6 millions d’unités |
|
Enfants de moins de 2 ans |
0,5 - 1 million d’unités 2 fois par jour |
2 millions d’unités |
Votre médecin peut décider d’adapter la dose en fonction de votre situation.
La durée de l'inhalation est fonction de la dose thérapeutique prescrite ; elle est environ de 5 à 10 minutes pour une inhalation de 1 M U.I. (solution reconstituée de 3 ml).
Si vous prenez également d'autres médicaments par voie inhalée, votre médecin vous indiquera l'ordre dans lequel vous devez les prendre.
COLISTIMETHATE SODIQUE AMDIPHARM est destiné à être administré par voie inhalée par nébulisation en utilisant l'appareil nébuliseur adapté prescrit par votre médecin.
Fréquence d’administration
1 à 3 prises journalières.
Durée du traitement
Pour être efficace, cet antibiotique doit être utilisé régulièrement aux doses prescrites, et aussi longtemps que votre médecin vous l'aura conseillé.
La disparition de la fièvre, ou de tout autre symptôme, ne signifie pas que vous êtes complètement guéri. L'éventuelle impression de fatigue n'est pas due au traitement antibiotique mais à l'infection elle-même. Le fait de réduire ou de suspendre votre traitement serait sans effet sur cette impression et retarderait votre guérison.
Mode d’administration
Ce médicament est administré par voie inhalée par la bouche.
NE PAS INJECTER- NE PAS AVALER.
La première administration de ce médicament sera effectuée sous surveillance avec contrôle notamment de la fonction respiratoire.
Si vous suivez différents traitements pour la mucoviscidose, respectez l'ordre suivant en consultant votre médecin:
· bronchodilatateur (médicament qui augmente le calibre des bronches),
· kinésithérapie respiratoire pour désencombrement des bronches,
· autres médicaments par inhalation (ex.: rhDNAse) si votre traitement habituel en comprend,
· puis ce médicament en dernier lieu.
Pour débuter ce traitement, il vous faut :
· un flacon de COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour solution pour inhalation par nébuliseur
· une ampoule de 3 mL de solution isotonique stérile de chlorure de sodium pour dissoudre la poudre*
· un nébuliseur approprié (par exemple PARI LC® system).
Préparation du médicament
La solution pour nébulisation doit être dissoute dans un solvant. La solution ainsi reconstituée va être administrée à l'aide d'un dispositif approprié comprenant un compresseur et un nébuliseur (appareil pour nébulisation).
Ne pas dissoudre COLISTIMETHATE SODIQUE AMDIPHARM avant le moment de l’utilisation (voir étape 5).
|
Etape 1 Prenez un flacon de COLISTIMETHATE SODIQUE AMDIPHARM et tapotez doucement le flacon en verre de telle sorte que la poudre se dépose au fond. Cela permet de vous assurer d'obtenir la dose appropriée de médicament. Ouvrir le flacon en soulevant le capuchon en plastique du dessus (Figure 1). |
|
|
Etape 2 Tirer le capuchon en plastique avec la bague métallique (Figure 2). |
|
|
Etape 3 Déchirer le capuchon en plastique avec un mouvement de torsion dans le sens de la flèche. Une bonne exécution vous permettra de déchirer l'ouverture de l'anneau de métal. Déchirer l'anneau métallique. Retirer l'anneau métallique du flacon (Figure 3). Jetez la bague et le capuchon. |
|
|
Etape 4 Retirez délicatement le bouchon (Figure 4). |
|
|
Etape 5 Ajouter 3 mL de solution isotonique stérile de chlorure de sodium dans le flacon. Afin d’éviter de faire mousser, agiter doucement le flacon jusqu’à ce que la poudre soit dissoute. N’utilisez pas COLISTIMETHATE SODIQUE AMDIPHARM si vous remarquez des particules visibles dans la solution après dissolution. |
|
La solution doit être vidée dans le nébuliseur. Une légère formation de mousse peut être observée.
Cette solution reconstituée est destinée à un usage unique. Cette solution ne contenant pas de conservateur, l'intégralité du contenu d'un flacon doit être utilisée immédiatement après ouverture. Les flacons ouverts ne doivent jamais être conservés pour être réutilisés.
A l'issue de votre traitement, vous devez jeter toute solution restant éventuellement dans le nébuliseur.
Information sur le dispositif d’inhalation
· Ce médicament est administré par inhalation avec un nébuliseur approprié (par exemple du type PARI LC STAR et le compresseur du type PARI TURBO BOY N. D'autres appareils ayant des caractéristiques similaires peuvent également être utilisés.
· Interrogez votre médecin ou votre kinésithérapeute pour tout conseil sur le matériel à utiliser et sur la manière correcte d'utiliser le médicament et le matériel nécessaire.
Pour des informations plus détaillées sur l’utilisation correcte du nébuliseur, pour le nettoyage et la désinfection, veuillez-vous référer au manuel d’utilisation du nébuliseur.
· L’inhalation doit se faire dans une pièce bien ventilée
Préparation
· Il est important que votre nébuliseur fonctionne correctement avant de commencer le traitement avec COLISTIMETHATE SODIQUE AMDIPHARM.
· Lisez attentivement les instructions pour l’utilisation du nébuliseur pour plus d’information sur la prise en main du système de nébulisation.
· Placez les composants de votre nébuliseur sur une surface propre et suivez les instructions du fabricant.
· Lavez-vous soigneusement les mains à l'eau et au savon.
Prise du médicament
· Mettez en marche le compresseur.
· Vérifiez que la vaporisation sortant de la pièce buccale soit régulière. Si rien ne sort, vérifiez que le tube soit bien branché et que le compresseur fonctionne correctement.
· Assis ou debout, tenez-vous droit de manière à pouvoir respirer normalement.
· Placez la pièce buccale entre les dents et sur la partie supérieure de la langue.
Respirez normalement, mais seulement par la bouche (une pince sur le nez peut être utile). Essayez de ne pas bloquer l'écoulement d'air avec la langue.
· Continuez jusqu'à ce que toute la préparation soit épuisée et que la vaporisation cesse.
· N'oubliez pas de nettoyer et de désinfecter le nébuliseur après le traitement.
· Si vous êtes interrompu, si vous avez besoin de tousser ou de vous reposer pendant le traitement, arrêtez le compresseur pour conserver le médicament.
Mettez à nouveau le compresseur en marche lorsque vous êtes prêt à recommencer.
Nettoyage du nébuliseur
· N'utilisez jamais le nébuliseur lorsque l'embout est bouché. Vous vous en rendrez compte s'il n'y a pas de vaporisation. Si cela se produit, remplacez le nébuliseur.
· Il est très important de toujours nettoyer soigneusement toutes les pièces du nébuliseur selon les instructions fournies par votre kinésithérapeute ou votre médecin après chaque traitement. Sinon, vous risquez une infection et une maladie du fait d'une contamination par le nébuliseur.
Désinfecter le nébuliseur
· Ne partagez jamais votre nébuliseur avec quelqu'un d'autre.
· Il est également très important de désinfecter le nébuliseur régulièrement tous les deux jours.
Entretien du compresseur
Observer les instructions du fabricant concernant l'entretien et l'utilisation du compresseur
Si vous avez pris plus de COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur que vous n’auriez dû
Signalez-le à un médecin ou un pharmacien.
Un surdosage peut se manifester par une difficulté respiratoire, une faiblesse musculaire ou un mauvais fonctionnement des reins. Si vous présentez ces troubles, consultez immédiatement votre médecin ou votre pharmacien, un traitement peut vous être proposé.
Si vous oubliez de prendre COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur
Informez-en votre médecin.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Effets sur l'appareil respiratoire
· Fréquent (pouvant affecter jusqu’à 1 personne sur 10) : difficulté à respirer, oppression thoracique, augmentation de la toux, augmentation des crachats pendant ou juste après l'aérosol. Prévenez immédiatement votre médecin.
· Peu fréquent (pouvant affecter jusqu’à 1 personne sur 100) : pharyngite, altération de la voix.
· Rare (pouvant affecter jusqu’à 1 personne sur 1 000) : crachat de sang. Prévenez immédiatement votre médecin.
Réactions allergiques
· Rare (pouvant affecter jusqu’à 1 personne sur 1 000) : éruption cutanée étendue ; allergie généralisée.
D'autres effets indésirables peuvent survenir en cas de mauvais fonctionnement rénal ou d'administration de doses très élevées de ce médicament :
Effets sur le rein
· Insuffisance de fonctionnement des reins.
Effets sur le système nerveux
· Sensations de picotement, de fourmillement autour de la bouche et/ou au niveau des mains et des pieds, désorientation, confusion ; paralysie respiratoire si anesthésie récente ou si prise de curarisants (médicaments utilisés en anesthésie) en même temps que ce médicament.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER COLISTIMETHATE SODIQUE AMDIPHARM 1 M U.I., poudre et solvant pour inhalation par nébuliseur ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le conditionnement. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas + 25°C.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Quantité correspondant en colistiméthate sodique à................................................................. 80 mg
Pour un flacon de poudre.
· Les autres composants sont : chlorure de sodium, eau pour préparations injectables.
Ce médicament se présente sous forme de poudre et solvant pour inhalation par nébuliseur.
Boîte de 1 flacon de poudre et 1 ampoule de 3 ml de solvant.
Boîte de 5 flacons de poudre et 5 ampoules de 3 ml de solvant.
Boîte de 15 flacons de poudre + 15 ampoules de 3 ml de solvant.
Boîte de 30 flacons de poudre + 30 ampoules de 3 ml de solvant.
Boîte de 60 flacons de poudre + 60 ampoules de 3 ml de solvant.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
AMDIPHARM LIMITED
NORTHWOOD CRESCENT
NORTHWOOD
DUBLIN 9 D09 V504
IRLANDE
Exploitant de l’autorisation de mise sur le marché
75 BOULEVARD HAUSSMANN
75008 PARIS
SANOFI S.r.l.
VIA VALCANELLO, 4
03012 ANAGNI (FR)
ITALIE
Xellia Pharmaceuticals Ltd.
SZÀLLÀS UTCA 1-3
1107 BUDAPEST
HONGRIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
CONSEILS D’EDUCATION SANITAIRE :
QUE SAVOIR SUR LES ANTIBIOTIQUES ?Les antibiotiques sont efficaces pour combattre les infections dues aux bactéries. Ils ne sont pas efficaces contre les infections dues aux virus.
Aussi, votre médecin a choisi de vous prescrire cet antibiotique parce qu’il convient précisément à votre cas et à votre maladie actuelle.
Les bactéries ont la capacité de survivre ou de se reproduire malgré l’action d’un antibiotique. Ce phénomène est appelé résistance : il rend certains traitements antibiotiques inactifs.
La résistance s’accroît par l’usage abusif ou inapproprié des antibiotiques.
Vous risquez de favoriser l’apparition de bactéries résistantes et donc de retarder votre guérison ou même de rendre inactif ce médicament, si vous ne respectez pas :
· la dose à prendre,
· les moments de prise,
· et la durée de traitement.
En conséquence, pour préserver l’efficacité de ce médicament :
1- N’utilisez un antibiotique que lorsque votre médecin vous l’a prescrit.
2- Respectez strictement votre ordonnance.
3- Ne réutilisez pas un antibiotique sans prescription médicale même si vous pensez combattre une maladie apparemment semblable.
4- Ne donnez jamais votre antibiotique à une autre personne, il n’est peut-être pas adapté à sa maladie.
5- Une fois votre traitement terminé, rapportez à votre pharmacien toutes les boîtes entamées pour une destruction correcte et appropriée de ce médicament.