Dernière mise à jour le 01/06/2026
CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : V09GA01
Ce médicament est un médicament radiopharmaceutique à usage diagnostique uniquement.
CARDIOMIBI contient une substance appelée tétrafluoroborate de tétrakis (1-isocyanide-2-méthoxy-2- méthylpropyl) cuivre (I) qui est utilisée pour évaluer le fonctionnement du cœur et le débit sanguin (perfusion du myocarde) en obtenant une image du cœur (scintigraphie), par exemple pour la détection d’un risque d’infarctus du myocarde ou lorsqu’une maladie entraîne une diminution de l’afflux sanguin vers le muscle cardiaque (ou une partie de celui-ci) (ischémie). CARDIOMIBI est également utilisé pour le diagnostic des anomalies du sein en complément d’autres méthodes diagnostiques lorsque les résultats n’ont pas été suffisamment concluants. CARDIOMIBI peut également être utilisé pour localiser les glandes parathyroïdiennes hyperactives (les glandes qui sécrètent l’hormone qui contrôle les taux de calcium dans le sang).
Une fois injecté, CARDIOMIBI s’accumule temporairement dans certaines régions du corps. Ce médicament radiopharmaceutique présente une radioactivité faible mais suffisante pour le détecter de l’extérieur du corps à l’aide de caméras spéciales. Votre médecin spécialiste de médecine nucléaire obtiendra alors une image (scintigraphie) de l’organe concerné qui lui fournira des informations utiles sur la structure et le fonctionnement de l’organe étudié, ou la localisation d’une tumeur par exemple.
L’utilisation de CARDIOMIBI entraîne l’exposition à de faibles niveaux de radioactivité. Votre médecin et le spécialiste de médecine nucléaire ont estimé que le bénéfice clinique que vous tirerez de la procédure réalisée avec le produit radiopharmaceutique dépasse le risque lié aux radiations.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 07/10/2009 | Inscription (CT) | Le service médical rendu est important par cette spécialité dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 07/10/2009 | Inscription (CT) | Absence d’amélioration du service médical rendu par rapport à CARDIOLITE. |
ANSM - Mis à jour le : 29/08/2019
CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 1 mg de
tétrafluoroborate de tétrakis (1-isocyanide-2-méthoxy-2- méthylpropyl) cuivre (I).
Le radionucléide n’est pas inclus dans la trousse.
Excipient à effet notoire :
0,009 mmol (0,2 mg) de sodium par flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
Trousse pour préparation radiopharmaceutique.
Ce produit consiste en une poudre lyophilisée blanche.
4.1. Indications thérapeutiques
Ce médicament est à usage diagnostique uniquement.
Il est indiqué chez l’adulte. Concernant la population pédiatrique, voir la rubrique 4.2.
Après préparation à l’aide d’une solution de pertechnétate (99mTc) de sodium, la solution de technétium (99mTc) sestamibi obtenue est indiquée pour :
· La détection et localisation des coronaropathies artérielles (angor et infarctus du myocarde) par scintigraphie de perfusion du myocarde.
· L’évaluation globale de la fonction ventriculaire par la technique du premier passage pour la détermination de la fraction d’éjection et/ou tomographie par émission monophotonique (TEMP) avec synchronisation à l’ECG pour l’évaluation de la fraction d’éjection ventriculaire gauche, des volumes et de la mobilité régionale des parois myocardiques.
· La détection du cancer du sein par scintigraphie mammaire lorsque la mammographie n’est pas concluante, n’est pas adaptée ou ne donne pas de résultat décisif.
· La localisation de foyers de tissu parathyroïdien hyperactifs chez les patients atteints d’une forme récidivante ou persistante d’hyperparathyroïdie primaire et secondaire et chez les patients atteints d’hyperparathyroïdie primaire devant bénéficier d’une première chirurgie des parathyroïdes.
4.2. Posologie et mode d'administration
Adultes et personnes âgées
La posologie peut varier selon les caractéristiques de la gamma-caméra et les modalités de reconstruction. Toute injection d’une activité supérieure aux activités diagnostiques de référence (NDR, Niveaux de Référence Diagnostiques) devrait être justifiée.
Les activités recommandées en injection intraveineuse chez un adulte de masse corporelle moyenne (70 kg) sont les suivantes :
Pour le diagnostic des défauts de perfusion coronarienne et des infarctus du myocarde :
400 - 900 MBq
Conformément aux recommandations européennes, les activités recommandées pour le diagnostic de l’ischémie myocardique sont:
· Protocole sur deux jours : 600 - 900 MBq/examen.
· Protocole sur un jour : 400 - 500 MBq pour la première injection, et trois fois plus pour la seconde injection.
Au total, l’activité administrée ne doit pas dépasser 2 000 MBq dans le cas d’un protocole d’examen sur un jour et 1 800 MBq si le protocole prévoit l’examen sur deux jours. Si l’examen a lieu sur un jour, les deux injections (à l’issue de l’épreuve de stimulation et au repos) doivent être pratiquées à deux heures d’intervalle au minimum mais l’ordre est indifférent. Après l’injection lors de l’épreuve de stimulation, le patient est encouragé à poursuivre l’activité physique pendant encore une minute (si possible).
Pour le diagnostic de l’infarctus du myocarde, une injection au repos est habituellement suffisante.
Pour le diagnostic de l’ischémie myocardique, deux injections (à l’issue de l’épreuve de stimulation et au repos) sont nécessaires afin de pouvoir différencier les hypofixations myocardiques transitoires des hypofixations persistantes.
Pour l’évaluation globale de la fonction ventriculaire
600 - 800 MBq injectés en embole.
Pour la scintigraphie mammaire
700 - 1 000 MBq injectés en intraveineux, généralement dans le bras opposé à la lésion.
Pour la localisation de foyers de tissu parathyroïdien hyperactifs
200 - 700 MBq injectés en embole. L’activité usuelle est comprise entre 500 et 700 MBq.
La posologie peut varier selon les caractéristiques de la gamma-caméra et les modalités de reconstruction. Toute injection d’une activité supérieure aux activités diagnostiques de référence (NDR, Niveaux de Référence Diagnostiques) devrait être justifiée.
Insuffisance rénale
Le niveau d’activité administré doit être adapté car une exposition accrue aux radiations est possible chez ces patients.
Insuffisance hépatique
De façon générale, la détermination de l’activité doit être adaptée chez les patients dont la fonction hépatique est altérée, en commençant habituellement par l’activité correspondant à la valeur basse de l’intervalle des posologies recommandées.
Population pédiatrique
L’utilisation de ce médicament chez l’enfant et l’adolescent doit être envisagée, avec prudence, à l’issue d’une évaluation soigneuse des besoins cliniques et du rapport bénéfices/risques dans cette population. Les activités administrées chez l’enfant et l’adolescent peuvent être calculées d’après les recommandations de la table d’activité pédiatrique de l’EANM (European Association of Nuclear Medicine) ; l’activité administrée chez l’enfant et l’adolescent peut être calculée en multipliant l’activité basale (fournie à des fins de calcul) par le coefficient correspondant à la masse corporelle, comme indiqué dans le tableau ci-dessous.
A [MBq]administrée = activité basale × coefficient multiplicateur
Pour la détection tumorale l’activité basale est de 63 MBq. Lors du protocole d’examen cardiaque sur deux jours, les activités basales minimales et maximales sont respectivement de 42 et 63 MBq, (au repos et à l’issue de l’épreuve de stimulation). Lors du protocole d’examen cardiaque sur un jour, l’activité basale est de 28 MBq au repos et de 84 MBq à l’issue de l’épreuve de stimulation.
L’activité minimale à injecter quel que soit l’examen d’imagerie est de 80 MBq.
|
Masse corporelle [kg] |
Coefficient multiplicateur
|
Masse corporelle [kg] |
Coefficient multiplicateur |
Masse corporelle [kg] |
Coefficient multiplicateur
|
|
3 |
1 |
22 |
5,29 |
42 |
9,14 |
|
4 |
1,14 |
24 |
5,71 |
44 |
9,57 |
|
6 |
1,71 |
26 |
6,14 |
46 |
10,00 |
|
8 |
2,14 |
28 |
6,43 |
48 |
10,29 |
|
10 |
2,71 |
30 |
6,86 |
50 |
10,71 |
|
12 |
3,14 |
32 |
7,29 |
52-54 |
11,29 |
|
14 |
3,57 |
34 |
7,72 |
56-58 |
12,00 |
|
16 |
4,00 |
36 |
8,00 |
60-62 |
12,71 |
|
18 |
4,43 |
38 |
8,43 |
64-66 |
13,43 |
|
20 |
4,86 |
40 |
8,86 |
68 |
14,00 |
Mode d’administration
Voie intraveineuse.
L’injection extravasculaire de ce médicament radioactif est à éviter absolument en raison du risque potentiel de lésion tissulaire.
Présentation multidose.
Précautions à prendre avant la manipulation ou l’administration du médicament
Ce médicament doit être reconstitué avant d’être administré au patient. Pour les instructions concernant la reconstitution et le contrôle de la pureté radiochimique du médicament avant administration, voir la rubrique 12.
Concernant la préparation du patient, voir la rubrique 4.4.
Acquisition des images
Imagerie cardiaque
L’acquisition est débutée 30 à 60 min environ après l’injection afin de permettre la clairance hépatobiliaire du produit. Un délai plus important peut être nécessaire lors de l’examen au repos ou lors de l’examen après stimulation pratiquée par administration des vasodilatateurs, en raison du risque d’activité sous-diaphragmatique élevée du technétium marqué au (99mTc). L’acquisition des images peut être réalisée jusqu’à 6 heures après l’injection car aucune variation significative de la concentration ou de la redistribution du traceur dans le myocarde n’a été démontrée. Le protocole d’examen peut être prévu sur un ou deux jours.
L’acquisition des images doit se faire de préférence selon le mode tomographique (tomographie par émission monophotonique, TEMP), avec ou sans synchronisation à l’ECG.
Pour la scintigraphie mammaire
Pour être optimale, l’acquisition des images mammaires est débutée 5 à 10 minutes après l’injection, la patiente étant placée en décubitus ventral, le sein examiné pendant librement. Le produit est administré dans une veine du bras opposé au sein présentant l’anomalie suspectée. Si l’atteinte est bilatérale, l’injection sera administrée idéalement dans une veine dorsale du pied.
Gamma-caméra classique
La patiente est ensuite repositionnée de façon à laisser pendre le sein opposé et une acquisition des images de profil est pratiquée pour ce sein. Ensuite, une image antérieure est réalisée, la patiente étant en décubitus dorsal, les deux bras derrière la tête.
Détecteur dédié à l’imagerie des seins
Si un détecteur dédié à l’imagerie des seins est utilisé, un protocole pertinent spécifique à l’appareil doit être suivi afin d’obtenir les meilleures performances d’imagerie possibles.
Pour l’imagerie des glandes parathyroïdes
La marche à suivre pour l’acquisition des images en cas d’hyperparathyroïdisme varie selon le protocole choisi. Les méthodes d’examen les plus utilisées sont la technique par soustraction et la technique en deux temps, qui peuvent être réalisées conjointement.
Pour la technique par soustraction, il est possible d’utiliser soit de l’iodure (123I) de sodium soit du pertechnétate (99mTc) de sodium pour l’imagerie de la glande thyroïde dans la mesure où ces produits radiopharmaceutiques font l’objet d’une rétention dans les tissus thyroïdiens fonctionnels. Cette image est soustraite de l’image obtenue avec le technétium (99mTc) sestamibi, et le tissu parathyroïdien hyperactif pathologique reste visible après la soustraction.
Lorsque l’iodure (123I) de sodium est utilisé, une activité de 10 à 20 MBq est administrée par voie orale. Les images du cou et du thorax peuvent être obtenues quatre heures après l’administration. Après l’acquisition des images avec l’iodure (123I) de sodium, 200 à 700 MBq de technétium (99mTc) sestamibi sont injectés et les images sont acquises 10 minutes après l’injection selon la technique de double acquisition avec 2 pics d’énergie gamma (140 keV pour le technétium (99mTc) et 159 keV pour l’iodure (123I)).
Lorsque le pertechnétate (99mTc) de sodium est utilisé, une activité de 40 à 150 MBq est injectée et les images du cou et du thorax sont acquises 30 minutes plus tard. Par la suite, 200 à 700 MBq de technétium (99mTc) sestamibi sont injectés et une seconde acquisition des images est effectuée 10 minutes plus tard.
Si la technique en deux temps est employée, 400 à 700 MBq de technétium (99mTc) sestamibi sont injectés 10 minutes avant de procéder à la première acquisition des images du cou et du médiastin. L’acquisition des images du cou et du médiastin est renouvelée après une période d’élimination de 1 à 2 heures.
Les images acquises en mode planaire peuvent être complétées par un examen TEMP ou TEMP/TDM lors des temps précoces ou tardifs.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Lors des examens de scintigraphie myocardique avec épreuve de stimulation, il faut prendre en compte les contre-indications générales applicables à la stimulation ergométrique ou à l’épreuve pharmacologique.
4.4. Mises en garde spéciales et précautions d'emploi
Potentiel de réactions d’hypersensibilité ou de réactions anaphylactiques
En cas de réaction d’hypersensibilité ou de réaction anaphylactique, l’administration du produit médicamenteux doit être immédiatement interrompue et un traitement par voie intraveineuse doit être débuté, si nécessaire. Afin de permettre une prise en charge rapide en cas d’urgence, il convient d’avoir à disposition immédiate les médicaments et le matériel nécessaires, notamment des sondes d’intubation trachéale et du matériel de ventilation.
Justification du rapport bénéfice/risque
Pour chaque patient, l’exposition aux radiations doit se justifier sur la base des bénéfices attendus. L’activité administrée doit, dans tous les cas, être déterminée en limitant autant que possible la dose de radiation résultante tout en permettant d’obtenir l’information diagnostique requise.
Insuffisance rénale ou hépatique
Le rapport bénéfice/risque doit être évalué avec soin chez ces patients car une exposition accrue aux radiations est possible (voir rubrique 4.2).
Population pédiatrique
Pour les informations concernant l’usage pédiatrique, voir rubrique 4.2.
L’indication doit être évaluée avec soin car la dose efficace par MBq est plus élevée que chez l’adulte (voir rubrique 11).
Préparation du patient
Le patient doit être bien hydraté avant le début de l’examen et invité à uriner aussi fréquemment que possible pendant les premières heures suivant l’examen afin de réduire l'exposition aux radiations.
Pour la scintigraphie cardiaque
Le patient doit, si possible, être à jeun depuis au moins quatre heures lors de l’examen. Il est recommandé que le patient absorbe une légère collation composée d’aliments lipidiques ou boive un ou deux verre(s) de lait après chaque injection et avant l’acquisition des images. Cette précaution augmente la clairance hépatobiliaire du technétium (99mTc) sestamibi, réduisant ainsi l’activité hépatique lors de l’acquisition des images.
Interprétation des images obtenues avec le technétium (99mTc) sestamibi
Interprétation de la scintigraphie mammaire
Les lésions mammaires d’un diamètre inférieur à 1 cm peuvent ne pas être systématiquement détectées par la scintigraphie mammaire car la sensibilité du technétium (99mTc) sestamibi pour la détection de ces lésions est faible. L’obtention d’un résultat négatif à l’examen n’exclut pas la présence d’un cancer du sein, en particulier dans le cas d’une lésion d’une aussi petite taille.
Après l’injection
Les contacts rapprochés avec les nourrissons et les femmes enceintes doivent être évités pendant les 24 heures suivant l’injection.
Mises en garde spécifiques
Lors des examens de scintigraphie myocardique avec épreuve de stimulation, il faut prendre en compte les contre-indications et précautions générales applicables à la stimulation ergométrique ou à l’épreuve pharmacologique.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu'il est pratiquement « sans sodium ».
Pour les précautions relatives au risque environnemental, voir rubrique 6.6.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les médicaments affectant la fonction myocardique et/ou le débit sanguin peuvent engendrer des résultats faux négatifs lors du diagnostic des coronaropathies. En particulier, les bêtabloquants et les inhibiteurs calciques réduisent la consommation d’oxygène et affectent donc également la perfusion et les bêtabloquants inhibent l’augmentation de la fréquence cardiaque et de la pression artérielle pendant l’épreuve de stimulation. En conséquence, les traitements co-administrés doivent être pris en compte lors de l’interprétation des résultats de la scintigraphie. Les recommandations des directives applicables concernant les épreuves de stimulation ergométrique ou pharmacologique doivent être observées.
Lorsque la technique par soustraction est utilisée pour l’imagerie des tissus parathyroïdiens hyperactifs, l’utilisation récente de produits de contraste radiologiques à base d’iodure, de médicaments utilisés pour le traitement de l’hyperthyroïdie ou de l’hypothyroïdie ou de plusieurs autres médicaments, est susceptible de réduire la qualité de l’imagerie thyroïdienne, voire de rendre la soustraction impossible. Pour la liste complète des interactions médicamenteuses éventuelles, voir le RCP de l’iodure (123I) de sodium ou du pertechnétate (99mTc) de sodium.
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Lorsque l’administration de médicaments radiopharmaceutiques est envisagée chez une femme en âge de procréer, il est important de déterminer si la patiente est ou non enceinte. Toute femme n’ayant pas eu ses règles doit être considérée comme enceinte jusqu’à preuve du contraire. En cas de doute, sur la présence éventuelle d’une grossesse (retard de règles, règles très irrégulières, etc.), d’autres techniques n’impliquant pas de rayons ionisants (s’il en existe) doivent être proposées à la patiente.
Grossesse
Les examens utilisant des radionucléides pratiqués chez les femmes enceintes exposent également le fœtus à une dose de radiation. Par conséquent, les examens ne doivent être pratiqués en cours de grossesse que s’ils sont essentiels et si les bénéfices attendus dépassent largement les risques encourus par la mère et le fœtus.
Avant toute administration de médicaments radiopharmaceutiques chez une femme qui allaite, il convient d’envisager la possibilité de retarder l’administration du radionucléide jusqu’à la fin de l’allaitement et de déterminer l’agent radiopharmaceutique le plus approprié, en gardant à l’esprit que la radioactivité passe dans le lait maternel.
Si l’administration du médicament est jugée nécessaire, l’allaitement doit être suspendu pendant 24 heures et le lait produit pendant cette période devra être éliminé.
Les contacts rapprochés avec les nourrissons doivent être évités pendant les 24 heures suivant l’injection.
Fertilité
Aucune étude sur la fertilité n’a été réalisée.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Le tableau suivant décrit les groupes de fréquence utilisés dans cette rubrique :
|
Très fréquent (≥ 1/10) |
|
Fréquent (≥ 1/100 à < 1/10) |
|
Peu fréquent (≥ 1/1 000 à < 1/100) |
|
Rare (≥ 1/10 000 à < 1/1 000) |
|
Très rare (< 1/10 000) |
|
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) |
Affections du système immunitaire :
Rare : sévères réactions d’hypersensibilité de type dyspnée, hypotension, bradycardie, asthénie et vomissements (généralement dans les deux heures suivant l’administration), œdème de Quincke. Autres réactions d’hypersensibilité (réactions allergiques affectant la peau et les muqueuses avec exanthème [prurit, urticaire, œdème], vasodilatation).
Très rare : d’autres réactions d’hypersensibilité ont été décrites chez des patients prédisposés.
Affections du système nerveux :
Peu fréquent : céphalées
Rare : crises convulsives (peu après l’administration), syncope.
Affections cardiaques :
Peu fréquent : douleur thoracique/angine de poitrine, anomalies de l’ECG.
Rare : arythmie.
Affections gastro-intestinales :
Peu fréquent : nausées
Rare : douleur abdominale.
Affections de la peau et des tissus sous-cutanés :
Rare : réactions locales au site d’injection, hypoesthésie et paresthésie, bouffées vasomotrices.
Fréquence indéterminée: érythème polymorphe.
Troubles généraux et anomalies au site d’administration :
Fréquent : immédiatement après l’injection, un goût métallique ou amer, associé dans certains cas à une sécheresse buccale et une altération de la perception olfactive, peut être ressenti.
Rare : fièvre, fatigue, étourdissements, douleur pseudo-arthritique transitoire, dyspepsie.
Autres troubles :
L’exposition aux rayons ionisants a été associée à l’induction de cancers et à l’apparition potentielle d’anomalies congénitales. La dose efficace étant de 16,4 mSv lorsque l’activité maximale recommandée de 2 000 MBq (500 MBq au repos et 1 500 MBq après épreuve de stimulation) est administrée dans le cadre d’un protocole sur 1 jour, la fréquence de survenue de ces réactions indésirables est indéterminée.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
En cas d'administration d'une dose excessive du technétium (99mTc) sestamibi, la dose de radiation absorbée par le patient doit être réduite, si possible, en augmentant l’élimination du radionucléide par des mictions et des défécations fréquentes. Il peut être utile d’estimer la dose efficace reçue.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : produit radiopharmaceutique à usage diagnostique ; Composés de technétium (99mTc), code ATC : V 09G A01.
Effets pharmacodynamiques
Aux concentrations chimiques administrées pour les examens diagnostiques, la solution de technétium (99mTc) sestamibi ne semble pas avoir d’activité pharmacodynamique.
5.2. Propriétés pharmacocinétiques
Après reconstitution à l’aide d’une solution injectable de pertechnétate (99mTc) de sodium, le complexe suivant se forme (technétium (99mTc) sestamibi) :
[99mTc (MIBI)6]+ où : MIBI = 2-méthoxy-isobutyl-isonitrile
Biodistribution
Le technétium (99mTc) sestamibi injecté par voie IV stricte se distribue rapidement dans les tissus : 5 minutes après l’injection, environ 8% seulement de l’activité injectée est encore présent dans le compartiment sanguin.La distribution physiologique du technétium (99mTc) sestamibi inclue les glandes salivaires, la thyroïde, le myocarde, le foie, la vésicule biliaire, le gros intestin, l’intestin grêle, les reins, la vessie, les plexus choroïdes et les muscles squelettiques, et occasionnellement dans les plaques aréolaires. Une fixation homogène de faible niveau dans le sein ou l'aisselle est normale.
Scintigraphie de perfusion du myocarde
Le technétium (99mTc) sestamibi est un complexe cationique qui diffuse passivement au travers des membranes capillaire et cellulaire. Au sein de la cellule, il est capté et retenu dans les mitochondries et sa rétention reflète la viabilité des cellules myocardiques. Après une injection intraveineuse, il est distribué dans le myocarde en fonction de la perfusion et de la viabilité du myocarde. La fixation myocardique, qui dépend du débit coronaire, est de 1,5% de l’activité injectée à l’issue d’une épreuve de stimulation et de 1,2% de celle injectée au repos. Cependant, les cellules endommagées de façon irréversible ne fixent plus le technétium (99mTc) sestamibi. L’hypoxie réduit le taux d’extraction myocardique. Sa redistribution est très limitée et des injections distinctes sont donc nécessaires pour les examens après épreuve de stimulation et au repos.
Pour la scintigraphie mammaire
La fixation tissulaire du technétium (99mTc) sestamibi dépend principalement de la vascularisation, laquelle est généralement accrue dans les tissus tumoraux. Le technétium (99mTc) sestamibi s’accumule dans diverses tumeurs et le plus notablement dans les mitochondries. Sa fixation est liée à l’augmentation du métabolisme dépendant de l’énergie et à la prolifération cellulaire. Son accumulation cellulaire est réduite lorsque les protéines de résistance multi-drogue (MDR) sont surexprimées.
Imagerie des tissus parathyroïdiens hyperactifs
Le technétium (99mTc) sestamibi se fixe aussi bien dans les tissus parathyroïdiens que dans les tissus thyroïdiens fonctionnels mais il est habituellement éliminé plus rapidement des tissus thyroïdiens normaux que des tissus parathyroïdiens anormaux.
L’élimination du technétium (99mTc) sestamibi se fait principalement par l’intermédiaire des reins et du système hépatobiliaire. L’activité du technétium (99mTc) sestamibi accumulée dans la vésicule biliaire est retrouvée dans l’intestin dans l’heure qui suit l’injection. Environ 27% de l’activité injectée est éliminé par voie rénale en 24 heures, et approximativement 33% est éliminé dans les selles en 48 heures. Les propriétés pharmacocinétiques chez les patients atteints d’insuffisance rénale ou hépatique n’ont pas été déterminées.
Demi-vie
La demi-vie biologique myocardique du technétium (99mTc) sestamibi est d’environ 7 heures au repos et après épreuve de stimulation. La demi-vie efficace, qui tient compte des demi-vies biologique et physique (décroissance radioactive), est approximativement de 3 heures pour le cœur et approximativement de 30 minutes pour le foie.
5.3. Données de sécurité préclinique
Lors des études de toxicité aiguë par voie intraveineuse chez la souris, le rat et le chien, la plus faible dose de produit reconstitué ayant entraîné des décès a été de 7 mg/kg (dose exprimée en tétrafluoroborate de tétrakis (2-méthoxy-2-méthylpropyl-1 isocyanide) cuivre (I)) chez la rate. Ceci correspond à 500 fois la dose maximale utilisée chez l’être humain (DMH), à savoir 0,014 mg/kg chez l’adulte (70 kg). Aucun effet lié à l’administration n’a été constaté chez le rat et le chien aux doses respectives de 0,42 mg/kg (30 fois la DMH) et de 0,07 mg/kg (5 fois la DMH) de produit reconstitué pendant 28 jours. Lors des administrations répétées, les premiers symptômes de toxicité sont apparus lors de l’administration de 150 fois la dose quotidienne pendant 28 jours.
L’administration extravasculaire chez l’animal a provoqué des inflammations aiguës avec œdème et hémorragies au site d’injection.
Aucune étude de toxicologie n’a été réalisée concernant les effets du médicament sur la reproduction.
Le tétrafluoroborate de tétrakis (2-méthoxy-2-méthylpropyl-1 isocyanide) cuivre (I) n’a présenté aucune activité génotoxique lors des tests d’Ames, de tests de mutation sur cellules de mammifères CHO/HPRT et de tests d’échange de chromatides sœurs. À des concentrations cytotoxiques, une augmentation du nombre d’aberrations chromosomiques a été observée lors du test in vitro sur les lymphocytes humains. Aucune activité génotoxique n’a été notée lors du test in vivo du micronucléus de souris à 9 mg/kg. Il n’y a pas eu d’études destinées à évaluer le potentiel cancérogène du produit radiopharmaceutique.
Chlorure stanneux dihydraté
Chlorhydrate de L-cystéine monohydraté
Citrate de sodium dihydraté
D-mannitol
Ce médicament ne doit pas être mélangé avec d’autres médicaments à l’exception de ceux mentionnés dans la rubrique 12.
1 an.
Après marquage : la stabilité chimique et physique en cours d'utilisation a été démontrée à 12 heures. Ne pas conserver à une température supérieure à 25°C après le marquage.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement, à moins que la méthode d'ouverture, de marquage et de dilution exclue le risque de contamination microbiologique.
S’il n’est pas utilisé immédiatement, les durées et conditions de conservation avant utilisation sont de la responsabilité de l’utilisateur.
6.4. Précautions particulières de conservation
Conserver au réfrigérateur (entre 2°C et 8ºC).
Lors du transport (7 jours au maximum), jusqu'à 35°C.
Pour les conditions de conservation du produit après radiomarquage du médicament, voir la rubrique 6.3.
Le stockage des médicaments radiopharmaceutiques doit être conforme aux réglementations nationales relatives aux produits radioactifs.
6.5. Nature et contenu de l'emballage extérieur
Flacon de 10 mL en verre (de type I), fermé au moyen d'un bouchon en caoutchouc chlorobutyle serti par une capsule en aluminium.
Les flacons sont emballés dans des boîtes en carton, et des boîtes de 3 ou 6 flacons sont disponibles.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Mises en garde générales
Les produits radiopharmaceutiques ne doivent être réceptionnés, utilisés et administrés que par des personnes autorisées dans des locaux spécialement équipés et habilités. Leur réception, leur stockage, leur utilisation, leur transfert et leur élimination sont soumis à la réglementation en vigueur et/ou aux autorisations appropriées des autorités compétentes.
Les produits radiopharmaceutiques doivent être préparés de manière à satisfaire à la fois aux normes de radioprotection et de qualité pharmaceutique. Les précautions appropriées d’asepsie doivent être prises.
Le contenu du flacon doit être utilisé exclusivement pour la préparation du technétium (99mTc) sestamibi et ne doit pas être administré directement au patient sans avoir fait l’objet de la procédure de préparation préalable.
Pour les instructions concernant la préparation extemporanée du médicament avant administration, voir rubrique 12.
Si l’intégrité de ce flacon est compromise à tout moment au cours de la préparation de ce produit, celui-ci ne doit pas être utilisé.
L’administration doit être réalisée de façon à limiter au maximum le risque de contamination du médicament et d’irradiation des opérateurs. L’utilisation de protections plombées adéquates est impérative.
Le contenu de la trousse n’est pas radioactif avant préparation extemporanée. Par contre, après ajout du pertechnétate (99mTc) de sodium, la préparation finale doit être placée dans une protection de plomb appropriée.
L’administration de produits radiopharmaceutiques présente des risques pour l'entourage du patient en raison de l’irradiation externe ou de la contamination par des traces d’urine, des vomissements ou tout autre liquide biologique. Par conséquent, il faut prendre des mesures de protection contre les radiations conformément aux réglementations nationales.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
NATIONAL CENTRE FOR NUCLEAR RESEARCH
ANDRZEJ SOŁTAN 7
05-400 OTWOCK
POLOGNE
Tél. : +48 22 718 07 00
Fax : +48 22 718 03 50
Courriel : polatom@polatom.pl
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 574 397 0 5 : 24,675 mg de poudre, boîte de 3.
· 34009 575 246 6 1 : 24,675 mg de poudre, boîte de 6.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Le technétium (99mTc) est produit à l’aide d’un générateur (99Mo/99mTc) et décroît en émettant des rayons gamma ayant une énergie moyenne de 140 keV et selon une période radioactive de 6,02 heures pour donner du technétium (99Tc) qui, au regard de sa longue période radioactive de 2,13 x 105 années, peut être considéré comme quasi-stable.
Les données répertoriées ci-dessous sont issues de l’ICRP 80 et ont été calculées sur la base des hypothèses suivantes : après injection intraveineuse, la substance quitte rapidement la circulation sanguine et se fixe principalement dans les tissus musculaires (y compris myocardique), le foie et les reins et, dans une moindre proportion, dans les glandes salivaires et la thyroïde. Lorsque la substance est injectée dans le cadre d’une épreuve d’effort, la captation par le cœur et les muscles squelettiques est considérablement augmentée, tandis que la captation par tous les autres organes et tissus est plus faible. La substance est excrétée à 75% par le foie et à 25% par les reins.
|
Organe |
Dose absorbée de radiations par unité d’activité administrée (mGy/MBq) (sujet au repos)
|
||||
|
|
Chez l’adulte |
À 15 ans |
À 10 ans |
À 5 ans |
À 1 an |
|
Surrénales |
0,0075 |
0,0099 |
0,015 |
0,022 |
0,038 |
|
Vessie |
0,011 |
0,014 |
0,019 |
0,023 |
0,041 |
|
Surfaces osseuses |
0,0082 |
0,010 |
0,016 |
0,021 |
0,038 |
|
Cerveau |
0,0052 |
0,0071 |
0,011 |
0,016 |
0,027 |
|
Sein |
0,0038 |
0,0053 |
0,0071 |
0,011 |
0,020 |
|
Paroi de la vésicule biliaire |
0,039 |
0,045 |
0,058 |
0,100 |
0,320 |
|
Tube gastro-intestinal : |
|
|
|
|
|
|
Estomac |
0,0065 |
0,0090 |
0,015 |
0,021 |
0,035 |
|
Intestin grêle |
0,015 |
0,018 |
0,029 |
0,045 |
0,080 |
|
Côlon |
0,024 |
0,031 |
0,050 |
0,079 |
0,015 |
|
(Côlon ascendant |
0,027 |
0,035 |
0,057 |
0,089 |
0,170) |
|
(Côlon descendant |
0,019 |
0,025 |
0,041 |
0,065 |
0,120) |
|
|
|
|
|
|
|
|
Cœur |
0,0063 |
0,0082 |
0,012 |
0,018 |
0,030 |
|
Reins |
0,036 |
0,043 |
0,059 |
0,085 |
0,150 |
|
Foie |
0,011 |
0,014 |
0,021 |
0,030 |
0,052 |
|
Poumons |
0,0046 |
0,0064 |
0,0097 |
0,014 |
0,025 |
|
Muscles |
0,0029 |
0,0037 |
0,0054 |
0,0076 |
0,014 |
|
|
|
|
|
|
|
|
Œsophage |
0,0041 |
0,0057 |
0,0086 |
0,013 |
0,023 |
|
Ovaires |
0,0091 |
0,012 |
0,018 |
0,025 |
0,045 |
|
Pancréas |
0,0077 |
0,010 |
0,016 |
0,024 |
0,039 |
|
Moelle rouge |
0,0055 |
0,0071 |
0,011 |
0,030 |
0,044 |
|
Glandes salivaires |
0,014 |
0,017 |
0,022 |
0,015 |
0,026 |
|
Peau |
0,0031 |
0,0041 |
0,0064 |
0,0098 |
0,019 |
|
|
|
|
|
|
|
|
Rate |
0,0065 |
0,0086 |
0,014 |
0,020 |
0,034 |
|
Testicules |
0,0038 |
0,0050 |
0,0075 |
0,011 |
0,021 |
|
Thymus |
0,0041 |
0,0057 |
0,0086 |
0,013 |
0,023 |
|
Thyroïde |
0,0053 |
0,0079 |
0,012 |
0,024 |
0,045 |
|
Utérus |
0,0078 |
0,010 |
0,015 |
0,022 |
0,038 |
|
|
|
|
|
|
|
|
Organes restants |
0,0031 |
0,0039 |
0,0060 |
0,0088 |
0,016 |
|
Dose efficace |
|
|
|
|
|
|
[mSv/MBq] |
0,0090 |
0,012 |
0,018 |
0,028 |
0,053 |
|
Organe |
Dose absorbée de radiations par unité d’activité administrée (mGy/MBq) (après stimulation) |
||||
|
|
Chez l’adulte |
À 15 ans |
À 10 ans |
À 5 ans |
À 1 an |
|
Surrénales |
0,0066 |
0,0087 |
0,013 |
0,019 |
0,033 |
|
Vessie |
0,0098 |
0,013 |
0,017 |
0,021 |
0,038 |
|
Surfaces osseuses |
0,0078 |
0,0097 |
0,014 |
0,020 |
0,036 |
|
Cerveau |
0,0044 |
0,0060 |
0,0093 |
0,014 |
0,023 |
|
Sein |
0,0034 |
0,0047 |
0,0062 |
0,0097 |
0,018 |
|
Paroi de la vésicule biliaire |
0,033 |
0,038 |
0,049 |
0,086 |
0,260 |
|
Tube gastro-intestinal : |
|
|
|
|
|
|
Estomac |
0,0059 |
0,0081 |
0,013 |
0,019 |
0,032 |
|
Intestin grêle |
0,012 |
0,015 |
0,024 |
0,037 |
0,066 |
|
Côlon |
0,019 |
0,025 |
0,041 |
0,064 |
0,120 |
|
(Côlon ascendant |
0,022 |
0,028 |
0,046 |
0,072 |
0,130) |
|
(Côlon descendant |
0,016 |
0,021 |
0,034 |
0,053 |
0,099) |
|
|
|
|
|
|
|
|
Cœur |
0,0072 |
0,0094 |
0,010 |
0,021 |
0,035 |
|
Reins |
0,026 |
0,032 |
0,044 |
0,063 |
0,110 |
|
Foie |
0,0092 |
0,012 |
0,018 |
0,025 |
0,044 |
|
Poumons |
0,0044 |
0,0060 |
0,0087 |
0,013 |
0,023 |
|
Muscles |
0,0032 |
0,0041 |
0,0060 |
0,0090 |
0,017 |
|
|
|
|
|
|
|
|
Œsophage |
0,0040 |
0,0055 |
0,0080 |
0,012 |
0,023 |
|
Ovaires |
0,0081 |
0,011 |
0,015 |
0,023 |
0,040 |
|
Pancréas |
0,0069 |
0,0091 |
0,014 |
0,021 |
0,035 |
|
Moelle rouge |
0,0050 |
0,0064 |
0,0095 |
0,013 |
0,023 |
|
Glandes salivaires |
0,0092 |
0,011 |
0,0015 |
0,0020 |
0,0029 |
|
Peau |
0,0029 |
0,0037 |
0,0058 |
0,0090 |
0,017 |
|
|
|
|
|
|
|
|
Rate |
0,0058 |
0,0076 |
0,012 |
0,017 |
0,030 |
|
Testicules |
0,0037 |
0,0048 |
0,0071 |
0,011 |
0,020 |
|
Thymus |
0,0040 |
0,0055 |
0,0080 |
0,012 |
0,023 |
|
Thyroïde |
0,0044 |
0,0064 |
0,0099 |
0,019 |
0,035 |
|
Utérus |
0,0072 |
0,0093 |
0,014 |
0,020 |
0,035 |
|
|
|
|
|
|
|
|
Organes restants |
0,0033 |
0,0043 |
0,0064 |
0,0098 |
0,018 |
|
Dose efficace |
|
|
|
|
|
|
[mSv/MBq] |
0,0079 |
0,010 |
0,016 |
0,023 |
0,045 |
La dose efficace a été calculée pour un délai entre deux mictions de 3,5 heures chez l’adulte.
Imagerie cardiaque
La dose efficace après administration de l’activité maximale recommandée de 2 000 MBq de technétium (99mTc) sestamibi chez un adulte de 70 kg est d’environ 16,4 mSv si le protocole sur un jour est appliqué, avec administration de 500 MBq au repos et de 1 500 MBq après épreuve de stimulation.
Lors de l’administration d’une activité de 2 000 MBq, la dose d’irradiation au niveau de l’organe cible, le myocarde, est de 14 mGy et les doses d’irradiation au niveau des organes critiques, la vésicule biliaire, les reins et le côlon ascendant, sont respectivement de 69, 57 et 46,5 mGy.
La dose efficace après administration de l’activité maximale recommandée de 1 800 MBq (900 MBq au repos et 900 MBq après épreuve de stimulation) de technétium (99mTc) sestamibi dans le cadre d’un protocole sur deux jours chez un adulte de 70 kg est d’environ 15,2 mSv.
Lors de l’administration d’une activité de 1 800 MBq, la dose d’irradiation au niveau de l’organe cible, le cœur, est de 12,2 mGy et les doses d’irradiation types au niveau des organes critiques, la vésicule biliaire, les reins et le côlon ascendant, sont respectivement de 64,8 55,8 et 44,1 mGy.
Pour la scintigraphie mammaire
La dose efficace après administration de l’activité maximale recommandée de 1 000 MBq de technétium (99mTc) sestamibi chez un adulte de 70 kg est d’environ 9 mSv.
Lors de l’administration d’une activité de 1 000 MBq, la dose d’irradiation type au niveau de l’organe cible, le sein, est de 3,8 mGy et les doses d’irradiation types au niveau des organes critiques, la vésicule biliaire, les reins et le côlon ascendant, sont respectivement de 39, 36 et 27 mGy.
Pour l’imagerie des glandes parathyroïdes
La dose efficace après administration de l’activité maximale recommandée de 700 MBq de technétium (99mTc) sestamibi chez un adulte de 70 kg est d’environ 6,3 mSv.
Lors de l’administration d’une activité de 700 MBq, la dose d’irradiation type au niveau de l’organe cible, la thyroïde, est de 3,7 mGy et les doses d’irradiation types au niveau des organes critiques, la vésicule biliaire, les reins et le côlon ascendant, sont respectivement de 27,3, 25,2 et 18,9 mGy.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Le prélèvement du produit doit être effectué dans des conditions d’asepsie. Les flacons ne doivent pas être ouverts avant la désinfection du bouchon, la solution doit être prélevée au travers du bouchon à l’aide d’une seringue à usage unique munie d’une protection de plomb adaptée et d’une aiguille stérile jetable ou à l’aide d’un système de dispensation automatisé agréé.
Si l’intégrité de ce flacon est compromise, le produit ne doit pas être utilisé.
Avant l'administration, la dilution du produit marqué avec une solution de chlorure de sodium (solution physiologique à 0,9 %) est possible.
Instructions pour la préparation du technétium (99mTc) sestamibi
A) Protocole par ébullition
La préparation du technétium (99mTc) sestamibi pour injection doit être réalisée en suivant la procédure aseptique suivante :
- Porter des gants imperméables tout au long de la procédure de préparation.
- Placer le flacon de lyophylisat dans un conteneur de protection contre les rayonnements blindé en plomb portant la date, l'heure de préparation, le volume et l'activité.
- À l’aide d’une seringue stérile munie d’une protection blindée en plomb (perçant le bouchon plastique), introduire 1 – 5 mL d’éluat de pertechnétate (99mTc) de sodium pour injection [produit à l'aide d'un générateur de radionucléide disposant d'une autorisation de mise sur le marché] d’activité maximale de 11 GBq (ou un volume d’éluat ajusté à la radioactivité désirée avec une solution saline physiologique) dans un flacon placé dans le conteneur blindé en plomb. Un volume d'au moins 5 mL de pertechnétate (99mTc) de sodium pour injection sera utilisé pour une activité maximale de 11 GBq.
- Sans enlever l’aiguille, soustraire un volume d’air équivalent au volume d’éluat injecté afin de rétablir la pression atmosphérique dans le flacon.
- Agiter le flacon jusqu’à dissolution complète du contenu (environ 1 minute).
- Enlever le flacon de sa protection de plomb et le placer à la verticale dans un bain-marie dûment blindé, de façon à éviter tout contact entre l’eau bouillante et la capsule en aluminium, et porter à ébullition pendant 10 – 12 minutes. Les 10 à 12 minutes sont décomptées à partir du moment où l’eau recommence à bouillir.
Remarque : le flacon doit rester vertical pendant toute la durée de l’ébullition. Veiller à ce que le bouchon dépasse le niveau de la surface de l’eau du bain-marie.
- Retirer le flacon du bain-marie, le placer dans un conteneur en plomb et le laisser refroidir à température ambiante (environ 15 minutes).
- Examiner le contenu du flacon afin de vérifier l’absence de particules ou de changement de couleur avant administration.
- En utilisant une technique aseptique, prélever le technétium (99mTc) sestamibi pour injection à l'aide d'une seringue stérile munie d’une protection blindée. Utiliser le radiopharmaceutique dans les douze (12) heures suivant sa préparation.
- La pureté radiochimique doit être vérifiée avant l'administration au patient en utilisant la méthode de radio-chromatographie en couche mince décrite ci-dessous ou la méthode décrite dans la monographie n° 1926 de la Pharmacopée Européenne [solution injectable de technétium (99mTc) sestamibi].
Remarque : dès lors qu’un flacon contenant une solution radioactive est chauffé, il existe un risque de fissuration et de contamination significative.
B) Procédure par thermocycleur
La préparation du technétium (99mTc) sestamibi pour injection doit être réalisée en suivant la procédure aseptique suivante :
- Porter des gants imperméables tout au long de la procédure de préparation.
- Placer le flacon de lyophylisat dans un conteneur de protection contre les rayonnements blindé en plomb portant la date, l'heure de préparation, le volume et l'activité.
- À l’aide d’une seringue stérile munie d’une protection blindée en plomb (perçant le bouchon plastique), introduire 1 – 5 mL d’éluat de pertechnétate (99mTc) de sodium pour injection [produit à l'aide d'un générateur de radionucléide disposant d'une autorisation de mise sur le marché] d’activité maximale de 11 GBq (ou un volume d’éluat ajusté à la radioactivité désirée avec une solution saline physiologique) dans un flacon placé dans le conteneur blindé en plomb. Un volume d'au moins 5 mL de pertechnétate (99mTc) de sodium pour injection sera utilisé pour une activité maximale de 11 GBq.
- Sans enlever l’aiguille, soustraire un volume d’air équivalent au volume d’éluat injecté afin de rétablir la pression atmosphérique dans le flacon.
- Agiter le flacon jusqu’à dissolution complète du contenu (environ 1 minute).
- Placer la protection de plomb dans le bloc à échantillon d’un thermocycleur. Tout en appuyant légèrement vers le bas, faire tourner la protection d'un quart de tour pour faire en sorte qu'elle soit bien insérée dans le bloc du thermocycleur.
- Appuyer sur le bouton de démarrage du thermocycleur pour lancer le programme (le thermocycleur procédera automatiquement au chauffage et au refroidissement du flacon et de son contenu). Veuillez consulter le mode d'emploi pour plus de détails.
- Examiner le contenu du flacon afin de vérifier l’absence de particules ou de changement de couleur avant administration.
- En utilisant une technique aseptique, prélever le technétium (99mTc) sestamibi pour injection à l'aide d'une seringue stérile munie d’une protection blindée. Utiliser le radiopharmaceutique dans les douze (12) heures suivant sa préparation.
- La pureté radiochimique doit être vérifiée avant l'administration au patient en utilisant la méthode de radio-chromatographie en couche mince décrite ci-dessous ou la méthode décrite dans la monographie n° 1926 de la Pharmacopée Européenne [solution injectable de technétium (99mTc) sestamibi].
Contrôle qualité
Procédure de contrôle quantificatif du technétium (99mTc) sestamibi par chromatographie en couche mince
1. Matériel.
1.1 Oxyde d'aluminium neutre de type T sur plaque en papier aluminium
1.2 Éthanol à >95%.
1.3 Détecteur de radiations adapté
1.4 Chambre chromatographique de petite taille
2. Procédure
· Déposer 2-5 µL de solution à tester à environ 1,5 cm de l’extrémité inférieure d’une plaque chromatographique en oxyde d’aluminium de 2 cm x 8 cm.
· Placer la plaque dans la chambre chromatographique contenant une couche d'environ 1 cm d'éthanol absolu.
· Développer le chromatogramme jusqu’à ce que le front de solvant migre à environ 6 cm de l’origine (environ 10 minutes).
· Retirer la plaque et la laisser sécher à l’air libre.
· Déterminer la distribution de la radioactivité sur la plaque en balayant le chromatogramme avec un détecteur de radiations adapté ou en coupant la plaque comme montré ci-après (en 3 pièces) et en mesurant la radioactivité dans chaque pièce à l’aide d’un détecteur de radiations adapté.
· Les zones radioactives se calculent en fonction de leur Rf :
o les formes réduites et/ou hydrolysées de Tc-99m restent à l'origine (Rf = 0,0 – 0,1)
o le pertechnétate 99mTcO4- libre (non lié) migre avec le solvant (Rf = 0,4 – 0,7).
o le complexe de technétium (99mTc) sestamibi migre avec le front de solvant (Rf = 0,8 – 1,0).
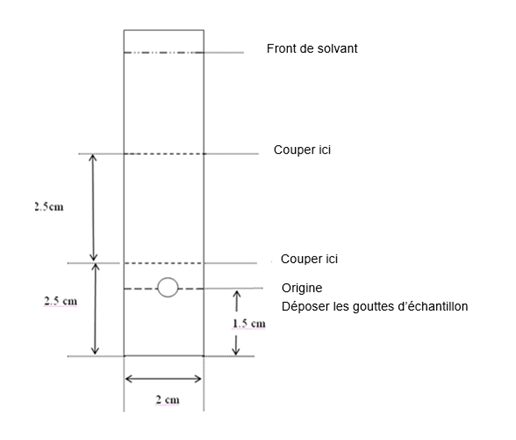
· Le taux de pureté radiochimique sera calculé comme suit : % Tc-99m sestamibi = activité de la partie supérieure (Rf 0,8 – 1,0) divisée par la somme des activités de toutes les parties multipliée par 100 :
Activ. Partie supérieure
% pureté radiochimique = ------------------------- x 100 %
Somme d'activ. de toutes les parties
· Le % de (99mTc) sestamibi doit être >94% ; si ce n’est pas le cas, la préparation doit être jetée.
Remarque : ne pas utiliser un produit dont la pureté radiochimique est inférieure à 94%.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM.
Médicament soumis à prescription médicale.
Les produits radiopharmaceutiques ne doivent être utilisés que par des personnes qualifiées. Ils ne peuvent être délivrés qu’à des praticiens ayant obtenu l’autorisation spéciale prévue à l’article R1333-24 du code de la Santé publique.
ANSM - Mis à jour le : 29/08/2019
CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique
Tétrafluoroborate de tétrakis (1-isocyanide-2-méthoxy-2- méthylpropyl) cuivre (I)
CETTE NOTICE EST DESTINEE AU PATIENT
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou le spécialiste de médecine nucléaire qui va réaliser votre examen.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou au spécialiste de médecine nucléaire. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique ?
3. Comment utiliser CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : V09GA01
Ce médicament est un médicament radiopharmaceutique à usage diagnostique uniquement.
CARDIOMIBI contient une substance appelée tétrafluoroborate de tétrakis (1-isocyanide-2-méthoxy-2- méthylpropyl) cuivre (I) qui est utilisée pour évaluer le fonctionnement du cœur et le débit sanguin (perfusion du myocarde) en obtenant une image du cœur (scintigraphie), par exemple pour la détection d’un risque d’infarctus du myocarde ou lorsqu’une maladie entraîne une diminution de l’afflux sanguin vers le muscle cardiaque (ou une partie de celui-ci) (ischémie). CARDIOMIBI est également utilisé pour le diagnostic des anomalies du sein en complément d’autres méthodes diagnostiques lorsque les résultats n’ont pas été suffisamment concluants. CARDIOMIBI peut également être utilisé pour localiser les glandes parathyroïdiennes hyperactives (les glandes qui sécrètent l’hormone qui contrôle les taux de calcium dans le sang).
Une fois injecté, CARDIOMIBI s’accumule temporairement dans certaines régions du corps. Ce médicament radiopharmaceutique présente une radioactivité faible mais suffisante pour le détecter de l’extérieur du corps à l’aide de caméras spéciales. Votre médecin spécialiste de médecine nucléaire obtiendra alors une image (scintigraphie) de l’organe concerné qui lui fournira des informations utiles sur la structure et le fonctionnement de l’organe étudié, ou la localisation d’une tumeur par exemple.
L’utilisation de CARDIOMIBI entraîne l’exposition à de faibles niveaux de radioactivité. Votre médecin et le spécialiste de médecine nucléaire ont estimé que le bénéfice clinique que vous tirerez de la procédure réalisée avec le produit radiopharmaceutique dépasse le risque lié aux radiations.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique ?
N’utilisez jamais CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique:
· si vous êtes allergique au tétrafluoroborate de tétrakis (1-isocyanide-2-méthoxy-2-méthylpropyl) cuivre (I) ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Faites attention avec CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique
· si vous êtes enceinte ou pensez que vous pourriez être enceinte ;
· si vous allaitez ;
· si vous présentez une affection des reins ou du foie.
Si vous êtes dans l’une des situations ci-dessus, vous devez en informer votre médecin spécialiste de médecine nucléaire. Votre médecin spécialiste de médecine nucléaire vous préviendra si vous devez prendre des précautions particulières après avoir utilisé ce médicament.
Adressez-vous à votre médecin spécialiste de médecine nucléaire si vous avez la moindre question.
Avant l’administration de CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique, vous devez
· être à jeun depuis au moins 4 heures si ce produit est utilisé pour capturer des images de votre cœur
· boire de l’eau en abondance afin de pouvoir uriner aussi souvent que possible pendant les heures suivant la procédure.
Enfants et adolescents
Si vous êtes âgé (e) de moins de 18 ans, signalez-le à votre médecin spécialiste de médecine nucléaire.
Autres médicaments et CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique
Divers médicaments, aliments et boissons peuvent avoir une influence négative sur les résultats de l’examen prévu. Il est donc recommandé que vous parliez avec votre médecin traitant afin de savoir ce que vous ne devez pas prendre avant l’examen et quand vous pourrez recommencer à prendre vos médicaments.
Informez également votre médecin spécialiste de médecine nucléaire si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, car ils pourraient interférer avec l’interprétation des images.
Veillez en particulier à prévenir votre médecin spécialiste de médecine nucléaire si vous prenez des médicaments qui agissent sur la fonction cardiaque et/ou le débit sanguin.
Interrogez votre médecin spécialiste de médecine nucléaire avant de prendre tout médicament.
CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique avec des aliments, boissons et de l’alcool
Sans objet.
S’il est possible que vous soyez enceinte, si vous avez un retard de règles ou si vous allaitez, vous devez en informer le médecin spécialiste de médecine nucléaire avant l’administration de CARDIOMIBI. En cas de doute, il est important de consulter le médecin spécialiste de médecine nucléaire.
Si vous êtes enceinte
Votre médecin spécialiste de médecine nucléaire vous administrera ce médicament uniquement si les bénéfices attendus dépassent les risques éventuels.
Si vous allaitez
Informez-en votre médecin spécialiste de médecine nucléaire, qui vous conseillera de suspendre l’allaitement jusqu’à ce que toute radioactivité ait disparu de votre corps. Il faut pour cela environ 24 heures. Le lait tiré pendant cette période doit être jeté. Demandez à votre médecin spécialiste de médecine nucléaire à quel moment vous pourrez reprendre l’allaitement.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin spécialiste en médecine nucléaire avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Il est considéré comme improbable que CARDIOMIBI ait un effet sur votre aptitude à conduire des véhicules ou à utiliser des machines.
CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu'il est pratiquement « sans sodium ».
3. COMMENT UTILISER CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique ?
Le médecin spécialiste de médecine nucléaire pratiquant l’examen déterminera la quantité de CARDIOMIBI devant être utilisée dans votre cas. Il s’agira de la plus faible quantité nécessaire pour obtenir les informations recherchées.
La quantité qu’il est habituellement recommandé d’administrer chez un adulte varie selon l’examen à pratiquer et peut aller de 200 à 2 000 MBq (méga-becquerel, l’unité utilisée pour exprimer la radioactivité).
Utilisation chez les enfants et les adolescents
Chez les enfants et les adolescents, la quantité à administrer sera ajustée en fonction du poids de l’enfant.
Administration de CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique et déroulement de l’examen
CARDIOMIBI est administré dans une veine du bras ou du pied (par voie intraveineuse).
Une ou deux injection(s) suffisent pour que le médecin puisse effectuer l’examen nécessaire.
Après l’injection, une boisson vous sera proposée et il vous sera demandé d’uriner immédiatement avant l’examen.
Le médecin spécialiste de médecine nucléaire vous indiquera si vous devez prendre des précautions particulières après avoir reçu ce médicament. Contactez votre médecin spécialiste de médecine nucléaire si vous avez la moindre question.
La solution prête à l’emploi vous sera injectée dans une veine avant la scintigraphie. Selon l’examen pratiqué, la scintigraphie pourra être réalisée dans les 5 à 10 minutes après l’injection ou jusqu’à 6 heures plus tard.
Dans le cas d’un examen cardiaque, deux injections peuvent être nécessaires, l’une au repos et l’autre à l’effort (au cours d’une activité physique ou d’une épreuve de stimulation pharmacologique, par ex.). Les deux injections seront pratiquées à un intervalle d’au moins deux heures, sans dépasser 2 000 MBq au total (protocole mené sur 1 jour). Le protocole pourra également se dérouler sur deux jours.
En cas de scintigraphie pour la recherche d’anomalies du sein, une injection de 750 à 1 100 MBq est administrée dans une veine de votre bras, du côté opposé au sein concerné, ou dans une veine de votre pied.
Pour la localisation des glandes parathyroïdiennes hyperactives, l’activité administrée est comprise entre 185 et 1 100 MBq, selon les méthodes utilisées.
Si le produit doit être utilisé pour obtenir des images de votre cœur, il vous sera demandé de ne rien manger pendant au moins 4 heures avant l’examen. Après l’injection, mais avant que la scintigraphie soit réalisée, il vous sera demandé, si possible, de prendre une légère collation riche en graisses ou de boire un ou deux verre(s) de lait afin de réduire la radioactivité dans votre foie et d’améliorer la qualité de l’image.
Durée de la procédure
Votre médecin spécialiste de médecine nucléaire vous informera de la durée habituelle de la procédure.
Après l’administration de CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique, vous devrez :
· éviter tout contact rapproché avec les enfants en bas âge et les femmes enceintes pendant les 24 heures suivant l’injection,
· uriner fréquemment afin d’éliminer le produit de votre corps.
Le médecin spécialiste de médecine nucléaire vous indiquera si vous devez prendre des précautions particulières après avoir reçu ce médicament. Contactez votre médecin spécialiste de médecine nucléaire si vous avez la moindre question.
Si vous avez reçu plus de CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique que vous n’auriez dû
Le surdosage est pratiquement impossible car vous recevrez uniquement une dose de CARDIOMIBI contrôlée avec précision par le médecin spécialiste de médecine nucléaire pratiquant l’examen. Cependant, en cas de surdosage, un traitement approprié vous sera administré. Le médecin spécialiste de médecine nucléaire chargé de l’examen pourra en particulier vous recommander de boire en abondance afin de faciliter l’élimination du produit par votre corps.
Si vous oubliez de prendre CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique
Sans objet.
Si vous arrêtez de prendre CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations au spécialiste de médecine nucléaire supervisant l'examen.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Dans de rares cas, des réactions allergiques ont été observées, celles-ci pouvant s’accompagner d’un essoufflement, d’une fatigue extrême, de nausées (habituellement dans les 2 heures suivant l’administration), d’un gonflement sous la peau pouvant affecter des régions telles que le visage et les membres (œdème de Quincke) et pouvant obstruer les voies respiratoires ou entraîner une dangereuse diminution de la pression artérielle (hypotension) et un ralentissement des battements cardiaques (bradycardie). Les médecins sont conscients de ce risque et tiennent à disposition le traitement d’urgence à utiliser en pareil cas. Des réactions cutanées localisées ont également été observées dans de rares cas, avec des démangeaisons, des poussées d’urticaire, des éruptions cutanées, un gonflement et une rougeur. Si vous ressentez l’un de ces symptômes, adressez-vous immédiatement à votre médecin spécialiste de médecine nucléaire.
Les autres effets indésirables possibles sont énumérés ci-dessous par ordre de fréquence :
|
Fréquence |
Effets indésirables possibles |
|
Fréquent : peut survenir chez au plus 1 personne sur 10 |
Goût métallique ou amer, altération de l’odorat et sécheresse de la bouche immédiatement après l’injection. |
|
Peu fréquent : peut survenir chez au plus 1 personne sur 100 |
Maux de tête, douleur dans la poitrine, anomalies de l’ECG et sensation de malaise. |
|
Rare : peut survenir chez au plus 1 personne sur 1 000 |
Anomalies du rythme cardiaque, réactions locales au site d’injection, douleur à l’estomac, fièvre, perte de connaissance, crises convulsives, étourdissement, bouffées vasomotrices, engourdissement ou picotement de la peau, fatigue, douleurs articulaires et estomac dérangé (dyspepsie). |
|
Fréquence indéterminée : ne peut être estimée sur la base des données disponibles |
Erythème polymorphe, une éruption cutanée étendue touchant la peau et les muqueuses. |
Ce produit radiopharmaceutique délivrera de faibles quantités de rayonnements ionisants, associées au risque minimal de cancer et d’anomalies congénitales.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en au spécialiste de médecine nucléaire qui supervise le traitement. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER CARDIOMIBI 1 mg, trousse pour préparation radiopharmaceutique ?
Vous n'aurez pas à conserver ce médicament. La responsabilité de la conservation de produit dans des locaux appropriés incombe au spécialiste. Le stockage des médicaments radiopharmaceutiques doit être conforme aux réglementations nationales relatives aux produits radioactifs.
Les informations suivantes sont destinées exclusivement au spécialiste :
N’utilisez pas ce médicament après la date de péremption indiquée sur l'étiquette. La date de péremption fait référence au dernier jour de ce mois.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient CARDIOMIBI 1 mg, trousse pour la préparation radiopharmaceutique
· La substance active est : tétrafluoroborate de tétrakis (1-isocyanide-2-méthoxy -2- méthylpropyl) cuivre (I)
Chaque flacon contient 1,0 mg de tétrafluoroborate de tétrakis (1-isocyanide-2-méthoxy-2- méthylpropyl) cuivre (I).
· Les autres composants sont : chlorure stanneux dihydraté, chlorhydrate de L-cystéine monohydraté, citrate de sodium dihydraté, D-mannitol.
Ce produit est une trousse pour préparation radiopharmaceutique.
CARDIOMIBI consiste en une poudre lyophilisée blanche qui doit être dissoute dans une solution et combinée avec du technétium radioactif avant injection. Après ajout de la substance radioactive, le pertechnétate (99mTc) de sodium, une solution de technétium (99mTc) sestamibi est formée. Cette solution est prête à être injectée.
Contenu de la trousse :
Flacon de 10 mL en verre, fermé au moyen d'un bouchon en caoutchouc chlorobutyle serti par une capsule en aluminium.
Les flacons sont emballés dans des boîtes en carton.
Présentation: 3 ou 6 flacons.
Titulaire de l’autorisation de mise sur le marché
NATIONAL CENTRE FOR NUCLEAR RESEARCH
ANDRZEJ SOŁTAN 7
05-400 OTWOCK
POLOGNE
TEL. : +48 22 718 07 00
FAX : +48 22 718 03 50
COURRIEL : POLATOM@POLATOM.PL
Exploitant de l’autorisation de mise sur le marché
CIS BIO INTERNATIONAL
ROUTE NATIONALE 306 SACLAY
BP 32
91192 GIF-SUR-YVETTE
NATIONAL CENTRE FOR NUCLEAR RESEARCH
ANDRZEJ SOŁTAN 7
05-400 OTWOCK
POLOGNE
OU
ROTOP PHARMAKA GMBH
BAUTZNER LANDSTRASSE 400
01328 DRESDEN
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Le Résumé des Caractéristiques du Produit (RCP) complet de CARDIOMIBI est fourni dans une partie détachable à la fin de la notice imprimée dans l'emballage du produit, afin d'offrir aux professionnels de santé d'autres informations de nature scientifique et pratique supplémentaires concernant l'administration et l'utilisation de ce produit radiopharmaceutique.
Veuillez vous reporter au RCP.