Dernière mise à jour le 01/06/2026
APREPITANT SANDOZ 80 mg, gélule
Indications thérapeutiques
Classe pharmacothérapeutique : Antiémétiques et antinauséeux - code ATC : A04AD12
La substance active contenue dans APREPITANT SANDOZ est l'aprépitant. APREPITANT SANDOZ appartient à un groupe de médicaments appelés « antagonistes des récepteurs de la neurokinine 1 (NK1) ». Le cerveau possède une zone spécifique qui contrôle les nausées et vomissements. APREPITANT SANDOZ agit en bloquant les signaux vers cette zone, réduisant ainsi les nausées et vomissements.
APREPITANT SANDOZ gélules est utilisé chez les adultes et les adolescents à partir de 12 ans, en association avec d’autres médicaments pour prévenir les nausées et les vomissements causés par une chimiothérapie (traitement du cancer) qui entraîne des nausées et vomissements importants et modérés (tels que cisplatine, cyclophosphamide, doxorubicine ou épirubicine).
Présentations
> plaquette(s) aluminium OPA : polyamide orienté PVC de 2 gélule(s)
Code CIP : 34009 301 394 6 2
Déclaration de commercialisation : 06/01/2020
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 14,84 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 15,86 €
- Taux de remboursement :65 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 03/05/2024
APREPITANT SANDOZ 80 mg, gélule
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque gélule contient 80 mg d’aprépitant.
Excipient à effet notoire : Chaque gélule contient 80 mg de saccharose.
Pour la liste complète des excipients, voir rubrique 6.1.
Gélule de taille 2, opaque avec un corps et une coiffe blancs, contenant des microgranules blancs à blanc cassé.
4.1. Indications thérapeutiques
APREPITANT SANDOZ 125 mg et APREPITANT SANDOZ 80 mg sont administrés dans le cadre d’un schéma thérapeutique (voir rubrique 4.2).
4.2. Posologie et mode d'administration
Adultes
APREPITANT SANDOZ est administré durant 3 jours dans le cadre d’un schéma thérapeutique comportant un corticostéroïde et un antagoniste 5-HT3. La dose recommandée est pour APREPITANT SANDOZ 125 mg par voie orale de une fois par jour une heure avant le début de la chimiothérapie à J1 et pour APREPITANT SANDOZ 80 mg par voie orale de une fois par jour à J2 et J3 le matin.
Les schémas thérapeutiques suivants sont recommandés pour la prévention des nausées et des vomissements associés à une chimiothérapie anticancéreuse émétisante chez les adultes :
Schéma thérapeutique dans le cadre d’une chimiothérapie hautement émétisante
|
|
J1 |
J2 |
J3 |
J4 |
|
Aprépitant |
125 mg par voie orale |
80 mg par voie orale |
80 mg par voie orale |
- |
|
Dexaméthasone |
12 mg par voie orale |
8 mg par voie orale |
8 mg par voie orale |
8 mg par voie orale |
|
Antagonistes 5-HT3 |
Dose standard des antagonistes 5-HT3. Voir le Résumé des Caractéristiques du Produit de l’antagoniste 5-HT3 choisi pour plus d’informations sur la posologie appropriée |
- |
- |
- |
La dexaméthasone doit être administrée 30 minutes avant le début de la chimiothérapie à J1 et le matin de J2 à J4. La dose de dexaméthasone tient compte des interactions entre les substances actives.
Schéma thérapeutique dans le cadre d’une chimiothérapie moyennement émétisante
|
|
J1 |
J2 |
J3 |
|
Aprépitant |
125 mg par voie orale |
80 mg par voie orale |
80 mg par voie orale |
|
Dexaméthasone |
12 mg par voie orale |
- |
- |
|
Antagonistes 5-HT3 |
Dose standard des antagonistes 5-HT3. Voir le Résumé des Caractéristiques du Produit de l’antagoniste 5-HT3 choisi pour plus d’informations sur la posologie appropriée |
- |
- |
La dexaméthasone doit être administrée 30 minutes avant le début de la chimiothérapie à J1. La dose de dexaméthasone tient compte des interactions entre les substances actives.
Population pédiatrique
Adolescents (âgés de 12 à 17 ans)
APREPITANT SANDOZ est administré durant 3 jours dans le cadre d’un schéma thérapeutique comportant un antagoniste 5-HT3. La dose recommandée de gélules d'APREPITANT SANDOZ est de 125 mg par voie orale à J1 et de 80 mg par voie orale à J2 et J3. APREPITANT SANDOZ est administré par voie orale une heure avant la chimiothérapie à J1, J2 et J3. Si aucune chimiothérapie n’est administrée à J2 et J3, APREPITANT SANDOZ doit être administré le matin. Voir le Résumé des Caractéristiques du Produit (RCP) de l'antagoniste 5-HT3 choisi pour des informations sur la posologie appropriée. Si un corticostéroïde, tel que la dexaméthasone, est co-administré avec APREPITANT SANDOZ, la dose de corticostéroïde doit être administrée à 50 % de la dose habituelle (voir rubriques 4.5 et 5.1).
La sécurité d’emploi et l'efficacité de APREPITANT SANDOZ 80 mg et de APREPITANT SANDOZ 125 mg n'ont pas été établies chez les enfants de moins de 12 ans. Aucune donnée n’est disponible. Pour le dosage approprié chez les enfants et les nourrissons âgés de 6 mois à moins de 12 ans, reportez-vous au RCP de la poudre pour suspension buvable.
Information générale
Les données d’efficacité en association avec d’autres corticostéroïdes et d’autres antagonistes 5-HT3 sont limitées. Pour plus d’informations concernant l'administration simultanée avec des corticostéroïdes, voir rubrique 4.5. Se référer au RCP de l’antagoniste 5-HT3 co-administré.
Populations particulières
Sujet âgé (≥ 65 ans)
Aucun ajustement posologique n’est nécessaire chez le sujet âgé (voir rubrique 5.2).
Sexe
Aucun ajustement posologique n’est nécessaire en fonction du sexe (voir rubrique 5.2).
Insuffisance rénale
Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuffisance rénale ou chez les patients présentant une insuffisance rénale au stade terminal nécessitant une hémodialyse (voir rubrique 5 2).
Insuffisance hépatique
Aucun ajustement posologique n’est nécessaire chez les patients ayant une insuffisance hépatique légère. Les données disponibles chez les patients ayant une insuffisance hépatique modérée sont limitées, et aucune donnée chez les patients ayant une insuffisance hépatique sévère n'est disponible.
APREPITANT SANDOZ doit être utilisé avec précaution chez ces patients (voir rubriques 4.4 et 5.2).
Mode d'administration
La gélule doit être avalée entière. L’aprépitant peut être pris avec ou sans aliments.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Co-administration avec le pimozide, la terfénadine, l’astémizole ou le cisapride (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Insuffisants hépatiques modérés à sévères
Les données chez les patients ayant une insuffisance hépatique modérée sont limitées, et aucune donnée chez les patients ayant une insuffisance hépatique sévère n'est disponible. L’aprépitant doit être utilisé avec précaution chez ces patients (voir rubrique 5.2).
Interactions avec le CYP3A4
APREPITANT SANDOZ doit être utilisé avec précaution chez les patients prenant de façon concomitante par voie orale des substances actives métabolisées principalement par le CYP3A4 et ayant une marge thérapeutique étroite, telles que la ciclosporine, le tacrolimus, le sirolimus, l'évérolimus, l'alfentanil, les alcaloïdes dérivés de l’ergot de seigle, le fentanyl et la quinidine (voir rubrique 4.5). De plus, l'administration concomitante avec l’irinotécan doit être envisagée avec une prudence toute particulière, cette association pouvant majorer sa toxicité.
Co-administration avec la warfarine (un substrat du CYP2C9)
Chez les patients traités au long cours par la warfarine, l’INR (International Normalised Ratio) doit être étroitement surveillé au cours du traitement par aprépitant et pendant 14 jours après chaque cure de 3 jours d’aprépitant (voir rubrique 4.5).
Co-administration avec les contraceptifs hormonaux
L’efficacité des contraceptifs hormonaux peut être réduite pendant l’administration d’aprépitant et au cours des 28 jours qui la suivent. Des méthodes alternatives de contraception non hormonale doivent être utilisées au cours du traitement par aprépitant et pendant les 2 mois qui suivent la dernière prise d’aprépitant (voir rubrique 4.5).
Excipient à effet notoire
APREPITANT SANDOZ contient du saccharose. Les patients présentant une intolérance au fructose, un syndrome de malabsorption du glucose et du galactose ou un déficit en sucrase/isomaltase (maladies héréditaires rares)
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Effet de l’aprépitant sur la pharmacocinétique d’autres substances actives
Inhibition du CYP3A4
En tant qu’inhibiteur modéré du CYP3A4, l’aprépitant (125 mg et 80 mg) peut entraîner une élévation des concentrations plasmatiques des substances actives administrées de façon concomitante et qui sont métabolisées par le CYP3A4. L’exposition totale de substrats du CYP3A4 administrés par voie orale peut augmenter jusqu’à 3 fois environ au cours du traitement de 3 jours par aprépitant ; l’effet attendu de l’aprépitant sur les concentrations plasmatiques des substrats du CYP3A4 administrés par voie intraveineuse est moindre. L’aprépitant ne doit pas être administré de façon concomitante avec le pimozide, la terfénadine, l'astémizole ou le cisapride (voir rubrique 4.3). L'inhibition du CYP3A4 par l'aprépitant pourrait entraîner une élévation des concentrations plasmatiques de ces substances actives, susceptible de provoquer des réactions graves ou de mettre en jeu le pronostic vital. La prudence s’impose lors de la co-administration d’APREPITANT SANDOZ et de substances actives administrées par voie orale, métabolisées principalement par le CYP3A4 et ayant une marge thérapeutique étroite, telles que la ciclosporine, le tacrolimus, le sirolimus, l'évérolimus, l'alfentanil, la diergotamine, l'ergotamine, le fentanyl et la quinidine (voir rubrique 4.4).
Corticostéroïdes
Dexaméthasone : La dose orale habituelle de dexaméthasone doit être réduite d’environ 50 % en cas de co-administration avec l’aprépitant selon le schéma posologique de 125 mg/80 mg. La dose de dexaméthasone au cours des essais cliniques portant sur les nausées et vomissements induits par une chimiothérapie a été choisie en tenant compte des interactions entre les substances actives (voir rubrique 4.2). L’administration de 125 mg d’aprépitant en association à 20 mg de dexaméthasone par voie orale à J1, et l’administration d’aprépitant 80 mg/jour en association à 8 mg de dexaméthasone par voie orale de J2 à J5, a entraîné une élévation de l’ASC de la dexaméthasone, un substrat du CYP3A4, de 2,2 fois à J1 et J5.
Méthylprednisolone : La dose habituelle de méthylprednisolone administrée par voie intraveineuse doit être réduite d’environ 25 %, et la dose orale habituelle de méthylprednisolone doit être réduite d’environ 50 % en cas de co-administration avec l’aprépitant selon le schéma posologique de 125 mg/80 mg. L’administration d’aprépitant selon le schéma posologique de 125 mg à J1 et 80 mg/jour à J2 et J3, a augmenté l’ASC de la méthylprednisolone, un substrat du CYP3A4, de 1,3 fois à J1 et de 2,5 fois à J3, lors de la co-administration de 125 mg de méthylprednisolone par voie intraveineuse à J1 et de 40 mg par voie orale à J2 et J3.
Au cours d’un traitement continu avec la méthylprednisolone, l’ASC de la méthylprednisolone peut diminuer ultérieurement dans les 2 semaines qui suivent l’initiation du traitement par aprépitant, à cause de l’effet inducteur de l’aprépitant sur le CYP3A4. On peut s’attendre à ce que cet effet soit plus prononcé avec la méthylprednisolone administrée par voie orale.
Médicaments chimiothérapeutiques
Lors d’études de pharmacocinétique, l’administration d’aprépitant à la posologie de 125 mg à J1 et de 80 mg/jour à J2 et J3, n’a pas modifié la pharmacocinétique du docétaxel administré par voie intraveineuse à J1 ou de la vinorelbine administrée par voie intraveineuse à J1 ou J8. L’effet d’APREPITANT SANDOZ sur la pharmacocinétique des substrats du CYP3A4 administrés par voie orale étant supérieur à celui sur la pharmacocinétique des substrats du CYP3A4 administrés par voie intraveineuse, une interaction avec les médicaments chimiothérapeutiques administrés par voie orale et métabolisés principalement ou partiellement par le CYP3A4 (par exemple, l’étoposide, la vinorelbine) ne peut être exclue. Il est recommandé d’être prudent et une surveillance supplémentaire peut être appropriée chez les patients recevant des médicaments métabolisés principalement ou partiellement par le CYP3A4 (voir rubrique 4.4). Depuis la commercialisation, des évènements de neurotoxicité, un effet indésirable potentiel de l’ifosfamide, ont été rapportés après une administration concomitante d’aprépitant et d’ifosfamide.
Immunosuppresseurs
Une augmentation transitoire modérée, suivie d'une légère diminution de l'exposition aux immunosuppresseurs métabolisés par le CYP3A4 (tels que la ciclosporine, le tacrolimus, l'évérolimus et le sirolimus) sont attendues au cours du traitement de 3 jours administré pour la prévention des nausées et des vomissements induits par la chimiothérapie (NVIC). La durée de traitement de 3 jours étant courte, et les variations de l’exposition étant limitées en fonction du temps, aucune réduction de la dose de l'immunosuppresseur n'est recommandée pendant ces 3 jours d'administration concomitante avec l’aprépitant.
Midazolam
De potentiels effets sur l’augmentation des concentrations du midazolam ou d’autres benzodiazépines métabolisées par le CYP3A4 (alprazolam, triazolam) doivent être envisagés en cas de co-administration de ces médicaments avec l’aprépitant (125 mg ou 80 mg).
L’aprépitant a augmenté l’ASC du midazolam, un substrat sensible du CYP3A4, de 2,3 fois à J1 et de 3,3 fois à J5, lorsqu’une dose orale unique de 2 mg de midazolam a été associée à J1 et à J5 au schéma posologique de 125 mg d’aprépitant à J1 et 80 mg/jour de J2 à J5.
Dans une autre étude réalisée avec le midazolam par voie intraveineuse, l’aprépitant a été administré à la posologie de 125 mg à J1 et 80 mg/jour à J2 et J3, et 2 mg de midazolam ont été administrés par voie intraveineuse avant l'administration d’aprépitant selon le schéma posologique de 3 jours ainsi qu’à J4, J8 et J15. L’aprépitant a augmenté l’ASC du midazolam de 25 % à J4 et a diminué l’ASC du midazolam de 19 % à J8 et de 4 % à J15. Ces effets n’ont pas été considérés comme cliniquement importants.
Dans une troisième étude réalisée avec le midazolam par voie intraveineuse et par voie orale, l’aprépitant a été administré à la posologie de 125 mg à J1 et 80 mg/j à J2 et J3, associé à 32 mg d’ondansétron à J1, à 12 mg de dexaméthasone à J1 et 8 mg de dexaméthasone de J2 à J4. Cette association (c’est-à-dire aprépitant, ondansétron et dexaméthasone) a diminué l’ASC du midazolam administré par voie orale de 16 % à J6, 9 % à J8, 7 % à J15 et 17 % à J22. Ces effets n’ont pas été considérés comme cliniquement importants.
Une étude supplémentaire a été réalisée avec administration intraveineuse de midazolam et d’aprépitant. 2 mg de midazolam ont été administrés par voie intraveineuse 1 heure après une prise unique d’aprépitant 125 mg par voie orale. L’ASC plasmatique du midazolam a été augmentée de 1,5 fois. Cet effet n’a pas été considéré comme cliniquement important.
Induction
En tant qu’inducteur léger du CYP2C9, du CYP3A4 et de la glucuronidation, l’aprépitant peut diminuer les concentrations plasmatiques des substrats éliminés par ces voies au cours des deux semaines suivant la mise en route du traitement. Cet effet peut n’apparaître qu’après la fin du traitement de 3 jours par aprépitant. Pour les substrats du CYP2C9 et du CYP3A4, l’induction est transitoire avec un effet maximum atteint 3 à 5 jours après la fin du traitement de 3 jours par aprépitant.
L’effet persiste pendant quelques jours, diminue ensuite lentement et est cliniquement non significatif deux semaines après la fin du traitement par aprépitant. Une induction légère de la glucuronidation est également constatée avec 80 mg d’aprépitant administrés par voie orale pendant 7 jours. Il n’y a pas de données concernant les effets sur le CYP2C8 et le CYP2C19. La prudence s’impose lors de l’administration, pendant cette période, de warfarine, d’acénocoumarol, de tolbutamide, de phénytoïne ou d’autres substances actives connues pour être métabolisées par le CYP2C9.
Warfarine
Chez les patients sous traitement chronique par la warfarine, le temps de Quick (INR) doit être surveillé étroitement au cours du traitement par aprépitant et pendant les 2 semaines suivant chaque cure de 3 jours d’aprépitant pour la prévention des nausées et vomissements induits par une chimiothérapie (voir rubrique 4.4). Lors de l’administration d’une dose unique de 125 mg d’aprépitant à J1, et de 80 mg/jour à J2 et J3, à des sujets sains stabilisés traités au long cours par la warfarine, il n’y a pas eu d’effet d’aprépitant sur l’ASC plasmatique des stéréoisomères R+ ou S- de la warfarine à J3 ; cependant, il y a eu une réduction de 34 % de la concentration résiduelle en stéréoisomère S- de la warfarine (un substrat du CYP2C9), accompagnée d’une diminution de 14 % de l’INR, 5 jours après la fin du traitement par aprépitant.
Tolbutamide
L’aprépitant, administré à la dose de 125 mg à J1 et de 80 mg/jour à J2 et J3, a abaissé l’ASC du tolbutamide (un substrat du CYP2C9) de 23 % à J4, de 28 % à J8 et de 15 % à J15, lors de l’administration d’une dose orale unique de 500 mg de tolbutamide avant l’administration d’aprépitant selon le schéma posologique de 3 jours et à J4, J8 et J15.
Contraceptifs hormonaux
L’efficacité des contraceptifs hormonaux peut être réduite pendant l'administration d’aprépitant et au cours des 28 jours qui la suivent. Des méthodes alternatives de contraception non hormonale doivent être utilisées au cours du traitement par aprépitant et pendant les 2 mois qui suivent la dernière prise d’aprépitant.
Dans une étude clinique, des doses uniques d’un contraceptif oral contenant de l’éthinylestradiol et de la noréthindrone ont été administrées de J1 à J21 avec de l’aprépitant pris selon le schéma posologique de 125 mg à J8 et 80 mg/jour à J9 et J10, associé à 32 mg d’ondansétron par voie intraveineuse à J8 et à la dexaméthasone par voie orale à la posologie de 12 mg à J8 et 8 mg/jour à J9, J10 et J11. Dans cette étude, il y a eu, de J9 à J21, une diminution allant jusqu’à 64 % des concentrations résiduelles d’éthinylestradiol et une diminution allant jusqu’à 60 % des concentrations résiduelles de noréthindrone.
Antagonistes 5-HT3
Au cours des études cliniques d’interaction, l’aprépitant n’a pas eu d’effet cliniquement significatif sur la pharmacocinétique de l’ondansétron, du granisétron ou de l’hydrodolasétron (le métabolite actif du dolasétron).
Effet d’autres médicaments sur la pharmacocinétique de l’aprépitant
La co-administration d’aprépitant et de substances actives inhibant l’activité du CYP3A4 (telles que le kétoconazole, l'itraconazole, le voriconazole, le posaconazole, la clarithromycine, la télithromycine, la néfazodone et les inhibiteurs de protéase) doit être envisagée avec précaution, une augmentation de plusieurs fois des concentrations plasmatiques d'aprépitant étant attendue avec cette association (voir rubrique 4.4).
La co-administration d’aprépitant et de substances actives induisant fortement l’activité du CYP3A4 (telles que la rifampicine, la phénytoïne, la carbamazépine, le phénobarbital) doit être évitée, une telle association entraînant des diminutions des concentrations plasmatiques de l'aprépitant et donc une diminution de l’efficacité d’aprépitant. La co-administration d’aprépitant et de préparations à base de plantes contenant du millepertuis (Hypericum perforatum) n’est pas recommandée.
Kétoconazole
Lors de l'administration d'une dose unique de 125 mg d’aprépitant à J5 d’un schéma posologique de 10 jours de 400 mg/jour de kétoconazole, un puissant inhibiteur du CYP3A4, l’ASC de l’aprépitant a augmenté d’environ 5 fois et la demi-vie terminale moyenne de l’aprépitant a augmenté d’environ 3 fois.
Rifampicine
Lors de l'administration d'une dose unique de 375 mg d’aprépitant à J9 d’un schéma posologique de 14 jours de 600 mg/jour de rifampicine, un puissant inducteur du CYP3A4, l’ASC de l’aprépitant a diminué de 91 % et la demi-vie terminale moyenne a diminué de 68 %.
Population pédiatrique
Les études d'interactions ont été réalisées uniquement chez l'adulte.
4.6. Fertilité, grossesse et allaitement
Contraception chez les hommes et les femmes
L’efficacité des contraceptifs hormonaux peut être réduite pendant l'administration d’aprépitant et au cours des 28 jours qui la suivent. Des méthodes alternatives de contraception non hormonale doivent être utilisées au cours du traitement par aprépitant et pendant les 2 mois qui suivent la dernière prise d'aprépitant (voir rubriques 4.4 et 4.5).
Grossesse
Il n'y a pas de données cliniques disponibles sur l'utilisation de l'aprépitant chez la femme enceinte. La toxicité potentielle de l’aprépitant sur la reproduction n’a pas été complètement décrite, car les niveaux d’exposition supérieurs à ceux obtenus chez l’homme en thérapeutique à la dose de 125 mg/80 mg n’ont pu être atteints dans les études chez l’animal. Ces études n’ont pas mis en évidence d’effets délétères directs ou indirects sur la grossesse, le développement embryonnaire ou foetal, l'accouchement ou le développement post-natal (voir rubrique 5.3). Les effets potentiels des altérations de la régulation de la neurokinine sur la reproduction ne sont pas connus. L’aprépitant ne doit pas être utilisé au cours de la grossesse sauf en cas de nécessité absolue.
Allaitement
L’aprépitant est excrété dans le lait des rates allaitantes. On ne sait pas si l’aprépitant est excrété dans le lait maternel humain ; par conséquent, il n’est pas recommandé d’allaiter au cours d’un traitement par aprépitant.
Fertilité
Les effets potentiels de l'aprépitant sur la fertilité n'ont pas été pleinement définis car les niveaux d'exposition supérieurs à l'exposition chez l'homme n'ont pu être atteints dans les études chez l'animal. Ces études de fertilité n'ont pas mis en évidence d'effets délétères directs ou indirects sur la procréation, la fertilité, le développement embryonnaire, ou foetal ou le nombre de spermatozoïdes et leur motilité (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Le profil de sécurité de l’aprépitant a été évalué chez environ 6 500 adultes dans plus de 50 essais cliniques et chez 184 enfants et adolescents dans 2 essais cliniques pédiatriques contrôlés.
Les effets indésirables les plus fréquents, rapportés avec une incidence supérieure chez les patients adultes traités par l’aprépitant comparés à ceux recevant un traitement standard dans le cadre d’une Chimiothérapie Hautement Emétisante (CHE), ont été : hoquet (4,6 % versus 2,9 %), élévation de l'alanine aminotransférase (ALAT) (2,8 % versus 1,1 %), dyspepsie (2,6 % versus 2,0 %), constipation (2,4 % versus 2,0 %), céphalées (2,0 % versus 1,8 %) et diminution de l'appétit (2,0 % versus 0,5 %). L’effet indésirable le plus fréquent, rapporté avec une incidence supérieure chez les patients traités par l'aprépitant comparés à ceux recevant un traitement standard dans le cadre d’une Chimiothérapie Moyennement Emétisante (CME), a été la fatigue (1,4 % versus 0,9 %).
Les effets indésirables les plus fréquents, rapportés avec une incidence supérieure chez les patients pédiatriques traités par l'aprépitant comparés à ceux recevant un traitement contrôle pendant leur chimiothérapie anticancéreuse émétisante, ont été : hoquet (3,3 % versus 0,0 %) et bouffées congestives (1,1 % versus 0,0 %).
Liste des effets indésirables présentée sous forme de tableau
Les effets indésirables suivants ont été observés avec une incidence supérieure chez les patients adultes ou pédiatriques traités par l’aprépitant comparés à ceux recevant un traitement standard d’après une analyse poolée des études cliniques réalisées avec des chimiothérapies hautement et moyennement émétisantes (CHE et CME), ou depuis la mise sur le marché. Les catégories de fréquences mentionnées dans le tableau sont basées sur les études menées chez les adultes ; les fréquences observées lors des études pédiatriques ont été similaires ou inférieures, sauf mention dans le tableau. Certains effets indésirables moins fréquents dans la population adulte n'ont pas été observés lors des études pédiatriques.
Définition des fréquences : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) et très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes de systèmes d’organes |
Effet indésirable |
Fréquence |
|
Infections et infestations |
candidose, infection à staphylocoques |
rare |
|
Affections hématologiques et du système lymphatique |
neutropénie fébrile, anémie |
peu fréquent |
|
Affections du système immunitaire |
réactions d’hypersensibilité incluant réactions anaphylactiques |
fréquence indéterminée |
|
Troubles du métabolisme et de la nutrition |
diminution de l’appétit |
fréquent |
|
polydipsie |
rare |
|
|
Affections psychiatriques |
anxiété |
peu fréquent |
|
désorientation, humeur euphorique |
rare |
|
|
Affections du système nerveux |
céphalées |
fréquent |
|
étourdissements, somnolence |
peu fréquent |
|
|
troubles cognitifs, léthargie, dysgueusie |
rare |
|
|
Affections oculaires |
conjonctivite |
rare |
|
Affections de l’oreille et du labyrinthe |
acouphènes |
rare |
|
Affections cardiaques |
palpitations |
peu fréquent |
|
bradycardie, troubles cardiovasculaires |
rare |
|
|
Affections vasculaires |
bouffées de chaleur/bouffées congestives |
peu fréquent |
|
Affections respiratoires, thoraciques et médiastinales |
hoquet |
fréquent |
|
douleur oro-pharyngée, éternuements, toux, rhinorrhée postérieure, irritation de la gorge |
rare |
|
|
Affections gastro-intestinales |
constipation, dyspepsie |
fréquent |
|
éructation, nausées, vomissements, reflux gastro-œsophagien, douleur abdominale, bouche sèche, flatulence |
peu fréquent |
|
|
ulcère duodénal perforé, stomatite, distension abdominale, selles dures, colite neutropénique |
rare |
|
|
Affections de la peau et du tissu sous-cutané |
rash, acné |
peu fréquent |
|
réaction de photosensibilité, hyperhidrose, séborrhée, lésions cutanées, éruption cutanée prurigineuse, syndrome de Stevens-Johnson/syndrome de Lyell |
rare |
|
|
prurit, urticaire |
fréquence indéterminée |
|
|
Affections musculo-squelettiques et systémiques |
faiblesse musculaire, spasmes musculaires |
rare |
|
Affections du rein et des voies urinaires |
dysurie |
peu fréquent |
|
pollakiurie |
rare |
|
|
Troubles généraux et anomalies au site d’administration |
fatigue |
fréquent |
|
asthénie, malaise |
peu fréquent |
|
|
œdème, gêne thoracique, trouble de la démarche |
rare |
|
|
Investigations |
élévation des ALAT |
fréquent |
|
élévation des ASAT, élévation des phosphatases alcalines sanguines |
peu fréquent |
|
|
présence de globules rouges dans les urines, diminution du taux de sodium sanguin, perte de poids, diminution du nombre de neutrophiles, présence de glucose dans les urines, augmentation de la diurèse |
rare |
Les nausées et vomissements étaient des paramètres d’efficacité au cours des 5 premiers jours suivant la chimiothérapie et n’étaient rapportés comme effets indésirables qu’ensuite.
Description de certains effets indésirables
Les profils des effets indésirables observés chez les adultes lors d’études d’extension à des cycles multiples de chimiothérapies dans le cadre de chimiothérapies hautement et moyennement émétisantes (CHE et CME), allant jusqu’à 6 cycles supplémentaires de chimiothérapie, ont été généralement similaires à ceux observés au cours du cycle 1.
Dans une étude clinique supplémentaire contrôlée versus comparateur actif, réalisée chez 1 169 patients adultes recevant de l'aprépitant et une chimiothérapie hautement émétisante (CHE), le profil des effets indésirables a été généralement similaire à celui observé au cours des autres études réalisées avec l'aprépitant dans le cadre de chimiothérapies hautement émétisantes (CHE).
D’autres effets indésirables ont été rapportés chez des patients adultes traités par l’aprépitant pour des nausées et vomissements post-opératoires (NVPO) avec une incidence supérieure à celle observée chez les patients traités par l’ondansétron : douleur abdominale haute, bruits intestinaux anormaux, constipation*, dysarthrie, dyspnée, hypoesthésie, insomnie, myosis, nausées, troubles sensoriels, gêne stomacale, subiléus*, baisse de l’acuité visuelle, respiration sifflante.
* rapporté chez des patients prenant une plus forte dose d'aprépitant.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En cas de surdosage, l’aprépitant doit être arrêté et des mesures générales symptomatiques ainsi qu’une surveillance clinique doivent être mises en oeuvre. En raison de l’activité antiémétique de l’aprépitant, les médicaments provoquant des vomissements peuvent ne pas être efficaces.
L’aprépitant ne peut être éliminé par hémodialyse.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antiémétiques et antinauséeux, code ATC : A04AD12
L’aprépitant est un antagoniste sélectif à haute affinité pour les récepteurs de la substance P neurokinine 1 (NK1) humaine.
Traitement de 3 jours par l'aprépitant chez les adultes
Au cours de deux études randomisées en double aveugle incluant un total de 1 094 patients adultes sous chimiothérapie avec une dose de cisplatine ≥ 70 mg/m2, l’aprépitant en association à un schéma posologique ondansétron/dexaméthasone (voir rubrique 4.2) a été comparé à un schéma posologique standard (placebo plus 32 mg d'ondansétron administré en intraveineux à J1 plus 20 mg de dexaméthasone par voie orale à J1 et 8 mg par voie orale deux fois par jour de J2 à J4). Bien qu’une dose intraveineuse de 32 mg d’ondansétron ait été utilisée dans les études cliniques, celle-ci n’est plus la dose recommandée. Voir le Résumé des Caractéristiques du Produit de l’antagoniste 5-HT3 choisi pour plus d’informations sur la posologie appropriée.
L’efficacité a été évaluée sur la base du critère composite suivant : réponse complète (définie par l’absence d’épisodes émétiques et l’absence de recours à un traitement de secours), principalement au cours du cycle 1. Les résultats ont été évalués individuellement pour chaque étude ainsi que pour les 2 études combinées.
Un résumé des résultats clés issus de l’analyse combinée des études est donné dans le Tableau 1.
Tableau 1
Dans le cadre d’une chimiothérapie hautement émétisante, pourcentage de patients adultes répondeurs par groupe et phase de traitement – cycle 1
|
CRITERES COMPOSITES |
Aprépitant (N= 521) % |
Traitement standard (N= 524) % |
Différences* % |
(IC 95 %) |
|
Réponse complète (pas de vomissements et pas de traitement de secours) |
||||
|
Total (0-120 heures) |
67,7 |
47,8 |
19,9 |
(14,0 ; 25,8) |
|
0-24 heures |
86,0 |
73,2 |
12,7 |
(7,9 ; 17,6) |
|
25-120 heures |
71,5 |
51,2 |
20,3 |
(14,5 ; 26,1) |
|
CRITERES INDIVIDUELS |
||||
|
Pas de vomissements (pas d’épisodes émétiques avec ou sans traitement de secours) |
||||
|
Total (0-120 heures) |
71,9 |
49,7 |
22,2 |
(16,4 ; 28,0) |
|
0-24 heures |
86,8 |
74,0 |
12,7 |
(8,0 ; 17,5) |
|
25-120 heures |
76,2 |
53,5 |
22,6 |
(17,0 ; 28,2) |
|
Pas de nausées significatives (VAS max < 25 mm sur une échelle de 0 à 100 mm) |
||||
|
Total (0-120 heures) |
72,1 |
64,9 |
7,2 |
(1,6 ; 12,8) |
|
25-120 heures |
74,0 |
66,9 |
7,1 |
(1,5 ; 12,6) |
*Les intervalles de confiance ont été calculés sans ajustement en fonction du sexe et des chimiothérapies concomitantes, lesquels avaient été pris en compte dans l’analyse primaire du risque relatif et des modèles logistiques.
Un patient dans le groupe aprépitant a été exclu de l'analyse globale et de celle de la phase retardée, ses données n'étant disponibles que pour la phase aiguë ; un patient recevant le traitement standard a été exclu de l'analyse globale et de celle de la phase aiguë, ses données n'étant disponibles que pour la phase retardée.
Dans l’analyse combinée, le délai estimé jusqu’au premier vomissement est donné par la courbe de Kaplan-Meier sur la Figure 1.
Figure 1
Pourcentage de patients adultes recevant une chimiothérapie hautement émétisante et indemnes de vomissements – cycle 1

Des différences statistiquement significatives dans l’efficacité ont également été observées individuellement dans chacune des 2 études.
Dans le cadre de ces 2 mêmes études cliniques, 851 patients adultes ont poursuivi une extension de l’évaluation lors des cycles ultérieurs allant jusqu’à 5 cycles supplémentaires de chimiothérapie. L’efficacité du schéma aprépitant s’est apparemment maintenue durant tous les cycles.
Dans une étude randomisée, en double aveugle, réalisée sur un total de 866 patients adultes (864 femmes, 2 hommes) recevant une chimiothérapie comprenant soit du cyclophosphamide 750-1 500 mg/m2, soit du cyclophosphamide 500-1 500 mg/m2 et de la doxorubicine (≤ 60 mg/m2) ou de l’épirubicine (≤ 100 mg/m2), l’aprépitant en association à un traitement ondansétron/dexaméthasone (voir rubrique 4.2) a été comparé à un traitement standard (placebo plus 8 mg d'ondansétron par voie orale (2 fois à J1 et toutes les 12 heures à J2 et J3) plus 20 mg de dexaméthasone par voie orale à J1).
L’efficacité a été évaluée sur la base du critère composite : réponse complète (définie par l’absence d’épisodes émétiques et l’absence de recours à un traitement de secours), principalement au cours du cycle 1.
Un résumé des résultats clés de l’étude est donné dans le Tableau 2.
Tableau 2
Dans le cadre d’une chimiothérapie moyennement émétisante, pourcentage de patients adultes répondeurs par groupe et phase de traitement - cycle 1
|
CRITERES COMPOSITES |
Aprépitant (N= 433) % |
Traitement standard (N= 424) % |
Différences* % |
(IC 95 %) |
|
Réponse complète (pas de vomissements et pas de traitement de secours) |
||||
|
Total (0-120 heures) |
50,8 |
42,5 |
8,3 |
(1,6 ; 15,0) |
|
0-24 heures |
75,7 |
69,0 |
6,7 |
(0,7 ; 12,7) |
|
25-120 heures |
55,4 |
49,1 |
6,3 |
(-0,4 ; 13,0) |
|
CRITERES INDIVIDUELS |
||||
|
Pas de vomissements (pas d’épisodes émétiques avec ou sans traitement de secours) |
||||
|
Total (0-120 heures) |
75,7 |
58,7 |
17,0 |
(10,8 ; 23,2) |
|
0-24 heures |
87,5 |
77,3 |
10,2 |
(5,1 ; 15,3) |
|
25-120 heures |
80,8 |
69,1 |
11,7 |
(5,9 ; 17,5) |
|
Pas de nausées significatives (VAS max < 25 mm sur une échelle de 0 à 100 mm) |
||||
|
Total (0-120 heures) |
60,9 |
55,7 |
5,3 |
(-1,3 ; 11,9) |
|
0-24 heures |
79,5 |
78,3 |
1,3 |
(-4,2 ; 6,8) |
|
25-120 heures |
65,3 |
61,5 |
3,9 |
(-2,6 ; 10,3) |
* Les intervalles de confiance ont été calculés sans ajustement en fonction de la tranche d’âge (< 55 ans, ≥ 55 ans) et du groupe d’investigateur, lesquels avaient été pris en compte dans l’analyse primaire du risque relatif et des modèles logistiques.
Un patient dans le groupe aprépitant a été exclu de l'analyse globale et de celle de la phase retardée, ses données n'étant disponibles que pour la phase aiguë.
Dans le cadre de cette même étude clinique, 744 patients adultes ont poursuivi une extension de l’évaluation lors des cycles ultérieurs allant jusqu’à 3 cycles supplémentaires de chimiothérapie. L’efficacité du schéma aprépitant s’est apparemment maintenue durant tous les cycles.
Dans une seconde étude clinique multicentrique, randomisée, en double aveugle, sur des groupes parallèles, l'aprépitant a été comparé au traitement standard chez 848 patients adultes (652 femmes, 196 hommes) recevant une chimiothérapie qui comportait une administration intraveineuse, quelle que soit la dose d'oxaliplatine, de carboplatine, d'épirubicine, d'idarubicine, d'ifosfamide, d'irinotécan, de daunorubicine, de doxorubicine ; du cyclophosphamide par voie intraveineuse (< 1 500 mg/m2) ; ou de la cytarabicine par voie intraveineuse (> 1g/m2). Les patients sous aprépitant recevaient une chimiothérapie pour divers types de tumeurs dont 52 % de cancers du sein, 21 % de cancers gastro-intestinaux y compris le cancer colorectal, 13 % de cancers pulmonaires et 6 % de cancers gynécologiques. L'aprépitant en association à un traitement ondansétron/dexaméthasone (voir rubrique 4.2) a été comparé au traitement standard (placebo associé à 8 mg d'ondansétron par voie orale (2 fois à J1 et toutes les 12 heures à J2 et J3) plus 20 mg de dexaméthasone par voie orale à J1).
L'efficacité était basée sur l'évaluation du critère primaire et du principal critère secondaire suivants : pas de vomissements pendant toute la période (de 0 à 120 heures après la chimiothérapie), évaluation de la sécurité d'emploi et de la tolérance de l'aprépitant pour le traitement des nausées et vomissements induits par une chimiothérapie (NVIC) ainsi que la réponse complète (pas de vomissements et pas de traitement de secours) pendant toute la période (0 à 120 heures après la chimiothérapie). De plus, le critère « Pas de nausées significatives pendant toute la période (0-120 heures après la chimiothérapie) » a été évalué à titre exploratoire et dans les phases aiguë et retardée sous forme d'analyse post-hoc.
Un résumé des résultats clés de l'étude est donné dans le tableau 3.
Tableau 3
Dans le cadre d’une chimiothérapie moyennement émétisante, pourcentage de patients adultes répondeurs par groupe et phase de traitement pour l'étude 2 – cycle 1
|
|
Aprépitant (N= 425) % |
Traitement standard (N= 406) % |
Différences* % |
(IC 95 %) |
|
Réponse complète (pas de vomissements et pas de traitement de secours) |
||||
|
Total (0-120 heures) |
68,7 |
56,3 |
12,4 |
(5,9 ; 18,9) |
|
0-24 heures |
89,2 |
80,3 |
8,9 |
(4,0 ; 13,8) |
|
25-120 heures |
70,8 |
60,9 |
9,9 |
(3,5 ; 16,3) |
|
Pas de vomissements (pas d’épisodes émétiques avec ou sans traitement de secours) |
||||
|
Total (0-120 heures) |
76,2 |
62,1 |
14,1 |
(7,9 ; 20,3) |
|
0-24 heures |
92,0 |
83,7 |
8,3 |
(3,9 ; 12,7) |
|
25-120 heures |
77,9 |
66,8 |
9,9 |
(5,1 ; 17,1) |
|
Pas de nausées significatives (VAS max < 25 mm sur une échelle de 0 à 100 mm) |
||||
|
Total (0-120 heures) |
73,6 |
66,4 |
7,2 |
(1,0 ; 13,4) |
|
0-24 heures |
90,9 |
86,3 |
4,6 |
(0,2 ; 9,0) |
|
25-120 heures |
74,9 |
69,5 |
5,4 |
(-0,7 ; 11,5) |
* Les intervalles de confiance ont été calculés sans ajustement en fonction du sexe et de la localisation de la tumeur, lesquels avaient été pris en compte dans l’analyse primaire utilisant des modèles logistiques.
Le bénéfice du traitement par l'aprépitant associé au traitement standard dans la population totale de l'étude est principalement dû aux résultats observés chez les patients faiblement contrôlés par le traitement standard tels que les femmes, même si les résultats sont supérieurs en nombre quels que soient l'âge, le type de tumeur ou le sexe. La réponse complète à l'aprépitant et au traitement standard a été atteinte chez respectivement 209/324 (65 %) et 161/320 (50 %) des femmes et chez 83/101 (82 %) et 68/87 (78 %) des hommes.
Population pédiatrique
Dans une étude clinique randomisée, en double aveugle, contrôlée versus comparateur actif, réalisée chez 302 enfants et adolescents (âgés de 6 mois à 17 ans) recevant une chimiothérapie moyennement ou hautement émétisante, le traitement par aprépitant a été comparé à un traitement contrôle pour la prévention des NVIC. L'efficacité de l'aprépitant a été évaluée sur un seul cycle (cycle 1). Les patients ont eu la possibilité de recevoir l’aprépitant en ouvert pour les cycles suivants (optionnel pour les cycles 2-6) ; cependant l'efficacité n'a pas été évaluée pour ces cycles optionnels. Le traitement par aprépitant pour les adolescents âgés de 12 à 17 ans (n = 47) était constitué de gélules d’aprépitant de 125 mg par voie orale à J1 et de 80 mg/jour à J2 et à J3, en association avec ondansétron à J1. Le traitement par aprépitant pour les enfants âgés de 6 mois à moins de 12 ans (n = 105) était constitué de poudre pour suspension buvable d’APREPITANT SANDOZ à 3,0 mg/kg (jusqu'à 125 mg) par voie orale à J1 et à 2,0 mg/kg (jusqu'à 80 mg) par voie orale à J2 et à J3, en association avec ondansétron à J1. Le traitement contrôle chez les adolescents âgés de 12 à 17 ans (n = 48) et les enfants âgés de 6 mois à moins de 12 ans (n = 102) se composait d’un placebo de l'aprépitant à J1, J2 et J3, en association avec ondansétron à J1. Les administrations d’aprépitant ou du placebo et d’ondansétron avaient lieu respectivement 1 heure et 30 minutes avant le début de la chimiothérapie. L’utilisation de la dexaméthasone par voie intraveineuse était autorisée dans le cadre du traitement antiémétique pour les patients pédiatriques dans les deux groupes d'âge, à la discrétion du médecin. Une réduction de la dose (50 %) de dexaméthasone était requise chez les patients pédiatriques recevant aprépitant. Aucune réduction de dose n’était requise chez les patients pédiatriques recevant le traitement contrôle. Parmi les patients pédiatriques, la dexaméthasone faisait partie du traitement au cours du cycle 1 chez 29 % de ceux recevant aprépitant et chez 28 % de ceux recevant le traitement contrôle.
L'action antiémétique d'aprépitant a été évaluée sur une période de 5 jours (120 heures) après l’initiation de la chimiothérapie à J1. Le critère d'évaluation principal était la réponse complète dans la phase retardée (25 à 120 heures après le début de la chimiothérapie) du cycle 1. Un résumé des résultats clés de l'étude est présenté dans le tableau 4.
Tableau 4
Nombre (%) de patients pédiatriques présentant une réponse complète et aucun vomissement par groupe de traitement et par phase – cycle 1 (population en intention de traiter)
|
|
Traitement par aprépitant n/m (%) |
Traitement contrôle n/m (%) |
|
CRITERE PRINCIPAL |
||
|
Réponse complète* - Phase retardée |
77/152 (50,7) |
39/150 (26,0) |
|
AUTRES CRITERES PREDEFINIS |
||
|
Réponse complète* - Phase aiguë |
101/152 (66,4) |
78/150 (52,0) |
|
Réponse complète* - Phases aiguë et retardée |
61/152 (40,1) |
30/150 (20,0) |
|
Pas de vomissement§ - Phases aiguë et retardée |
71/152 (46,7) |
32/150 (21,3) |
|
*Réponse complète = Pas de vomissement ni de haut-le-coeur ni de nausées et pas de recours à des traitements de secours. p < 0,01 par rapport au traitement contrôle p < 0,05 par rapport au traitement contrôle §Pas de vomissement = pas de vomissement ni de haut-le-coeur ni de nausées n/m = Nombre de patients présentant une réponse souhaitée / nombre de patients inclus à cet instant. Phase aiguë : 0 à 24 heures après l'initiation de la chimiothérapie. Phase retardée : 25 à 120 heures après l'initiation de la chimiothérapie. Phases aiguë et retardée : 0 à 120 heures après l'initiation de la chimiothérapie. |
||
Le délai estimé jusqu’au premier vomissement après l’initiation de la chimiothérapie était plus long avec le traitement par l'aprépitant (le délai estimé médian jusqu’au premier vomissement était de 94,5 heures) par rapport au groupe de traitement contrôle (le délai estimé médian jusqu’au premier vomissement était de 26,0 heures), comme illustré par la courbe de Kaplan-Meier en Figure 2.
Figure 2
Délai jusqu’au premier épisode de vomissement à partir du début de l'administration de la chimiothérapie dans la population pédiatrique pendant les phases aiguë et retardée – cycle 1 (population en intention de traiter)
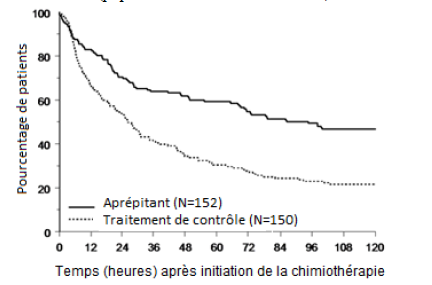
Une analyse de l'efficacité au cycle 1 dans les sous-groupes a démontré que, indépendamment de la catégorie d'âge, du sexe, de l'utilisation de la dexaméthasone pour la prophylaxie antiémétique, et du potentiel émétogène de la chimiothérapie, le traitement par aprépitant a permis un meilleur contrôle que le traitement contrôle selon les critères d'évaluation de réponse complète.
5.2. Propriétés pharmacocinétiques
Absorption
La biodisponibilité absolue moyenne de l’aprépitant par voie orale est de 67 % pour la gélule de 80 mg et de 59 % pour la gélule de 125 mg. Le pic moyen de concentration plasmatique (Cmax) de l’aprépitant est survenu aux environs de la 4ème heure (tmax). L’administration orale de la gélule avec un petit déjeuner standard d’environ 800 Kcal a entraîné une augmentation de 40 % de l’ASC de l’aprépitant. Cette augmentation n’est pas jugée pertinente sur le plan clinique.
La pharmacocinétique de l’aprépitant est non linéaire sur l’éventail des doses cliniques. Chez le jeune adulte sain, l’augmentation de l’ASC0-∞ a été de 26 % supérieure à la proportionnalité de la dose, pour des doses uniques de 80 et de 125 mg administrées non à jeun.
Après administration orale d’une dose unique de 125 mg d’aprépitant à J1 et de 80 mg une fois par jour à J2 et J3, l’ASC0-24h (moyenne ± ET) a été de 19,6 ± 2,5 μgh/mL et de 21,2 ± 6,3 μgh/mL à J1 et J3 respectivement. La Cmax a été de 1,6 ± 0,36 μg/mL et de 1,4 ± 0,22 μg/mL à J1 et J3 respectivement.
Distribution
L’aprépitant se lie fortement aux protéines, avec une moyenne de 97 %. La moyenne géométrique du volume apparent de distribution à l’état d’équilibre (Vdss) est d’environ 66 L chez l’homme.
Biotransformation
L’aprépitant subit un métabolisme important. Chez le jeune adulte sain, l’aprépitant représente environ 19 % de la radioactivité mesurée au niveau du plasma durant les 72 heures qui suivent l’administration d’une dose intraveineuse unique de 100 mg de fosaprépitant, une prodrogue de l’aprépitant, marqué au [14C], ce qui indique une présence substantielle de métabolites au niveau du plasma. Douze métabolites de l’aprépitant ont été identifiés dans le plasma humain. Le métabolisme de l’aprépitant intervient largement via l’oxydation au niveau du cycle de la morpholine et de ses chaînes latérales, les métabolites qui en résultent n’étant que faiblement actifs. Les études réalisées in vitro sur des microsomes hépatiques humains indiquent que l’aprépitant est tout d’abord métabolisé au niveau du CYP3A4, et potentiellement dans une moindre proportion par les CYP1A2 et CYP2C19.
Elimination
L’aprépitant n’est pas excrété sous forme inchangée dans les urines. Les métabolites sont excrétés dans les urines et, par voie biliaire, dans les fèces. Après administration à des sujets sains d’une dose intraveineuse unique de 100 mg de fosaprépitant, une prodrogue de l’aprépitant, marqué au [14C], 57 % de la radioactivité ont été récupérés dans les urines et 45 % dans les fèces.
La clairance plasmatique de l’aprépitant est dose-dépendante et décroît avec l’augmentation de la dose, allant de 60 à 72 mL/min environ dans la fourchette des doses thérapeutiques. La demi-vie terminale varie d'environ 9 à 13 heures.
Pharmacocinétique chez des populations particulières
Sujet âgé
Après administration orale d’une dose unique de 125 mg d’aprépitant à J1 et de 80 mg une fois par jour de J2 à J5, l’ASC0-24h de l’aprépitant a été supérieure de 21 % à J1 et de 36 % à J5 chez les sujets âgés (≥ 65 ans) comparés aux jeunes adultes. La Cmax a été supérieure de 10 % à J1 et de 24 % à J5 chez les sujets âgés comparés aux jeunes adultes. Ces différences ne sont pas considérées comme étant cliniquement significatives. Aucun ajustement posologique d’aprépitant n’est nécessaire chez les patients âgés.
Sexe
Après administration orale d’une dose unique de 125 mg d’aprépitant, la Cmax de l’aprépitant a été supérieure de 16 % chez les femmes comparées aux hommes. La demi-vie de l’aprépitant a été inférieure de 25 % chez les femmes comparées aux hommes, et son tmax survient approximativement au même moment. Ces différences ne sont pas considérées comme étant cliniquement significatives. Aucun ajustement posologique d’aprépitant n’est nécessaire en fonction du sexe.
Insuffisance hépatique
Une insuffisance hépatique légère (classe A de Child-Pugh) n’affecte pas la pharmacocinétique de l’aprépitant de façon cliniquement significative. Aucun ajustement posologique n’est nécessaire chez les patients en insuffisance hépatique légère. On ne peut pas tirer de conclusions concernant l’influence d’une insuffisance hépatique modérée (classe B de Child-Pugh) sur la pharmacocinétique de l’aprépitant à partir des données actuellement disponibles. On ne dispose d’aucune donnée clinique ni pharmacocinétique chez des patients en insuffisance hépatique sévère (classe C de Child-Pugh).
Insuffisance rénale
Une dose unique de 240 mg d’aprépitant a été administrée à des patients en insuffisance rénale sévère (ClCr < 30 mL/min) et à des patients atteints de néphropathie à un stade terminal nécessitant une hémodialyse.
Chez les patients en insuffisance rénale sévère, l’ASC0-∞ de l’aprépitant total (lié ou non aux protéines) a diminué de 21 % et la Cmax a diminué de 32 % comparées à des sujets sains. Chez les patients atteints de néphropathie à un stade terminal et sous hémodialyse, l’ASC0-∞ de l’aprépitant total a diminué de 42 % et la Cmax a diminué de 32 %. En raison d’une baisse modeste de la liaison protéique de l’aprépitant chez les patients atteints de néphropathie, l’ASC de l'aprépitant non lié et pharmacologiquement actif n’est pas affectée de façon significative chez les patients insuffisants rénaux comparés aux sujets sains. Une hémodialyse réalisée entre 4 et 48 heures après la prise n’a eu aucun effet significatif sur la pharmacocinétique de l’aprépitant ; moins de 0,2 % de la dose a été récupéré au niveau du dialysat.
Aucun ajustement posologique d’aprépitant n’est nécessaire chez les patients en insuffisance rénale ni chez les patients atteints d’une néphropathie à un stade terminal sous hémodialyse.
Population pédiatrique
Dans le cadre d'un schéma thérapeutique de 3 jours, l’administration des gélules d'aprépitant (125/80/80 mg) chez des patients adolescents (âgés de 12 à 17 ans) a conduit à une ASC0-24h supérieure à 17 μgh/mL à J1 avec des concentrations (Cmin) à l’issue de J2 et J3 supérieures à 0,4 μg/mL chez une majorité de patients. Le pic de concentration plasmatique (Cmax) médian était d'environ 1,3 μg/mL à J1, atteint au bout de 4 heures environ. Dans le cadre d'un schéma thérapeutique de 3 jours, l’administration d’aprépitant en poudre pour suspension buvable (3/2/2 mg/kg) chez des patients âgés de 6 mois à moins de 12 ans a conduit à une ASC0-24h supérieure à 17 μgh/mL à J1 avec des concentrations (Cmin) à l’issue de J2 et J3 supérieures à 0,1 μg/mL chez une majorité de patients. Le pic de concentration plasmatique (Cmax) médian était d'environ 1,2 μg/mL à J1, atteint entre 5 et 7 heures.
Une analyse pharmacocinétique de population de l'aprépitant chez des patients pédiatriques (âgés de 6 mois à 17 ans) suggère que le sexe et l’origine ethnique n’ont aucun effet cliniquement significatif sur la pharmacocinétique de l'aprépitant.
Relation effet/dose
A l’aide d’un traceur hautement spécifique du récepteur de la NK1, des études par tomographie par émission de positrons (TEP) menées auprès de jeunes hommes sains ont montré que l’aprépitant pénètre dans le cerveau et se lie aux récepteurs de la NK1 de façon dose et concentration plasmatiques dépendante. Les concentrations plasmatiques de l’aprépitant obtenues avec le schéma posologique de 3 jours d’aprépitant chez les adultes permettent d’envisager un taux de liaison aux récepteurs cérébraux de la NK1 supérieur à 95 %.
5.3. Données de sécurité préclinique
Dans une étude de toxicité juvénile chez des rats traités entre le 10ème et le 63ème jour après la naissance, l’aprépitant a entrainé une ouverture vaginale prématurée chez les femelles à partir de la dose de 250 mg/kg 2 fois par jour, et une séparation retardée du prépuce chez les mâles à partir de la dose de 10 mg/kg 2 fois par jour. Il n’y avait pas de marge de sécurité pour une exposition clinique pertinente. Aucun effet lié au traitement sur l’accouplement, la fertilité ou la survie embryonnaire/fœtale, ni aucune modification pathologique des organes reproducteurs n’ont été observés. Dans une étude de toxicité juvénile chez les chiens traités entre le 14ème et le 42ème jour après la naissance, une diminution du poids des testicules et de la taille des cellules de Leydig a été observée chez les mâles à la dose de 6 mg/kg/jour, et une augmentation du poids de l'utérus, une hypertrophie de l'utérus et du col de l’utérus, ainsi qu’un œdème des tissus vaginaux ont été observés chez les femelles à la dose de 4 mg/kg/jour. Il n’y avait pas de marge de sécurité pour une exposition clinique pertinente à l’aprépitant. Pour un traitement à court terme et à la posologie recommandée, la pertinence clinique de ces observations est considérée comme peu probable.
Saccharose
Cellulose microcristalline Sphère 500 (E 460)
Hydroxypropylcellulose (HPC-SL) (E 463)
Laurilsulfate de sodium
Enveloppe de la gélule
Gélatine
Dioxyde de titane (E 171)
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Plaquette (Aluminium-OPA/Aluminium/PVC) contenant une gélule de 80 mg.
Plaquette (Aluminium-OPA/Aluminium /PVC) contenant deux gélules de 80 mg.
3 plaquettes (Aluminium-OPA/Aluminium /PVC) contenant chacune une gélule de 80 mg.
5 plaquettes (Aluminium-OPA/Aluminium /PVC) contenant chacune une gélule de 80 mg.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
49 AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 394 5 5 : 1 gélule sous plaquette (Aluminium-OPA/Aluminium/PVC).
· 34009 301 394 6 2 : 2 gélules sous plaquette (Aluminium-OPA/Aluminium/PVC).
· 34009 550 524 0 1 : 3 plaquettes (Aluminium-OPA/Aluminium/PVC) contenant 1 gélule.
34009 550 524 1 8 : 5 plaquettes (Aluminium-OPA/Aluminium/PVC) contenant 1 gélule.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
à compléter ultérieurement par le titulaire
10. DATE DE MISE A JOUR DU TEXTE
à compléter ultérieurement par le titulaire
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 03/05/2024
APREPITANT SANDOZ 80 mg, gélule
Aprépitant
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit ou pour votre enfant. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ou votre enfant ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que APREPITANT SANDOZ 80 mg, gélule et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre APREPITANT SANDOZ 80 mg, gélule ?
3. Comment prendre APREPITANT SANDOZ 80 mg, gélule ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver APREPITANT SANDOZ 80 mg, gélule ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE APREPITANT SANDOZ 80 mg, gélule ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Antiémétiques et antinauséeux - code ATC : A04AD12
La substance active contenue dans APREPITANT SANDOZ est l'aprépitant. APREPITANT SANDOZ appartient à un groupe de médicaments appelés « antagonistes des récepteurs de la neurokinine 1 (NK1) ». Le cerveau possède une zone spécifique qui contrôle les nausées et vomissements. APREPITANT SANDOZ agit en bloquant les signaux vers cette zone, réduisant ainsi les nausées et vomissements.
APREPITANT SANDOZ gélules est utilisé chez les adultes et les adolescents à partir de 12 ans, en association avec d’autres médicaments pour prévenir les nausées et les vomissements causés par une chimiothérapie (traitement du cancer) qui entraîne des nausées et vomissements importants et modérés (tels que cisplatine, cyclophosphamide, doxorubicine ou épirubicine).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE APREPITANT SANDOZ 80 mg, gélule ?
Ne prenez jamais APREPITANT SANDOZ 80 mg, gélule :
· si vous ou votre enfant êtes allergique à l’aprépitant ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· avec des médicaments contenant du pimozide (utilisé pour traiter les maladies psychiatriques), de la terfénadine et de l’astémizole (utilisés pour le rhume des foins et d’autres maladies allergiques), du cisapride (utilisé pour traiter des problèmes digestifs). Informez le médecin si vous ou l’enfant prenez ces médicaments car le traitement doit être modifié avant de commencer à prendre ou donner APREPITANT SANDOZ.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre ou de donner à votre enfant APREPITANT SANDOZ 80 mg, gélule.
Avant de débuter le traitement par APREPITANT SANDOZ, informez le médecin si vous ou l’enfant avez une maladie du foie car le foie est important pour dégrader le médicament dans le corps. C'est pourquoi le médecin peut avoir à surveiller l'état de votre foie ou du foie de l’enfant.
Enfants et adolescents
Ne donnez pas APREPITANT SANDOZ 80 mg et 125 mg gélules aux enfants âgés de moins de 12 ans car les gélules de 80 mg et 125 mg n’ont pas été étudiées chez cette population.
Autres médicaments et APREPITANT SANDOZ 80 mg, gélule
APREPITANT SANDOZ peut interférer avec d’autres médicaments, aussi bien pendant qu’après le traitement par aprépitant (la substance active de APREPITANT SANDOZ). Certains médicaments ne doivent pas être pris avec APREPITANT SANDOZ (tels que pimozide, terfénadine, astémizole et cisapride) ou nécessitent un ajustement de leur posologie (voir également « Ne prenez jamais APREPITANT SANDOZ »).
La prise simultanée par vous ou votre enfant d'APREPITANT SANDOZ et d'autres médicaments, y compris ceux listés ci-dessous, peut avoir une influence sur les effets d'APREPITANT SANDOZ ou d'autres médicaments. Informez le médecin ou le pharmacien si vous ou l’enfant prenez l’un des médicaments suivants :
· les contraceptifs, pouvant inclure les pilules, les patchs cutanés, les implants et certains dispositifs intra-utérins (DIU) qui libèrent des hormones, peuvent ne pas fonctionner correctement lorsqu'ils sont utilisés avec APREPITANT SANDOZ. Une méthode de contraception non hormonale alternative ou complémentaire doit être utilisée pendant le traitement par APREPITANT SANDOZ et jusqu'à 2 mois après la prise d'APREPITANT SANDOZ,
· la ciclosporine, le tacrolimus, le sirolimus, l'évérolimus (immunosuppresseurs),
· l’alfentanil, le fentanyl (utilisés pour traiter la douleur),
· la quinidine (utilisée pour traiter le battement irrégulier du coeur),
· l’irinotécan, l’étoposide, la vinorelbine, l’ifosfamide (médicaments utilisés pour traiter le cancer),
· les médicaments contenant des alcaloïdes dérivés de l’ergot de seigle, tels que l’ergotamine et la diergotamine (utilisés pour le traitement des migraines),
· la warfarine, l’acénocoumarol (fluidifiants du sang ; des examens sanguins peuvent être nécessaires),
· la rifampicine, la clarithromycine, la télithromycine (antibiotiques utilisés pour traiter les infections),
· la phénytoïne (un médicament utilisé pour traiter les convulsions),
· la carbamazépine (utilisée pour traiter la dépression et l'épilepsie),
· le midazolam, le triazolam, le phénobarbital (médicaments utilisés pour calmer ou pour aider à dormir),
· le millepertuis (une préparation à base de plantes utilisée pour traiter la dépression),
· les inhibiteurs de protéase (utilisés pour traiter les infections VIH),
· le kétoconazole sauf en shampoing (utilisé pour traiter le syndrome de Cushing - lorsque le corps produit un excès de cortisol),
· l'itraconazole, le voriconazole, le posaconazole (antifongiques),
· la néfazodone (utilisée pour traiter la dépression),
· les corticoïdes (tels que la dexaméthasone et la méthylprednisolone),
· les anxiolytiques (tel que l'alprazolam),
· le tolbutamide (un médicament utilisé pour traiter le diabète).
Informez le médecin si vous ou l’enfant prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Ce médicament ne doit pas être utilisé pendant la grossesse sauf en cas de nécessité établie. Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
Pour toute information concernant les contraceptifs, voir « Autres médicaments et APREPITANT SANDOZ ».
On ne sait pas si APREPITANT SANDOZ est excrété dans le lait maternel ; par conséquent, l’allaitement n’est pas recommandé pendant le traitement par ce médicament. Il est important d’informer le médecin si vous allaitez ou souhaitez allaiter, avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Il est à noter que des étourdissements et une somnolence peuvent survenir chez certains sujets après la prise d'APREPITANT SANDOZ. Si vous ou l’enfant ressentez des étourdissements ou une somnolence, il convient d’éviter de conduire, de faire du vélo ou d'utiliser des machines ou des outils après la prise de ce médicament (voir « Quels sont les effets indésirables éventuels »).
APREPITANT SANDOZ 80 mg, gélule contient du saccharose
Les gélules d’APREPITANT SANDOZ contiennent du saccharose. Si le médecin a dit que vous ou l’enfant aviez une intolérance à certains sucres, contactez le médecin avant de prendre ce médicament.
3. COMMENT PRENDRE APREPITANT SANDOZ 80 mg, gélule ?
La dose orale recommandée d’APREPITANT SANDOZ est :
Jour 1 :
· une gélule de 125 mg 1 heure avant le début de votre séance de chimiothérapie
et
Jour 2 et Jour 3 :
· une gélule de 80 mg chaque jour.
· si aucune chimiothérapie n’est administrée, prendre APREPITANT SANDOZ le matin.
· si une chimiothérapie est administrée, prendre APREPITANT SANDOZ 1 heure avant le début de la séance de chimiothérapie.
APREPITANT SANDOZ peut être pris avec ou sans aliments. Avalez la gélule entière avec du liquide.
Si vous avez pris plus de APREPITANT SANDOZ 80 mg, gélule que vous n’auriez dû
Ne prenez pas plus de gélules que ne l’a recommandé votre médecin. Si vous ou l’enfant avez pris trop de gélules, contactez immédiatement votre médecin.
Si vous oubliez de prendre APREPITANT SANDOZ 80 mg, gélule
Si vous ou l’enfant avez oublié une dose, demandez conseil à votre médecin.
Si vous arrêtez de prendre APREPITANT SANDOZ 80 mg, gélule
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations au médecin ou au pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Arrêtez de prendre APREPITANT SANDOZ et consultez immédiatement un médecin si vous ou l’enfant constatez un des effets indésirables suivants, pouvant être grave, et pour lequel vous ou l’enfant pouvez avoir besoin d'un traitement médical en urgence :
· urticaire, éruption cutanée, démangeaisons, difficultés à respirer ou à avaler (fréquence indéterminée, ne pouvant être estimée sur la base des données disponibles) : ce sont des signes d'une réaction allergique.
D'autres effets indésirables ont été rapportés et sont mentionnés ci-dessous.
Les effets indésirables fréquents (pouvant affecter jusqu'à 1 personne sur 10) sont :
· constipation, indigestion,
· maux de tête,
· fatigue,
· perte de l'appétit,
· hoquet,
· augmentation de la quantité d'enzymes hépatiques dans le sang.
Les effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) sont :
· étourdissements, somnolence,
· acné, éruption cutanée,
· anxiété,
· éructation, nausées, vomissements, brûlures d’estomac, douleurs d’estomac, bouche sèche,
· flatulences,
· mictions douloureuses ou avec sensation de brûlures plus fréquentes,
· faiblesse, sensation générale d’inconfort,
· bouffées de chaleur/rougeurs sur le visage ou sur la peau,
· battements du cœur rapides ou irréguliers,
· fièvre avec risque accru d’infection, diminution du nombre de globules rouges dans le sang.
Les effets indésirables rares (pouvant affecter jusqu’à 1 personne sur 1 000) sont :
· difficulté à penser, manque d’énergie, altération du goût,
· sensibilité de la peau au soleil, transpiration excessive, peau grasse, plaies sur la peau, éruption cutanée avec démangeaisons, syndrome de Stevens-Johnson/syndrome de Lyell (rare réaction sévère de la peau),
· euphorie (sentiment de joie intense), désorientation,
· infection bactérienne, infection fongique,
· constipation sévère, ulcère de l’estomac, inflammation de l’intestin grêle et du côlon, lésions buccales, ballonnements,
· mictions plus fréquentes, mictions plus abondantes que la normale, présence de sucre ou de sang dans les urines,
· gêne thoracique, œdème, modification de la façon de marcher,
· toux, mucus dans l’arrière-gorge, irritation de la gorge, éternuements, maux de gorge,
· écoulements et démangeaisons au niveau des yeux,
· bourdonnements d’oreille,
· spasmes musculaires, faiblesse musculaire,
· soif excessive,
· ralentissement de la fréquence cardiaque, maladie du cœur et des vaisseaux sanguins,
· diminution des globules blancs dans le sang, taux faibles de sodium dans le sang, perte de poids.
Déclaration des effets secondaires
Si vous ou votre enfant ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER APREPITANT SANDOZ 80 mg, gélule ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l'emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne sortez pas la gélule de la plaquette avant utilisation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient APREPITANT SANDOZ 80 mg, gélule
· La substance active est l’aprépitant. Chaque gélule contient 80 mg d’aprépitant.
· Les autres composants sont : saccharose, cellulose microcristalline (E 460), hydroxypropylcellulose (E 463), laurilsulfate de sodium, gélatine, dioxyde de titane (E 171).
Qu’est-ce que APREPITANT SANDOZ 80 mg, gélule et contenu de l’emballage extérieur
La gélule de 80 mg est opaque avec un corps et une coiffe blancs, contenant des microgranules blancs à blanc cassé. APREPITANT SANDOZ 80 mg, gélule est disponible dans les conditionnements suivants :
· Plaquette (Aluminium-OPA/Aluminium/PVC) contenant une gélule de 80 mg.
· Plaquette (Aluminium-OPA/Aluminium/PVC) contenant deux gélules de 80 mg.
· 3 plaquettes (Aluminium-OPA/Aluminium/PVC) contenant chacune une gélule de 80 mg.
· 5 plaquettes (Aluminium-OPA/Aluminium/PVC) contenant chacune une gélule de 80 mg.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
49 AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
Exploitant de l’autorisation de mise sur le marché
49 AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
RONTIS HELLAS MEDICAL AND PHARMACEUTICAL PRODUCTS S.A.
LARISSA INDUSTRIAL AREA
P.O. BOX 3012
41500 LARISSA
GRECE
Ou
LEK PHARMACEUTICALS D.D.
VEROVSKOVA ULICA 57
1526 LJUBLJANA
SLOVENIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
à compléter ultérieurement par le titulaire
La dernière date à laquelle cette notice a été révisée est :
à compléter ultérieurement par le titulaire
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).