Dernière mise à jour le 29/06/2026
RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale
Indications thérapeutiques
Ryaltris contient deux substances actives : le furoate de mométasone et l’olopatadine.
· Le furoate de mométasone appartient à une famille de médicaments appelés les corticoïdes (corticostéroïdes), qui réduisent l’inflammation souvent associée à la rhinite allergique.
· L’olopatadine appartient à une famille de médicaments appelés les antihistaminiques. Les antihistaminiques agissent en bloquant les effets de substances comme l’histamine, que l’organisme produit lors d’une réaction allergique. Ils réduisent donc les symptômes de la rhinite allergique.
Ryaltris est utilisé pour traiter les symptômes de la rhinite allergique saisonnière modérée à sévère (portant également le nom de rhume des foins) et de la rhinite perannuelle chez les adultes et les adolescents âgés de 12 ans ou plus.
La rhinite allergique saisonnière (rhume des foins) est une réaction allergique qui survient à certaines périodes de l’année et qui est causée par l’inhalation du pollen des arbres, des herbacées, des mauvaises herbes ainsi que des moisissures et des spores de champignons.
La rhinite perannuelle survient tout au long de l’année et ses symptômes peuvent être causés par une sensibilité à une multitude de choses, notamment les acariens, les poils (ou les squames) d’animaux, les plumes et certains aliments.
Ryaltris soulage les symptômes des allergies, comme le nez qui coule, les éternuements et le nez qui démange ou le nez bouché.
Présentations
> 1 flacon polyéthylène haute densité (PEHD) de 30 mL avec pompe doseuse (240 pulvérisations)
Code CIP : 34009 302 405 9 5
Déclaration de commercialisation : 28/03/2023
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 10,23 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 11,25 €
- Taux de remboursement :30 %
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Insuffisant | Avis du 31/08/2022 | Inscription (CT) | Le service médical rendu par RYALTRIS (association chlorhydrate d’olopatadine/furoate de mométasone), suspension pour pulvérisation nasale, est : • insuffisant chez les patients pour lesquels une monothérapie par antihistaminique (oral ou intranasal) ou corticoïde intranasal est considérée comme suffisante pour jus-tifier une prise en charge par la solidarité nationale au regard des alternatives dispo-nibles. |
| Modéré | Avis du 31/08/2022 | Inscription (CT) | Le service médical rendu par RYALTRIS (association chlorhydrate d’olopatadine/furoate de mométasone), suspension pour pulvérisation nasale, est : - modéré uniquement chez les adultes et les adolescents âgés de 12 ans et plus, pour le traitement des symptômes nasaux modérés à sévères de la rhinite allergique, unique-ment en deuxième intention, lorsqu’une monothérapie par antihistaminique ou par cor-ticoïde intranasal n’est pas considérée comme suffisante. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 31/08/2022 | Inscription (CT) | RYALTRIS (association chlorhydrate d’olopatadine/furoate de mométasone) n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux associations d’un d’anti-histaminique et d’un corticoïde intranasal dans la prise en charge dans la prise en charge des symp-tômes nasaux modérés à sévères de la rhinite allergique, en deuxième intention, lorsqu’une mono-thérapie par antihistaminique ou par corticoïde intranasal n’est pas considérée comme suffisante. |
ANSM - Mis à jour le : 04/06/2025
RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Une dose délivrée (la dose qui sort du pulvérisateur) contient une quantité de furoate de mométasone monohydraté équivalente à 25 microgrammes de furoate de mométasone et une quantité d’olopatadine équivalente à 600 microgrammes d’olopatadine.
Excipient à effet notoire :
Chaque pulvérisation contient 0,02 mg de chlorure de benzalkonium.
Pour la liste complète des excipients, voir rubrique 6.1
Suspension pour pulvérisation nasale.
Suspension blanche et homogène.
4.1. Indications thérapeutiques
Ryaltris est indiqué, chez les adultes et les adolescents âgés de 12 ans et plus, pour le traitement des symptômes nasaux modérés à sévères de la rhinite allergique.
4.2. Posologie et mode d'administration
Adultes et adolescents (12 ans et plus)
La dose habituelle recommandée est de deux pulvérisations dans chaque narine, deux fois par jour (matin et soir).
Enfants de moins de 12 ans
L’utilisation de Ryaltris est déconseillée chez les enfants de moins de 12 ans, car la sécurité d’emploi et l’efficacité n’ont pas été établies dans cette tranche d’âge.
Sujets âgés
Aucun ajustement posologique n’est nécessaire dans cette population.
Insuffisance rénale et hépatique
Bien qu’aucune donnée ne soit disponible chez les patients atteints d’insuffisance rénale ou hépatique, aucune adaptation posologique ne devrait être nécessaire dans ces populations compte tenu de l’absorption, du métabolisme et de l’élimination des substances actives (voir rubrique 5.2).
Mode d’administration
Ryaltris doit uniquement être administré par voie nasale.
Avant d’administrer la première dose, bien agiter le flacon et réaliser 6 pressions de la pompe (jusqu’à l’obtention d’une pulvérisation uniforme). Si la pompe n’a pas été utilisée pendant 14 jours ou plus, elle doit être réamorcée par 2 pressions jusqu’à obtention d’une pulvérisation uniforme, avant toute nouvelle utilisation.
Agiter le flacon pendant au moins 10 secondes avant chaque utilisation. Après utilisation, essuyer soigneusement l’embout du pulvérisateur à l’aide d’un mouchoir propre en tissu ou en papier et remettre le capuchon en place, de manière à éviter que l’embout ne se bouche. Le flacon doit être jeté quand le nombre de doses indiqué a été atteint, ou dans les 2 mois suivant la première utilisation.
Hypersensibilité aux substances actives ou à l’un des excipients mentionnés à la rubrique 6.1.
Ryaltris ne doit pas être utilisé en présence d’une infection localisée non traitée au niveau de la muqueuse nasale, telle que l’herpès.
Du fait de l’effet inhibiteur des corticoïdes sur la cicatrisation des plaies, les patients ayant récemment subi une chirurgie nasale ou un traumatisme nasal doivent s’abstenir d’utiliser un corticoïde nasal jusqu’à la cicatrisation.
4.4. Mises en garde spéciales et précautions d'emploi
Effets locaux au niveau des voies nasales
Des cas d’ulcération nasale et de perforation de la cloison nasale ont été rapportés chez des patients après l’administration intranasale d’antihistaminiques.
Des cas de perforation de la cloison nasale ont été rapportés après l’administration intranasale de corticoïdes.
Les patients qui utilisent Ryaltris pendant plusieurs mois ou une période plus longue doivent être régulièrement examinés afin de détecter toute modification de la muqueuse nasale.
Ryaltris n’est pas recommandé en cas de perforation de la cloison nasale (voir rubrique 4.8).
Des cas d’épistaxis ont été rapportés chez certains patients après administration intranasale d’antihistaminiques et de corticoïdes (voir rubrique 4.8).
Lors des études cliniques portant sur l’administration intranasale du furoate de mométasone, des infections localisées du nez et du pharynx par Candida albicans se sont produites. Quand une telle infection se produit, elle peut nécessiter d’être prise en charge à l’aide d’un traitement local approprié et l’arrêt du traitement par Ryaltris. Les patients qui utilisent Ryaltris pendant plusieurs mois ou une période plus longue doivent être régulièrement examinés afin de détecter toute présence d'infection par Candida ou d’autres signes d’effets indésirables sur la muqueuse nasale.
Troubles visuels
Des troubles visuels sont susceptibles d’être signalés en cas d’utilisation de corticoïdes systémiques et topiques (y compris par voie intranasale). Si un patient présente des symptômes tels qu’une vision floue ou d’autres troubles visuels, il faudra envisager de l’orienter vers un ophtalmologiste qui évaluera les causes potentielles des troubles visuels, par exemple, une cataracte, un glaucome ou des maladies rares telles que la choriorétinopathie séreuse centrale (CRSC), qui ont été rapportés après l’utilisation de corticoïdes systémiques et topiques.
Des réactions d’hypersensibilité, dont des cas de respiration sifflante, peuvent se produire après l’administration intranasale de furoate de mométasone monohydraté et de chlorhydrate d’olopatadine. Interrompre l’administration de Ryaltris si de telles réactions se produisent (voir rubrique 4.8).
Les personnes qui utilisent des médicaments immunosuppresseurs, tels que les corticoïdes, sont plus sensibles aux infections que les personnes en bonne santé. La varicelle et la rougeole, par exemple, peuvent avoir une évolution plus grave, voire fatale, chez les enfants ou les adultes sensibles qui utilisent des corticoïdes. Une prudence particulière s’impose pour éviter toute exposition chez les enfants ou les adultes qui n’ont pas eu ces maladies ou qui n’ont pas été convenablement immunisés contre celles-ci. On ne sait pas dans quelle mesure la dose, la voie d’administration et la durée d’administration des corticoïdes influencent le risque d’apparition d’une infection généralisée.
Les corticoïdes doivent être utilisés avec prudence, voire évités complètement, chez les patients qui présentent des infections tuberculeuses actives ou asymptomatiques des voies respiratoires, des infections fongiques ou bactériennes localisées ou systémiques non traitées, des infections virales ou parasitaires systémiques, ou un herpès simplex oculaire en raison du risque d’aggravation de ces infections.
Effets systémiques des corticoïdes
Les effets systémiques potentiels peuvent inclure le syndrome de Cushing, des caractéristiques cushingoïdes, une suppression surrénalienne, un retard de croissance chez les enfants et les adolescents, la cataracte, le glaucome et plus rarement, une série d’effets psychologiques ou comportementaux, notamment une hyperactivité psychomotrice, des troubles du sommeil, de l’anxiété, de la dépression ou de l’agressivité (en particulier chez les enfants).
Lorsque les corticoïdes à administration intranasale sont utilisés à des posologies plus élevées que les doses recommandées ou s’ils sont utilisés chez des personnes sensibles aux posologies recommandées, des effets corticoïdes systémiques tels que l’hypercorticisme et la suppression surrénalienne risquent d’apparaître. Si de tels changements apparaissent, la posologie de Ryaltris doit être interrompue lentement, conformément aux procédures acceptées pour l’arrêt de la corticothérapie par voie orale. L’utilisation concomitante de corticoïdes administrés par voie nasale et d’autres corticoïdes inhalés est susceptible d’augmenter le risque de signes ou de symptômes d’hypercorticisme et/ou de suppression de l’axe HPA.
S’il existe des preuves en faveur de l’utilisation de doses supérieures aux doses recommandées, il faudra envisager une couverture corticoïde systémique supplémentaire pendant les périodes de stress ou de chirurgie élective.
Le remplacement d’un corticoïde systémique par un corticoïde topique peut s’accompagner de signes d'insuffisance surrénalienne, et certains patients peuvent présenter des symptômes de sevrage (par ex. douleurs articulaires et/ou musculaires, lassitude et dépression). Les patients précédemment traités de façon prolongée par des corticoïdes systémiques et dont le traitement est remplacé par des corticoïdes topiques doivent faire l’objet d’une surveillance attentive visant à déceler une insuffisance surrénalienne aiguë en réponse au stress. Chez les patients qui souffrent d’asthme ou d’autres affections cliniques nécessitant un traitement systémique au long cours par des corticoïdes systémiques, une réduction trop rapide des corticoïdes systémiques risque d’entraîner une exacerbation grave de leurs symptômes.
Comme les autres antihistaminiques, l’olopatadine peut provoquer de la somnolence chez les mêmes patients lorsqu’elle est absorbée par voie systémique.
Les patients doivent être incités à la prudence pour ce qui est de pratiquer des activités dangereuses nécessitant une grande vigilance et coordination motrice, comme l’utilisation de machines ou la conduite d’un véhicule, après l’administration de Ryaltris. L’utilisation concomitante de Ryaltris avec de l’alcool ou d’autres dépresseurs du système nerveux central (SNC) doit être évitée en raison du risque de potentialisation de la diminution de la vigilance et de l’insuffisance de la performance du SNC.
Des cas de somnolence ont été rapportés suite à l’administration de Ryaltris lors des études cliniques (voir rubrique 4.8).
Effets antihistaminiques
L’utilisation concomitante d’olopatadine (par ex. des collyres) ou d’autres médicaments antihistaminiques administrés par voie nasale, oculaire ou orale peut augmenter le risque d’effets indésirables antihistaminiques.
Population pédiatrique
Il est recommandé de mesurer régulièrement la taille des enfants qui reçoivent un traitement prolongé par des corticoïdes à administration intranasale. Si la croissance est ralentie, le traitement doit être réévalué dans le but de réduire si possible la dose de corticoïde à administration intranasale, jusqu’à la dose la plus faible à laquelle le contrôle efficace des symptômes est maintenu. En outre, il faudra envisager d’orienter le patient vers un pédiatre.
Excipients :
Chaque pulvérisation de Ryaltris contient 0,02 mg de chlorure de benzalkonium. Le chlorure de benzalkonium peut provoquer une irritation ou un gonflement à l’intérieur du nez, surtout en cas d’utilisation prolongée.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée avec Ryaltris.
Toutes les interactions médicamenteuses découlant de l’association d’olopatadine et de furoate de mométasone devraient refléter celles des composants individuels, car aucune interaction pharmacocinétique entre l’olopatadine et le furoate de mométasone n’a été constatée lorsque de l’administration concomitante.
Olopatadine :
Aucune interaction entre l’olopatadine et d’autres médicaments n’est attendue (voir rubrique 5.2).
Furoate de mométasone :
On s’attend à ce que le traitement concomitant par des inhibiteurs du CYP3A, dont des produits contenant du cobicistat, augmente le risque d’effets indésirables systémiques. Cette association doit être évitée sauf si le bénéfice l’emporte sur le risque accru d’effets indésirables corticoïdes systémiques, auquel cas les patients doivent faire l’objet d’une surveillance afin de déceler la présence d’effets indésirables corticoïdes systémiques.
4.6. Fertilité, grossesse et allaitement
Grossesse
Furoate de mométasone :
Il n’existe pas de données ou il existe des données limitées sur l'utilisation du furoate de mométasone chez la femme enceinte. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3).
Olopatadine :
Il n’existe pas de données ou il existe des données limitées sur l'utilisation de l’olopatadine intranasale chez la femme enceinte.
Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction après une administration systémique (voir rubrique 5.3).
Ryaltris ne doit pas être utilisé pendant la grossesse à moins que le bénéfice potentiel pour la mère ne justifie le risque potentiel pour la mère, le fœtus ou le nouveau-né. Les nouveau-nés dont les mères ont reçu des corticoïdes pendant la grossesse doivent faire l’objet d’une surveillance attentive afin de déceler la survenue potentielle d’un hyposurrénalisme.
Allaitement
Furoate de mométasone :
On ne sait pas si le furoate de mométasone est excrété dans le lait maternel.
Olopatadine :
Les données disponibles chez l’animal montrent une excrétion d’olopatadine dans le lait suite à l’administration orale (pour plus d’informations, voir rubrique 5.3). Un risque pour le nouveau-né/nourrisson ne peut pas être exclu.
Une décision doit être prise pour savoir s’il convient d’arrêter l’allaitement ou d’arrêter/de ne pas instaurer le traitement par Ryaltris en prenant en compte le bénéfice de l’allaitement pour l’enfant et le bénéfice du traitement pour la femme.
Fertilité
Il n’existe que des données limitées en ce qui concerne la fertilité.
Il n’existe aucune donnée clinique concernant l’effet du furoate de mométasone sur la fertilité. Les études menées chez l’animal ont mis en évidence une toxicité sur la reproduction, mais aucun effet sur la fertilité.
Il n’existe aucune donnée clinique concernant l’effet de l’olopatadine sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés pendant le traitement avec Ryaltris étaient la dysgueusie (un goût désagréable spécifique à la substance), l’épistaxis et une gêne nasale.
Liste tabulée des effets indésirables
Les effets indésirables suivants ont été rapportés lors des études cliniques et du suivi post-commercialisation et sont classifiés conformément à la convention ci-après : très fréquents (≥ 1/10), fréquents (≥ 1/100 à < 1/10), peu fréquents (≥ 1/1 000 à < 1/100), rares (≥ 1/10 000 à < 1/1 000), très rares (< 1/10 000) ou fréquence indéterminée (ne peut pas être estimée à partir des données disponibles).
|
Fréquence |
Fréquents |
Peu fréquents |
Rares |
Fréquence indéterminée |
|
Classe de système d’organes |
||||
|
Infections et infestations |
|
|
Vaginose bactérienne |
Pharyngite* Infection des voies respiratoires hautes* |
|
Affections du système immunitaire |
|
|
|
Hypersensibilité, notamment réactions anaphylactiques, angiœdème, bronchospasme et dyspnée* |
|
Affections psychiatriques |
|
|
Anxiété Dépression Insomnie |
|
|
Affections du système nerveux |
Dysgueusie (goût désagréable) |
Étourdissements Céphalées Somnolence |
Léthargie Migraine |
|
|
Affections oculaires |
|
|
Vision floue Sécheresse oculaire Gêne oculaire |
Cataracte* Glaucome* Hypertension intraoculaire* |
|
Affections de l’oreille et du labyrinthe |
|
|
Otalgie |
|
|
Affections respiratoires, thoraciques et médiastinales |
Épistaxis Gêne nasale |
Sécheresse nasale |
Inflammation nasale Trouble de la muqueuse nasale Douleur oropharyngée Éternuements Irritation de la gorge |
Perforation de la cloison nasale* |
|
Affections gastro-intestinales |
|
Sécheresse buccale Douleurs abdominales Nausées |
Constipation Glossalgie |
|
|
Troubles généraux et anomalies au site d’administration |
|
Fatigue |
|
|
|
Lésions, intoxications et complications liées aux procédures |
|
|
Lacération |
|
*signalé avec l’utilisation des corticoïdes.
Des effets systémiques de certains corticoïdes à administration nasale peuvent se produire, en particulier lorsqu’ils sont administrés à doses élevées pendant des périodes prolongées (voir rubrique 4.4).
Population pédiatrique
Un retard de croissance a été rapporté chez des enfants qui recevaient des corticoïdes par voie nasale. Un retard de croissance peut être également possible chez les adolescents (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
On ne prévoit pas de réactions de surdosage avec la voie d’administration intranasale.
Aucune donnée n’est disponible chez l’homme en ce qui concerne le surdosage par ingestion accidentelle ou délibérée.
L’inhalation ou l’administration orale de doses excessives de corticoïdes peut donner lieu à la suppression de la fonction de l’axe HPA.
Il n’existe pas d’antidotes spécifiques connus aux principes actifs de Ryaltris.
En cas de surdosage, il faudra mettre en œuvre une surveillance et un traitement symptomatique appropriés du patient.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action et effets pharmacodynamiques
Ryaltris contient du chlorhydrate d’olopatadine et du furoate de mométasone, qui ont différents modes d’action et présentent des effets synergiques en termes de l’amélioration des symptômes de rhinite allergique.
L’olopatadine est un agent antiallergique / antihistaminique sélectif puissant qui exerce ses effets par le biais de plusieurs mécanismes d’action distincts. Il bloque l’action de l’histamine (le médiateur principal de la réponse allergique chez l’homme).
Le furoate de mométasone est un glucocorticoïde topique qui possède des propriétés anti-inflammatoires.
Il est probable qu’une partie importante du mécanisme responsable des effets antiallergiques et anti-inflammatoires du furoate de mométasone réside dans sa capacité d’inhiber la libération des médiateurs des réactions allergiques. Le furoate de mométasone inhibe de façon importante la libération des leucotriènes par les leucocytes des patients allergiques. Dans des cultures cellulaires, le furoate de mométasone s'est révélé extrêmement puissant pour inhiber la synthèse et la libération de l’IL-1, l’IL-5, l’IL-6 et du TNFα ; il est également un inhibiteur puissant de la production des leucotriènes. En outre, il est un inhibiteur extrêmement puissant de la production des cytokines Th2, de l’IL-4 et de l’IL-5, par les lymphocytes T CD4+ humains.
Efficacité et sécurité clinique
Lors de 2 études cliniques (GSP 301-301 et GSP 301-304) menées chez des adultes et des adolescents âgés de 12 ans ou plus souffrant de rhinite allergique, deux pulvérisations de Ryaltris dans chaque narine deux fois par jour ont amélioré les symptômes nasaux (à savoir, rhinorrhée, congestion nasale, éternuements et démangeaisons nasales) comparé au placebo, au chlorhydrate d’olopatadine seul et au furoate de mométasone seul. Le début de l'action a été observé dans les 15 minutes, il est défini comme le premier moment après le début du traitement où Ryaltris a montré un changement statistiquement significatif et cliniquement pertinent par rapport à l’état initial pour le score total instantané des symptômes nasaux (à savoir rhinorrhée, congestion nasale, éternuements et démangeaisons nasales) par rapport au placebo. Les résultats de ces deux études cliniques sont récapitulés dans le Tableau 1 et le Tableau 2 ci-dessous.
Tableau 1 : Variation moyenne par rapport aux valeurs initiales des scores totaux reflétant les symptômes nasaux sur 2 semaines* chez des adultes et des adolescents âgés d’au moins 12 ans souffrant de rhinite allergique saisonnière lors de l’étude GSP 301-301 (ensemble d’analyse intégral)
|
Valeurs initiales |
Variation par rapport aux valeurs initiales |
Différence liée à l’effet du traitement par RYALTRIS |
||||
|
Traitement |
N |
Moyenne |
Moyenne des MC |
Moyenne des MC |
IC à 95 % |
Valeur p |
|
Ryaltris |
299 |
10,1 |
‑3,48 |
-- |
-- |
-- |
|
Placebo |
283 |
10,2 |
‑2,50 |
‑0,98 |
(‑1,38 ; ‑0,57) |
< 0,0001 |
|
Chlorhydrate d’olopatadine |
294 |
10,3 |
-2,87 |
-0,61 |
(-1,01 ; -0,21) |
0,0029 |
|
Furoate de mométasone |
294 |
10,2 |
-3,09 |
-0,39 |
(-0,79 ; 0,01) |
0,0587 |
Tableau 2 : Variation moyenne par rapport aux valeurs initiales des scores totaux reflétant les symptômes nasaux sur 2 semaines* chez des adultes et des adolescents âgés d’au moins 12 ans souffrant de rhinite allergique saisonnière lors de l’étude GSP 301-304 (ensemble d’analyse intégral)
|
Valeurs initiales |
Variation par rapport aux |
Différence liée à l’effet du traitement par RYALTRIS |
||||
|
Traitement |
N |
Moyenne |
Moyenne des MC |
Moyenne des MC |
IC à 95 % |
Valeur p |
|
Ryaltris |
291 |
10,09 |
‑3,52 |
-- |
-- |
-- |
|
Placebo |
290 |
10,32 |
‑2,44 |
‑1,09 |
(‑1,49 ; ‑0,69) |
< 0,001 |
|
Chlorhydrate d’olopatadine |
290 |
10,16 |
‑3,08 |
‑0,44 |
(‑0,84 ; ‑0,05) |
0,028 |
|
Furoate de mométasone |
293 |
10,20 |
‑3,05 |
‑0,47 |
(‑0,86 ; ‑0,08) |
0,019 |
*Moyenne des rTNSS (matin et soir) pour chaque jour (score maximum = 12) sur la période de traitement de 2 semaines.
les valeurs p sont nominales
IC = intervalle de confiance ; MC = moindres carrés ; rTNSS = réduction du score total des symptômes nasaux
5.2. Propriétés pharmacocinétiques
Après administration intranasale répétée de 2 pulvérisations par narine de Ryaltris (2400 microgrammes d’olopatadine et 100 microgrammes de furoate de mométasone) deux fois par jour à des patients souffrant de rhinite allergique saisonnière, la moyenne (± écart-type) de l’exposition plasmatique maximale (Cmax) était de 19,80 ± 7,01 ng/mL pour l’olopatadine et de 9,92 ± 3,74 pg/mL pour le furoate de mométasone, et l’exposition moyenne pour l’ensemble du schéma posologique (ASCtau) était de 88,77 ± 23.87 ng*h/mL pour l’olopatadine et de 58,40 ± 27,00 pg*h/mL pour le furoate de mométasone. Le délai médian d’apparition de l’exposition maximale après administration d’une dose unique était d’1 heure pour l’olopatadine et pour le furoate de mométasone.
Rien ne semblait indiquer l’existence d’interactions pharmacocinétiques entre le furoate de mométasone et le chlorhydrate d’olopatadine.
Distribution
La liaison de l’olopatadine aux protéines a été rapportée comme étant modérée, à environ 55 % dans le sérum humain, et n’était pas dépendante de la concentration du médicament sur la plage de 0,1 à 1000 ng/mL. L’olopatadine se lie principalement à l’albumine sérique humaine.
In vitro, la liaison aux protéines pour le furoate de mométasone était de 98 % à 99 % dans la plage de concentrations de 5 à 500 ng/mL.
Biotransformation
La petite quantité de furoate de mométasone susceptible d’être ingérée et absorbée subit un important métabolisme hépatique de premier passage.
L’olopatadine n’est pas métabolisée de manière importante. Deux métabolites, le mono-déméthyl et le N-oxyde, ont été détectés à faibles concentrations dans les urines.
Les études in vitro ont montré que l’olopatadine n’inhibait pas les réactions métaboliques qui font intervenir les isoenzymes 1A2, 2C8, 2C9, 2C19, 2D6, 2E1 et 3A4 du cytochrome P-450.
Ces résultats indiquent qu’il est improbable que l’olopatadine soit à l’origine d’interactions avec d’autres substances actives administrées de façon concomitante.
Élimination
Le furoate de mométasone absorbé est métabolisé de façon importante, et les métabolites sont excrétés dans les urines et dans la bile. Après administration nasale à des volontaires en bonne santé, la demi-vie du furoate de mométasone dans le plasma était d’environ 18 à 20 heures.
Les études pharmacocinétiques indiquent qu’après administration orale, la demi-vie de l’olopatadine dans le plasma était d’environ 8 à 12 heures, et que l’élimination se faisait essentiellement par excrétion rénale. Environ 60 à 70 % de la dose étaient récupérés dans les urines sous forme de substance active.
Après administration nasale à des volontaires en bonne santé, la demi-vie de l’olopatadine dans le plasma était d’environ 6 à 7 heures.
Insuffisance hépatique
Olopatadine :
Aucun effet cliniquement pertinent de l'insuffisance hépatique n’est attendu sur la pharmacocinétique de l’olopatadine, car celle-ci est essentiellement excrétée sous forme inchangée dans les urines (voir rubrique 4.2).
Furoate de mométasone :
Une étude effectuée avec du furoate de mométasone inhalé chez des adultes présentant une insuffisance hépatique légère, modérée ou sévère, a montré que les concentrations plasmatiques maximales de furoate de mométasone semblent augmenter avec la sévérité de l’insuffisance hépatique. Toutefois, le nombre de niveaux détectables était faible (voir rubrique 4.2).
Insuffisance rénale
Olopatadine :
L’olopatadine étant excrétée dans les urines essentiellement sous forme de substance active inchangée, l’insuffisance rénale altère la pharmacocinétique de l’olopatadine pour produire une ASC0-∞ plasmatique 8 fois plus importante chez les patients qui présentent une insuffisance rénale sévère (clairance moyenne de la créatinine de 13,0 ml/min) comparé à des adultes en bonne santé. Après administration d’une dose orale de 10 mg à des patients sous hémodialyse (absence de débit urinaire), les concentrations plasmatiques d’olopatadine étaient sensiblement inférieures le jour de l’hémodialyse par rapport au jour sans hémodialyse, ce qui suggère que l’olopatadine peut être éliminée par l’hémodialyse.
Furoate de mométasone :
Compte tenu de la très faible contribution de la voie urinaire à l’élimination corporelle totale du furoate de mométasone, les effets de l’insuffisance rénale sur la pharmacocinétique du furoate de mométasone n’ont pas été étudiés (voir rubrique 4.2).
Sujets âgés
Des études comparant la pharmacocinétique de doses orales de 10 mg d’olopatadine chez des personnes jeunes (âge moyen 21 ans) et des personnes âgées (âge moyen 74 ans) n’a pas mis en évidence de différences notables au niveau des concentrations plasmatiques (ASC), de la liaison aux protéines ou de l’excrétion urinaire de la molécule mère inchangée et de ses métabolites.
5.3. Données de sécurité préclinique
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité, cancérogenèse, et des fonctions de reproduction et de développement, n’ont pas révélé de risque particulier pour l’homme.
Les études chez l'animal ont mis en évidence une réduction de la croissance des petits allaités de femelles recevant des doses systémiques d’olopatadine bien supérieures au niveau maximum recommandé pour l’utilisation intranasale chez l’homme. L’olopatadine a été détectée dans le lait de rates allaitantes après une administration orale.
Furoate de mométasone :
Aucun effet toxicologique propre à l’exposition au furoate de mométasone n’a été démontré. Tous les effets observés sont typiques de cette classe de composés et sont liés aux effets pharmacologiques exagérés des glucocorticoïdes.
Les études précliniques démontrent que le furoate de mométasone n’exerce pas d’activité androgénique, antiandrogénique, œstrogénique ou antiœstrogénique, mais qu’il présente, comme les autres glucocorticoïdes, une certaine activité anti-utérotrophique et retarde l’ouverture vaginale dans des modèles animaux à des doses orales élevées de 56 mg/kg/jour et de 280 mg/kg/jour.
Comme les autres glucocorticoïdes, le furoate de mométasone a présenté un potentiel clastogène in vitro à concentrations élevées. Cependant, aucun effet mutagène ne peut être attendu aux doses thérapeutiques pertinentes.
Lors d’études de la fonction de reproduction, l’administration sous-cutanée de furoate de mométasone à 15 microgrammes/kg a prolongé la gestation et une mise bas prolongée et difficile a eu lieu, accompagnée d’une réduction de la survie de la progéniture, ainsi qu’une réduction du poids corporel ou du gain de poids corporel des petits. Il n’y a eu aucun effet sur la fertilité.
Comme les autres glucocorticoïdes, le furoate de mométasone est un tératogène chez les rongeurs et le lapin. Les effets constatés étaient des malformations ombilicales chez le rat, des becs-de-lièvre chez la souris et des agénésies de la vésicule biliaire, des hernies ombilicales et des pattes avant fléchies chez le lapin. Ont également été notés des réductions des gains de poids corporels maternels, des effets sur la croissance fœtale (poids corporel fœtal réduit et/ou retard d’ossification) chez le rat, le lapin et la souris, ainsi qu’une réduction de la survie de la progéniture chez la souris.
Le potentiel carcinogène du furoate de mométasone inhalé (aérosol avec gaz propulseur composé de CFC et tensioactif) à des concentrations de 0,25 à 2,0 microgrammes/l a été étudié lors d’études de 24 mois chez la souris et le rat. Des effets typiques liés aux glucocorticoïdes, notamment plusieurs lésions non néoplasiques, ont été constatés. Aucune relation dose-réponse notable n’a été détectée pour aucun des types de tumeurs.
Ryaltris Pulvérisateur nasal
Une étude de toxicité en administration intranasale répétée de Ryaltris à des rats pendant une période maximum de 13 semaines n’a mis en évidence aucun nouvel effet indésirable comparativement aux composants individuels.
Evaluation du risque environnemental (ERE)
Les études sur l’évaluation du risque environnemental ont montré que le furoate de mométasone peut présenter un risque pour l’environnement aquatique (voir rubrique 6.6).
3 ans
Durée de conservation après ouverture (après la première utilisation) : 2 mois
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
La suspension pour pulvérisation nasale est contenue dans un flacon en polyéthylène haute densité blanc, avec dispositif d’actionnement manuel en polypropylène de la pompe doseuse du pulvérisateur. Le dispositif d’actionnement est muni d’un capuchon violet en PEHD.
Présentations :
1 flacon de 20 ml de 56 pulvérisations,
1 flacon de 20 ml de 120 pulvérisations,
1 flacon de 30 ml de 240 pulvérisations,
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Ce médicament peut présenter un risque pour l’environnement (voir rubrique 5.3).
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
GLENMARK PHARMACEUTICALS S.R.O.
Hvezdova 1716/2b
Prague 4, 140 78
République tchèque
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 405 9 5 : 29 g (240 pulvérisations) de suspension en flacon (PEHD) de 30 mL
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 04/06/2025
RYaltris 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale
Furoate de mométasone/Olopatadine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d’utiliser RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale?
3. Comment prendre RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale ET DANS QUELS CAS EST-IL UTILISE ?
Ryaltris contient deux substances actives : le furoate de mométasone et l’olopatadine.
· Le furoate de mométasone appartient à une famille de médicaments appelés les corticoïdes (corticostéroïdes), qui réduisent l’inflammation souvent associée à la rhinite allergique.
· L’olopatadine appartient à une famille de médicaments appelés les antihistaminiques. Les antihistaminiques agissent en bloquant les effets de substances comme l’histamine, que l’organisme produit lors d’une réaction allergique. Ils réduisent donc les symptômes de la rhinite allergique.
Ryaltris est utilisé pour traiter les symptômes de la rhinite allergique saisonnière modérée à sévère (portant également le nom de rhume des foins) et de la rhinite perannuelle chez les adultes et les adolescents âgés de 12 ans ou plus.
La rhinite allergique saisonnière (rhume des foins) est une réaction allergique qui survient à certaines périodes de l’année et qui est causée par l’inhalation du pollen des arbres, des herbacées, des mauvaises herbes ainsi que des moisissures et des spores de champignons.
La rhinite perannuelle survient tout au long de l’année et ses symptômes peuvent être causés par une sensibilité à une multitude de choses, notamment les acariens, les poils (ou les squames) d’animaux, les plumes et certains aliments.
Ryaltris soulage les symptômes des allergies, comme le nez qui coule, les éternuements et le nez qui démange ou le nez bouché.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’utiliser RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale ?
· si vous êtes allergique au furoate de mométasone ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6 ;
· si vous avez une infection non traitée dans le nez. L’utilisation de Ryaltris pendant que vous avez une infection non traitée dans le nez, comme de l’herpès, peut aggraver cette infection. Vous devez attendre que cette infection ait disparu avant de commencer à utiliser le pulvérisateur nasal ;
· si vous avez récemment subi une opération du nez ou si vous avez subi une blessure au nez. Vous ne devez pas utiliser le pulvérisateur nasal tant que votre nez n’a pas cicatrisé.
Avertissements et précautions
Adressez-vous à votre médecin ou votre pharmacien avant de prendre Ryaltris.
· si vous avez ou si vous avez déjà eu la tuberculose ;
· si vous souffrez de toute autre infection ;
· si vous prenez d’autres médicaments stéroïdes, soit par voie orale, soit pas injection.
Adressez-vous à votre médecin ou à votre pharmacien pendant que vous utilisez Ryaltris
· si vous avez des difficultés à lutter contre les infections (car votre système immunitaire ne fonctionne pas bien) et si vous entrez en contact avec une personne souffrant de la rougeole ou de la varicelle. Vous devez éviter d’entrer en contact avec toute personne souffrant de ces infections.
· si vous avez une infection du nez ou de la gorge;
· si vous utilisez ce médicament pendant plusieurs mois ou plus longtemps;
· si vous avez une irritation persistante du nez ou de la gorge;
· si vous avez une vision floue ou d’autres troubles visuels.
Lorsqu’un pulvérisateur nasal de stéroïde est utilisé à doses élevées pendant des périodes prolongées, des effets indésirables peuvent survenir du fait de l’absorption du médicament dans l’organisme. Ces effets indésirables peuvent inclure : perte de poids, fatigue, faiblesse musculaire, faible taux de sucre dans le sang (hypoglycémie), envies de sel, douleurs articulaires, dépression et brunissement de la peau. Si cela se produit, votre médecin pourra recommander un autre médicament en périodes de stress ou de chirurgie élective (non urgente).
Si vous n’êtes pas sûr(e) de savoir si ce qui précède vous concerne personnellement, consultez votre médecin ou votre pharmacien avant de prendre Ryaltris.
Enfants et adolescents
Ryaltris est déconseillé chez les enfants de moins de 12 ans.
La prise de Ryaltris de façon prolongée peut occasionner des retards de croissance chez les enfants et les adolescents. Le médecin contrôlera régulièrement la taille de votre enfant et veillera à ce qu’il ou elle prenne la dose efficace la plus faible possible.
Autres médicaments et RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Si vous prenez d’autres médicaments stéroïdes pour une allergie, par voie orale ou par injection, votre médecin pourra vous conseiller d’arrêter immédiatement de les prendre une fois que vous commencez à utiliser Ryaltris.
Si vous prenez d’autres médicaments contenant de l’olopatadine ou d’autres antihistamines, administrés par voie orale ou locale (gouttes pour le nez ou collyre), votre médecin pourra vous conseiller d’arrêter immédiatement de les prendre une fois que vous commencez à utiliser Ryaltris.
Certains médicaments peuvent accroître les effets de Ryaltris et votre médecin souhaitera peut-être vous surveiller attentivement si vous prenez ces médicaments (notamment certains médicaments pour le VIH : ritonavir, cobicistat).
RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale avec des aliments, boissons et de l’alcool
Sans objet.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
RYALTRIS ne doit pas être utilisé pendant la grossesse sauf si votre médecin le juge approprié.
Si vous prenez RYALTRIS, votre médecin discutera avec vous pour savoir si vous devez allaiter votre enfant en prenant en considération le bénéfice que vous tirerez de votre traitement et le bénéfice de l’allaitement pour votre enfant. Vous ne devez pas faire les deux à la fois.
Conduite de véhicules et utilisation de machines
Dans de très rares cas, vous pouvez présenter des étourdissements, de la léthargie, de la fatigue et de la somnolence. Si cela se produit, vous devez vous abstenir de conduire ou d’utiliser des machines. Veuillez noter que la consommation d’alcool peut accentuer ces effets.
RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale contient du chlorure de benzalkonium
Chaque pulvérisation de ce médicament contient 0,02 mg de chlorure de benzalkonium. Le chlorure de benzalkonium peut provoquer une irritation ou un gonflement à l’intérieur du nez, surtout s’il est utilisé de façon prolongée.
3. COMMENT UTILISER RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale ?
Éviter tout contact avec les yeux.
Adultes et adolescents (12 ans et plus)
La dose habituelle est de deux pulvérisations dans chaque narine, le matin et le soir.
Utilisation chez les enfants de moins de 12 ans
Ce médicament est déconseillé chez les enfants de moins de 12 ans.
Mode d’administration
Le pulvérisateur doit être utilisé par voie nasale.
Lisez attentivement et respectez strictement les instructions suivantes.
Agitez le flacon pendant au moins 10 secondes avant chaque utilisation.
Entre les utilisations de Ryaltris, le capuchon de protection violet doit toujours être maintenu fermement en place sur l’embout nasal blanc.
Votre flacon pour pulvérisation nasale Ryaltris
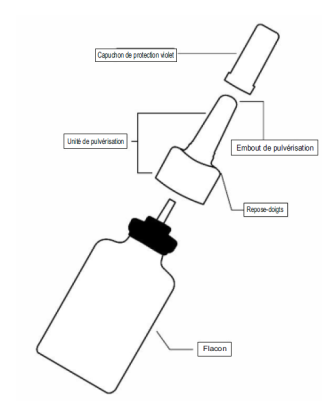
Préparation du flacon pour pulvérisation nasale
|
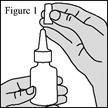
1. Agitez le flacon pendant au moins 10 secondes, puis retirez le capuchon de protection violet (voir figure 1). 2. Si vous utilisez le pulvérisateur pour la première fois, vous devrez « amorcer » le flacon en actionnant la pompe pour libérer une pulvérisation dans l’air. 3. Maintenez fermement le flacon pour pulvérisation nasale en position verticale avec l’index et le majeur de part et d’autre de l’applicateur du pulvérisateur nasal (sur les repose-doigts) avec le pouce reposant sur la base rainurée du flacon. 4. Dirigez l’embout loin de vous puis réalisez 6 pressions de la pompe pour déclencher celle-ci jusqu’à l’apparition d’une fine brume (voir figure 2). 5. Votre pompe est à présent amorcée et prête à l’emploi. 6. Si vous n’avez pas utilisé le pulvérisateur pendant 14 jours ou plus, vous devrez bien agiter le flacon et « réamorcer » le flacon en réalisant 2 pressions de la pompe du pulvérisateur ou jusqu’à production d’une fine brume. |
|
|
|
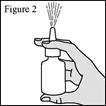
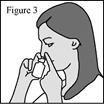
Comment utiliser votre pulvérisateur nasal
|
1. Agitez le flacon pendant au moins 10 secondes avant chaque utilisation (matin et soir). 2. Mouchez-vous doucement pour vider vos narines. 3. Maintenez fermement le flacon en position verticale avec l’index et le majeur de part et d’autre de l’applicateur du pulvérisateur nasal (sur les repose-doigts) avec le pouce reposant sur la base rainurée du flacon. 4. Pincez une narine avec le doigt et introduisez avec précaution l’embout du pulvérisateur dans l’autre narine, en le dirigeant légèrement vers la paroi extérieure du nez (voir figure 3). 5. Inclinez légèrement la tête vers l’avant. Réalisez une pression rapide et ferme sur les repose-doigts pour activer la pompe. 6. Inspirez (inhalez) doucement par le nez tout en administrant une pulvérisation. Expirez ensuite par la bouche (voir figure 4). 7. Répétez les étapes ci-dessus et administrez une deuxième pulvérisation dans la même narine. 8. Répétez ces étapes pour administrer 2 pulvérisations dans l’autre narine. 9. Afin d’éviter que l’embout ne se bouche après chaque utilisation, essuyez soigneusement l’embout du pulvérisateur à l’aide d’un mouchoir ou d’un chiffon propre et sec (voir figure 5). 10. Maintenez l’unité de pulvérisation et remettez le capuchon de protection violet sur l’embout jusqu’à entendre un « déclic » (voir figure 6). |
|
|
|

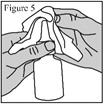
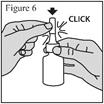
Comment nettoyer votre pulvérisateur nasal
Si l’embout se bouche, veuillez procéder de la manière décrite dans les étapes suivantes :
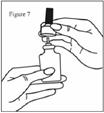
· Retirez l’embout de pulvérisation en le tirant doucement vers le haut (voir la figure 7). Retirez le capuchon de protection violet et placez seulement l’embout de pulvérisation dans de l’eau chaude et laissez-le tremper.
· N’essayez pas de déboucher l’applicateur nasal en y introduisant une aiguille ou un autre objet pointu, car cela endommagerait l’applicateur et vous n’obtiendriez pas la dose correcte de médicament.
· Après avoir laissé tremper l’embout de pulvérisation pendant 15 minutes, rincer le ainsi que le capuchon de protection violet avec de l’eau chaude et laissez-les sécher complètement.
· Remettez le capuchon de protection violet sur l’embout de pulvérisation et remettez l’ensemble en place sur le flacon.
· Après avoir suivi les instructions pour déboucher votre embout de pulvérisation encrassé, voir la rubrique « Préparation du flacon pour pulvérisation nasale » ci-dessus et réamorcez celui-ci en effectuant 2 pulvérisations. Remettez le capuchon de protection violet en place et votre Ryaltis est prêt à être utilisé.
· Répétez les étapes précédentes pour déboucher l’embout si nécessaire.
Si vous avez pris plus de RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale que vous n’auriez dû
Il est peu probable que vous ayez des problèmes, mais si vous craignez d’avoir utilisé des doses supérieures à la dose recommandée pendant une période prolongée, contactez votre médecin.
Si vous utilisez des stéroïdes pendant une période prolongée ou en grandes quantités, ils peuvent dans de rares cas affecter certaines hormones. Chez l’enfant, cela peut avoir des répercussions sur la croissance et le développement.
Si vous oubliez de prendre RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale
Utilisez votre pulvérisateur nasal dès que vous vous en rendez compte, puis prenez la dose suivante à l’heure habituelle. Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale
Il est très important que vous utilisiez votre pulvérisateur nasal de façon régulière. N’arrêtez pas votre traitement, même si vous vous sentez mieux, sauf si votre médecin vous l’a indiqué.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Des réactions immédiates d’hypersensibilité (allergiques) peuvent se produire après avoir utilisé ce produit. Ces réactions peuvent être sévères. Vous devez arrêter d’utiliser Ryaltris et consulter immédiatement un médecin ou vous rendre sur-le-champ à l’hôpital le plus proche si vous ressentez des symptômes tels que : gonflement du visage, de la langue ou du pharynx, difficultés à avaler, urticaire, respiration sifflante ou difficultés à respirer.
Effets fréquents (peuvent toucher jusqu’à 1 personne sur 10) :
· Un goût amer dans la bouche
· Saignement de nez
· Légère irritation à l’intérieur du nez.
Effets peu fréquents (peuvent toucher jusqu’à 1 personne sur 100) :
· Étourdissements
· Maux de tête
· Somnolence
· Sécheresse nasale
· Sécheresse buccale
· Douleurs abdominales
· Nausées
· Fatigue
Effets rares (peuvent toucher jusqu’à 1 personne sur 1 000) :
· Vaginose bactérienne (infection bactérienne du vagin)
· Anxiété, dépression, insomnie
· Léthargie, migraine
· Sécheresse oculaire, vision floue, gêne oculaire
· Otalgie (douleur à l’oreille)
· Maux de gorge
· Éternuements
· Irritation de la gorge
· Constipation
· Langue douloureuse
· Gonflement et ulcères à l’intérieur du nez
Fréquence indéterminée (ne peut pas être estimée à partir des données disponibles)
· Augmentation de la pression à l’intérieur de l'œil (glaucome) et/ou cataracte entraînant des troubles visuels
· Dommages au niveau de la cloison nasale qui sépare les narines
· Difficulté à respirer et/ou respiration sifflante
· Infection des voies respiratoires
Des effets indésirables systémiques (effets indésirables concernant l’ensemble du corps) peuvent se produire lorsque ce médicament est utilisé à doses élevées de manière prolongée. Ces effets sont bien moins susceptibles de se produire si vous utilisez un pulvérisateur nasal de stéroïdes que si vous prenez des stéroïdes par voie orale. Ces effets peuvent varier d’un patient à l’autre.
Les stéroïdes à administration nasale peuvent altérer la production normale des hormones dans l’organisme, en particulier si vous utilisez des doses élevées de manière prolongée. Chez les enfants et les adolescents, cet effet indésirable peut entraîner un retard de croissance.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon et sur la boîte après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Ne pas congeler.
Le flacon doit être utilisé dans les 2 mois après la première ouverture.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient RYALTRIS 25 microgrammes/600 microgrammes/dose, suspension pour pulvérisation nasale
· Les substances actives sont : le furoate de mométasone (sous forme monohydratée) et l’olopatadine (sous forme de chlorhydrate).
Une dose délivrée (la dose qui sort du pulvérisateur) contient une quantité de furoate de mométasone monohydraté équivalente à 25 microgrammes de furoate de mométasone et une quantité d’olopatadine équivalente à 600 microgrammes d’olopatadine.
· Les autres composants sont :
Cellulose microcristalline (E 460), carmellose sodique (E 466), phosphate de sodium dibasique heptahydraté (E 339), chlorure de sodium, chlorure de benzalkonium, Glycérol, édétate disodique, polysorbate 80 (E 433), acide chlorhydrique (E 507), hydroxyde de sodium (E 524) et eau pour préparations injectables.
Ryaltris est une suspension blanche et homogène.
Ryaltris est contenu dans un flacon en polyéthylène haute densité blanc, avec dispositif d’actionnement manuel de la pompe doseuse de pulvérisation en polypropylène. Le dispositif d’actionnement est protégé par un capuchon violet en PEHD.
Présentations :
1 flacon de 20 ml de 56 pulvérisations,
1 flacon de 20 ml de 120 pulvérisations,
1 flacon de 30 ml de 240 pulvérisations,
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
GLENMARK PHARMACEUTICALS S.R.O.
Hvezdova 1716/2b
Prague 4, 140 78
République tchèque
Exploitant de l’autorisation de mise sur le marché
MENARINI FRANCE
1-7, rue du Jura
94633 Rungis Cedex
Glenmark Pharmaceuticals s.r.o.
Fibichova 143
566 17 Vysoke Myto
République tchèque
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).