Dernière mise à jour le 01/06/2026
BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose
Indications thérapeutiques
GLUCOCORTICOÏDE PAR VOIE INHALEE – Code ATC : R03BA02 (R : Système respiratoire).
ANTIASTHMATIQUE
BUDESONIDE ZENTIVA contient la substance active budésonide. Il appartient à une classe médicamenteuse appelée corticostéroïdes qui possèdent une action anti-inflammatoire réduisant les gonflements et l’irritation dans les voies respiratoires (par exemple, nez, poumons) soulageant ainsi les difficultés respiratoires.
BUDESONIDE ZENTIVA nébulisation s'administre par voie inhalée à l'aide d'un appareil de nébulisation.
Il est indiqué dans le traitement de l’asthme chez les adultes, les adolescents et les enfants lorsque l’utilisation des inhalateurs pressurisés ou à poudre sèche ne peuvent être utilisés ou sont inadaptés.
Attention : ce médicament n’est pas un bronchodilatateur.
Il ne permet pas le traitement de la crise d’asthme.
Présentations
> 20 récipient(s) unidose(s) polyéthylène basse densité (PEBD) de 2 ml en sachets polytéréphtalate (PET) aluminium polyéthylène de (4x5)
Code CIP : 34009 302 023 4 0
Déclaration de commercialisation : 23/03/2021
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 24,20 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 25,22 €
- Taux de remboursement :65 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 04/06/2024
BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Budésonide micronisé............................................................................................................. 1 mg
Pour un récipient unidose de 2 ml.
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension pour inhalation par nébuliseur.
4.1. Indications thérapeutiques
BUDESONIDE ZENTIVA, suspension pour inhalation par nébuliseur en récipient unidose, est indiqué :
· en traitement de fond de l’asthme chez les adultes, les adolescents et les enfants lorsque les inhalateurs pressurisés ou à poudre sèche ne peuvent être utilisés ou sont inadaptés.
4.2. Posologie et mode d'administration
La dose initiale sera déterminée selon la sévérité de l'asthme avant traitement et sera ensuite ajustée en fonction des résultats individuels.
Adulte : 0,5 mg à 4 mg par jour.
Enfant : 0,25 mg à 2 mg par jour. La dose de 2 mg est réservée au traitement de l'asthme sévère.
Les administrations sont habituellement réparties en 2 séances de nébulisation par jour. Une dose quotidienne allant jusqu’à 1mg peut être administrée en une seule séance de nébulisation par jour.
La suspension contenue dans l’unidose est stérile. La quantité inutilisée restant dans l’unidose entamée doit être jetée.
Après plusieurs jours (ou semaines) de traitement initial lorsque l’état clinique est amélioré, que les symptômes ont régressé et que l’asthme est contrôlé, la posologie minimale efficace devra être recherchée. Dans ce but, la dose quotidienne pourra être administrée en une séance de nébulisation par jour, si ce rythme d’administration favorise la compliance au traitement.
En cas de déstabilisation de l’asthme, la dose et le nombre de prises devront être ré-augmentés.
Chez les enfants de moins de 5 ans présentant des épisodes récurrents de sifflements bronchiques, l'administration de budésonide inhalé en cure de 2 à 3 mois peut être envisagée dans le but de déterminer s'il s'agit d'un asthme. Une amélioration significative des symptômes sous traitement corticoïde et leur réapparition lors de l'arrêt du traitement doit faire évoquer le diagnostic d'asthme. Le traitement par BUDESONIDE ZENTIVA, nébulisation devra être arrêté si aucun bénéfice clinique n’est observé dans les 2-3 mois. Sauf si le diagnostic d'asthme est confirmé, le traitement par budésonide ne doit pas être maintenu plus de 3 mois pour éviter une exposition prolongée injustifiée (voir rubrique 4.4.et 4.8).
Mode d’administration
Cette suspension de budésonide doit être administrée par voie inhalée à l’aide d’un appareil pour nébulisation (nébuliseur) à air comprimé (ou pneumatique). Les générateurs ultrasoniques ne sont pas recommandés car ils peuvent ne pas être adaptés pour une administration correcte de BUDESONIDE ZENTIVA.
NE PAS INJECTER – NE PAS AVALER
La suspension de BUDESONIDE ZENTIVA est prête à l’emploi.
Les patients doivent être informés qu'ils doivent suivre attentivement les instructions du fabricant pour l’utilisation et l'entretien de l'appareil de nébulisation.
Un volume de remplissage de 2 à 4 ml convient pour la plupart des nébuliseurs. Si nécessaire, compléter au volume recommandé avec du sérum physiologique stérile.
Le mélange obtenu est pulsé par débit d’air ou oxygène (6 à 8 litres par minutes) pendant environ 10 à 15 minutes durant lesquelles le patient respire à son rythme habituel.
La technique d’utilisation par le patient doit être vérifiée régulièrement.
Après inhalation, la suspension inutilisée restant dans la cuve de l’appareil doit être jetée.
Il convient de recommander au patient de se rincer la bouche après chaque séance de nébulisation afin de diminuer le risque de candidose oro-pharyngée et de se rincer le visage à l’eau en cas d’utilisation d’un masque facial afin de diminuer le risque d'irritations locales cutanées au niveau du visage.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Si, en dépit d’un traitement bien conduit, une dyspnée paroxystique survient, on doit avoir recours à un bronchodilatateur bêta2 mimétique par voie inhalée d’action rapide et de courte durée pour traiter les symptômes aigus. Il conviendra d’en informer le patient et de lui préciser qu’une consultation médicale immédiate est nécessaire si, dans ce cas, le soulagement habituellement obtenu n’est pas rapidement observé après inhalation du bronchodilatateur bêta2 mimétique.
Si un patient développe en quelques jours une augmentation rapide de sa consommation en bronchodilatateurs bêta2 mimétiques d’action rapide et de courte durée par voie inhalée, on doit craindre (surtout si les valeurs du débit-mètre de pointe s’abaissent et/ou deviennent irrégulières) une décompensation de sa maladie et la possibilité d’une évolution vers un asthme aigu grave (état de mal asthmatique). Le médecin devra également prévenir le patient de la nécessité dans ce cas, d’une consultation immédiate. La conduite thérapeutique devra alors être réévaluée.
Le patient doit être averti que l’amélioration de son état clinique ne doit pas conduire à une modification de son traitement, en particulier à l’arrêt de la corticothérapie par voie inhalée, sans avis médical.
En cas d’infection bronchique ou de bronchorrhée abondante, un traitement approprié est nécessaire afin de favoriser la diffusion optimale du produit dans les voies respiratoires.
En cas de déstabilisation de l’asthme, ou de contrôle insuffisant des exacerbations d’asthme malgré des doses maximales de corticoïdes par voie inhalée, un traitement par corticothérapie par voie générale en cure courte doit être envisagé. Il est alors nécessaire de maintenir la corticothérapie inhalée associée au traitement par voie générale.
La corticothérapie par voie inhalée peut entraîner des effets systémiques, en particulier lors de traitements à fortes doses ou prolongés. La survenue de ces effets avec la voie inhalée est beaucoup moins probable qu’au cours d’une corticothérapie orale. Les effets systémiques possibles sont : syndrome de Cushing ou tableau cushingoïde, amincissement cutané, hématomes sous cutanés, insuffisance surrénalienne, retard de croissance chez les enfants et les adolescents, diminution de la densité osseuse, cataracte et glaucome et plus rarement, troubles psychologiques et du comportement comprenant hyperactivité psychomotrice, troubles du sommeil, anxiété, dépression ou agressivité (en particulier chez l’enfant). Il est important de toujours rechercher la posologie minimale efficace de corticoïdes inhalés permettant d'obtenir le contrôle des symptômes d'asthme.
Il convient de garder en mémoire les effets potentiels sur la densité minérale osseuse en particulier chez les patients recevant de fortes doses de corticoïdes par voie inhalée au long cours et présentant des facteurs de risque d’ostéoporose. Il n’a pas été mis en évidence d’effets significatifs sur la densité minérale osseuse au cours d’études cliniques à long terme effectuées chez des enfants recevant en moyenne 400 µg/j (dose nominale) de budésonide ou chez des adultes recevant 800 µg/j (dose nominale) de budésonide. Aucune donnée n’est disponible concernant l’effet à des doses plus élevées.
Les patients ayant nécessité de fortes doses de corticostéroïdes en urgence ou ayant reçu une corticothérapie inhalée au long cours aux posologies maximales recommandées, peuvent développer une insuffisance surrénalienne. Ces patients sont susceptibles de présenter des signes et des symptômes d’insuffisance surrénalienne lors de situation de stress sévère. Les signes d’une insuffisance surrénale aigue peuvent être non spécifiques : anorexie, douleurs abdominales, perte de poids, fatigue, céphalées, nausées, vomissements, perte de connaissance, convulsions, hypotension et hypoglycémie. Une corticothérapie de supplémentation devra être envisagée dans les situations susceptibles de déclencher un stress ou en cas de chirurgie programmée.
L’administration conjointe de corticoïdes par voie inhalée chez les patients sous corticothérapie orale au long cours (patients corticodépendants) ne dispense pas des précautions nécessaires lors d’une réduction des doses de corticoïdes par voie orale. Celles-ci seront diminuées très progressivement et le sevrage devra être effectué sous surveillance médicale attentive (à la recherche de l’apparition de signes d’insuffisance surrénale aiguë ou subaiguë) qui peut persister pendant une période prolongée après l’arrêt de la corticothérapie générale.
Lors du remplacement d'une corticothérapie orale par une corticothérapie inhalée, l’effet systémique du corticoïde est diminué ce qui peut entrainer la réapparition de symptômes allergiques (tels que rhinite, eczéma) et/ou rhumatologiques (telles que douleurs musculaires et articulaires). Un traitement spécifique devra être instauré. Une insuffisance cortico-surrénalienne doit être suspectée si, dans de rares cas, les symptômes suivants surviennent : fatigue, céphalée, nausée et vomissements. Une augmentation temporaire des doses de corticoïdes oraux peut alors parfois être nécessaire.
En cas de tuberculose pulmonaire active ou quiescente, d’infection mycosique pulmonaire, l’instauration d’une surveillance étroite et d’un traitement adapté s’impose.
La corticothérapie inhalée peut entrainer une candidose oropharyngée pouvant nécessiter un traitement antifongique et un arrêt de la corticothérapie inhalée (voir également rubrique 4.2).
Comme avec les autres produits inhalés, un bronchospasme peut survenir se manifestant par une majoration des sibilants, une dyspnée et une toux immédiatement après la prise du médicament. Le bronchospasme sera traité avec un bronchodilatateur d’action rapide qui devra être administré immédiatement. Le traitement par BUDESONIDE ZENTIVA devra être arrêté immédiatement et la conduite thérapeutique sera réévaluée pour envisager, si nécessaire, les alternatives thérapeutiques.
Des troubles visuels peuvent apparaitre lors d’une corticothérapie par voie systémique ou locale. En cas de vision floue ou d’apparition de tout autre symptôme visuel apparaissant au cours d’une corticothérapie, un examen ophtalmologique est requis à la recherche notamment d’une cataracte, d’un glaucome, ou d’une lésion plus rare telle qu’une choriorétinopathie séreuse centrale, décrits avec l’administration de corticostéroïdes par voie systémique ou locale.
En cas d'insuffisance hépatique, l'élimination des corticoïdes est réduite et en conséquence expose les patients à des concentrations systémiques plus élevées et une augmentation du risque d'effets systémiques. La prudence est requise en cas d'insuffisance hépatique.
L’attention des sportifs sera attirée sur le fait que cette spécialité contient un principe actif pouvant induire une réaction positive des tests pratiqués lors des contrôles antidopage.
Population pédiatrique
Il a été observé un ralentissement initial léger mais généralement transitoire de la croissance (environ 1 cm), qui apparait habituellement pendant la 1ère année de traitement. Des études à long terme en pratique clinique suggèrent que les enfants et les adolescents traités par du budésonide inhalé atteignent en moyenne leur taille adulte prédite. Toutefois, dans une étude clinique à long terme menée en double aveugle, dans laquelle la dose administrée de budésonide inhalé n’était généralement pas ajustée à la dose minimale efficace, les enfants et les adolescents traités par du budésonide inhalé ont atteint une taille adulte en moyenne de 1,2 cm de moins que ceux randomisés sous placebo. La croissance des enfants recevant une corticothérapie inhalée à long terme doit être surveillée régulièrement. En cas de ralentissement de la croissance, le traitement devra être réévalué en vue de réduire les doses du corticoïde inhalé.
Il conviendra de soigneusement peser les bénéfices attendus d’une corticothérapie face aux risques éventuels de ralentissement de la croissance. L’avis d’un spécialiste pneumo-pédiatre peut être requis.
La décision d’instaurer un traitement inhalé par budésonide chez l’enfant jusqu’à 5 ans présentant des épisodes récurrents de sifflements bronchiques doit tenir compte de la sévérité et de la fréquence des épisodes de sibilances. Un suivi régulier est essentiel afin de réévaluer la réponse au traitement. Si aucun bénéfice clinique n’est observé sous traitement dans les 2-3 mois, ou si le diagnostic d’asthme n’est pas confirmé, le traitement par budésonide doit être arrêté afin d’éviter une exposition prolongée non justifiée aux corticoïdes inhalés et les risques associés de retard de croissance (voir rubrique 4.2 et rubrique 4.8.).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Associations à prendre en compte
+ ritonavir, kétoconazole, itraconazole
Augmentation des concentrations plasmatiques du budésonide par diminution de son métabolisme hépatique par l’inhibiteur enzymatique, avec risque d’apparition d’un syndrome cushingoïde.
Le budésonide est principalement métabolisé par le cytochrome CYP P450 3A4. Une augmentation significative des taux sanguins de budésonide peut être observée avec les inhibiteurs puissants du CYP3A4 (ex : kétoconazole, itraconazole, voriconazole, posaconazole, clarithromycine, télithromycine, néfazodone et inhibiteurs des protéases du VIH). La prise concomitante de ces médicaments doit être évitée. Si cette association ne peut être évitée, un intervalle de temps suffisamment long devra être respecté entre l’administration de l’inhibiteur du CYP3A4 et celle du budésonide (voir rubrique 5.2.).
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données disponibles sur un grand nombre de grossesses n’ont pas révélé d’augmentation du risque tératogène associé à l’utilisation du budésonide inhalé. Chez l’animal, les glucocorticoïdes induisent des malformations (voir rubrique 5.3). Toutefois ces observations ne semblent pas pertinentes chez la femme enceinte aux doses thérapeutiques.
Il est important pour le fœtus et la mère de maintenir un traitement adéquat de l'asthme pendant la grossesse.
Le budésonide pourra être administré pendant la grossesse si le bénéfice attendu chez la mère l’emporte sur les risques encourus par le fœtus.
Le budésonide est excrété dans le lait maternel. Toutefois, une étude de pharmacocinétique a montré qu'après administration de budésonide inhalé aux doses de 200 ou 400 µg deux fois par jour, l'exposition systémique au budésonide chez les enfants allaités était négligeable. A doses thérapeutiques, il n’est pas attendu de retentissement sur l’enfant allaité.
Le budésonide peut être utilisé au cours de l’allaitement si nécessaire.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les évènements indésirables imputables au budésonide sont présentés ci-après par classe-organe et par fréquence. Les fréquences sont définies telles que : très fréquents (≥ 1/10), fréquents (≥ 1/100 et < 1/10), peu fréquents (≥ 1/1000 et < 1/100), rares (≥ 1/10 000 et < 1/1000) et très rares (< 1/10 000) et indéterminée (la fréquence de survenue ne peut être estimée d’après les données disponibles).
|
Classe organe |
Fréquence |
Evènement indésirable |
|
Infections et infestations |
Fréquent |
Candidose oropharyngée |
|
Troubles du système immunitaire |
Rare |
Réactions d’hypersensibilité immédiate et retardée, telles que : rash cutané, dermatite de contact, urticaire, angioedème et réaction anaphylactique |
|
Troubles endocriniens |
Rare |
Inhibition des fonctions surrénaliennes, Retard de croissance* |
|
Troubles oculaires |
Peu fréquent Peu fréquent Inconnue |
Cataracte Vision floue (voir rubrique 4.4) Glaucome |
|
Troubles psychiatriques |
Peu fréquent Rare Inconnue |
Anxiété, dépression Impatiences, nervosité, Troubles du comportement (principalement chez les enfants) Troubles du sommeil, Hyperactivité psychomotrice, Agressivité |
|
Troubles du système nerveux |
Peu fréquent |
Tremblements |
|
Troubles respiratoires, thoraciques et médiastinaux |
Fréquent
|
Toux, raucité de la voix, irritation pharyngée
Bronchospasme |
|
Troubles cutanés et du tissu sous-cutané |
Rare |
Ecchymoses |
|
Troubles musculo-squelettiques et du tissu conjonctif |
Peu fréquent |
Contractures musculaires |
Description de l’effet indésirable : irritation cutanée au niveau du visage
Des cas d'irritation du visage ont été décrits lors de l'utilisation d'un masque facial pour la nébulisation. Il est recommandé de rincer le visage à l'eau après une séance de nébulisation utilisant un masque facial.
Effets systémiques
Occasionnellement, des signes et symptômes d’effets secondaires systémiques liés aux glucocorticoïdes peuvent survenir lors de l’utilisation de glucocorticoïdes inhalés (voir rubrique 4.4).
* Population pédiatrique
Compte-tenu du risque de ralentissement de croissance dans la population pédiatrique, la croissance des enfants et des adolescents doit être surveillée régulièrement (voir rubrique 4.4.).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Un surdosage aigu en budésonide inhalé ne devrait pas avoir d’impact clinique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Le budésonide, en inhalation, exerce une action anti inflammatoire marquée sur la muqueuse bronchique.
Chez l'adulte, l'effet freinateur du budésonide sur l'axe hypophysosurrénalien ne se manifeste qu'à une posologie supérieure ou égale à 1600 µg/24 heures.
5.2. Propriétés pharmacocinétiques
Chez l’adulte, la biodisponibilité systémique du budésonide après une administration de budésonide suspension pour inhalation par nébuliseur au moyen d’un nébuliseur à air comprimé est d’environ 15% de la dose nominale et 40 à 70% de la dose effectivement délivrée aux patients. Une faible fraction de la quantité de budésonide circulant provient du médicament dégluti. La concentration maximale atteinte environ 10 à 30 minutes après le début de la nébulisation est d’environ de 4 nmol/L pour une dose administrée de 2 mg.
Distribution
Le budésonide a un volume de distribution de 3 litres/kg. La fixation aux protéines plasmatiques est d’environ 85 à 90%.
Biotransformation
Le budésonide subit un important effet de premier passage hépatique (90%) avec transformation en métabolites pratiquement dénués d’activité glucocorticoïde. L’activité glucocorticoïde des métabolites principaux, 6β-hydroxybudésonide et 16α-hydroxyprednisolone, est inférieure à 1%.
Le budésonide est principalement métabolisé par le cytochrome CYP450 3A.
Élimination
Les métabolites du budésonide sont excrétés dans les urines, en partie sous forme conjuguée. Le budésonide est retrouvé en quantité négligeable sous forme inchangée.
La clairance plasmatique du budésonide est élevée (environ 1,2 L/min) et la demi-vie plasmatique après administration intraveineuse est de 2 à 3 heures.
Linéarité/non-linéarité
La cinétique du budésonide est linéaire aux doses thérapeutiques préconisées.
Population pédiatrique
La clairance plasmatique du budésonide est d’environ 0,5 L/min chez les enfants de 4 à 6 ans asthmatiques. Elle est environ 50% plus élevée que chez l’adulte. La demi-vie terminale du budésonide après inhalation est environ de 2,3 heures chez l’enfant asthmatique. Elle est approximativement la même chez l’adulte sain.
Après administration de budésonide suspension pour inhalation par nébuliseur à l'aide d'un nébuliseur à air comprimé (Pari LC Jet Plus muni d’un compresseur Pari Master), la biodisponibilité systémique chez des enfants asthmatiques âgés de 4 à 6 ans est d’environ 6% de la dose nominale et 26% de la dose délivrée soit une biodisponibilité systémique environ 50% de celle observée chez l’adulte sain. L’exposition systémique du budésonide (Cmax et ASC) après administration par nébulisation d’une dose unique de 1 mg à des enfants âgés de 4 à 6 ans est comparable à celle observée chez des adultes sains.
Interaction pharmacocinétique avec les inhibiteurs du CYP450 3A4 :
Les concentrations plasmatiques de budésonide ont été 6 fois plus importantes lors de l’administration concomitante de budésonide par voie orale (dose unique de 3 mg) et de kétoconazole 200 mg une fois par jour. Lorsque le kétoconazole était administré 12 heures après le budésonide, les concentrations plasmatiques du budésonide n’étaient augmentées que d'un facteur 3, traduisant une interaction pharmacocinétique moindre lorsque les produits sont administrés à distance. Des données limitées avec le budésonide administrés à forte dose indiquent également une augmentation significative des taux plasmatiques de budésonide (en moyenne d’un facteur 4) lors de l'administration concomitante d’itraconazole 200 mg en une prise par jour et de budésonide inhalé (en une dose unique de 1000 µg).
5.3. Données de sécurité préclinique
Des malformations ont été observées au cours des études de reproduction menées chez l’animal avec les glucocorticoïdes tels que le budésonide (fente palatine, malformation squelettique). Toutefois, ces résultats expérimentaux observés chez l’animal ne sont pas extrapolables à l’homme aux doses recommandées.
3 ans.
3 mois après ouverture du sachet protecteur.
6.4. Précautions particulières de conservation
La suspension contenue dans l'unidose étant stérile, toute suspension inutilisée restant dans l'unidose doit être jetée après utilisation.
6.5. Nature et contenu de l'emballage extérieur
Récipient unidose (PE) de 2 ml. Boîte de 20 unidoses réparties en 4 sachets (PET/Al/PE) de 5 unidoses.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
35 rue du val de marne
75013 paris
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 023 4 0 : 2 ml en récipient unidose (LDPE). Boîte de 4 sachets de 5 unidoses chacun.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 04/06/2024
BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose
Budésonide
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, qu'il soit mentionné ou non dans cette notice, parlez-en à votre médecin ou votre pharmacien. Voir rubrique 4.
1. Qu'est-ce que BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose ?
3. Comment prendre BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose ET DANS QUELS CAS EST-IL UTILISE ?
GLUCOCORTICOÏDE PAR VOIE INHALEE – Code ATC : R03BA02 (R : Système respiratoire).
ANTIASTHMATIQUE
BUDESONIDE ZENTIVA contient la substance active budésonide. Il appartient à une classe médicamenteuse appelée corticostéroïdes qui possèdent une action anti-inflammatoire réduisant les gonflements et l’irritation dans les voies respiratoires (par exemple, nez, poumons) soulageant ainsi les difficultés respiratoires.
BUDESONIDE ZENTIVA nébulisation s'administre par voie inhalée à l'aide d'un appareil de nébulisation.
Il est indiqué dans le traitement de l’asthme chez les adultes, les adolescents et les enfants lorsque l’utilisation des inhalateurs pressurisés ou à poudre sèche ne peuvent être utilisés ou sont inadaptés.
Attention : ce médicament n’est pas un bronchodilatateur.
Il ne permet pas le traitement de la crise d’asthme.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose ?
· si vous êtes allergique (hypersensible) à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
EN CAS DE DOUTE, IL EST INDISPENSABLE DE DEMANDER L’AVIS DE VOTRE MEDECIN.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre BUDESONIDE ZENTIVA 1 mg/2ml, suspension pour inhalation par nébuliseur en récipient unidose
Faites attention avec BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose.
Mises en garde
En cas de survenue de crises d’asthme il faut utiliser un autre médicament « bronchodilatateur bêta2 mimétique par voie inhalée à action rapide et de courte durée » que votre médecin aura prescrit à cet effet. Le soulagement habituellement obtenu avec le bronchodilatateur bêta2 mimétique par voie inhalée doit alors être observé rapidement.
EN CAS D’ECHEC CONSULTER IMMEDIATEMENT UN MEDECIN.
Si la dose habituellement efficace de ce médicament devient insuffisante, si les crises ou les épisodes de gêne respiratoire deviennent plus fréquents, il faut craindre une aggravation de l’asthme, consultez rapidement votre médecin qui réévaluera le traitement.
Contactez votre médecin en cas de vision floue ou d’autres troubles visuels.
Précautions d’emploi
Ce produit, actif en inhalation doit atteindre l’extrémité des petites bronches. En cas d’encombrement des voies respiratoires (par des mucosités abondantes) ou d’infection, son efficacité peut être diminuée. Il convient de consulter rapidement votre médecin afin qu’il instaure un traitement adapté.
EN CAS DE DOUTE NE PAS HESITER A DEMANDER L’AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN.
Autres médicaments et BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose avec des aliments et boissons
Sans objet.
Ce médicament dans les conditions normales d’utilisation peut être utilisé pendant la grossesse et l’allaitement.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Sportifs
Attention cette spécialité contient un principe actif pouvant induire une réaction positive des tests pratiqués lors des contrôles antidopage.
Conduite de véhicules et utilisation de machines
Le budésonide par voie inhalée n’a pas d'effet sur l’aptitude à conduire des véhicules et à utiliser des machines.
BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose contient :
Sans objet.
3. COMMENT PRENDRE BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose ?
Posologie
La posologie est strictement individuelle et sera adaptée par votre médecin en fonction de la sévérité de l’asthme.
Adultes : de 0,5 mg à 4 mg par jour (soit 1/2 à 4 ampoules pour nébulisation par jour).
Enfant: 0,25 mg à 2 mg par jour (soit 1/4 à 2 ampoules par jour). La dose maximale de 2 mg est réservée au traitement de l'asthme sévère.
Les administrations sont habituellement réparties en 2 séances de nébulisation par jour. Une dose quotidienne allant jusqu’à 1mg peut être administrée en une seule séance de nébulisation par jour.
Après plusieurs jours (ou semaines) de ce traitement lorsque l’état clinique est amélioré, que les symptômes ont régressé et que l’asthme est contrôlé, votre médecin peut prescrire une seule séance de nébulisation par jour. Si les symptômes (toux, sifflement ) augmentent, la dose et le nombre de séances d’aérosolthérapie pourront être réaugmentés.
DANS TOUS LES CAS SE CONFORMER STRICTEMENT A L’ORDONNANCE DE VOTRE MEDECIN.
EN CAS DE PERSISTANCE DES TROUBLES, CONSULTER IMPERATIVEMENT VOTRE MEDECIN.
Mode et voie d’administration
Voie inhalée exclusivement
NE PAS AVALER – NE PAS INJECTER
Cette suspension de budésonide doit être administrée par voie inhalée à l’aide d’un appareil pour nébulisation (nébuliseur) La nébulisation est une technique d’inhalation qui fait appel à un appareil générateur d’aérosol utilisant de l’air comprimé ou de l’oxygène (6 à 8 litres/minute).
L’appareillage complet se compose :
I. d’une source de pression, le compresseur, qui sert à pulvériser la suspension médicamenteuse ;
II. du nébuliseur proprement dit, récipient en plastique dans lequel on verse la suspension à nébuliser ;
III. d’un masque facial, relié au nébuliseur par une tubulure, à adapter au visage de l’enfant (ou d’un embout buccal pour les enfants plus grands).
Les nébuliseurs munis de générateurs ultrasoniques ne sont pas recommandés car ils peuvent ne pas être adaptés pour une administration correcte de BUDESONIDE ZENTIVA.
COMMENT UTILISER BUDESONIDE ZENTIVA SUSPENSION POUR INHALATION PAR NEBULISEUR (accompagné des schémas correspondants)
L’efficacité de ce médicament est en partie dépendante du bon usage de l’appareil de nébulisation.
Lire très attentivement le mode d’emploi de l’appareil. Respectez les instructions du fabricant pour l'utilisation et l'entretien de l'appareil de nébulisation.
Au besoin, n’hésitez pas à demander à votre médecin ou à votre pharmacien de vous fournir des explications détaillées. Si vous êtes est hospitalisé, regardez bien le personnel soignant afin de bien connaître ces quelques gestes simples.
1ère étape : préparation (voir schémas)
|
1. Se laver les mains avec de l’eau et du savon. |
2. Prendre une unidose de BUDESONIDE ZENTIVA suspension pour inhalation par nébuliseur.
3. Ouvrir l’unidose.
4. Verser la quantité prescrite* dans la cuve du nébuliseur.
Un volume de remplissage de 2 à 4 ml convient pour la plupart des nébuliseurs. Si nécessaire, compléter au volume recommandé avec de sérum physiologique stérile.
* Les unidoses sont marquées sur une de leurs faces par un trait noir correspondant à un volume de 1 ml lorsqu’elles sont retournées (ceci permet d’ajuster la dose). Lorsqu’on ne doit utiliser qu’1 ml, vider le contenu de l’unidose jusqu’à ce que le liquide atteigne le trait.
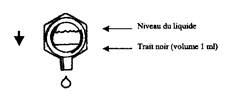
2ème étape : pendant la nébulisation (voir schémas)
5. Adapter le masque sur le visage.
6. Mettre l’appareil en marche.
7. Les enfants doivent être distraits pendant la durée de leur nébulisation, afin qu’il respire au rythme habituel. La nébulisation ne devra pas excéder 10 à 15 min.
3ème étape : après la nébulisation (voir schémas)
8. Ne pas oublier de rincer à l’eau la bouche et le pourtour buccal. En cas d’utilisation d’un masque facial, rincer le visage à l’eau. Ne pas appliquer de crème grasse sur le visage.
9. Après la séance de nébulisation, la suspension inutilisée restant dans la cuve du nébuliseur doit être jetée.
10. Laver à l’eau savonneuse le réservoir du nébuliseur et le masque.
11. Rincer puis sécher le tout soigneusement
12. Nettoyer régulièrement la tubulure et le filtre à air d’arrivée au compresseur
Après chaque nébulisation, pensez à l’entretien du matériel.
La suspension étant stérile, la quantité non utilisée dans l’unidose entamée doit être jetée
Fréquence d’administration
Se conformer à l’ordonnance de votre médecin.
Durée du traitement
Le traitement de l’asthme est quotidien. Ce médicament doit être utilisé très régulièrement et aussi longtemps que le médecin l’aura conseillé.
Si vous avez pris plus de BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Dans tous les cas, se conformer à l’ordonnance de votre médecin. Ne pas augmenter ou diminuer la dose sans l’avis de votre médecin ou de votre pharmacien.
Si vous oubliez de prendre BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose :
Si vous arrêtez de prendre BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose :
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
De rares cas de réactions allergiques ont été signalés (survenant chez moins de 1 personne sur 1000). Elles se manifestent par la brusque apparition d'un gonflement du visage, de la langue et/ou gorge), de difficulté pour avaler ou pour respirer, d'éruption cutanée (urticaire) et/ou sensation de malaise après l'administration de BUDESONIDE ZENTIVA. Dans ce cas, vous devez cesser de prendre BUDESONIDE ZENTIVA et consulter votre médecin immédiatement.
En cas de gêne persistante dans la bouche ou dans la gorge, de modification de voix, de voix rauque, avertir votre médecin, ne pas modifier ou arrêter le traitement sans son avis.
Une candidose buccale peut parfois apparaître. Elle ne nécessite pas nécessairement un arrêt de ce traitement mais peut être la mise en route d’un traitement spécifique. Elle peut être prévenue en se rinçant la bouche après inhalation du produit.
Des cas d’irritation du visage après l’utilisation d’un masque facial ont été décrits. Il est recommandé de rincer le visage à l’eau après utilisation d’un masque facial.
Les corticoïdes inhalés à forte dose ou administrés durant une longue durée peuvent interférer sur la fonction des glandes surrénales, pouvant se manifester par des symptômes tels que : fatigue, perte de poids, nausées et diarrhées persistantes, causés par un mauvais fonctionnement de ces glandes.
Si vous ressentez ces symptômes, consulter votre médecin.
Autres effets indésirables possibles :
Peu fréquent (survient chez 1 patient sur 100 à 1 patient sur 1000) :
· cataracte (opacification du cristallin de l’œil) ;
· contractures musculaires, tremblements ;
· anxiété, dépression ;
· vision floue.
Rares (survient chez 1 patient sur 1000 à 1 patient sur 10 000) :
· bronchospasme (réduction du calibre des bronches) se traduisant par une gêne respiratoire et un sifflement bronchique. Si ces symptômes apparaissent brusquement après la nébulisation, ne renouvelez pas la prise de BUDESONIDE ZENTIVA et contactez votre médecin.
· nervosité, agitation, troubles du comportement (principalement observés chez l’enfant) ;
· ecchymoses ;
· réactions cutanées allergiques.
Très rare (survient chez moins d’1 patients sur 10 000) :
· possibilité de glaucome (augmentation de la pression à l’intérieur de l’œil).
Fréquence indéterminée (la fréquence de survenue n'est pas connue) :
· difficulté pour s’endormir ou trouble du sommeil, état d’hyperexcitation ou d’irritabilité ;
· glaucome.
Effet possible chez l’enfant ou l’adolescent
Un ralentissement de croissance peut être observé chez l’enfant et l’adolescent lors d’un traitement au long cours et à forte dose par BUDESONIDE ZENTIVA.
Le médecin surveillera la croissance de l’enfant.
EN CAS DE DOUTE, NE PAS HESITER A PREVENIR VOTRE MEDECIN.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER BUDESONIDE ZENTIVA 1 mg/2 ml, suspension pour inhalation par nébuliseur en récipient unidose ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte. La date de péremption fait référence au dernier jour de ce mois.
Une fois le sachet protecteur ouvert, les unidoses doivent être maintenues à l'abri de la lumière dans le sachet et utilisées dans les 3 mois.
La suspension étant stérile, l'unidose entamée doit être jetée après utilisation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Budésonide ............................................................................................................................ 1 mg
Pour un récipient unidose de 2 ml.
· Les autres composants sont :
Edetate disodique, chlorure de sodium, polysorbate 80, acide citrique anhydre, citrate de sodium, eau pour préparations injectables.
Suspension pour inhalation par nébuliseur.
Boîte de 20 récipients unidoses de 2 ml répartis dans 4 sachets de 5 unidoses.
Titulaire de l’autorisation de mise sur le marché
35 rue du val de marne
75013 paris
Exploitant de l’autorisation de mise sur le marché
zentiva france
35 rue du val de marne
75013 paris
PHARMACEUTICAL MANUFACTURING SITE
NUCLEO INDUSTRIALE, CONTRADA CANFORA
84084 FISCIANO (SA)
ITALIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).