Dernière mise à jour le 29/06/2026
GENOTONORM 5,3 mg, poudre et solvant pour solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique – Code ATC : H01AC01
GENOTONORM est une hormone de croissance recombinante humaine (également appelée somatropine). Elle a une structure similaire à celle de l’hormone de croissance humaine naturelle qui est nécessaire à la croissance des os et des muscles. Elle permet aussi aux tissus graisseux et musculaire de votre organisme de se développer dans des proportions convenables. Le terme « recombinante » signifie qu’elle n’est pas fabriquée à partir d’un tissu humain ou animal.
Chez les enfants, GENOTONORM est utilisé dans le traitement des troubles suivants :
· Si vous ne grandissez pas correctement et que vous ne produisez pas assez de votre propre hormone de croissance.
· Si vous souffrez d’un syndrome de Turner. Le syndrome de Turner est une anomalie chromosomique observée chez les filles qui peut avoir des conséquences sur la croissance – votre médecin vous aura indiqué si vous en êtes atteinte.
· Si vous souffrez d’une insuffisance rénale chronique (reins). Les reins perdent leur capacité à fonctionner normalement ce qui peut avoir des conséquences sur la croissance.
· Si vous êtes atteint d’un syndrome Prader-Willi (anomalie chromosomique). L’hormone de croissance vous aidera à grandir si vous êtes encore en période de croissance et améliorera également votre composition corporelle (diminution des graisses et augmentation de la masse musculaire).
· Si vous étiez petit(e) ou d’un poids trop faible à la naissance. Si vous n’avez pas pu conserver une croissance normale ou rattraper le retard de croissance à l’âge de quatre ans ou plus, l’hormone de croissance pourra vous aider à grandir.
Chez l’adulte, GENOTONORM est indiqué en cas d’insuffisance en hormone de croissance. Celle-ci peut survenir à l’âge adulte, ou peut être acquise depuis l’enfance.
Si vous avez été traité par GENOTONORM pour insuffisance en hormone de croissance pendant votre enfance, le déficit en hormone de croissance sera réévalué lorsque la croissance staturale sera achevée. Si l’insuffisance sévère en hormone de croissance est confirmée, votre médecin vous proposera de continuer le traitement par GENOTONORM.
Ce traitement doit vous être prescrit après confirmation du diagnostic par un médecin spécialisé et expérimenté dans la prise en charge des patients souffrant d’insuffisance en hormone de croissance.
Présentations
> 1 cartouche(s) bicompartimentée(s) en verre
Code CIP : 349 755-0 ou 34009 349 755 0 9
Déclaration de commercialisation : 19/11/1999
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 122,75 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 123,77 €
- Taux de remboursement :100%
- Chez l'enfant :
. retard de croissance lié à un déficit en hormone de croissance
. retard de croissance lié à un syndrome de Turner
. retard de croissance due à une insuffisance rénale chronique
. retard de croissance chez les enfants de petite taille à la naissance, qui n'ont pas rattrapé ce retard à l'âge de 4 ans ou plus, selon certains critères de taille et de poids
. Syndrome de Prader-Willi
- Chez l'adulte :
. déficit sévère en hormone de croissance, acquis à l'âge adulte avec déséquilibre hormonal associé
. déficit sévère en hormone de croissance, acquis dans l'enfance ; JOURNAL OFFICIEL ; 29/01/97
> 1 cartouche(s) bicompartimentée(s) en verre dans stylo pré-rempli GoQuick
Code CIP : 497 418-2 ou 34009 497 418 2 3
Déclaration de commercialisation : 03/06/2013
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 122,75 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 123,77 €
- Taux de remboursement :100%
> 5 cartouche(s) bicompartimentée(s) en verre dans stylo pré-rempli GoQuick
Code CIP : 497 419-9 ou 34009 497 419 9 1
Déclaration de commercialisation : 03/06/2013
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 603,29 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 604,31 €
- Taux de remboursement :100%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Modéré | Avis du 10/04/2024 | Réévaluation suite à résultats étude post-inscript | Le service médical rendu par GENOTONORM (somatropine) est modéré dans l’indication du retard de croissance (taille actuelle < -3 DS et taille des parents ajustée < -1 DS) chez les enfants nés petits pour l’âge gestationnel avec un poids et/ou une taille de naissance < -2 DS, n’ayant pas rattrapé leur retard de croissance (vitesse de croissance < 0 DS au cours de la dernière année) à l’âge de 4 ans ou plus. |
| Modéré | Avis du 18/09/2019 | Nouvel examen suite au dépôt de nouvelles données | Le service médical rendu par ces spécialités reste modéré dans le traitement substitutif chez l’adulte ayant un déficit en hormone de croissance. |
| Faible | Avis du 22/07/2015 | Renouvellement d'inscription (CT) | Le service médical rendu par GENOTONORM reste faible chez les enfants nés petits pour l’âge gestationnel avec un poids et/ou une taille de naissance < - 2 DS, n'ayant pas rattrapé leur retard de croissance (vitesse de croissance < 0 DS au cours de la dernière année) à l'âge de 4 ans ou plus et dont la taille pour l’âge chronologique est inférieure ou égale à - 3 DS et à - 1 DS par rapport à la taille attendue en fonction des tailles des parents. |
| Important | Avis du 22/07/2015 | Renouvellement d'inscription (CT) | Le service médical rendu par GENOTONORM reste important dans : • le traitement à long terme des enfants atteints d'un retard de croissance lié à un déficit en hormone de croissance normale endogène, • le traitement de la petite taille chez les enfants atteints du syndrome de Turner, confirmé par analyse chromosomique, • le traitement du retard de croissance chez l'enfant pré-pubère atteint d'une insuffisance rénale chronique, • le syndrome de Prader Willi confirmé par un test génétique approprié. |
| Modéré | Avis du 22/07/2015 | Renouvellement d'inscription (CT) | Le service médical rendu par GENOTONORM dans le traitement substitutif chez le sujet adulte présentant un déficit en hormone de croissance sévère reste modéré. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 15/02/2012 | Inscription (CT) | Ces spécialités sont des compléments de gamme qui n'apportent pas d'amélioration du service médical rendu (ASMR V). |
| V (Inexistant) | Avis du 07/12/2011 | Réévaluation SMR | GENOTONORM n'apporte pas d'amélioration du service médical rendu (ASMR V) dans la stratégie thérapeutique chez l'enfant né petit pour l'âge gestationnel. |
| IV (Mineur) | Avis du 07/12/2011 | Réévaluation SMR | GENOTONORM apporte une amélioration du service médical rendu mineure (ASMR IV) dans la prise en charge du syndrome de Turner, de l'insuffisance rénale et du syndrome de Prader Willi. |
Autres informations
- Titulaire de l'autorisation : PFIZER HOLDING FRANCE
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière annuelle
- prescription réservée aux spécialistes et services ENDOCRINOLOGIE
- prescription réservée aux spécialistes et services MALADIES METABOLIQUES
- prescription réservée aux spécialistes et services PEDIATRIE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 467 985 2
ANSM - Mis à jour le : 22/05/2024
GENOTONORM 5,3 mg, poudre et solvant pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
*produite dans des cellules d’Escherichia coli par la technique de l’ADN recombinant.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable.
Dans la cartouche à double compartiment, il y a une poudre blanche dans le compartiment avant et une solution limpide dans le compartiment arrière.
4.1. Indications thérapeutiques
Retard de croissance lié à un déficit somatotrope.
Retard de croissance lié à un syndrome de Turner.
Retard de croissance lié à une insuffisance rénale chronique.
Retard de croissance (taille actuelle < -2,5 DS et taille parentale ajustée < -1 DS) chez les enfants nés petits pour l'âge gestationnel avec un poids et/ou une taille de naissance < -2 DS, n'ayant pas rattrapé leur retard de croissance (vitesse de croissance < 0 DS au cours de la dernière année) à l'âge de 4 ans ou plus.
Syndrome de Prader-Willi (SPW), afin d'améliorer la croissance et la composition corporelle. Le diagnostic de SPW doit être confirmé par le test génétique approprié.
Chez l'adulte
Traitement substitutif chez les adultes présentant un déficit somatotrope sévère.
Déficit acquis à l’âge adulte : Les patients qui présentent un déficit somatotrope sévère associé à des déficits hormonaux multiples résultant d’une pathologie hypothalamique ou hypophysaire connue et ayant au moins un autre déficit hormonal hypophysaire, excepté la prolactine. Un test dynamique approprié sera pratiqué afin de diagnostiquer ou d'exclure un déficit en hormone de croissance.
Déficit acquis dans l’enfance : Chez les patients qui présentent un déficit somatotrope acquis dans l'enfance d’origine congénitale, génétique, acquise ou idiopathique. La capacité de sécrétion en hormone de croissance doit être réévaluée chez les patients ayant un déficit acquis dans l’enfance une fois leur croissance staturale achevée. Chez les patients présentant une forte probabilité de déficit somatotrope persistant, c'est-à-dire d’origine congénitale ou secondaire à une pathologie hypothalamo-hypophysaire ou un traumatisme hypothalamo-hypophysaire, un dosage d’Insulin-like growth factor (IGF-I) < -2DS, mesuré au moins quatre semaines après l’arrêt du traitement par hormone de croissance, doit être considéré comme une preuve suffisante d’un déficit somatotrope sévère.
Tous les autres patients auront besoin d’un dosage d’IGF-I et d’un test de stimulation à l’hormone de croissance.
4.2. Posologie et mode d'administration
La posologie et le schéma d’administration doivent être adaptés à chaque patient.
L’injection doit être sous-cutanée et le point d’injection devra varier pour éviter l’apparition de lipoatrophies.
Retard de croissance lié à un déficit somatotrope chez l’enfant : en général, la posologie recommandée est de 0,025 à 0,035 mg/kg de poids corporel par jour ou de 0,7 à 1,0 mg/m2 de surface corporelle par jour. Des doses plus élevées peuvent être utilisées.
Lorsque le déficit somatotrope acquis dans l’enfance persiste à l’adolescence, le traitement doit être continué jusqu’au développement somatique complet (concernant la composition corporelle, la densité osseuse). Pour le suivi, l’atteinte d’un pic normal de densité osseuse définie par un T-score > -1 (c’est-à-dire standardisé pour un pic normal de densité osseuse mesuré par absorptiométrie à rayons X en double énergie prenant en compte le sexe et l’ethnie) est un des objectifs thérapeutiques durant la période de transition. Pour des recommandations sur le dosage, voir ci-dessous la rubrique adulte.
Syndrome de Prader-Willi, afin d'améliorer la croissance et la composition corporelle chez l'enfant : en général, la posologie recommandée est de 0,035 mg/kg de poids corporel par jour soit 1,0 mg/m2 de surface corporelle par jour. La dose quotidienne ne devra pas dépasser 2,7 mg. Les enfants dont la vitesse de croissance est inférieure à 1 cm par an et dont les épiphyses sont presque soudées ne devront pas être traités.
Retard de croissance dans le syndrome de Turner : la posologie recommandée est de 0,045 à 0,050 mg/kg de poids corporel par jour, soit 1,4 mg/m2 de surface corporelle par jour.
Retard de croissance lié à une insuffisance rénale chronique : la posologie recommandée est de 0,045 à 0,050 mg/kg de poids corporel par jour (1,4 mg/m² de surface corporelle par jour)). Des doses plus élevées peuvent être utilisées si la vitesse de croissance est trop faible. Il est possible qu’un ajustement de la posologie soit nécessaire après 6 mois de traitement.
Retard de croissance chez les enfants nés petits pour l'âge gestationnel : la posologie habituellement recommandée est de 0,035 mg/kg de poids corporel par jour (1 mg/m² de surface corporelle par jour) jusqu'à ce que la taille finale soit atteinte (voir rubrique 5.1).
Le traitement devra être interrompu après la première année de traitement si la vitesse de croissance est inférieure à +1 DS. Le traitement devra être interrompu si la vitesse de croissance est < 2 cm/an et, si une confirmation est nécessaire, l'âge osseux est > 14 ans (pour les filles) et > 16 ans (pour les garçons), correspondant à la soudure des épiphyses.
Doses recommandées chez l’enfant :
|
Indication |
mg/kg de poids corporel (dose par jour) |
mg/m2 de surface corporelle (dose par jour) |
|
Déficit en hormone de croissance |
0,025 - 0,035 |
0,7 - 1,0 |
|
Syndrome de Prader-Willi |
0,035 |
1,0 |
|
Syndrome de Turner |
0,045 - 0,050 |
1,4 |
|
Insuffisance rénale chronique |
0,045 - 0,050 |
1,4 |
|
Enfants nés petits pour l'âge gestationnel |
0,035 |
1,0 |
Déficit en hormone de croissance chez l’adulte :
Chez les patients qui continuent le traitement par hormone de croissance après un déficit somatotrope acquis dans l’enfance, la dose recommandée pour redémarrer est de 0,2 à 0,5 mg par jour. La dose doit être progressivement augmentée ou diminuée en fonction des besoins individuels du patient, déterminés par le taux d’IGF-I.
Chez les patients qui ont un déficit somatotrope acquis à l’âge adulte, le traitement doit débuter avec une faible dose, de 0,15 à 0,3 mg par jour. La dose peut être augmentée progressivement en fonction des besoins individuels du patient, déterminés par le taux d'IGF‑I.
Dans les deux cas, le traitement doit conduire à obtenir des concentrations d’IGF-I, en fonction de l'âge, ne dépassant pas la limite de 2 DS. Les patients dont le taux d'IGF-I est normal au début du traitement devront recevoir de l'hormone de croissance jusqu'à atteindre un taux d'IGF-I dans les limites supérieures de la normale, sans excéder 2 DS. La réponse clinique, de même que les effets indésirables peuvent également guider l'adaptation de la posologie.
Certains patients qui ont un déficit somatotrope ne normalisent pas leur taux d’IGF-I, malgré une bonne réponse clinique. Ils n’ont donc pas besoin d’une escalade de dose. La dose d’entretien excède rarement 1,0 mg par jour. Les femmes peuvent nécessiter des doses plus élevées que les hommes ; les hommes montrant une sensibilité croissante à l’IGF-1 au fil du temps. Cela signifie qu’il y a un risque que les femmes, notamment celles qui reçoivent un traitement oestrogénique par voie orale, soient sous-traitées alors que les hommes sont sur-traités.
Par conséquent, la bonne adaptation de la dose d'hormone somatotrope devra être contrôlée tous les 6 mois. La sécrétion physiologique d'hormone de croissance diminuant avec l'âge, une réduction de la posologie est nécessaire. Chez les patients de plus de 60 ans, le traitement doit être initié à une dose de 0,1 à 0,2 mg par jour. Cette posologie doit être augmentée progressivement en fonction des besoins individuels du patient. La dose minimale efficace devra être utilisée. La dose d’entretien chez ces patients excède rarement 0,5 mg par jour.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
La somatropine ne doit pas être utilisée en présence d’une preuve quelconque d’activité d’une tumeur. Les tumeurs intracrâniennes doivent être inactives et tout traitement anti-tumoral doit être terminé avant de commencer un traitement par l’hormone de croissance. Le traitement doit être interrompu en présence d’une preuve de croissance tumorale.
GENOTONORM ne doit pas être utilisé pour améliorer la croissance des enfants dont les épiphyses sont soudées.
Les patients présentant un état critique aigu, souffrant de complications secondaires à une intervention chirurgicale à cœur ouvert, une intervention chirurgicale abdominale, un polytraumatisme, une insuffisance respiratoire aiguë ou à une situation similaire ne doivent pas être traités par GENOTONORM (pour les patients recevant un traitement de substitution, se reporter à la rubrique 4.4).
4.4. Mises en garde spéciales et précautions d'emploi
L’apparition d’une myosite constitue un effet secondaire très rare qui peut être lié à la présence du conservateur métacrésol. En cas de myalgie ou de douleur exagérée au niveau du site d’injection une myosite doit être suspectée et si elle est confirmée, une spécialité GENOTONORM ne contenant pas de métacrésol doit être utilisée.
La dose maximale quotidienne recommandée ne doit pas être dépassée (voir rubrique 4.2).
Sensibilité à l’insuline
La somatropine peut réduire la sensibilité à l’insuline. Chez les patients atteints de diabète sucré, la dose d’insuline peut nécessiter un ajustement après la mise en place d’un traitement par la somatropine. Les patients atteints de diabète, d’intolérance au glucose ou tout autre facteur de risque de diabète doivent être étroitement surveillés pendant le traitement par la somatropine.
Fonction thyroïdienne
L’hormone de croissance augmente la conversion extrathyroïdienne de T4 en T3 ce qui peut entraîner une diminution de la concentration sérique en T4 et une augmentation de la concentration sérique en T3. Bien que les taux périphériques d’hormones thyroïdiennes restent dans les fourchettes de référence chez la majorité des sujets sains, une hypothyroïdie peut en théorie se développer chez les sujets ayant une hypothyroïdie infraclinique. En conséquence, une surveillance de la fonction thyroïdienne doit être effectuée chez tous les patients. Chez les patients souffrant d’un hypopituitarisme et recevant un traitement substitutif, l’effet potentiel du traitement par l’hormone de croissance sur la fonction thyroïdienne doit être étroitement surveillé.
Hypoadrénalisme
L'initiation du traitement par la somatropine peut entraîner une inhibition de la 11βHSD-1 et réduire les concentrations sériques de cortisol. Chez les patients traités par la somatropine, une insuffisance surrénale centrale (secondaire) non diagnostiquée auparavant peut être découverte et un traitement substitutif par glucocorticoïde peut être nécessaire. De plus, les patients traités par glucocorticoïde pour une insuffisance surrénalienne préalablement diagnostiquée peuvent nécessiter une augmentation de leurs doses d'entretien ou de stress, après le début du traitement par la somatropine (voir rubrique 4.5).
Utilisation avec un traitement œstrogénique oral
Si une femme traitée par somatropine débute un traitement œstrogénique par voie orale, il peut être nécessaire d'augmenter la dose de somatropine pour maintenir les taux sériques d'IGF-1 dans l’intervalle normal pour l'âge. Inversement, si une femme sous somatropine interrompt un traitement œstrogénique oral, il se peut que la dose de somatropine doive être réduite pour éviter un excès d'hormone de croissance et / ou des effets indésirables (voir rubrique 4.5).
En cas de déficit somatotrope secondaire à un traitement antitumoral, il est recommandé de surveiller les signes éventuels de récidive du processus tumoral. Parmi les patients ayant survécu à une tumeur pendant l’enfance, il a été rapporté un risque accru de tumeur secondaire chez les patients traités par la somatropine suite à leur première tumeur. Les tumeurs secondaires les plus fréquentes étaient les tumeurs intracrâniennes, et en particulier les méningiomes, chez les patients traités pour leur tumeur initiale par des radiations à la tête.
Chez les patients présentant des troubles endocriniens y compris ceux relatifs à un déficit en hormone de croissance, la survenue d'une épiphysiolyse de la hanche peut être plus fréquente que dans la population générale. Tout enfant présentant une claudication au cours du traitement par la somatropine devra être examiné.
Hypertension intracrânienne bénigne
En cas de céphalées sévères ou répétées, de troubles visuels, de nausées et/ou de vomissements, il est recommandé d'effectuer un fond d'œil afin de dépister un éventuel œdème papillaire. Si celui-ci est confirmé, un diagnostic d'hypertension intracrânienne bénigne devra être considéré et s'il y a lieu, le traitement par la somatropine devra être interrompu. L'état actuel des connaissances ne permet pas de recommander la poursuite du traitement par l’hormone de croissance chez des patients ayant une hypertension intracrânienne résolue. Si le traitement par l'hormone de croissance est ré-instauré, une surveillance attentive de la survenue de symptômes d'hypertension intracrânienne est nécessaire.
Leucémie
Des cas de leucémie ont été rapportés chez un petit nombre de patients présentant un déficit en hormone de croissance, certains d’entre eux ayant été traités par la somatropine. Toutefois, il n'a pas été démontré que l’incidence de la leucémie est augmentée chez les patients sans facteurs de risque receveurs d'hormone de croissance.
Anticorps
Comme pour tous les produits contenant de la somatropine, un faible pourcentage de patients peut développer des anticorps contre GENOTONORM. GENOTONORM entraine la formation d'anticorps chez environ 1 % des patients. La capacité de liaison de ces anticorps est faible et il n'y a pas d'effet sur le taux de croissance. La recherche d’anticorps à la somatropine doit être réalisée chez tout patient non répondeur.
Patients âgés
Chez les patients âgés de plus de 80 ans, l’expérience est limitée. Les patients âgés peuvent être plus sensibles à l’action de GENOTONORM, et peuvent donc être plus sujets à l’apparition d’effets indésirables.
Etat critique aigu
Les effets de GENOTONORM sur l'évolution d'un état critique ont été étudiés dans deux études contrôlées versus placebo chez 522 patients adultes présentant des complications secondaires à une intervention chirurgicale à cœur ouvert, une intervention chirurgicale abdominale, un polytraumatisme ou une insuffisance respiratoire aiguë. La mortalité était plus élevée chez les patients traités par 5,3 ou 8 mg de GENOTONORM par jour comparativement aux patients recevant le placebo, soit 42 % contre 19 %. Compte tenu de ces résultats, ces patients ne devront pas être traités par GENOTONORM.
Etant donné l’absence d’information disponible sur la sécurité d’un traitement substitutif par l’hormone de croissance chez les patients présentant un état critique aigu, les bénéfices de la poursuite du traitement dans cette situation doivent être mis en balance avec les risques potentiels.
Chez tous les patients qui développent un état critique aigu autre ou similaire, le bénéfice possible d’un traitement par GENOTONORM doit être mis en balance avec le risque potentiel.
Pancréatite
Bien que rare, il faut prendre en considération l’éventualité d’une pancréatite chez les patients, et particulièrement les enfants, traités par la somatropine qui présentent une douleur abdominale.
Syndrome de Prader-Willi
Chez les patients présentant un syndrome de Prader-Willi, le traitement devra toujours être associé à un régime hypocalorique.
Des cas de décès associés à l’utilisation de l’hormone de croissance ont été rapportés chez des enfants atteints d’un syndrome de Prader-Willi et qui présentaient un ou plusieurs des facteurs de risque suivants : obésité sévère (c’est-à-dire les enfants dont le rapport poids/taille excède 200%), antécédents d’insuffisance respiratoire ou d’apnée du sommeil, ou infection respiratoire non spécifiée. Les patients avec un ou plusieurs de ces facteurs pourraient présenter un risque accru.
Avant de débuter le traitement par la somatropine chez les patients atteints d’un syndrome de Prader-Willi, une recherche des signes d’obstruction des voies aériennes supérieures, d'apnée du sommeil ou d’infection respiratoire devra être effectuée.
Si lors de la recherche d'obstruction des voies aériennes supérieures, des anomalies sont observées, alors les enfants devront être orientés vers un spécialiste en Oto-Rhino-Laryngologie (ORL) pour le traitement et l’éradication des troubles respiratoires avant d’initier le traitement par l’hormone de croissance.
La recherche d'apnée du sommeil devra être effectuée avant l’instauration du traitement par l’hormone de croissance par des méthodes reconnues telles que la polysomnographie ou l’oxymétrie durant la nuit, et surveillée si celle-ci est suspectée.
Si lors du traitement par l’hormone de croissance, les patients présentent des signes d’obstruction des voies aériennes supérieures (incluant la survenue ou l’aggravation d’un ronflement), le traitement devra être interrompu, et un nouvel examen ORL devra être réalisé.
Tous les patients atteints d’un syndrome de Prader-Willi devront être suivis si une apnée du sommeil est suspectée.
Les patients devront être suivis pour les signes d’infections respiratoires, qui devront être diagnostiqués aussi précocement que possible et traités efficacement.
Tous les patients atteints d’un syndrome de Prader-Willi devront également faire l’objet d’un contrôle pondéral efficace avant et pendant le traitement par l’hormone de croissance.
Une scoliose est fréquemment observée chez les patients présentant un syndrome de Prader-Willi. Chez tous les enfants, la scoliose est susceptible d'évoluer lors d'une croissance rapide. Les signes de scoliose devront être recherchés au cours du traitement.
L'expérience d'un traitement au long cours par l’hormone de croissance chez l’adulte et les patients présentant un syndrome de Prader-Willi est limitée.
Enfants nés petits pour l’âge gestationnel
Chez les enfants nés petits pour l'âge gestationnel, les autres causes ou traitements pouvant expliquer un retard de croissance doivent être exclus avant de commencer le traitement.
Chez les enfants nés petits pour l'âge gestationnel, il est recommandé de mesurer l'insulinémie et la glycémie à jeun avant de commencer le traitement puis annuellement. Chez les patients ayant un risque accru de diabète (antécédents familiaux de diabète, obésité, insulino-résistance sévère, acanthosis nigricans), un test d'hyperglycémie provoquée par voie orale doit être réalisé. Si un diabète clinique apparaît, l'hormone de croissance ne devra pas être administrée.
Chez les enfants nés petits pour l'âge gestationnel, il est recommandé de mesurer le taux d'IGF-I avant d'initier le traitement, et par la suite 2 fois par an. Si sur des mesures répétées, les taux d'IGF-I sont supérieurs à + 2 DS comparés aux valeurs standard pour l'âge et le stade pubertaire, le ratio IGF-I/IGFBP-3 devrait être pris en considération pour l'ajustement de la dose.
L'expérience en ce qui concerne l'initiation du traitement juste avant la puberté chez les enfants nés petits pour l'âge gestationnel est limitée. Par conséquent, il n'est pas recommandé d'initier le traitement juste avant la puberté.
L'expérience chez les patients atteints du syndrome de Silver-Russell est limitée.
Une partie du gain de taille chez les enfants nés petits pour l'âge gestationnel traités par l'hormone de croissance pourrait disparaître si le traitement est arrêté avant que la taille finale ne soit atteinte.
Insuffisance rénale chronique
Dans le cas d'une insuffisance rénale chronique, la fonction rénale devra être diminuée de 50 % par rapport à la normale. Afin de confirmer le retard de croissance, la croissance aura dû être suivie au préalable pendant 1 an avant de mettre en route le traitement. Au cours de cette période, un traitement de l’insuffisance rénale chronique (incluant le contrôle de l’acidose, de l’hyperparathyroïdie et de l’état nutritionnel) devra avoir été instauré et devra être maintenu pendant la durée du traitement. Le traitement devra être interrompu en cas de transplantation rénale.
Il n’existe pas à ce jour, de données disponibles sur la taille définitive des patients atteints d’insuffisance rénale chronique traités par GENOTONORM.
Teneur en sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose. Les patients suivant un régime hyposodé peuvent être informés que ce médicament est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Un traitement concomitant par des glucocorticoïdes inhibe les effets stimulants de la croissance des produits contenant de la somatropine. Le traitement substitutif par glucorticoïdes des patients présentant un déficit en hormone adrénocorticotrope (ACTH) doit être ajusté avec précaution afin d’éviter tout effet inhibiteur sur la croissance. Par conséquent, chez les patients traités par les glucocorticoïdes, une surveillance de la croissance doit être effectuée afin d’évaluer l'impact potentiel du traitement par les glucocorticoïdes sur la croissance.
L'hormone de croissance diminue la conversion de la cortisone en cortisol et peut mettre en évidence une insuffisance surrénale centrale non encore diagnostiquée ou rendre inefficaces de faibles doses des glucocorticoïdes (voir rubrique 4.4).
Selon les résultats d’une étude d’interaction réalisée chez des adultes atteints d’un déficit en hormone de croissance, l’administration de somatropine peut augmenter la clairance des composés métabolisés par les isoenzymes du cytochrome P450. En particulier, la clairance des composés métabolisés par le cytochrome P450 3A4 (exemples : hormones sexuelles stéroïdes, corticostéroïdes, anticonvulsivants et ciclosporine) peut être augmentée entraînant une diminution des taux plasmatiques de ces composés. La conséquence clinique de cet effet est inconnue.
Se reporter à la rubrique 4.4. concernant le diabète et les troubles thyroïdiens.
Chez les femmes sous traitement œstrogénique substitutif par voie orale, une dose plus élevée d'hormone de croissance peut être nécessaire pour atteindre l’objectif du traitement (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études chez l'animal sont insuffisantes en ce qui concerne les effets sur la grossesse, le développement embryofœtal, la parturition ou le développement postnatal (voir rubrique 5.3). Aucune étude clinique sur les grossesses exposées n'est disponible. Ainsi, les produits contenant de la somatropine ne sont pas recommandés pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Allaitement
Aucune étude clinique n’a été réalisée avec des produits contenant de la somatropine chez les femmes qui allaitent. On ne sait pas si la somatropine passe dans le lait maternel, cependant l’absorption gastro-intestinale de la protéine chez l’enfant est très improbable. Une attention particulière doit donc être apportée lorsque les produits contenant de la somatropine sont administrés aux femmes qui allaitent.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
GENOTONORM n’a aucune influence sur l’aptitude à conduire des véhicules et à utiliser des machines.
La fréquence de ces effets indésirables est liée à la dose administrée et à l’âge des patients ; elle peut être inversement liée à l’âge des patients lors de l’apparition du déficit en hormone de croissance. Chez les enfants de tels effets indésirables ne sont pas fréquents.
Chez environ 1 % des patients GENOTONORM a entraîné la formation d’anticorps. La capacité de liaison de ces anticorps est faible et aucune modification clinique n’a été associée à leur présence, voir rubrique 4.4.
Liste tabulée des effets indésirables
Le tableau 1 montre les effets indésirables par Classe de Système d’Organe et par fréquence, chez les enfants et les adultes selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Tableau 1 : Liste tabulée des effets indésirables |
||||||
|
Classe de Système d’Organe |
Très fréquent (³ 1/10) |
Fréquent (³ 1/100 à < 1/10) |
Peu fréquent (³ 1/1 000 à < 1/100) |
Rare (³ 1/10 000 à < 1/1 000) |
Très rare (< 1/10 000) |
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
|
|
(Enfants) Leucémie |
|
|
|
|
Troubles du métabolisme et de la nutrition |
|
|
|
|
|
(Adultes et enfants) Diabète sucré de type 2 |
|
Affections du système nerveux |
|
(Adultes) Paresthésie*
(Adultes) Syndrome du canal carpien |
(Enfants) Hypertension intracrânienne bénigne
(Enfants) Paresthésie*
|
|
|
(Adultes) Hypertension intracrânienne bénigne
(Adultes et enfants) Céphalée |
|
Affections de la peau et du tissu sous-cutané |
|
|
(Enfants) Éruption cutanée** Prurit** Urticaire** |
|
|
(Adultes) Éruption cutanée** Prurit** Urticaire** |
|
Affections musculo-squelettiques et du tissu conjonctif |
(Adultes) Arthralgie* |
(Adultes) Myalgie*
(Adultes) Rigidité musculo-squelettique*
(Enfants) Arthralgie* |
(Enfants) Myalgie* |
|
|
(Enfants) Rigidité musculo-squelettique* |
|
Affections des organes de reproduction et du sein |
|
|
(Adultes et enfants) Gynécomastie |
|
|
|
|
Troubles généraux et anomalies au site d’administration |
(Adultes) Œdème périphérique*
|
(Enfants) Réaction au site d’injection$ |
(Enfants) Œdème périphérique*
|
|
|
(Adultes et enfants) Œdème facial*
(Adultes) Réaction au site d’injection$ |
|
Investigations |
|
|
|
|
|
(Adultes et enfants) Cortisolémie diminuée |
|
* En règle générale, ces effets indésirables sont d’intensité légère à modérée, surviennent au cours des premiers mois de traitement et disparaissent spontanément ou avec une diminution de la dose. L’incidence de ces effets indésirables est liée à la dose administrée, à l’âge des patients, et il se peut qu’elle soit inversement liée à l’âge des patients à l’apparition du déficit en hormone de croissance. ** Effets Indésirables du Médicament (EIM) identifiés après la commercialisation. $ Des réactions transitoires au site d’injection ont été rapportées chez l’enfant. Signification clinique indéterminée. Rapportée chez des enfants souffrant d’un déficit en hormone de croissance traités par somatropine, cependant, l’incidence semble similaire à celle retrouvée chez les enfants ne présentant pas de déficit en hormone de croissance. |
||||||
Diminution des taux de cortisol sérique
Une diminution des taux de cortisol sérique a été rapportée avec la somatropine ; cette diminution peut être liée à la modification des protéines de transport ou à une augmentation de la clairance hépatique. Il est possible que la signification clinique de ces observations soit limitée. Toutefois, la corticothérapie de substitution devra être optimisée avant d’instaurer le traitement par GENOTONORM.
Syndrome de Prader-Willi
Lors de la surveillance après commercialisation, de rares cas de mort subite ont été rapportés chez les patients présentant un syndrome de Prader-Willi et traités par la somatropine, bien que la relation de causalité n’ait pas été démontrée.
Leucémie
Des cas de leucémie ont été rapportés chez les enfants atteints d’un déficit en hormone de croissance, dont certains étaient traités par somatropine et inclus dans l’expérience post-commercialisation. Cependant, il n’y a pas de preuve d’une augmentation du risque de leucémie sans facteurs prédisposants, tels que les radiations de la tête et du cerveau.
Épiphysiolyse fémorale supérieure et maladie de Legg-Calve-Perthes
Des cas d’épiphysiolyse fémorale supérieure et de maladie de Legg-Calve-Perthes ont été rapportés chez des enfants traités par hormone de croissance recombinantes. L’épiphysiolyse fémorale supérieure est plus fréquente en cas de troubles endocriniens, et la maladie de Legg-Calve-Perthes est plus fréquente en cas d’insuffisance staturale. Mais on ne sait pas si ces deux pathologies sont ou non plus fréquentes pendant le traitement par somatropine. Leur diagnostic doit être envisagé chez un enfant présentant une gêne ou une douleur au niveau de la hanche ou du genou.
Autres effets indésirables médicamenteux
D’autres effets indésirables médicamenteux peuvent être considérés comme des effets de classe de la somatropine, tels qu’une éventuelle hyperglycémie due à une baisse de la sensibilité à l’insuline, une diminution du taux de thyroxine libre et une hypertension intracrânienne bénigne.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Le surdosage aigu peut conduire initialement à une hypoglycémie, puis secondairement à une hyperglycémie.
Un surdosage peut, à long terme, conduire à des signes et symptômes similaires aux effets connus de l'excès d'hormone de croissance chez l'homme.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Hormones de l’anté-hypophyse et analogues, code ATC : H01AC01.
La somatropine est une hormone métabolique puissante jouant un rôle important dans le métabolisme des lipides, des glucides et des protéines. La somatropine stimule la croissance linéaire et augmente la vitesse de croissance chez les enfants présentant un déficit en hormone de croissance. Chez les adultes, comme chez les enfants, la somatropine maintient la composition corporelle normale en augmentant la rétention azotée, en stimulant la croissance du muscle squelettique, et en mobilisant les graisses corporelles. Le tissu adipeux viscéral est très sensible à la somatropine. La somatropine augmente la lipolyse et diminue l’entrée des triglycérides dans les réserves lipidiques de l’organisme. La somatropine augmente les concentrations sériques d’IGF-I et IGFBP-3 (Insulin-like Growth Factor Binding Protein 3).
Par ailleurs, les actions suivantes ont été mises en évidence :
· Métabolisme lipidique : La somatropine est un inducteur des récepteurs hépatiques du LDL cholestérol, et modifie le profil des lipides et des lipoprotéines sériques. En général l’administration de somatropine chez les patients ayant un déficit en hormone de croissance entraîne une diminution des LDL et des apolipoprotéines B sériques. Une diminution du cholestérol total sérique peut aussi être observée.
· Métabolisme glucidique : La somatropine augmente le taux d’insuline mais la glycémie à jeun est généralement inchangée. Les enfants ayant un hypopituitarisme ont parfois des épisodes d’hypoglycémie lorsqu'ils sont à jeun qui peuvent être corrigés par l’administration de somatropine.
· Métabolisme hydroélectrolytique : Le déficit en hormone de croissance s’accompagne d’une diminution des volumes plasmatique et extracellulaire, qui augmentent rapidement avec un traitement par la somatropine. La somatropine entraîne une rétention sodée, potassique et du phosphore.
· Métabolisme osseux : La somatropine stimule le renouvellement osseux. Le contenu minéral osseux et la densité osseuse au niveau des sites de charge corporelle augmentent après une administration à long terme de somatropine à des patients ayant un déficit en hormone de croissance et ayant une ostéopénie.
· Capacité physique : La force musculaire et la capacité à l’exercice physique sont améliorées après un traitement à long terme avec la somatropine. La somatropine augmente aussi le débit cardiaque, mais le mécanisme n’a pas encore été élucidé. Une diminution de la résistance périphérique vasculaire peut contribuer à cet effet.
Lors d’études cliniques sur des enfants nés petits pour l'âge gestationnel, des dosages de 0,033 et 0,067 mg/kg/jour ont été utilisés jusqu'à la taille finale.
Pour 56 patients traités en continu et qui ont atteint (ou presque) leur taille finale, la variation moyenne de taille depuis le début du traitement est + 1,90 DS (0,033 mg/kg/jour) et + 2,19 DS (0,067 mg/kg/jour). Les données de la littérature suggèrent une croissance tardive de 0,5 DS chez les enfants nés petits pour l'âge gestationnel non traités et sans rattrapage spontané précoce de croissance.
5.2. Propriétés pharmacocinétiques
La biodisponibilité de GENOTONORM administré par voie sous-cutanée est environ de 80 % à la fois chez les sujets sains et les patients ayant un déficit en hormone de croissance. Le taux plasmatique des valeurs de Cmax et tmax est de 13 à 35 ng/mL et 3 à 6 heures respectivement après une dose sous-cutanée de 0,035 mg/kg de GENOTONORM.
Elimination
La demi-vie terminale moyenne de GENOTONORM après administration intraveineuse chez des patients adultes ayant un déficit en hormone de croissance est d’environ 0,4 heure. Cependant, après une administration sous-cutanée, la demi-vie est de 2 à 3 heures. La différence observée est probablement liée à une absorption lente à partir du site d’injection après administration sous-cutanée.
Sous-populations
La biodisponibilité absolue de GENOTONORM semble similaire chez l’homme et la femme après administration sous-cutanée.
Les informations concernant la pharmacocinétique de la somatropine chez des populations âgées et chez des enfants, dans différentes races et chez des patients atteints d’insuffisance rénale, hépatique ou cardiaque sont soit inexistantes soit incomplètes.
5.3. Données de sécurité préclinique
Une augmentation de la fragilité des chromosomes a été observée dans une étude in-vitro sur des lymphocytes prélevés chez des patients après un traitement de longue durée par la somatropine et après addition d’un médicament radiomimétique, la bléomycine. La signification clinique de cette observation n’est pas connue.
Dans une autre étude, aucune augmentation d’anomalies chromosomiques n’a été retrouvée dans les lymphocytes des patients traités au long cours par la somatropine.
Solvant (compartiment arrière) : eau pour préparations injectables, mannitol (E421), métacrésol.
La stabilité chimique et physique a été démontrée pendant 28 jours entre 2 °C et 8 °C.
D’un point de vue microbiologique, une fois reconstitué, le médicament peut être conservé 28 jours entre 2 °C et 8 °C.
Toutes autres durées et conditions de conservation sont de la responsabilité de l’utilisateur.
6.4. Précautions particulières de conservation
A conserver au réfrigérateur (entre 2 °C et 8 °C) ou jusqu’à un mois maximum à une température ne dépassant pas 25 °C. Conserver la cartouche à double compartiment/le stylo prérempli dans l’emballage extérieur à l’abri de la lumière.
Après reconstitution
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler. Conserver la cartouche à double compartiment/le stylo prérempli dans l’emballage extérieur à l'abri de la lumière.
Pour les conditions de conservation du médicament reconstitué, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre et 1 mL de solvant en cartouche à double compartiment (verre de type I) séparés par un piston en caoutchouc (bromobutyl).
La cartouche est scellée à une extrémité par un disque en caoutchouc (bromobutyl) et une capsule en aluminium et à l’autre extrémité par un bouchon en caoutchouc (bromobutyl). La cartouche à double compartiment est fournie pour une utilisation avec le dispositif d’injection réutilisable GENOTONORM Pen ou insérée dans le stylo jetable multidose prérempli GoQuick
Les GENOTONORM Pen ont un code couleur et doivent être utilisés avec les cartouches à double compartiment de même code couleur afin de délivrer la dose appropriée. Le GENOTONORM Pen 5,3 (bleu) doit être utilisé avec la cartouche 5,3 mg (bleue).
Le stylo prérempli GoQuick 5,3 mg a un code couleur bleu.
Boîte de 1 x 5,3 mg, 5 x 5,3 mg, 1 x 5,3 mg stylo prérempli, 5 x 5,3 mg stylos préremplis.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Ne reconstituer la poudre qu’avec le solvant fourni.
La solution est préparée en vissant le dispositif d’injection ou les différentes parties du stylo prérempli GoQuick de sorte que le solvant se mélange à la poudre dans la cartouche à double compartiment. Dissoudre lentement la poudre en l’inclinant doucement d’avant en arrière.
Ne pas agiter vigoureusement car ceci peut entraîner la dénaturation de la substance active. La solution reconstituée est pratiquement incolore ou légèrement opalescente.
La solution injectable reconstituée doit être examinée avant utilisation et seules les solutions limpides exemptes de particules doivent être utilisées.
Des instructions détaillées pour la préparation et l’administration du produit reconstitué GENOTONORM sont fournies dans la notice, au niveau de la rubrique 3, paragraphe « Injections de GENOTONORM » ainsi que dans les instructions d’utilisation fournies avec le dispositif utilisé.
Lors de l’utilisation du dispositif d’injection, l’aiguille doit être mise en place avant reconstitution.
Instructions d’élimination :
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur. Les stylos préremplis GoQuick vides ne doivent jamais être remplis à nouveau et doivent être jetés de façon appropriée.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
23-25, AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 349 755 0 9 : Poudre et solvant en cartouche à double compartiment (verre de type I) munie de pistons (caoutchouc bromobutyl) et d’une capsule (aluminium) avec un disque (caoutchouc bromobutyl) ; Boîte de 1.
· 34009 342 014 5 5 : Poudre et solvant en cartouche à double compartiment (verre de type I) munie de pistons (caoutchouc bromobutyl) et d’une capsule (aluminium) avec un disque (caoutchouc bromobutyl) ; Boîte de 5.
· 34009 497 418 2 3 : Poudre et solvant en cartouche à double compartiment (verre de type I) séparée par un piston en caoutchouc (bromobutyl) et insérée dans un stylo prérempli GoQuick 5,3 mg (couleur bleu) ; Boîte de 1.
· 34009 497 419 9 1 : Poudre et solvant en cartouche à double compartiment (verre de type I) séparée par un piston en caoutchouc (bromobutyl) et insérée dans un stylo prérempli GoQuick 5,3 mg (couleur bleu) ; Boîte de 5.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Prescription initiale hospitalière annuelle réservée aux spécialistes en pédiatrie et/ou en endocrinologie et maladies métaboliques exerçant dans les services spécialisés en pédiatrie et/ou en endocrinologie et maladies métaboliques.
ANSM - Mis à jour le : 22/05/2024
GENOTONORM 5,3 mg, poudre et solvant pour solution injectable
Somatropine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que GENOTONORM 5,3 mg, poudre et solvant pour solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser GENOTONORM 5,3 mg, poudre et solvant pour solution injectable ?
3. Comment utiliser GENOTONORM 5,3 mg, poudre et solvant pour solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver GENOTONORM 5,3 mg, poudre et solvant pour solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE GENOTONORM 5,3 mg, poudre et solvant pour solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique – Code ATC : H01AC01
GENOTONORM est une hormone de croissance recombinante humaine (également appelée somatropine). Elle a une structure similaire à celle de l’hormone de croissance humaine naturelle qui est nécessaire à la croissance des os et des muscles. Elle permet aussi aux tissus graisseux et musculaire de votre organisme de se développer dans des proportions convenables. Le terme « recombinante » signifie qu’elle n’est pas fabriquée à partir d’un tissu humain ou animal.
Chez les enfants, GENOTONORM est utilisé dans le traitement des troubles suivants :
· Si vous ne grandissez pas correctement et que vous ne produisez pas assez de votre propre hormone de croissance.
· Si vous souffrez d’un syndrome de Turner. Le syndrome de Turner est une anomalie chromosomique observée chez les filles qui peut avoir des conséquences sur la croissance – votre médecin vous aura indiqué si vous en êtes atteinte.
· Si vous souffrez d’une insuffisance rénale chronique (reins). Les reins perdent leur capacité à fonctionner normalement ce qui peut avoir des conséquences sur la croissance.
· Si vous êtes atteint d’un syndrome Prader-Willi (anomalie chromosomique). L’hormone de croissance vous aidera à grandir si vous êtes encore en période de croissance et améliorera également votre composition corporelle (diminution des graisses et augmentation de la masse musculaire).
· Si vous étiez petit(e) ou d’un poids trop faible à la naissance. Si vous n’avez pas pu conserver une croissance normale ou rattraper le retard de croissance à l’âge de quatre ans ou plus, l’hormone de croissance pourra vous aider à grandir.
Chez l’adulte, GENOTONORM est indiqué en cas d’insuffisance en hormone de croissance. Celle-ci peut survenir à l’âge adulte, ou peut être acquise depuis l’enfance.
Si vous avez été traité par GENOTONORM pour insuffisance en hormone de croissance pendant votre enfance, le déficit en hormone de croissance sera réévalué lorsque la croissance staturale sera achevée. Si l’insuffisance sévère en hormone de croissance est confirmée, votre médecin vous proposera de continuer le traitement par GENOTONORM.
Ce traitement doit vous être prescrit après confirmation du diagnostic par un médecin spécialisé et expérimenté dans la prise en charge des patients souffrant d’insuffisance en hormone de croissance.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER GENOTONORM 5,3 mg, poudre et solvant pour solution injectable ?
N’utilisez jamais GENOTONORM 5,3 mg, poudre et solvant pour solution injectable
· si vous êtes allergique à la somatropine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous avez une tumeur active (cancer). Les tumeurs doivent être inactives et vous devez avoir terminé votre traitement anti-tumoral avant de commencer votre traitement par GENOTONORM.
· si vous êtes gravement malade (par exemple, si vous souffrez de complications suite à une intervention chirurgicale à cœur ouvert ou abdominale, d’insuffisance respiratoire aiguë, d’un traumatisme accidentel ou d’affections similaires). Si vous êtes sur le point de subir une intervention majeure ou que vous venez de la subir ou êtes sur le point d’aller à l’hôpital quelle qu’en soit la cause, informez-en votre médecin et rappelez aux autres médecins que vous consultez que vous prenez de l’hormone de croissance.
· si GENOTONORM a été prescrit pour stimuler votre croissance mais vous avez déjà arrêté de grandir (épiphyses soudées).
Avertissements et précautions
Faites attention avec GENOTONORM 5,3 mg, poudre et solvant pour solution injectable et prévenez votre médecin si vous êtes concerné par un des faits suivants :
· Si vous êtes sujet à risque de développer un diabète, votre médecin devra surveiller votre taux de glucose dans le sang (glycémie) lors du traitement par GENOTONORM.
· Si vous êtes diabétique, vous devrez contrôler votre taux de glucose dans le sang au cours du traitement par GENOTONORM et discuter des résultats avec votre médecin afin de déterminer s’il convient de changer la dose de votre traitement contre le diabète.
· Après le début du traitement par GENOTONORM, certains patients peuvent avoir besoin de débuter un traitement substitutif par des hormones thyroïdiennes.
· Si vous recevez un traitement à base d’hormones thyroïdiennes, il sera peut-être nécessaire d’adapter la posologie des hormones thyroïdiennes.
· Si vous prenez de l’hormone de croissance pour stimuler votre croissance et que vous boitez ou si vous commencez à boiter à cause de douleurs dans votre hanche au cours de votre traitement par l’hormone de croissance, vous devez en informer votre médecin.
· Si vous présentez une hypertension intracrânienne (avec des symptômes tels que des maux de tête sévères, des troubles visuels ou des vomissements) vous devez en informer votre médecin.
· Si votre médecin confirme que vous avez développé une inflammation des muscles proche du site d’injection à cause de la présence d’un conservateur, le métacrésol, vous devrez utiliser un produit GENOTONORM sans métacrésol.
· Si vous recevez un traitement par GENOTONORM pour une insuffisance en hormone de croissance suite à un ancien processus tumoral, vous devez continuer à être surveillé pour vérifier que la tumeur ou tout autre cancer ne réapparaisse pas.
· Si vous présentez une douleur abdominale s’aggravant, vous devez en informer votre médecin.
· L’expérience de ce traitement chez les patients âgés de plus de 80 ans est limitée. Les personnes âgées peuvent être plus sensibles à l’action de GENOTONORM, et peuvent donc être plus sujets à l’apparition d’effets indésirables.
· L’attention des sportifs sera attirée sur le fait que cette spécialité contient un principe actif pouvant induire une réaction positive des tests pratiqués lors des contrôles antidopages.
Enfants souffrant d’insuffisance rénale chronique (rein) :
· Votre médecin devra vérifier votre fonction rénale et votre vitesse de croissance avant d’initier GENOTONORM. Le traitement que vous recevez pour votre insuffisance rénale chronique doit être poursuivi. Le traitement par GENOTONORM devra être interrompu en cas de transplantation rénale.
Enfants atteints du syndrome de Prader-Willi :
· Votre médecin vous indiquera quel est le régime hypocalorique à suivre pour contrôler votre poids.
· Votre médecin recherchera des signes d’obstruction des voies aériennes supérieures, d’apnée du sommeil (lorsque votre respiration s’interrompt au cours du sommeil), ou d’infections respiratoires avant de débuter votre traitement par GENOTONORM.
· Au cours du traitement, si vous présentez des signes d’obstruction des voies aériennes supérieures (y compris survenue ou aggravation des ronflements), votre médecin devra vous examiner et il est possible qu’il interrompe votre traitement par GENOTONORM.
· Au cours du traitement, votre médecin pratiquera un examen afin de rechercher des signes de scoliose (type de déformation de la colonne vertébrale) .
· Au cours du traitement, si vous développez une infection pulmonaire, informez-en votre médecin afin qu’il/elle puisse traiter cette infection.
Enfants nés petits pour l’âge gestationnel :
· Si vous êtes né(e) petit(e) pour l’âge gestationnel et si vous avez entre 9 et 12 ans, demandez conseil à votre médecin concernant la puberté et le traitement par ce médicament.
· Votre médecin contrôlera vos taux de glucose et d’insuline dans le sang avant de commencer le traitement puis tous les ans pendant le traitement.
· Le traitement devra être poursuivi jusqu’à l’arrêt de votre croissance staturale.
Adressez-vous à votre médecin ou pharmacien avant d’utiliser la somatropine.
Si vous avez un traitement substitutif par des glucocorticoïdes, vous devez consulter votre médecin régulièrement, car la dose des glucocorticoïdes peut nécessiter un ajustement.
Autres médicaments et GENOTONORM
Informez votre médecin ou votre pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament, y compris un médicament obtenu sans ordonnance.
Vous devez informer votre médecin si vous utilisez:
· des médicaments destinés à traiter le diabète,
· des hormones thyroïdiennes,
· des hormones surrénaliennes de synthèse (corticoïdes),
· des œstrogènes pris par voie orale ou d'autres hormones sexuelles,
· de la ciclosporine (un médicament qui affaiblit le système immunitaire après une greffe),
· des médicaments anti-épileptiques (anticonvulsivants).
Votre médecin pourra adapter la posologie de ces médicaments ou la dose de GENOTONORM.
GENOTONORM avec les aliments et boissons
Sans objet.
Vous ne devez pas utiliser GENOTONORM si vous êtes enceinte, si vous pensez être enceinte ou si vous essayez de le devenir.
Demandez conseil à votre médecin avant de prendre ce médicament si vous allaitez.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Ce médicament n’altère pas votre capacité à conduire des véhicules ou à utiliser des machines.
GENOTONORM contient du sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER GENOTONORM 5,3 mg, poudre et solvant pour solution injectable ?
La dose dépend de votre taille, de l’affection pour laquelle vous êtes traité(e) et de votre réponse au traitement. Chaque personne réagit différemment.
Votre médecin vous indiquera la dose de GENOTONORM qui vous est nécessaire soit en fonction de votre poids en kilogrammes (kg) soit en fonction de votre surface corporelle exprimée en mètres carrés (m2), ainsi que le calendrier de votre traitement. Ne modifiez pas la posologie et le calendrier du traitement sans consulter votre médecin.
Enfants présentant un déficit en hormone de croissance :
0,025 à 0,035 mg/kg de poids corporel par jour ou 0,7 à 1,0 mg/m2 de surface corporelle par jour. Des doses plus élevées peuvent être utilisées. Lorsque le déficit en hormone de croissance se poursuit à l’adolescence, GENOTONORM doit être continué jusqu'au développement physique complet.
Enfants atteints d’un syndrome de Turner :
0,045 à 0,050 mg/kg de poids corporel par jour ou 1,4 mg/m2 de surface corporelle par jour.
Enfants atteints d’insuffisance rénale (rein) :
0,045 à 0,050 mg/kg de poids corporel par jour ou 1,4 mg/m2 de surface corporelle par jour. Des doses plus élevées peuvent être nécessaires si le taux de croissance est trop faible. Un ajustement de la posologie peut s’avérer nécessaire après 6 mois de traitement.
Enfants atteints d’un syndrome de Prader-Willi:
0,035 mg/kg de poids corporel par jour ou 1,0 mg/m2 de surface corporelle par jour. La posologie quotidienne ne devra pas dépasser 2,7 mg. Le traitement ne devra pas être utilisé chez des enfants qui ont pratiquement arrêté de grandir après la puberté.
Enfants nés petits pour l’âge gestationnel et ayant des troubles de la croissance:
0,035 mg/kg de poids corporel par jour ou 1,0 mg/m2 de surface corporelle par jour. Il est important de continuer le traitement jusqu’à ce que la taille finale soit atteinte. Le traitement devra être arrêté au bout de la première année si vous ne répondez pas au traitement ou si vous avez atteint votre taille définitive ou arrêté de grandir.
Adultes présentant un déficit en hormone de croissance :
Si vous poursuivez GENOTONORM après un traitement durant l’enfance, le traitement doit être initié à une dose de 0,2 à 0,5 mg par jour. Ce dosage peut être augmenté ou diminué progressivement en fonction des résultats des examens sanguins ainsi que de la réponse clinique et des effets indésirables.
Si votre déficit en hormone de croissance a débuté alors que vous étiez adulte, la dose recommandée pour débuter le traitement est de 0,15 à 0,3 mg par jour. La dose peut être augmentée progressivement en fonction des résultats des examens sanguins ainsi que de la réponse clinique et des effets indésirables. La dose d’entretien quotidienne excède rarement 1,0 mg par jour. Les femmes peuvent avoir besoin de doses plus élevées que les hommes.
La posologie devra être contrôlée tous les 6 mois. Chez les patients de plus de 60 ans, le traitement doit être initié à une dose de 0,1 à 0,2 mg par jour. Cette posologie doit être augmentée progressivement en fonction des besoins individuels du patient. La dose d’entretien quotidienne chez ces patients excède rarement 0,5 mg par jour. Conformez vous aux instructions données par votre médecin.
Injections de GENOTONORM
GENOTONORM est destiné à être injecté par voie sous-cutanée. L’injection se fait à l’aide d’une aiguille courte dans le tissu graisseux juste sous la peau. Votre médecin doit vous avoir déjà montré comment utiliser GENOTONORM. Pratiquez l’injection de GENOTONORM exactement comme votre médecin vous a indiqué de l’effectuer. En cas de doute demandez conseil à votre médecin ou à votre pharmacien.
Les instructions d’utilisation du stylo prérempli GoQuick sont fournies dans l’emballage du stylo prérempli.
Les instructions d’utilisation de GENOTONORM cartouche à double compartiment avec le GENOTONORM Pen sont fournies avec le dispositif.
Veuillez lire les instructions d’utilisation avant d’utiliser le médicament.
Lorsque vous utilisez un stylo prérempli ou un dispositif d’injection, l’aiguille doit être vissée avant de mélanger. Une nouvelle aiguille doit être utilisée à chaque injection. Les aiguilles ne doivent pas être réutilisées.
· Préparation de l’injection:
Vous pouvez sortir GENOTONORM du réfrigérateur une demi-heure avant l’heure d’injection. Ceci permet de le réchauffer légèrement et augmente le confort de l’injection.
Le stylo prérempli GoQuick® inclut une cartouche à double compartiment qui contient à la fois l’hormone de croissance et le solvant. L’hormone de croissance et le solvant sont mélangés en faisant tourner le porte-cartouche (voir les différentes étapes décrites dans les instructions d’utilisation). Un dispositif séparé n’est pas nécessaire.
GENOTONORM sous forme de cartouche à double compartiment contient à la fois l’hormone de croissance et le solvant qui doivent être utilisés dans un dispositif GENOTONORM. L’hormone de croissance et le solvant contenus dans la cartouche à double compartiment peuvent être mélangés en vissant celle-ci dans le dispositif GENOTONORM Pen.
Pour le stylo prérempli GoQuick comme pour la cartouche à double compartiment, dissoudre la poudre en l’inclinant doucement vers l’arrière jusqu’à dissolution complète de la poudre.
Lorsque vous mélangez GENOTONORM, veillez à NE PAS AGITER la solution. Mélangez-la doucement. Le fait d’agiter la solution pourrait faire mousser l’hormone de croissance et endommager la substance active. Examinez la solution et n’effectuez pas l’injection si la solution est trouble ou si elle contient des particules.
· Injection de GENOTONORM :
N’oubliez pas de vous laver les mains et nettoyez votre peau en premier lieu.
Injectez l’hormone de croissance à peu près à la même heure tous les jours. Au moment du coucher est une heure qui convient car il est facile de s’en souvenir. Il est également naturel d’avoir un taux d’hormone de croissance plus élevé la nuit.
La plupart des personnes effectuent leur injection dans la cuisse ou la fesse. Faites l’injection à l’endroit que vous a indiqué votre médecin. Il peut se produire un amincissement des tissus graisseux de la peau au site de l’injection. Pour éviter cela, utilisez un endroit différent à chaque injection. Ceci donne le temps à votre peau et à la région située sous la peau de se reconstituer avant d’effectuer une autre injection au même endroit.
N’oubliez pas de remettre GENOTONORM au réfrigérateur aussitôt après l’injection.
Si vous avez utilisé plus de GENOTONORM 5,3 mg, poudre et solvant pour solution injectable que vous n’auriez dû
Si vous injectez beaucoup plus d’hormone de croissance que vous n’auriez dû, prévenez immédiatement votre médecin ou votre pharmacien. Votre taux de glucose dans le sang peut chuter trop bas et ultérieurement peut remonter trop haut. Vous pouvez vous sentir faible, en sueur, endormi(e) ou ne pas vous sentir « vous-même » et il est possible que vous vous évanouissiez.
Si vous oubliez d’utiliser GENOTONORM 5,3 mg, poudre et solvant pour solution injectable
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Il est préférable d’utiliser votre hormone de croissance régulièrement. S’il vous arrive d’oublier une dose, effectuez votre injection le lendemain comme si de rien n’était. Notez toutes les injections oubliées et parlez-en à votre médecin lors du prochain bilan de santé.
Si vous arrêtez d’utiliser GENOTONORM 5,3 mg, poudre et solvant pour solution injectable
Demandez conseil à votre médecin ou à votre pharmacien avant d’arrêter d’utiliser GENOTONORM.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables très fréquents et les effets indésirables fréquents chez les adultes peuvent survenir dans les premiers mois du traitement et ils peuvent disparaître spontanément ou après réduction de la dose.
Les effets indésirables très fréquents (pouvant affecter plus de 1 personne sur 10) sont les suivants :
Chez les adultes :
· Douleurs articulaires,
· Rétention d'eau (qui se traduit par un gonflement des doigts ou des chevilles).
Les effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10) sont les suivants :
Chez les enfants:
· Douleurs articulaires,
· Rougeurs temporaires, démangeaisons ou douleurs au site d’injection.
Chez les adultes:
· Engourdissements / picotements,
· Douleur ou sensation de brûlure dans les mains ou les avant-bras (Syndrome du Canal Carpien,
· Raideur des bras et des jambes, douleurs musculaires.
Les effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) sont les suivants :
Chez les enfants :
· Leucémie (ceci a été rapporté chez un petit nombre de patients atteints d’un déficit en hormone de croissance, dont certains étaient traités par somatropine. Cependant, il n'y a pas de preuve d’une augmentation de l’incidence de la leucémie chez les personnes recevant de l’hormone de croissance sans facteurs prédisposants),
· Augmentation de la pression intra-crânienne (qui se traduit par des symptômes tels que des maux de tête sévères, des troubles visuels ou des vomissements),
· Engourdissements / picotements,
· Eruption cutanée,
· Démangeaisons,
· Boursouflures associées à des démangeaisons sur la peau,
· Douleurs musculaires,
· Augmentation mammaire (gynécomastie),
· Rétention d’eau (qui se traduit par un gonflement des doigts ou des chevilles pendant une courte période au début du traitement).
Chez les adultes :
· Augmentation mammaire (gynécomastie).
Fréquence indéterminée : ne peut être estimée sur la base des données disponibles
· Diabète de type 2,
· Œdème facial,
· Maux de tête,
· Diminution des taux de l’hormone cortisol dans le sang.
Chez les enfants :
· Raideur des bras et des jambes.
Chez les adultes :
· Augmentation de la pression intracrânienne (qui se traduit par des symptômes tels que des maux de tête sévères, des troubles visuels ou des vomissements),
· Démangeaisons,
· Boursouflures associées à des démangeaisons sur la peau,
· Rougeur, démangeaisons ou douleur au site d’injection.
Formation d’anticorps dirigés contre l’hormone de croissance injectée, mais ils ne semblent pas bloquer l’action de l’hormone de croissance.
La peau autour du site d’injection peut devenir rugueuse ou bosselée mais ceci ne devrait pas se produire si vous effectuez les injections à un endroit différent chaque fois.
Un effet indésirable très rare pouvant survenir en raison de la présence d’un conservateur le métacrésol est une inflammation des muscles proches du site d’injection.
Si votre médecin vous confirme qu’il s’agit bien de ce phénomène, vous devrez utiliser un produit GENOTONORM sans métacrésol.
De rares cas de mort subite ont été observés chez des patients atteints d’un syndrome de Prader-Willi. Aucun lien n’a néanmoins été fait entre ces observations et le traitement par GENOTONORM.
En cas de survenu de gêne ou de douleur dans la hanche ou le genou pendant le traitement par GENOTONORM, votre médecin peut envisager une épiphysiolyse fémorale supérieure ou la maladie de Legg-Calve-Perthes.
Les autres effets secondaires possibles liés à votre traitement par l’hormone de croissance peuvent inclure les effets suivants.Vous (ou votre enfant) pouvez développer une augmentation du taux de sucre dans le sang ou une diminution des taux d’hormones thyroidiennes. Pour le savoir, votre médecin peut faire des analyses, et, au besoin, il prescrira le traitement adéquat. Rarement, une inflammation du pancréas a été signalée chez des patients traités par l’hormone de croissance.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER GENOTONORM 5,3 mg, poudre et solvant pour solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage après {MM/AAAA}. La date de péremption fait référence au dernier jour de ce mois.
Avant reconstitution
A conserver au réfrigérateur (entre 2 °C et 8 °C). Conserver la cartouche à double compartiment dans l’emballage extérieur à l’abri de la lumière.
Avant ouverture, le produit peut être sorti du réfrigérateur, sans y être remis, pour une durée maximale de 1 mois, à une température ne dépassant pas 25 °C mais passé ce délai, il doit être jeté.
Après reconstitution
A conserver au réfrigérateur (entre 2 °C et 8 °C) pendant au maximum 28 jours. Ne pas congeler. Conserver le stylo prérempli GoQuick dans l’emballage extérieur de GoQuick ou la cartouche à double compartiment dans la boite de GENOTONORM Pen à l’abri de la lumière.
N’utilisez pas ce médicament si vous remarquez la présence de particules ou si la solution est trouble.
Ne pas congeler ou exposer GENOTONORM au gel. S’il a été congelé, ne pas l’utiliser.
Ne jetez jamais les aiguilles ou les cartouches partiellement utilisées ou vides dans votre poubelle habituelle. Lorsque vous avez fini de vous servir d’une aiguille, vous devez la jeter dans un récipient approprié pour objets piquants/tranchants de sorte que personne ne puisse l’utiliser ou se piquer. Vous devez vous procurer des poubelles spéciales pour objets pointus auprès de l’hôpital ou du centre de soins.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient GENOTONORM 5,3 mg, poudre et solvant pour solution injectable
· La substance active est la somatropine*
· Une cartouche contient 5,3 mg de somatropine*.
· Après reconstitution, la concentration de somatropine* est de 5,3 mg par mL.
· Les autres composants de la poudre sont : glycine (E640), mannitol (E421), phosphate monosodique anhydre (E339) et phosphate disodique anhydre (E339) (voir rubrique 2 « GENOTONORM contient du sodium »).
· Les composants du solvant sont : eau pour préparations injectables, mannitol (E421) et métacrésol.
* Produite dans des cellules d’Escherichia coli par la technique de l’ADN recombinant
GENOTONORM se présente sous la forme d’une poudre et d’un solvant pour solution injectable, en cartouche à double compartiment contenant la poudre dans un des compartiments et le solvant dans l’autre (5,3 mg/mL). La cartouche peut être incluse dans un stylo prérempli. Boîte de 1 ou 5 stylo(s) préremplis, ou 1, 5 ou 20 cartouche(s).
Tous les dosages et présentations peuvent ne pas être commercialisés.
La poudre est blanche et le solvant est limpide.
Vous pouvez utiliser les cartouches dans un stylo d’injection spécifique de GENOTONORM. Les cartouches de GENOTONORM ont un code couleur et doivent être utilisées avec les GENOTONORM PEN de même code couleur afin de délivrer la dose correcte.
La cartouche de GENOTONORM Pen 5,3 mg (bleue) doit être utilisée avec le GENOTONORM Pen 5,3 mg (bleu).
Les instructions d’utilisation du dispositif sont incluses dans l’emballage du dispositif. Vous devez demander à votre médecin un dispositif d’injection si vous n’en avez pas déjà un.
Titulaire de l’autorisation de mise sur le marché
23, 25, AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
Exploitant de l’autorisation de mise sur le marché
23-25, AVENUE DU DOCTEUR LANNELONGUE
75014 PARIS
PFIZER MANUFACTURING BELGIUM NV
RIJKSWEG 12
2870 PUURS-SINT-AMANDS
BELGIQUE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen et au Royaume-Uni (Irlande du Nord) sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
|
INSTRUCTIONS D’UTILISATION DU GENOTONORM GOQUICK®
|
|
Lisez attentivement ces instructions avant d’utiliser votre stylo GoQuick.
Si vous avez d’autres questions à propos de votre traitement GENOTONORM ou de la dose à utiliser, demandez plus d’informations à votre médecin ou à votre infirmière.
A propos du GoQuick
GoQuick est un stylo pour injection prérempli, multidose, jetable. Il contient 5,3 mg de somatropine. Votre stylo peut administrer des doses allant de 0,1 mg à 1,5 mg de GENOTONORM. Chaque clic du sélecteur de dose noir modifie la dose par tranche de 0,05 mg. GENOTONORM dans votre stylo est mélangé une seule fois, lorsque vous utilisez un nouveau stylo. Vous n’avez jamais besoin de changer de cartouche. Quand votre stylo est vide, il vous suffit d’utiliser un nouveau stylo.
Votre stylo contient un sélecteur de dose. La dose est réglée une seule fois sur un nouveau stylo. Votre stylo vous permet ensuite de préparer la même dose à chaque injection. Cela vous évitera de dépasser la dose réglée. |
|
Information importante · Ne mélanger pas la poudre et le liquide de votre stylo s’il n’y a pas d’aiguille sur votre stylo. · Ne conservez pas votre stylo avec une aiguille fixée. Du Genotonorm peut fuir de votre stylo et des bulles d’air peuvent se former dans la cartouche. Retirez toujours l’aiguille et remettez le capuchon de l’aiguille ou le protège-aiguille avant de le ranger. · Faites attention à ne pas laisser tomber votre stylo. Si vous faites tomber votre stylo et qu’une partie de celui-ci semble cassée ou endommagée, ne l’utilisez pas. Contactez votre médecin ou votre infirmière pour obtenir un nouveau stylo. Si vous faites tomber votre stylo et qu’il n’est ni endommagé ni cassé, veuillez réamorcer le stylo comme décrit à l’étape 6 (Réglage et utilisation d’un nouveau stylo GoQuick). · Nettoyez votre stylo à l’aide d’un chiffon humide. Ne mettez pas le stylo sous l’eau. · Utilisez toujours une nouvelle aiguille à chaque injection. Ne partagez pas les aiguilles de votre stylo. · La graduation du volume restant située sur le côté du porte-cartouche vous indique le volume de GENOTONORM restant dans votre stylo. Conservation et mise au rebut · Conservez votre stylo au réfrigérateur (entre 2 °C et 8 °C) dans l’emballage extérieur, à l’abri de la lumière. Ne l’exposez pas au gel et ne le congelez pas. · N’utilisez pas votre stylo après la date de péremption. · 28 jours après le mélange, jetez (éliminez) votre stylo même s’il reste du médicament. · Voir au verso de cette notice comment conserver votre stylo GoQuick. · Jetez (éliminez) votre stylo conformément à la réglementation en vigueur. Demandez à votre médecin ou à votre infirmière si vous ne savez pas comment faire. Parties de votre stylo GoQuick
Les aiguilles de stylo ne sont pas fournies avec votre stylo GoQuick. Vous devrez vous procurer des aiguilles de stylo d’une longueur maximale de 8 mm dans votre pharmacie. · Aiguilles à utiliser avec votre stylo GoQuick : o 31G ou 32G (Becton Dickinson and Company) o 31G ou 32G (Novo Nordisk®) o 32,5G ou 34G (Terumo)
|
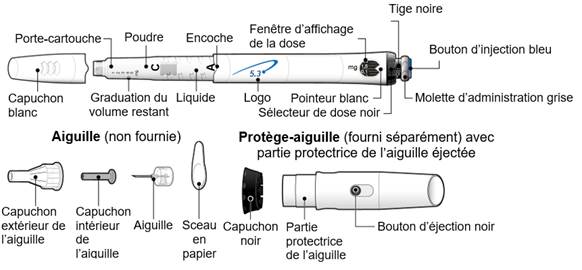
Réglage et utilisation d’un nouveau stylo GoQuick
Etape 1 Préparation

· Lavez-vous et séchez-vous les mains.
· Rassemblez le matériel suivant sur une surface plane et propre :
o un nouveau stylo GoQuick ;
o une nouvelle aiguille (non fournie) ;
o un récipient approprié pour objets piquants/tranchants (non fourni).
· Vérifiez la date de péremption sur l’étiquette de votre stylo. N’utilisez pas votre stylo si la date de péremption est dépassée.
Etape 2 Choix du site d’injection
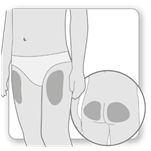
· Choisissez et nettoyez un site d’injection selon les recommandations de votre médecin ou de votre infirmière. Choisissez un site différent à chaque fois que vous pratiquez une auto-injection. Chaque nouvelle injection doit être effectuée à au moins 2 cm du site que vous avez utilisé précédemment.
· Evitez les zones osseuses, contusionnées, rouges, douloureuses ou indurées, ainsi que les zones de la peau présentant des cicatrices ou des affections cutanées.
Etape 3 Fixation d’une nouvelle aiguille
· Retirez le capuchon blanc de votre stylo.
· Prenez une nouvelle aiguille et retirez le film protecteur en papier.
· Poussez doucement l’aiguille sur votre stylo, puis vissez-la. Ne serrez pas trop.
Remarque : veillez à ne pas fixer l’aiguille de biais. Cela pourrait entraîner une fuite de votre stylo.
· Laissez les deux capuchons sur l’aiguille.
Etape 4 Mélange de GENOTONORM
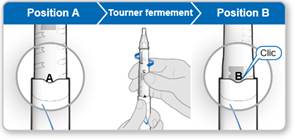
· Tenez votre stylo avec l’aiguille pointée vers le haut et le repère « A » face à vous.
· Vissez fermement le porte-cartouche dans votre stylo jusqu’à ce que le repère « B » clique dans l’encoche.
· Inclinez doucement votre stylo d’un côté puis de l’autre pour faciliter la dissolution complète de la poudre. Ne secouez pas. Les secousses pourraient endommager l’hormone de croissance.
· Vérifiez que le liquide dans la cartouche est limpide et que toute la poudre est dissoute.
o Si le liquide est trouble ou si de la poudre reste visible, inclinez doucement votre stylo d’un côté puis de l’autre plusieurs fois.
o Si le liquide est toujours trouble ou si vous voyez de la poudre, n’utilisez pas le stylo et réessayez avec un nouveau stylo.
Etape 5 Elimination de l’air de votre stylo
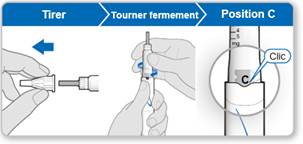
· Retirez le capuchon extérieur de l’aiguille. Conservez-le pour retirer l’aiguille après l’injection.
· Laissez le capuchon intérieur de l’aiguille.
Remarque : un capuchon intérieur de l’aiguille doit apparaître après le retrait du capuchon extérieur. Si ce n’est pas le cas, essayez à nouveau de fixer l’aiguille.
· Tenez votre stylo avec l’aiguille pointée vers le haut.
· Tapotez légèrement sur le porte-cartouche pour chasser l’air qui pourrait s’y trouver.
· Tournez fermement le porte-cartouche dans votre stylo jusqu’à ce que le repère « C » clique dans l’encoche.
o Du liquide peut apparaître autour du capuchon intérieur de l’aiguille. C’est normal.
Etape 6 Amorce de votre stylo
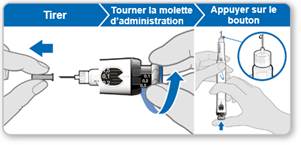
L’amorçage élimine l’air restant en poussant une petite quantité de liquide hors de votre stylo. La dose d’amorçage est de 0,1 mg et est différente de la dose prescrite par votre médecin ou votre infirmière. N’amorcez votre stylo que la première fois que vous l’utilisez.
· Retirez le capuchon intérieur de l’aiguille et jetez-le.
Attention : ne touchez pas l’aiguille pour éviter toute piqûre.
· Vérifiez que 0,1 mg apparaît dans la fenêtre d’affichage de dose.
· Tournez à fond la molette d’administration grise dans le sens des flèches jusqu’à ce qu’elle s’arrête de cliquer.
· Tenez votre stylo avec l’aiguille pointée tout droit vers le haut.
· Appuyez sur le bouton d’injection bleu jusqu’au bout.
· Vérifiez la présence de liquide à l’extrémité de l’aiguille. Si le liquide sort, votre stylo est amorcé.
o Si le liquide ne sort pas, répétez les étapes d’amorçage jusqu’à encore deux fois.
o Si le liquide ne sort toujours pas, n’utilisez pas votre stylo. Contactez votre médecin ou votre infirmière pour obtenir des conseils.
Etape 7 Réglage et préparation de votre dose
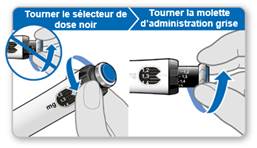
La première fois que vous utiliserez votre stylo, vous réglerez la dose prescrite par votre médecin ou votre infirmière.
Vous n’aurez plus besoin de régler la dose jusqu’à ce que vous commenciez à utiliser un nouveau stylo ou que votre médecin ou votre infirmière vous dise de le faire.
· Tournez le sélecteur de dose noir dans le sens inverse des aiguilles d’une montre jusqu’à ce que la dose soit alignée avec le pointeur blanc dans la fenêtre d’affichage de dose. Faites attention à ne pas utiliser la molette d’administration grise.
o Si vous avez dépassé votre dose avec le pointeur blanc, tournez en arrière le sélecteur de dose noir pour régler la dose correcte.
Remarque : si vous ne pouvez pas tourner le sélecteur de dose noir, appuyez sur le bouton d’injection bleu jusqu’à ce qu’il s’arrête de cliquer. Essayez ensuite de régler à nouveau votre dose. Du liquide sortira de l’aiguille.
· Tournez à fond la molette d’administration grise dans le sens des flèches jusqu’à ce qu’elle s’arrête de cliquer.
Etape 8 Vérification de votre dose

La dose sur la tige noire doit être alignée sur le pointeur blanc.
· Vérifiez que la dose que vous avez prélevée sur la tige noire correspond à la dose que vous avez réglée dans la fenêtre d’affichage de la dose.
o Si les doses correspondent, votre stylo est prêt à effectuer l’injection.
o Si les doses ne correspondent pas, assurez-vous d’avoir tourné la molette d’administration grise dans le sens des flèches jusqu’à ce qu’elle ne clique plus.
Etape 9 Réalisation de votre injection de GENOTONORM
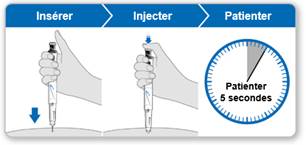
· Maintenez votre stylo sur le site d’injection.
· Insérez l’aiguille directement dans la peau.
· Appuyez à fond sur le bouton d’injection bleu jusqu’à ce qu’il arrête de cliquer.
· Patientez 5 secondes pour vous assurer que la dose complète a été injectée. Continuez à appuyer légèrement sur le bouton d’injection bleu pendant que vous comptez.
· Une fois les 5 secondes écoulées, retirez l’aiguille directement de la peau.
Remarque : si une goutte de liquide apparaît au niveau du site d’injection ou à l’extrémité de l’aiguille, essayez lors de votre prochaine injection d’appuyer plus longtemps sur le bouton d’injection bleu avant de retirer l’aiguille de votre peau.
Étape 10 Retrait de l’aiguille
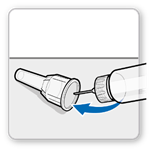
· Recouvrez soigneusement l’aiguille avec le capuchon extérieur de l’aiguille.
Attention : ne touchez pas l’aiguille pour éviter toute piqûre.
· Utilisez le capuchon de l’aiguille pour dévisser l’aiguille.
· Jetez (éliminez) l’aiguille dans un récipient approprié pour objets piquants/tranchants.
· Placez le capuchon blanc sur votre stylo.
· Conservez votre stylo au réfrigérateur jusqu’à la prochaine injection.
Utilisation courante (quotidienne) du stylo GoQuick
Etape 1 Préparation

· Lavez-vous et séchez-vous les mains.
· Rassemblez le matériel suivant sur une surface plane et propre :
o un stylo GoQuick déjà mélangé ;
o une nouvelle aiguille (non fournie) ;
o un récipient approprié pour objets piquants/tranchants (non fourni).
· Vérifiez la date de péremption sur l’étiquette de votre stylo. N’utilisez pas votre stylo si la date de péremption est dépassée.
N’utilisez pas votre stylo 28 jours après la première utilisation.
Etape 2 Choix du site d’injection
· Choisissez et nettoyez un site d’injection selon les recommandations de votre médecin ou de votre infirmière. Choisissez un site différent à chaque fois que vous pratiquez une auto-injection. Chaque nouvelle injection doit être effectuée à au moins 2 cm du site que vous avez utilisé précédemment.
· Evitez les zones osseuses, contusionnées, rouges, douloureuses ou indurées, ainsi que les zones de la peau présentant des cicatrices ou des affections cutanées.
Etape 3 Fixation d’une nouvelle aiguille
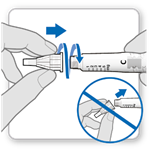
· Retirez le capuchon blanc de votre stylo.
· Prenez une nouvelle aiguille et retirez le film protecteur en papier.
· Poussez doucement l’aiguille sur votre stylo, puis vissez-la. Ne serrez pas trop.
Remarque : veillez à ne pas fixer l’aiguille de biais. Cela pourrait entraîner une fuite de votre stylo.
· Retirez les capuchons de l’aiguille.
o Conservez le capuchon extérieur de l’aiguille pour retirer l’aiguille après votre injection.
Etape 4 Préparation de votre dose
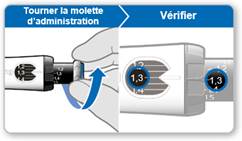
· Tournez à fond la molette d’administration grise dans le sens des flèches jusqu’à ce qu’elle s’arrête de cliquer.
· La dose sur la tige noire doit être alignée sur le pointeur blanc.
· Vérifiez que la dose que vous avez prélevée sur la tige noire correspond à la dose que vous avez réglée dans la fenêtre d’affichage de la dose.
o Si les doses correspondent, votre stylo est prêt à effectuer l’injection.
Remarque : si la dose que vous avez prélevée est plus petite, votre stylo ne contient pas une dose complète de GENOTONORM.
Suivez les instructions de votre médecin ou de votre infirmière quand il ne reste pas une dose complète dans votre stylo. Ou contactez votre médecin ou votre infirmière pour obtenir des conseils.
Etape 5 Réalisation de votre injection de GENOTONORM
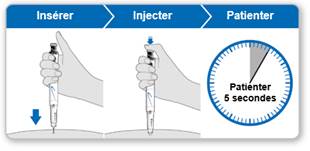
· Maintenez votre stylo sur le site d’injection.
· Insérez l’aiguille directement dans la peau.
· Appuyez à fond sur le bouton d’injection bleu jusqu’à ce qu’il arrête de cliquer.
· Patientez 5 secondes pour vous assurer que la dose complète a été injectée. Continuez à appuyer légèrement sur le bouton d’injection bleu pendant que vous comptez.
· Une fois les 5 secondes écoulées, retirez l’aiguille directement de la peau.
Remarque : si une goutte de liquide apparaît au niveau du site d’injection ou à l’extrémité de l’aiguille, essayez lors de votre prochaine injection d’appuyer plus longtemps sur le bouton d’injection bleu avant de retirer l’aiguille de votre peau.
Etape 6 Retrait de l’aiguille
· Recouvrez soigneusement l’aiguille avec le capuchon extérieur de l’aiguille.
Attention : ne touchez pas l’aiguille pour éviter toute piqûre.
· Utilisez le capuchon de l’aiguille pour dévisser l’aiguille.
· Jetez (éliminez) l’aiguille dans un récipient approprié pour objets piquants/tranchants.
· Placez le capuchon blanc sur votre stylo.
· Conservez votre stylo au réfrigérateur jusqu’à la prochaine injection.
Utilisation du protège-aiguille (en option)
Le protège-aiguille est un élément optionnel fourni séparément pour cacher l’aiguille pendant l’injection.
Fixer le protège-aiguille :
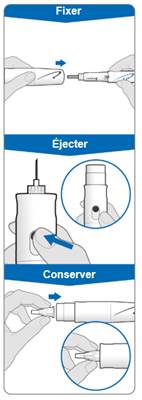 Fixez le protège-aiguille après l’étape 5 (Réglage et utilisation d’un nouveau stylo GoQuick) pour éviter toute piqûre d’aiguille.
Fixez le protège-aiguille après l’étape 5 (Réglage et utilisation d’un nouveau stylo GoQuick) pour éviter toute piqûre d’aiguille.
· Retirez le capuchon noir du protège-aiguille.
o Si la partie protectrice de l’aiguille se déplace, remettez-la en place sur le protège-aiguille jusqu’au clic.
· Alignez le logo noir du protège-aiguille avec le logo bleu de votre stylo. Poussez soigneusement le protège-aiguille sur votre stylo jusqu’à ce qu’il soit inséré correctement.
· Après l’étape 6 (Réglage et utilisation d’un nouveau stylo GoQuick), appuyez sur le bouton noir pour éjecter la partie protectrice du protège-aiguille.
· Suivez les instructions décrites à l’étape 7 (Réglage et utilisation d’un nouveau stylo GoQuick).
Pour retirer l’aiguille avec le protège-aiguille en place :
· Placez le capuchon extérieur de l’aiguille au niveau de la partie protectrice de l’aiguille.
· Utilisez le capuchon extérieur de l’aiguille pour pousser la partie protectrice de l’aiguille jusqu’à ce qu’elle se verrouille.
· Utilisez le capuchon de l’aiguille pour dévisser l’aiguille et jetez-la (éliminez-la) dans un récipient approprié pour objets piquants/tranchants.
· Laissez le protège-aiguille sur votre stylo.
· Placez le capuchon noir sur le protège-aiguille. Conservez votre stylo au réfrigérateur.
Pour retirer le protège-aiguille :
· Retirez d’abord l’aiguille, puis retirez délicatement le protège-aiguille du stylo.
· Ne jetez pas le protège-aiguille. Il pourra être utilisé avec votre prochain stylo.