Dernière mise à jour le 29/06/2026
MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable
Indications thérapeutiques
Ce médicament se présente sous la forme d'une solution à injecter dans l'œil.
Il contient 3 substances actives :
· le tropicamide, qui appartient à un groupe de médicaments bloquant le passage de l’influx nerveux à travers des nerfs particuliers (connus sous le nom des anticholinergiques) ;
· la phényléphrine (sous forme de chlorhydrate de phényléphrine), qui appartient à un groupe de médicaments mimant les effets des influx nerveux transmis à travers les nerfs particuliers (qui appartient au groupe des alpha-sympathomimétiques) ;
· la lidocaïne (sous forme de chlorhydrate de lidocaïne monohydraté), qui appartient à un groupe de médicaments appelés anesthésiques locaux de type amide.
Pourquoi MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable est-il utilisé
Ce médicament est utilisé uniquement chez l'adulte.
Il doit être administré par un chirurgien ophtalmologiste par injection dans l'œil au début d'une opération de la cataracte (opacification du cristallin) afin d'obtenir un élargissement (mydriase) de la pupille de l'œil et une anesthésie dans votre l'œil au cours de l'opération.
Présentations
> 20 kits contenant 1 x 0,6 ml ampoule stérile et 1 aiguille-filtre de 5 µm stérile sous plaquette (papier/PVC)
Code CIP : 34009 550 555 0 1
Déclaration de commercialisation : 09/10/2019
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 10/07/2019 | Inscription (CT) | Le service médical rendu par MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable est important dans l’indication de l’AMM. |
| Important | Avis du 20/07/2016 | Inscription (CT) | Le service médical rendu par MYDRANE 2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable, est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 10/07/2019 | Inscription (CT) | Ces présentations sont des compléments de gamme qui n’apportent pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
| V (Inexistant) | Avis du 20/07/2016 | Inscription (CT) | MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable, n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres mydriatiques en collyres et insert ophtalmiques. |
Autres informations
- Titulaire de l'autorisation : Laboratoires THEA
- Conditions de prescription et de délivrance :
- liste I
- prescription réservée aux spécialistes et services OPHTALMOLOGIE
- réservé à l'usage HOSPITALIER
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 508 240 8
ANSM - Mis à jour le : 19/03/2024
MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Tropicamide......................................................................................................................... 0,2 mg
Chlorhydrate de phényléphrine.............................................................................................. 3,1 mg
Chlorhydrate de lidocaïne monohydraté.................................................................................. 10 mg
pour 1 ml
Une dose de 0,2 ml de solution contient 0,04 mg de tropicamide, 0,62 mg de chlorhydrate de phényléphrine et 2 mg de chlorhydrate de lidocaïne monohydraté.
Excipient à effet notoire : sodium (0,59 mg par dose ; voir rubrique 4.4).
Pour la liste complète des excipients, voir rubrique 6.1.
Solution limpide légèrement marron-jaune pratiquement exempte de particules visibles.
pH : 6,9 - 7,5
Osmolalité : 290 – 350 mosmol/kg.
4.1. Indications thérapeutiques
MYDRANE est indiqué chez l'adulte uniquement.
4.2. Posologie et mode d'administration
Voie intracamérulaire. Une ampoule à usage unique par œil.
MYDRANE doit être administré par un chirurgien ophtalmologiste.
MYDRANE doit être utilisé uniquement chez les patients ayant déjà présenté, lors d’une évaluation préopératoire, une dilatation satisfaisante de la pupille suite à l'administration d'agents mydriatriques topiques.
Adultes
Injecter lentement par voie intracamérulaire 0,2 ml de MYDRANE en une seule injection au début de l'intervention chirurgicale.
Populations particulières
Sujets âgés
L’adaptation posologique n'est pas nécessaire.
Population pédiatrique
L'efficacité et la tolérance de MYDRANE chez l'enfant âgé de 0 à 18 ans n'ont pas été établies.
Insuffisants rénaux
Etant donné la faible dose utilisée et la très faible exposition systémique (voir rubrique 5.2), aucune adaptation posologique n'est nécessaire (voir rubrique 4.4).
Insuffisants hépatiques
Etant donné la faible dose utilisée et la très faible exposition systémique (voir rubrique 5.2), aucune adaptation posologique n'est nécessaire.
Mode d’administration
Voie intracamérulaire.
La procédure d'administration suivante doit être suivie :
1. Cinq minutes avant de réaliser la procédure antiseptique préopératoire et la première incision, une à deux gouttes de collyre anesthésique doivent être instillées dans l’œil.
2. Au début de l'intervention chirurgicale, 0,2 ml de MYDRANE doit être injecté lentement, en une seule injection intracamérulaire, par un chirurgien ophtalmologiste, par l’incision latérale ou principale.
Pour les instructions relatives à la manipulation du médicament avant administration, voir rubrique 6.6.
· Hypersensibilité connue aux anesthésiques de type amide.
· Hypersensibilité connue aux dérivés atropiniques.
4.4. Mises en garde spéciales et précautions d'emploi
Mises en garde spéciales
La dose recommandée est de 0,2 ml de MYDRANE ; aucune dose supplémentaire ne doit être injectée étant donné qu’aucun effet additionnel significatif n'a été démontré et qu’une augmentation de la perte en cellules endothéliales a été observée (voir rubrique 4.9).
Aucune toxicité sur l'endothélium cornéen n'a été associée à l'utilisation de MYDRANE à la dose recommandée ; néanmoins, en raison de données limitées, ce risque ne peut être exclu.
Il n'existe pas d’expérience clinique avec MYDRANE chez :
· les patients présentant un diabète insulino-dépendant ou non contrôlé ;
· les patients présentant une pathologie cornéenne, notamment ceux présentant une atteinte de l'endothélium cornéen ;
· les patients ayant des antécédents d'uvéite ;
· les patients présentant des anomalies de la pupille ou un traumatisme oculaire ;
· les patients à l’iris très sombre,
· les patients subissant une chirurgie de la cataracte combinée à une greffe de cornée.
Il n'existe aucune expérience avec MYDRANE chez les patients à risque de syndrome de l'iris hypotonique. Chez ce type de patients, une stratégie de dilatation progressive de la pupille à l'aide de collyres mydriatiques peut s'avérer bénéfique.
Il n’existe pas d'expérience clinique avec MYDRANE au cours de la chirurgie de la cataracte chez les patients traités par mydriatiques topiques et pour qui la contraction de la pupille (voire un myosis) se produit pendant la chirurgie.
L'utilisation de MYDRANE n'est pas recommandée dans la chirurgie de la cataracte combinée à une vitrectomie, en raison des effets vasoconstricteurs de la phényléphrine.
MYDRANE n'est pas recommandé chez les sujets présentant une chambre antérieure peu profonde ou des antécédents de glaucome aigu à angle étroit.
L’utilisation de MYDRANE chez des patients présentant une chambre antérieure peu profonde, des antécédents de glaucome aigu à angle étroit et/ou une dilatation de la pupille insuffisante, peut augmenter le risque à la fois de syndrome de l’iris hypotonique et d’iridocèle.
Précautions d'emploi
Après administration de MYDRANE, les concentrations systémiques des substances actives étaient indétectables ou très faibles (voir rubrique 5.2). Etant donné que les effets systémiques de la phényléphrine et de la lidocaïne sont dose-dépendants, il est peu probable que ces effets surviennent lors de l'utilisation de MYDRANE. Cependant, dans la mesure où ce risque ne peut être exclu, il est important de noter que :
· La phényléphrine a une activité sympathomimétique qui pourrait affecter les patients présentant une hypertension, des troubles cardiaques, une hyperthyroïdie, une athérosclérose ou des troubles prostatiques, et chez tous les sujets présentant une contre-indication à l'usage systémique d'amines vasopressives.
· La lidocaïne doit être utilisée avec précaution chez les patients atteints d'épilepsie, de myasthénie, de troubles de la conduction cardiaque, d'insuffisance cardiaque congestive, de bradycardie, de choc sévère, d’insuffisance respiratoire ou d’insuffisance rénale avec une clairance de la créatinine inférieure à 10 ml/minute.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d'interaction n'a été réalisée avec MYDRANE.
Dans la mesure où l'exposition systémique attendue est très faible (voir rubrique 5.2), des interactions systémiques sont peu probables.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas de données suffisamment pertinentes sur l'utilisation de la phényléphrine et du tropicamide chez la femme enceinte. Les études effectuées chez l’animal sont insuffisantes pour permettre de conclure sur des effets sur la grossesse, le développement embryonnaire ou fœtal, l'accouchement ou le développement post-natal.
Bien que les études menées chez l'animal n'aient mis en évidence aucun effet délétère pour le fœtus, la lidocaïne traverse la barrière placentaire et ne doit donc pas être administrée au cours de la grossesse.
Bien que l'absorption systémique attendue soit négligeable, une faible exposition systémique ne peut être exclue.
En conséquence, MYDRANE ne doit pas être utilisé au cours de la grossesse.
Allaitement
Aucune donnée n'est disponible concernant l'excrétion de la phényléphrine ou du tropicamide dans le lait maternel. Comme la phényléphrine est faiblement absorbée oralement, l'absorption éventuelle par le nourrisson peut être estimée négligeable. Cependant, le tropicamide n'est pas recommandé au cours de l'allaitement, car, bien qu’une exposition systémique négligeable soit attendue, les nourrissons peuvent être très sensibles aux anticholinergiques.
De faibles quantités de lidocaïne sont excrétées dans le lait maternel ; une réaction allergique peut donc survenir chez le nourrisson.
En conséquence, MYDRANE ne doit pas être utilisé au cours de l'allaitement.
Fertilité
Aucune information n'est disponible sur les effets potentiels de MYDRANE sur la fertilité chez l'homme ou la femme.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Des effets indésirables ont été rapportés après l’administration de MYDRANE au cours des essais cliniques (voir rubrique 5.1). La plupart étaient des troubles oculaires d'intensité légère à modérée.
Résumé du profil de sécurité :
La rupture de la capsule postérieure et l’œdème maculaire cystoïde sont des complications bien connues survenant pendant ou après une chirurgie de la cataracte. Elles surviennent peu fréquemment (moins d’un cas pour 100 patients).
Tableau récapitulatif des effets indésirables :
Les événements indésirables sont classés selon les catégories de fréquence suivantes : Très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Les effets indésirables rapportés pendant les essais cliniques sont présentés par classe de système d'organes (SOC) dans le tableau suivant et par ordre décroissant de gravité dans chaque catégorie de fréquence :
|
Classe de systèmes d'organes |
Fréquence |
Effet indésirable |
|
Affections du système nerveux |
Peu fréquent |
Céphalées |
|
Affections oculaires |
Peu fréquent |
Kératite, œdème maculaire cystoïde, pression intra-oculaire augmentée, rupture de la capsule postérieure, hyperhémie oculaire |
|
Affections vasculaires |
Peu fréquent |
Hypertension |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Effets systémiques
En raison de l'administration en une seule injection et de la faible absorption systémique attendue, le risque d’effets systémiques dû à un surdosage de MYDRANE est considéré comme minimal.
Les symptômes d'un surdosage en phényléphrine par voie ophtalmique sont plus susceptibles de résulter d'une absorption systémique et incluent fatigue extrême, transpiration, vertiges, ralentissement du rythme cardiaque et coma.
En raison de l’apparition rapide et de la courte durée des réactions toxiques sévères à la phényléphrine le traitement est essentiellement symptomatique. Une injection immédiate d'un agent bloquant alpha-adrénergique à action rapide tel que la phentolamine (à la dose de 2 à 5 mg par voie intraveineuse) a été recommandée.
Les symptômes d'un surdosage de tropicamide par voie ophtalmique incluent maux de têtes, accélération du rythme cardiaque, sécheresse de la bouche et de la peau, somnolence inhabituelle et bouffées vasomotrices.
Aucun effet systémique du tropicamide n'est attendu. En cas de surdosage avec des effets locaux tels qu'une mydriase prolongée, il convient d'administrer de la pilocarpine ou de la physostigmine à 0,25 %.
En cas d'absorption excessive de lidocaïne dans la circulation sanguine, les symptômes peuvent inclure des effets sur le SNC (convulsions, perte de connaissance et potentiellement arrêt respiratoire) et des réactions cardiovasculaires (hypotension, dépression myocardique, bradycardie et potentiellement arrêt cardiaque).
La prise en charge de la toxicité systémique par la lidocaïne consiste à faire cesser les convulsions et à assurer une ventilation adéquate avec de l'oxygène, si nécessaire par ventilation (respiration) assistée ou contrôlée.
Effets locaux
Un surdosage peut entraîner une perte de cellules endothéliales cornéennes (voir rubriques 4.4 et 5.1).
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
MYDRANE est une solution injectable par voie intracamérulaire combinant deux agents mydriatiques de synthèse (le tropicamide, un anticholinergique, et la phényléphrine, un alpha-sympathomimétique) et un anesthésique local (le chlorhydrate de lidocaïne monohydraté).
Mécanisme d’action
La phényléphrine est un agent sympathomimétique à action directe. Elle provoque une mydriase par la stimulation des récepteurs alpha-adrénergiques du muscle dilatateur de la pupille (la contraction du muscle dilatateur qui en résulte entraîne la dilatation pupillaire). Elle n'induit quasiment aucun effet cycloplégique.
Le tropicamide est un agent parasympatholytique qui agit en se liant aux récepteurs muscariniques M4 des muscles oculomoteurs et en les bloquant. Il empêche le sphincter de l'iris et le muscle ciliaire de répondre à la stimulation cholinergique, entraînant une dilatation de la pupille et une paralysie du muscle ciliaire (cycloplégie).
La lidocaïne est un anesthésique local de type amide. Elle agit en inhibant les flux ioniques nécessaires à l'initiation et à la conduction des impulsions nerveuses, stabilisant ainsi la membrane neuronale.
Effets pharmacodynamiques
Bien que le tropicamide en monothérapie produise à la fois une mydriase et une cycloplégie, un effet mydriatique supplémentaire est obtenu lors de l'utilisation simultanée d'agents sympathomimétiques tels que la phényléphrine. Ce type d'association synergique est communément prescrit afin d'obtenir une dilatation maximale de la pupille en vue de l'extraction du cristallin lors d'une chirurgie de la cataracte.
Au cours d'un essai clinique de phase II, en moyenne à 95 % de la dilatation pupillaire mesurée avant l'injection de substance viscoélastique a été obtenue dans les 30 secondes suivant l'injection unique de 200 µl de MYDRANE par voie intracamérulaire. Les mesures de la taille de la pupille observées au cours des essais cliniques de phases II et III sont présentées dans le tableau suivant (patients ayant reçu une injection unique de 200 µl de MYDRANE par voie intracamérulaire) :
|
Etude de phase II, n = 24 |
Etude de phase III, n = 181 |
|||
|
Dans les 30 secondes suivant l'injection de MYDRANE |
Après injection de MYDRANE et injection du viscoélastique |
Après injection de MYDRANE et injection du viscoélastique |
Juste avant injection de l'implant intra-oculaire |
|
|
Taille de la pupille (mm) Moyenne (ET) Médiane |
6,7 (0,7) 6,7 |
7,7 (0,7) 7,7 |
7,8 (0,8) 7,8 |
7,9 (0,9) 7,9 |
Au cours d'un essai de phase III, après injection unique de 200 µl de MYDRANE et injection du viscoélastique (juste avant le capsulorhexis), la taille de la pupille était supérieure ou égale à 7 mm chez 86,7 % des patients. Dans ces essais de phases II et III, la mydriase obtenue sous MYDRANE s'est avérée stable jusqu'à la fin de l'intervention chirurgicale.
La normalisation de la taille de la pupille est connue pour intervenir après 5 à 7 heures.
Efficacité et sécurité clinique
Efficacité clinique
Les effets mydriatiques et anesthésiques de MYDRANE ont été évalués au cours d'une étude de phase III multicentrique randomisée, ouverte, par rapport à un traitement topique standard (phényléphrine et tropicamide) chez 555 patients subissant une chirurgie de la cataracte et présentant un diamètre de la pupille ≥ 7 mm après administration d'agents mydriatiques topiques. Un collyre à base de tétracaïne à 1 % était instillé 5 minutes et 1 minute avant le début de l'intervention dans les deux groupes de traitement.
Mydriase :
La non-infériorité de MYDRANE par rapport au traitement de référence (tropicamide en collyre à 0,5 % et phényléphrine en collyre à 10 %, à raison d'une goutte de chaque produit instillée 3 fois avant l'intervention chirurgicale) a été démontrée pour les critères d'efficacité primaire et co-primaire dans la population en intention de traiter modifiée [mITT] (voir tableau ci-dessous) :
|
Population mITT |
MYDRANE |
Traitement de référence |
Différence (%) entre les groupes (MYDRANE - Référence) |
|
[IC à 95 %] |
|||
|
Critère primaire d'efficacité Nombre (%) de patients répondeurs* IC à 95 % |
N=268 265 (98,9) [96,8 ; 99,8] |
N=281 266 (94,7) [91,3 ; 97,0] |
4,2 [-4,2 ; 12,6] |
|
Critère co-primaire d'efficacité Nombre (%) de patients répondeurs** IC à 95 % |
N=250
246 (98,4) [96,0 ; 99,6] |
N=261
246 (94,3) [90,7 ; 96,7] |
4,1 [-4,5 ; 12,8] |
|
* Patient répondeur défini comme un patient chez qui le capsulorhexis a été réalisé sans utiliser de traitement mydriatique supplémentaire. ** Patient répondeur défini comme un patient chez qui le capsulorhexis a été réalisé sans utiliser de traitement mydriatique supplémentaire et chez qui la taille de la pupille juste avant le capsulorhexis était ≥ 5,5 mm. |
|||
Au cours de l'essai de phase III, dans le groupe MYDRANE (N = 268), 197 patients ont reçu une injection intracamérulaire unique de 200 µl et 71 une injection supplémentaire de 100 µl sans qu’un effet additionnel significatif ait été démontré alors qu’une augmentation de la perte en cellules endothéliales était observée (voir rubrique 4.9).
Le tableau ci-dessous présente l'analyse des données des patients ayant reçu une seule injection de 200 µl par voie intracamérulaire, chez qui le capsulorhexis a été réalisé sans utiliser de traitement mydriatique supplémentaire et chez qui la taille de la pupille avant le capsulorhexis était > 6 mm.
|
|
MYDRANE 200 µl |
Traitement de référence |
Différence (%) entre les groupes (MYDRANE 200 µl - Référence) |
|
[IC à 95 %] |
|||
|
N Nombre (%) de patients sans traitement mydriatique supplémentaire et dont la taille de la pupille juste avant le capsulorhexis est > 6 mm. IC à 95 % |
N=181
[97,0 ; 100,0] |
N=261
[90,7 ; 96,7] |
[-4,3 ; 14,6] |
Anesthésie :
Avant injection de l'implant intra-oculaire, le confort du patient s'est révélé supérieur de manière statistiquement significative sous MYDRANE (p = 0,034), et aucune différence statistiquement significative entre les groupes n'a été observée aux autres temps d'évaluation au cours de l'intervention chirurgicale (avant injection de substance viscoélastique, capsulorhexis et injection de céfuroxime).
5.2. Propriétés pharmacocinétiques
Aucune donnée de pharmacocinétique oculaire n'est disponible pour MYDRANE.
Après injection de MYDRANE par voie intracamérulaire chez 15 patients subissant une chirurgie de la cataracte, les concentrations plasmatiques en principes actifs à 2, 12 et 30 minutes après l'injection ont été comparées à celles obtenues sous traitement topique standard (phényléphrine en collyre à 10 % et tropicamide en collyre à 0,5 %). Concernant le tropicamide, tous les patients du groupe MYDRANE se situaient en-dessous de la limite de quantification (< 0,1 ng/ml), contrairement aux patients du groupe de référence, qui se situaient tous au-dessus de cette limite. Le taux de phényléphrine (limite de quantification < 0,1 ng/ml) était indétectable chez tous les patients du groupe MYDRANE à l'exception de 2 patients (maximum : 0,59 ng/ml). En comparaison, tous les patients du groupe de référence présentaient un taux de phényléphrine supérieur à cette limite (maximum : 1,42 ng/ml). Les concentrations plasmatiques de lidocaïne ont été mesurées chez tous les patients traités par MYDRANE, la concentration la plus élevée obtenue étant 1,45 ng/ml (bien inférieure aux valeurs associées à des effets systémiques, entre 1 500 et 5 000 µg/ml).
5.3. Données de sécurité préclinique
Des signes d'intolérance oculaire n'ont été observés qu'avec les formulations plus concentrées de ces trois substances (à 5 fois ou plus la concentration de MYDRANE). A la concentration maximale testée (10 fois plus élevée), des augmentations de l'épaisseur cornéenne et des atteintes oculaires sévères ont été observées chez un animal euthanasié le jour 3.
Aucune étude n'a été menée sur la toxicité systémique de l'association à dose fixe de phényléphrine, tropicamide et lidocaïne.
Néanmoins, la tolérance ophtalmique de chacune des trois substances étant considérée établie et MYDRANE étant administré exclusivement par injection intracamérulaire unique, aucun risque particulier lié à l'utilisation de cette association n'est attendu.
De même, aucune étude n'a été menée sur la pharmacologie de sécurité, la génotoxicité et la toxicité sur la reproduction de chacune des trois substances composant l'association fixe. Chez le rat, l'administration de phényléphrine (12,5 mg/kg par voie sous-cutanée) a entraîné une réduction du flux sanguin dans l'utérus (réduction de 86,8 % en 15 minutes environ), indiquant des effets fœtotoxiques et co-tératogènes. Concernant la lidocaïne, aucun effet tératogène n'a été observé lors des études sur le développement embryonnaire et fœtal chez le rat et le lapin. Une embryotoxicité et une réduction de la survie post-natale n'ont été mises en évidence qu'à des doses toxiques pour la mère. La lidocaïne n'a également montré aucun potentiel génotoxique.
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Un blister en papier/PVC contenant une ampoule stérile de 1 ml en verre (type I) brun remplie de 0,6 ml de solution injectable. Des aiguilles-filtre de 5 microns stériles sont fournies conditionnées dans des blisters individuels séparés.
Boîte de 1, 20 ou 100 ampoules stériles fournies respectivement avec 1, 20 et 100 aiguilles-filtre de 5 microns stériles.
Kit d’un blister en papier/PVC contenant une ampoule stérile de 1 ml en verre (type I) brun remplie de 0,6 ml de solution injectable et une aiguille-filtre de 5 microns stérile.
Boîte de 1, 20 ou 100 kits (c’est-à-dire blister contenant une ampoule stérile et une aiguille-filtre stérile).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
A usage unique pour un œil seulement.
Utiliser immédiatement après la première ouverture de l'ampoule.
Uniquement pour la présentation en kit (c'est-à-dire blister contenant une ampoule et une aiguille) : coller l'étiquette détachable du blister sur le dossier du patient.
Avertissement : Ne pas utiliser si la plaquette ou la partie détachable sont détériorées ou endommagées. Procéder à l'ouverture uniquement dans des conditions d'asepsie. Le contenu du blister est garanti stérile.
La solution doit être inspectée visuellement et ne doit être utilisée que si elle est limpide, légèrement marron-jaune et pratiquement exempte de particules visibles.
MYDRANE doit être administré exclusivement par injection intracamérulaire par un chirurgien ophtalmologiste dans les conditions d'asepsie recommandées pour une chirurgie de la cataracte.
Respecter les instructions suivantes lors de la préparation du produit en vue d'une injection intracamérulaire :
1. Inspecter la plaquette avant ouverture afin de vérifier son intégrité. Décoller la partie détachable pour ouvrir la plaquette dans des conditions aseptiques afin de garantir la stérilité du contenu.
2. Ouvrir l'ampoule stérile contenant le médicament. Instructions à suivre pour ouvrir l'ampoule au niveau de l’unique point de cassure (OPC) : Tenir la partie inférieure de l'ampoule avec le pouce pointant sur le point coloré. Saisir la tête de l'ampoule avec l'autre main en positionnant le pouce sur le point coloré et presser pour casser l'ampoule au niveau du point de cassure situé en dessous du point coloré.
3. Assembler l'aiguille-filtre de 5 microns stérile (fournie) sur une seringue stérile. Retirer le capuchon protecteur de l'aiguille-filtre de 5 microns stérile et aspirer au moins 0,2 ml de solution injectable de l'ampoule dans la seringue.
4. Déconnecter l'aiguille de la seringue et assembler la seringue sur une canule appropriée pour la chambre antérieure.
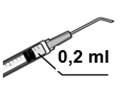
5. Expulser l’air de la seringue avec précaution. Ajuster la dose à 0,2 ml. La seringue est prête pour l’injection.
6. Injecter lentement le volume de 0,2 ml contenu dans la seringue dans la chambre antérieure de l'œil en une seule injection par l’incision latérale ou principale.
7. Après utilisation, éliminer la solution restante de manière appropriée. Ne pas la conserver en vue d’une utilisation ultérieure.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur. Jeter les aiguilles utilisées dans un conteneur pour objets piquants.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
12 RUE LOUIS BLERIOT
63017 CLERMONT-FERRAND CEDEX 2
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 550 153 8 3 : 0,6 ml de solution injectable en ampoule en verre brun sous plaquette (papier/ PVC) fournie avec 1 aiguille-filtre de 5 µm; boîte de 1
· 34009 550 153 9 0 : 0,6 ml de solution injectable en ampoule en verre brun sous plaquette (papier/ PVC) fournie avec 1 aiguille-filtre de 5 µm; boîte de 20
· 34009 550 154 1 3 : 0,6 ml de solution injectable en ampoule en verre brun sous plaquette (papier/ PVC) fournie avec 1 aiguille-filtre de 5 µm; boîte de 100
· 34009 550 554 8 8 : 1 kit contenant 1 x 0,6 ml ampoule stérile et 1 aiguille-filtre de 5 µm stérile sous plaquette (papier/PVC).
· 34009 550 555 0 1 : 20 kits contenant 1 x 0,6 ml ampoule stérile et 1 aiguille-filtre de 5 µm stérile sous plaquette (papier/PVC)
· 34009 550 555 1 8 : 100 kits contenant 1 x 0,6 ml ampoule stérile et 1 aiguille-filtre de 5 µm stérile sous plaquette (papier/PVC)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament réservé à l’usage hospitalier.
Prescription réservée aux spécialistes en ophtalmologie.
ANSM - Mis à jour le : 19/03/2024
MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable
tropicamide / chlorhydrate de phényléphrine / chlorhydrate de lidocaïne monohydraté
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable ?
3. Comment utiliser MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Ce médicament se présente sous la forme d'une solution à injecter dans l'œil.
Il contient 3 substances actives :
· le tropicamide, qui appartient à un groupe de médicaments bloquant le passage de l’influx nerveux à travers des nerfs particuliers (connus sous le nom des anticholinergiques) ;
· la phényléphrine (sous forme de chlorhydrate de phényléphrine), qui appartient à un groupe de médicaments mimant les effets des influx nerveux transmis à travers les nerfs particuliers (qui appartient au groupe des alpha-sympathomimétiques) ;
· la lidocaïne (sous forme de chlorhydrate de lidocaïne monohydraté), qui appartient à un groupe de médicaments appelés anesthésiques locaux de type amide.
Pourquoi MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable est-il utilisé
Ce médicament est utilisé uniquement chez l'adulte.
Il doit être administré par un chirurgien ophtalmologiste par injection dans l'œil au début d'une opération de la cataracte (opacification du cristallin) afin d'obtenir un élargissement (mydriase) de la pupille de l'œil et une anesthésie dans votre l'œil au cours de l'opération.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable?
· si vous êtes allergique au tropicamide, au chlorhydrate de phényléphrine et/ou au chlorhydrate de lidocaïne monohydraté ou à l'un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6 ;
· Si vous êtes allergique aux anesthésiques de type amide (articaïne, bupivacaïne, mepivacaïne, prilocaïne, ropivacaïne).
· si vous êtes allergique aux dérivés atropiniques
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable.
L'utilisation de MYDRANE n'est pas recommandée :
· si vous devez subir une opération de la cataracte combinée à un certain type de chirurgie de l'œil (vitrectomie) ;
· si la partie antérieure (chambre antérieure) de votre œil est peu profonde ;
· si vous avez des antécédents d'augmentation aiguë de la pression intraoculaire (glaucome aigu à angle étroit).
Adressez-vous à votre médecin en particulier :
· si vous avez une pression artérielle élevée (hypertension) ;
· si vous présentez un épaississement de la paroi de vos artères (athérosclérose) ;
· si vous êtes atteint d'une maladie cardiaque et en particulier si cela entraîne un trouble du rythme cardiaque ;
· si vous présentez une contre-indication aux médicaments augmentant la pression sanguine (amines vasopressives : épinéphrine, norépinéphrine, dopamine, dobutamine) administrés par voie générale ;
· si votre glande thyroïde est hyperactive (hyperthyroïdie) ;
· si vous avez des problèmes de prostate ;
· si vous êtes épileptique ;
· si vous avez des problèmes de foie ou de reins ;
· si vous avez des problèmes respiratoires ;
· si vous avez une perte de la fonction musculaire ou une faiblesse musculaire (myasthénie).
Enfants
Sans objet.
Autres médicaments et MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
Ce médicament ne doit pas être utilisé :
· en cas de grossesse ;
· en cas d'allaitement.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
MYDRANE a une influence modérée sur l'aptitude à conduire des véhicules et à utiliser des machines. Par conséquent, vous ne devez pas conduire et/ou utiliser de machines jusqu'à ce que votre vue redevienne normale.
MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable contient du sodium
Ce médicament contient moins de 1mmol (23 mg) de sodium par dose, c'est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable ?
Posologie et mode d’administration
1. L'injection de MYDRANE doit être effectuée par un chirurgien ophtalmologiste sous anesthésie locale au début de l'opération de la cataracte.
2. La dose recommandée est de 0,2 ml de solution en une seule injection. Aucune dose supplémentaire ne doit être injectée dans la mesure où aucun effet additionnel n’a été démontré et qu’une augmentation de la perte en cellules endothéliales (cellules de la couche recouvrant la surface postérieure de la cornée) a été observée.
La même dose doit être utilisée chez l'adulte et la personne âgée.
Si vous avez utilisé plus de MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable que vous n’auriez dû
Ce médicament doit être administré par un chirurgien ophtalmologiste.
Il est peu probable qu’il vous soit administré un surdosage.
Un surdosage peut entraîner une perte accrue de cellules endothéliales cornéennes (cellules couvrant la surface postérieure de la cornée).
Si vous oubliez d’utiliser MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable
Si vous arrêtez d’utiliser MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable
Sans objet.
Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez plus d'informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Principales complications graves bien connues survenant pendant et après la chirurgie de la cataracte :
Peu fréquent : pouvant affecter jusqu'à 1 personne sur 100
· Traumatisme chirurgical du cristallin (rupture de la capsule postérieure),
· Gonflement de la rétine (œdème cystoïde maculaire),
Dans ce cas, veuillez consulter un médecin de toute urgence.
Autres effets indésirables :
Peu fréquent : pouvant affecter jusqu'à 1 personne sur 100
· Maux de tête,
· Gonflement de la cornée (kératite), augmentation de la pression dans l’œil, rougeur de l’œil (hyperhémie oculaire),
Augmentation de la pression artérielle (hypertension)
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte, la plaquette et l'ampoule. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précaution particulière de conservation.
A usage unique pour un œil seulement. Ce médicament doit être utilisé immédiatement après la première ouverture de l'ampoule.
Ne jetez aucun médicament au tout-à-l'égout ou avec les ordures ménagères. Demandez à votre pharmacien d'éliminer les médicaments que vous n'utilisez plus. Ces mesures contribueront à protéger l'environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml, solution injectable
· Les substances actives sont :
Tropicamide................................................................................................................... 0,2 mg
Chlorhydrate de phényléphrine........................................................................................ 3,1 mg
Chlorhydrate de lidocaïne monohydraté............................................................................. 10 mg
pour 1 ml
Une dose de 0,2 ml de solution contient 0,04 mg de tropicamide, 0,62 mg de chlorhydrate de phényléphrine et 2 mg de chlorhydrate de lidocaïne monohydraté.
· Les autres composants sont :
Chlorure de sodium, phosphate disodique dodécahydraté, phosphate disodique dihydraté, édétate disodique et eau pour préparations injectables.
MYDRANE se présente sous la forme d'une solution injectable limpide, légèrement marron-jaune, pratiquement exempte de particules visibles, disponible en ampoule stérile de 1 ml en verre brun. Chaque ampoule stérile contient 0,6 ml de solution injectable et est présentée seule ou avec une aiguille-filtre de 5 microns dans une plaquette en papier/PVC scellé.
Chaque boîte contient 1, 20 ou 100 ampoules stériles avec aiguille(s)-filtre stérile(s) de 5 microns à part ou dans le même blister. Les aiguilles-filtre de 5 microns doivent être utilisées uniquement lors de l'aspiration du contenu de l'ampoule dans la seringue. Tous les composants sont à usage unique.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
12 RUE LOUIS BLERIOT
63017 CLERMONT-FERRAND CEDEX 2
Exploitant de l’autorisation de mise sur le marché
37 RUE GEORGES BESSE
63100 CLERMONT-FERRAND
RUE PAUL LANGEVIN
37170 CHAMBRAY LES TOURS
OU
LABORATOIRES THEA
12 RUE LOUIS BLERIOT
63017 CLERMONT-FERRAND CEDEX 2
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{MM/AAAA}
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Incompatibilités
Aucune incompatibilité n'a été rapportée en cas d'utilisation des substances actives de MYDRANE avec les produits les plus couramment utilisés dans la chirurgie de la cataracte dans la littérature et au cours des essais cliniques. Un test d'interaction pharmaceutique a permis de confirmer ces données pour les substances viscoélastiques courantes.
Avertissements
Ne pas utiliser si la plaquette est détériorée ou endommagée. Procéder à l'ouverture uniquement dans des conditions d'asepsie. Le contenu de la plaquette avant ouverture est garanti stérile.
Comment préparer et administrer MYDRANE
Solution à usage unique pour injection par voie intracamérulaire exclusivement.
MYDRANE doit être administré exclusivement par injection intraoculaire dans la chambre antérieure de l'œil (injection intracamérulaire) par un chirurgien ophtalmologiste dans les conditions d'asepsie recommandées pour une chirurgie de la cataracte.
Avant injection par voie intracamérulaire, la solution doit être inspectée visuellement et ne doit être utilisée que si elle est limpide, légèrement marron-jaune et pratiquement exempte de particules visibles.
La dose recommandée est de 0,2 ml de MYDRANE ; aucune dose supplémentaire ne doit être injectée dans la mesure où aucun effet additionnel significatif n'a été démontré et qu’une augmentation de la perte en cellules endothéliales a été observée.
Le produit doit être utilisé immédiatement après ouverture de l'ampoule et ne doit pas être réutilisé pour l'autre œil ou pour un autre patient.
Uniquement pour la présentation en kit (c'est-à-dire blister contenant une ampoule et une aiguille) : coller l'étiquette détachable du blister sur le dossier du patient.
Respecter les instructions suivantes lors de la préparation de MYDRANE en vue d'une administration intracamérulaire : |
||||
|
1. Inspecter la plaquette avant ouverture afin de vérifier son intégrité. Décoller la partie détachable pour ouvrir la plaquette dans des conditions aseptiques afin de garantir la stérilité du contenu. |
|||
|
|
2. Ouvrir l'ampoule stérile contenant le médicament. Instructions à suivre pour ouvrir l'ampoule au niveau de l’unique point de cassure (OPC) : Tenir la partie inférieure de l'ampoule avec le pouce pointant sur le point coloré. Saisir la tête de l'ampoule avec l'autre main en positionnant le pouce sur le point coloré et presser pour casser l'ampoule au niveau du point de cassure situé en dessous du point coloré. |
|||
|
|
3. Assembler l'aiguille-filtre de 5 microns stérile (fournie) sur une seringue stérile. Ôter le capuchon protecteur de l'aiguille-filtre de 5 microns stérile et aspirer au moins 0,2 ml de solution injectable de l'ampoule dans la seringue. |
|||
|
|
4. Déconnecter l'aiguille de la seringue et assembler la seringue sur une canule appropriée pour la chambre antérieure. |
|||
|
|
5. Expulser l’air de la seringue avec précaution. Ajuster la dose à 0,2 ml. La seringue est prête pour l’injection. 6. Injecter lentement le volume de 0,2 ml contenu dans la seringue dans la chambre antérieure de l'œil en une seule injection par l’incision latérale ou principale. |
|||
|
Après utilisation, éliminer la solution restante. Ne pas la conserver en vue d’une utilisation ultérieure. |
||||




Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur. Eliminer les aiguilles usagées dans un conteneur pour objets piquants.