Dernière mise à jour le 01/06/2026
CLOTTAFACT 1,5 g, poudre et solvant pour solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : B02BB01
CLOTTAFACT est un médicament qui appartient à la classe des antihémorragiques. La substance active est le fibrinogène humain, une protéine naturellement présente dans l’organisme. Le rôle de cette protéine est de participer à une coagulation normale du sang et d’empêcher les saignements trop longs.
CLOTTAFACT est utilisé dans les situations suivantes :
· Comme traitement dans la prise en charge de saignements importants (hémorragies) spontanés, provoqués par un traumatisme ou en cas de chirurgie, chez les adultes, les adolescents et les enfants ayant une maladie héréditaire entraînant un taux de fibrinogène dans le sang en dessous de la normale (hypofibrinogénémie constitutionnelle), un mauvais fonctionnement du fibrinogène (dysfibrinogénémie constitutionnelle) ou une absence de fibrinogène (afibrinogénémie constitutionnelle).
· Comme traitement complémentaire dans la prise en charge de saignements très importants ne pouvant être arrêtés (hémorragie sévère incontrôlée) chez les patients ayant un taux de fibrinogène dans le sang inférieur à la normale (hypofibrinogénémie acquise) causé par une :
o Augmentation de la consommation du fibrinogène en cas de saignements incontrôlés menaçant la vie du patient lors de complications d’accouchement, de chirurgie ou de traumatisme.
o Diminution de la production du fibrinogène par le foie en cas de maladie grave du foie (insuffisance hépatique sévère ou secondaire à un traitement par la L-Asparaginase).
Présentations
> 1 flacon(s) en verre - 1 flacon(s) en verre de 100 ml avec dispositif(s) de transfert avec filtre(s) avec matériel(s) de perfusion avec filtre(s)
Code CIP : 574 971-9 ou 34009 574 971 9 4
Déclaration de commercialisation : 09/11/2009
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 18/11/2020 | Modification des conditions d'inscription (CT) | Le service médical rendu par CLOTTAFACT (fibrinogène humain) est important dans hypo-, dys- ou afibrinogénémie constitutionnelle, chez les adultes, les adolescents et les enfants, en cas de chirurgie. |
| Important | Avis du 24/06/2009 | Inscription (CT) | Le service médical rendu par cette spécialité est important dans les déficits constitutionnels en fibrinogène et dans les hypofibrinogénémies profondes, acquises au cours des hémorragies. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| I (Majeur) | Avis du 18/11/2020 | Modification des conditions d'inscription (CT) | L’extension d’utilisation en cas de chirurgie n’est pas susceptible de modifier l'appréciation précédente de la Commission dans la prise en charge des patients atteints d’une hypo-, dys- ou afibrinogénémie constitutionnelle : CLOTTAFACT apporte une amélioration du service médical rendu majeure (de niveau I) dans la prise en charge de ces patients. |
| I (Majeur) | Avis du 24/06/2009 | Inscription (CT) | Dans les hypo-, dys- ou afibrinogénémies constitutionnelles, en l'absence d'alternative thérapeutique validée par une AMM, CLOTTAFACT apporte une amélioration du service médical rendu majeure (de niveau I) dans la prise en charge des patients. |
| IV (Mineur) | Avis du 24/06/2009 | Inscription (CT) | Dans les hypofibrinogénémies acquises au cours des hémorragies, les données présentées dans le dossier ne concernent que les hémorragies du post-partum. De plus, le nombre de patientes incluses et analysées est restreint et la majorité des patientes figurant dans l'analyse d'efficacité avait à l'inclusion un taux de fibrinogène supérieur à 1g/l. Dans ces conditions, étant donné le caractère limité des données produites, la commission considère que CLOTTAFACT n'apporte qu'une amélioration du service médical rendu mineure (de niveau IV) dans la prise en charge des patients présentant une hypofibrinogénémie acquise au cours des hémorragies. |
Autres informations
- Titulaire de l'autorisation : LFB-BIOMEDICAMENTS
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 442 145 3
ANSM - Mis à jour le : 21/05/2025
CLOTTAFACT 1,5 g, poudre et solvant pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon de CLOTTAFACT contient 1,5 g de fibrinogène humain.
Après reconstitution avec 100 mL de solvant (eau pour préparations injectables), CLOTTAFACT contient 15 mg/mL de fibrinogène humain.
L’activité est déterminée selon la monographie de la Pharmacopée Européenne pour le fibrinogène humain.
Produit à partir de plasma de donneurs humains.
Excipients à effet notoire : Ce médicament contient au maximum 69 mg (3 mmol) de sodium par flacon, cela équivaut à 3,45% de l’apport journalier maximal de 2 g de sodium recommandé par l’OMS pour un adulte.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable.
Poudre ou solide friable blanc ou jaunâtre dans un flacon.
Solvant : solution transparente et incolore.
Ce produit reste stable après un traitement thermique à sec dans un intervalle de pH de 6,5-7,5.
L’osmolalité de ce produit est entre 450-550 mOsmol/kg.
4.1. Indications thérapeutiques
· Hypo-, dys- ou afibrinogénémie constitutionnelle, chez les adultes, les adolescents et les enfants présentant une hémorragie spontanée ou post-traumatique ou en cas de chirurgie.
· En tant que traitement complémentaire dans la prise en charge d'une hémorragie sévère incontrôlée dans le cadre d'une hypofibrinogénémie acquise telle que :
o Augmentation de la consommation du fibrinogène associée à un saignement incontrôlé menaçant le pronostic vital dans les complications obstétricales, en situation chirurgicale ou en traumatologie.
o Altération de la synthèse hépatique du fibrinogène en cas d’insuffisance hépatique sévère ou secondaire à un traitement par la L-Asparaginase.
4.2. Posologie et mode d'administration
Le traitement devra être initié sous la surveillance d’un médecin spécialiste des troubles de l’hémostase.
Posologie
Le dosage et la durée de la substitution dépendent de la sévérité des troubles, de la localisation et de l'étendue du saignement et de l’état clinique du patient.
La concentration plasmatique de fibrinogène (fonctionnel) doit être mesurée initialement, et surveillée régulièrement, pour déterminer la posologie et la fréquence d’administration adaptée à chaque individu. L’état clinique du patient et les autres thérapies de substitution utilisées feront l’objet d’une surveillance continue.
Les valeurs normales de fibrinogène circulant sont en moyenne de 1,5 à 4,5 g/L. La valeur critique de fibrinogène en dessous de laquelle une hémorragie peut apparaître est approximativement de 0,5-1,0 g/L.
En cas d’intervention chirurgicale majeure, une surveillance étroite du traitement par des tests de coagulation est nécessaire.
Déficits constitutionnels
L'objectif de la première injection est d'atteindre un taux de fibrinogène circulant supérieur à 1 g/L. Ce taux est à maintenir jusqu’au contrôle et à la stabilisation de l’hémostase, puis un taux au-dessus de 0,5 g/L est recommandé jusqu’à cicatrisation complète.
En cas de procédure chirurgicale ou de traitement d’un épisode de saignement non-chirurgical, la dose doit être calculée comme suit :
Quantité à injecter (g) = (taux à obtenir (g/L) - taux basal (g/L)) ´ 1/ taux de récupération (g/L)/(g/kg)´ poids corporel (kg).
Le ratio « 1/taux de récupération » est déterminé à partir du taux de récupération du patient* (voir section 5.2), ou si le taux de récupération est inconnu :
· 0,053 (g/kg)/(g/L) pour les enfants et adolescents <40 kg
· 0,043 (g/kg)/(g/L) pour les adultes et adolescents ³40 kg.
* Exemple pour le calcul du taux de récupération du patient et de la dose
Dans le cas d’un patient de 60 kg avec un taux de fibrinogène indétectable en basal et à 1,20 g/L 1 heure après l’injection de 0,060 g/kg de CLOTTAFACT :
· Le calcul du taux de récupération du patient est :
1,20 (g/L) / 0,060 (g/kg) = 20,0 (g/L)/(g/kg)
· Le calcul de la dose pour une augmentation du taux à 1,0 g/L est :
1,0 g/L x 1 / 20,0 (g/L)/(g/kg) [ou 0,050 (g/kg)/(g/L)] x 60 kg = 3 g.
En cas d’urgence si le taux basal de fibrinogène n’est pas disponible, la dose initiale recommandée est de 0,05 g/kg administré en intraveineux chez les adultes et adolescents ³40 kg, et doit être majorée de 20-25% chez les enfants et adolescents <40 kg.
La posologie ultérieure (doses et fréquence des injections) sera adaptée à l'état clinique et au suivi biologique du patient.
La demi-vie biologique du fibrinogène est de 3-4 jours. Ainsi, en l’absence de dégradation, un traitement répété avec le fibrinogène humain n’est habituellement pas nécessaire. Compte tenu de l’accumulation survenant en cas d’administration répétée dans un but prophylactique, la dose et la fréquence doivent être déterminées en fonction des objectifs thérapeutiques du médecin pour chaque patient.
Population pédiatrique
Les données montrent que le taux de récupération et la demi-vie in vivo chez les enfants et les adolescents ayant un poids corporel <40 kg sont inférieurs à ceux des adultes et des adolescents ayant un poids corporel ³40 kg (voir la rubrique 5.2). Par conséquent, lorsqu’il est inconnu, il convient d’utiliser des taux de récupération adaptés pour calculer la dose de CLOTTAFACT dans les groupes de poids corporel respectifs, en considérant qu’un poids corporel inférieur à 40 kg couvre la tranche d’âge allant de la naissance à environ 12 ans. La posologie (doses et fréquence des injections) doit être adaptée en fonction de la réponse clinique individuelle.
Déficits acquis
Généralement, une dose initiale de 1 à 2 g est administrée, et éventuellement répétée. Dans les hémorragies aigues sévères obstétricales, des quantités plus importantes de fibrinogène (4 à 8 g) peuvent être nécessaires.
En cas d’urgence dans les hémorragies aiguës, si la concentration de fibrinogène n’est pas disponible, une dose initiale sera administrée et les doses suivantes seront adaptées aux concentrations disponibles dans l’intervalle.
Population pédiatrique
La posologie déterminée en fonction du poids corporel et de la situation clinique est généralement de 0,02 à 0,03 g/kg.
Mode d’administration
Perfusion ou injection intraveineuse.
CLOTTAFACT est une poudre, à reconstituer extemporanément avec de l'eau pour préparations injectables.
CLOTTAFACT doit être injecté lentement par voie intraveineuse, en une seule fois, immédiatement après reconstitution.
En situation clinique stable, le débit d’administration de CLOTTAFACT ne doit pas dépasser
4 mL/min. En situation d’hémorragie aiguë sévère, le débit d’administration peut atteindre 20 mL/min.
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.2 et 6.6.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Pour améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement consignés.
Thrombo-embolie
Il existe un risque de thrombose, chez les patients traités pour un déficit acquis ou constitutionnel avec du fibrinogène humain particulièrement à forte dose ou à doses répétées. Les patients traités par du fibrinogène humain devront être surveillés étroitement pour détecter les signes ou symptômes de thrombose.
Le bénéfice potentiel du traitement par du fibrinogène humain devra être évalué en fonction du risque thromboembolique dans les situations suivantes : patients avec antécédents de maladie coronarienne ou infarctus du myocarde, avec une maladie hépatique, en péri ou post opératoire, chez les nouveau-nés ou les patients à risque d’événements thromboemboliques ou de coagulopathie intra vasculaire disséminée. Une étroite surveillance devra être observée.
L’hypofibrinogénémie acquise est associée à des concentrations plasmatiques basses de tous les facteurs (pas uniquement le fibrinogène) et inhibiteurs de coagulation. Par conséquent, l’utilisation de produits sanguins contenant des facteurs de coagulation devra être prise en considération. Une surveillance stricte de la coagulation est nécessaire.
Dans le traitement des hémorragies aiguës sévères, la prescription de CLOTTAFACT doit s’effectuer en association avec les mesures de réanimation adaptées à la situation clinique et biologique du patient.
Réactions allergiques ou anaphylactiques
En cas d'allergie ou de réaction de type anaphylactique l'administration devra être interrompue immédiatement. En cas de choc anaphylactique un traitement symptomatique adapté devra être instauré.
Agents transmissibles
Les mesures habituelles de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection clinique des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de plasma ainsi que la mise en œuvre dans le procédé de fabrication d’étapes efficaces pour l’inactivation/élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents et aux autres types d’agents infectieux. Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC).
Les mesures prises peuvent être d’efficacité limitée vis-à-vis des virus non-enveloppés tels que le virus de l’hépatite A (VHA) et le parvovirus B19. Une infection par le parvovirus B19 peut être grave pour les femmes enceintes (infection foetale) et les personnes immunodéficientes ou atteintes d'une augmentation de l'érythropoïèse (par exemple, d'une anémie hémolytique).
Une vaccination appropriée (hépatites A et B) des patients recevant régulièrement du fibrinogène est recommandée.
Immunogénicité
En cas de traitement substitutif dans d’autres déficits constitutionnels avec des facteurs de coagulation, l’apparition d’anticorps a pu être observée mais aucun cas n’a été rapporté à ce jour avec le fibrinogène.
Taux de sodium
CLOTTAFACT contient au maximum 3 mmol (ou 69 mg) de sodium/flacon. Ceci est à prendre en compte chez les patients suivant un régime hyposodé strict.
Population pédiatrique
Les mêmes mises en garde et précautions d’emploi s’appliquent à la population pédiatrique.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction médicamenteuse avec CLOTTAFACT n'est connue à ce jour.
4.6. Fertilité, grossesse et allaitement
Grossesse
L'innocuité du fibrinogène humain au cours de la grossesse n'a pas été évaluée par des essais cliniques contrôlés.
Néanmoins, l’expérience clinique dans les traitements des complications obstétricales suggère qu'aucun effet néfaste n'est attendu soit sur l'évolution de la grossesse, soit sur le développement du fœtus ou du nouveau-né.
L'innocuité du fibrinogène humain au cours de l'allaitement n'a pas été évaluée par des essais cliniques contrôlés.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
CLOTTAFACT n’a aucun effet sur l’aptitude à conduire des véhicules et à utiliser des machines.
Au total, 443 patients ont été exposés à CLOTTAFACT au cours des études cliniques.
Parmi 47 patients atteints d’un déficit constitutionnel en fibrinogène dans le cadre de trois études cliniques interventionnelles et d’une étude clinique non-interventionnelle de tolérance post autorisation de mise sur le marché (post-AMM), 39 effets indésirables ont été rapportés chez 14/47 patients (29,8%) ayant reçu un total de 631 injections de CLOTTAFACT.
Parmi 396 patients atteints d’un déficit acquis en fibrinogène dans le cadre de deux études cliniques interventionnelles et d’une étude non-interventionnelle de tolérance post-AMM, 8 effets indésirables ont été rapportés chez 5/396 patients (1,3%) ayant reçu un total de 473 injections de CLOTTAFACT.
Au total, 47 effets indésirables ont été rapportés chez 19/443 patients (4,3%) ayant reçu un total de 1104 injections de CLOTTAFACT. L’effet indésirable le plus fréquemment rapporté après l’administration de CLOTTAFACT est l’apparition de céphalées, qui sont survenues dans 0,9% des injections (10/1104), toutes d’intensité légère à modérée, survenues dans les 48 heures suivant l’injection et résolues sans séquelles.
Liste tabulée des effets indésirables
Les effets indésirables associés à CLOTTAFACT, obtenus dans le cadre d’études cliniques et de la surveillance post-AMM, sont présentés dans le tableau ci-dessous.
Les effets indésirables sont présentés conformément à la classification MedDRA (Classes de système d’organes et termes préférentiels). Les fréquences ont été estimées par injection selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000).
Tableau 1 : Effets indésirables observés dans la population adulte et pédiatrique au cours des études cliniques et de la surveillance post-AMM
|
Système - Organe |
Effets indésirables |
Fréquence par injection |
|
Affections du système immunitaire |
Réactions allergiques/anaphylactiques (dont choc anaphylactique, pâleur, vomissements, toux, baisse de la tension artérielle, frissons, urticaire, dermatite allergique) |
Peu fréquent* |
|
Affections du système nerveux |
Céphalées Étourdissements |
Peu fréquent Rare |
|
Affections de l’oreille et du labyrinthe |
Acouphènes |
Rare |
|
Affections vasculaires |
Evènements thromboemboliques (dont thrombose veineuse profonde, thrombose veineuse superficielle, embolie pulmonaire) (voir rubrique 4.4) |
Peu fréquent |
|
Affections respiratoires, thoraciques et médiastinales |
Asthme |
Rare |
|
Affections gastro-intestinales |
Vomissements |
Rare |
|
Affections de la peau et du tissu sous-cutané |
Sueurs nocturnes Eruption érythémateuse Erythème Irritation cutanée |
Peu fréquent Rare Rare Rare |
|
Troubles généraux et anomalies au site d’administration |
Hyperhidrose Sensation de chaleur |
Rare Rare |
*« fréquent » dans la population pédiatrique
Pour la sécurité vis-à-vis des agents transmissibles, voir rubrique 4.4.
Population pédiatrique
Parmi les 47 patients inclus dans l’analyse de la tolérance pour le déficit constitutionnel en fibrinogène, 26 avaient moins de 18 ans, dont 5 âgés de 12 à 17 ans, 11 âgés de 6 à 11 ans et 10 âgés de moins de 6 ans. La fréquence, le type et la sévérité des effets indésirables sont similaires chez les adultes et les enfants, à l’exception des réactions de type allergique/anaphylactique qui sont survenues plus fréquemment (chez 2 nourrissons âgés de 1 et 5 ans). Parmi les 396 patients inclus dans l’analyse de la tolérance pour le déficit acquis en fibrinogène, 2 avaient moins de 18 ans (entre 12 et 17 ans). Le profil de sécurité ne diffère pas entre les adultes et les enfants.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antihémorragiques, code ATC : B02BB01.
Le fibrinogène humain (facteur I de coagulation) injecté se transforme en fibrine sous l'action de la thrombine, et, en présence de facteur XIII activé (FXIIIa) et d'ions calcium, forme un réseau de fibrine tridimensionnel, stable et élastique.
L’administration de fibrinogène humain induit une augmentation du taux plasmatique de fibrinogène et peut momentanément corriger l’altération de la coagulation des patients présentant un déficit en fibrinogène.
Trois études cliniques multicentriques, réalisées en ouvert, non contrôlées (une chez l’adulte, une chez l’adulte, l’adolescent et l’enfant et une chez l’enfant) ont évalué la pharmacologie clinique, la sécurité et l’efficacité de CLOTTAFACT dans le déficit congénital en fibrinogène. De plus, une étude de tolérance post-AMM, non-interventionnelle, a inclus des adultes et patients pédiatriques. La partie « pharmacologie clinique » de chaque étude clinique (voir section 5.2) a inclus un total de 31 patients atteints d’afibrinogénémie recevant une dose unique fixe de 0,06 g/kg de CLOTTAFACT. Une normalisation des tests d’exploration globale de la coagulation (temps de céphaline activé [TCA] et temps de Quick [TQ]) a été obtenue avec un taux de fibrinogène circulant supérieur ou égal à 0,5 g/L.
La variation de la fermeté maximale du caillot (MCF) entre avant et 1 heure après injection était de 6,3 mm chez les patients <40 kg de poids corporel et de 10,0 mm pour les patients ³40 kg de poids corporel.
Population adulte
Dans l’ensemble des études dans le déficit congénital en fibrinogène, 19 patients ³18 ans avec un âge médian de 30 ans (extrêmes 19-78 ans) ont été inclus dans des études d’efficacité soit pour un traitement à la demande soit pour chirurgie, et parmi lesquels 18 étaient atteints d’afibrinogénémie et un était atteint de dysfibrinogénémie. CLOTTAFACT a été administré pour :
· 74 épisodes hémorragiques non chirurgicaux chez 12 patients (dont 6 épisodes majeurs chez 3 patients),
· 24 interventions chirurgicales chez 8 patients (dont 8 interventions chirurgicales lourdes chez 5 patients).
La plupart (94,9%) des événements (93/98) ont été pris en charge avec une dose unique de CLOTTAFACT (0,050 g/kg pour épisodes hémorragiques et 0,055 g/kg pour interventions chirurgicales).
Population pédiatrique
L’efficacité clinique au cours des études interventionnelles a été analysée chez 20 patients de moins de 18 ans (entre 1 et 17 ans), atteints d’afibrinogénémie, qui ont reçu CLOTTAFACT dans 80 situations soit pour un traitement à la demande ou soit pour chirurgie.
Quatorze patients ont été traités avec CLOTTAFACT pour 55 épisodes de saignement et 15 patients ont été traités pour couvrir 25 chirurgies. La dose médiane par injection était de 0,064 g/kg en cas de saignement (poids moyen de 30 kg de la population traitée) et 0,069 g/kg en chirurgie (poids moyen de 26 kg de la population traitée). La majorité (90,0%) des situations (72/80) était résolue avec une seule dose de CLOTTAFACT.
Dans une étude post-AMM, 9 patients (incluant 4 enfants) ont reçu une prophylaxie au long cours pendant au moins 12 mois, à raison d’une dose médiane de 0,059 g/kg avec un intervalle médian de 7,6 jours entre deux administrations.
Déficits acquis
Chez les patientes présentant un syndrome hémorragique sévère du post-partum immédiat (hémorragies de la délivrance), une administration de 3 g de CLOTTAFACT a induit une augmentation du pouvoir coagulant plasmatique mesuré par thromboelastographie. Ce traitement a permis un contrôle de l'hémorragie et a évité de recourir aux traitements invasifs (embolisation ou ligature des artères utérines, hystérectomie) dans 75% des cas.
5.2. Propriétés pharmacocinétiques
Dans le plasma, la demi-vie biologique du fibrinogène est de 3 à 4 jours.
Le médicament est administré par voie intraveineuse et est immédiatement disponible dans le plasma à une concentration correspondant à la dose administrée.
Dans une étude de pharmacologie clinique, 14 patients adultes et adolescents atteints d’afibrinogénémie ont été évalués pendant 14 jours. Après la perfusion d’une dose de 0,06 g/kg de CLOTTAFACT, la moyenne géométrique (coefficient de variation géométrique (%)) du taux de récupération in vivo était de 93,6 % (21). La concentration de fibrinogène mesurée en activité était maximale après 1 heure (1,4 g/L) (24), suivie par une lente décroissance exponentielle en une phase atteignant le seuil critique de fibrinogène de 0,5 g/L en environ 3 à 4 jours. Une analyse non-compartimentale a permis de déterminer la moyenne géométrique (coefficient de variation géométrique (%)) de l’aire sous la courbe de 0 à l’infini à 114 g.h/L (23) et celle de la demi-vie à 69,3 heures (22). Le fibrinogène humain est largement retenu dans le compartiment vasculaire : la moyenne géométrique (coefficient de variation géométrique (%)) du volume de distribution apparent était de 50,7 mL/kg (17).
Un modèle de pharmacocinétique de population intégrant des relations allométriques (poids corporel) a été développé à partir des données recueillies chez 31 patients atteints d’afibrinogénémie âgés de 1 à 48 ans : les paramètres dérivés du modèle sont présentés dans le Tableau 2. Les enfants et adolescents <40 kg de poids corporel ont présenté une clairance plus élevée, une demi-vie plus courte et une récupération plus faible mesurée 1 heure après injection que les adolescents et les adultes ³40 kg.
Tableau 2 : Résumé des paramètres pharmacocinétiques de CLOTTAFACT (fibrinogène mesuré en activité) dérivés de l’analyse pharmacocinétique de population après injection de 0,06 g/kg, et du taux de récupération à 1 heure par groupe d’âge et poids corporel.
Moyenne géométrique (CV géométrique (%))
|
|
Patients |
Patients |
Enfants |
Enfants |
Adolescents |
Adultes |
|
|
Nombre de patients |
12 |
19 |
6 |
8 |
3 |
14 |
|
|
AUC0-∞ (g.h/L) |
81 (23) |
133 (26) |
74 (15) |
93 (23) |
144 (30) |
136 (26) |
|
|
Cl (mL/h/kg) |
0,74 (23) |
0,45 (25) |
0,81 (15) |
0,65 (23) |
0,43 (27) |
0,44 (25) |
|
|
t1/2 (h) |
49,0 (12) |
66,7 (19) |
46,6 (10) |
52,1 (10) |
64,2 (10) |
69,3 (20) |
|
|
MRT (h) |
70,7 (12) |
96,2 (13) |
67,3 (10) |
75,2 (10) |
92,6 (10) |
100,0 (20) |
|
|
Vss (mL/kg) |
52,2 (16) |
43,2 (17) |
54,4 (10) |
48,6 (19) |
39,5 (19) |
43,8 (18) |
|
|
|
|
|
|
|
|
|
|
|
Concentration de fibrinogène |
1,15 (20) |
1,40 (22) |
1,12 (14) |
1,21 (24) |
1,56 (27) |
1,38 (23) |
|
|
Taux de récupération (g/L per g/kg) |
19,1 (20) |
23,3 (21) |
18,7 (14) |
20,1 (24) |
25,4 (24) |
23,1 (22) |
|
|
AUC0-∞: Aire sous la courbe de 0 à l’infini, Cl: clairance, t1/2 : demi-vie d’élimination, MRT: Temps de séjour moyen, Vss: volume de distribution à l’état d’équilibre, CV = coefficient de variation |
|
||||||
Chez les patientes présentant un syndrome hémorragique sévère du post-partum immédiat, une administration de 3 g de CLOTTAFACT conduit à une augmentation d'environ 20% des concentrations plasmatiques de fibrinogène, soit un taux de récupération de l'ordre de 14,0 [g/L]/[g/kg].
5.3. Données de sécurité préclinique
Etant donné la nature du produit, aucune étude de cancérogénicité n'a été réalisée. Aucune étude de reproduction animale n'a été réalisée, le fibrinogène étant un constituant normal du corps humain.
Poudre : chlorhydrate d’arginine, isoleucine, chlorhydrate de lysine, glycine, citrate de sodium.
Solvant : eau pour préparations injectables.
Après reconstitution, le produit doit être utilisé immédiatement.
6.4. Précautions particulières de conservation
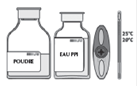
A conserver à une température ne dépassant pas 25°C. Ne pas congeler.
A conserver dans l'emballage extérieur d’origine et à l'abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre en flacon (verre incolore de type I) munis d’un bouchon (bromobutyle) + 100 mL de solvant en flacon (verre de type II) munis d’un bouchon (bromobutyle) et d’une capsule de protection accompagnés d’un système de transfert muni d'un évent à filtre stérilisant et un nécessaire de perfusion muni d’un filtre de 15 µm- boîte de 1.
6.6. Précautions particulières d’élimination et de manipulation
Reconstitution :
Respecter les règles d’asepsie habituelles.
|
· Si nécessaire, amener les deux flacons (poudre et solvant) à température ambiante.
|
|
|
|
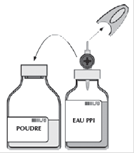
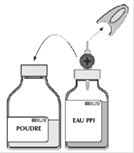
· Retirer la capsule protectrice du flacon de solvant (eau pour préparations injectables) et du flacon de poudre. · Désinfecter la surface de chaque bouchon. |
|
|
· Retirer le capuchon protecteur translucide du système de transfert et insérer à fond le biseau, ainsi dégagé, au centre du bouchon du flacon de solvant en opérant simultanément un mouvement de rotation.
|
|
|
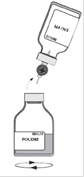
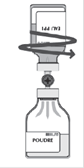
· Retirer le deuxième capuchon protecteur de l'autre extrémité du système de transfert. · Retourner le flacon de solvant et enfoncer rapidement l’extrémité libre du biseau au centre du bouchon du flacon de poudre afin de permettre le transfert du solvant vers la poudre. · Veiller à ce que le biseau soit toujours immergé dans le solvant pour éviter un cassage précoce du vide.
|
|
|
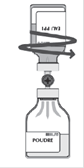
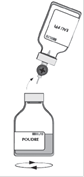
· Pendant le transfert, diriger le jet de solvant sur toute la surface de la poudre et sur la paroi du flacon en opérant un mouvement de rotation horizontale. Veiller à ce que la totalité du solvant soit transférée. · A la fin du transfert, le vide est automatiquement cassé (air stérile).
|
|
|
· Retirer le flacon vide (solvant) avec le système de transfert. · Agiter modérément par un mouvement de rotation doux pour éviter la formation de mousse, jusqu'à dissolution complète de la poudre. |



Le produit reconstitué doit être examiné visuellement avant administration, afin de s'assurer qu'il ne contient pas de particules. La solution reconstituée présente une opalescence plus ou moins prononcée. Ne pas utiliser de solution trouble ou contenant un dépôt.
Après reconstitution, le produit doit être utilisé immédiatement.
Administration :
CLOTTAFACT doit être exclusivement administré par voie intraveineuse, en une seule fois et immédiatement après reconstitution sans dépasser 4 mL/min. En situations d’hémorragies sévères, le débit d’administration peut atteindre 20 mL/min.
L’utilisation d’un nécessaire de perfusion muni d’un filtre de 15 µm, tel que celui fourni dans l’emballage, est obligatoire.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
3, AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
[TEL, FAX, E-MAIL : à compléter ultérieurement par le titulaire]
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’ANSM.
Liste I
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 21/05/2025
CLOTTAFACT 1,5 g, poudre et solvant pour solution injectable
Fibrinogène humain
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que CLOTTAFACT et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser CLOTTAFACT ?
3. Comment utiliser CLOTTAFACT ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver CLOTTAFACT ?
6. Contenu de l’emballage et autres informations
1. QU’EST-CE QUE CLOTTAFACT 1,5 g, poudre et solvant pour solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : B02BB01
CLOTTAFACT est un médicament qui appartient à la classe des antihémorragiques. La substance active est le fibrinogène humain, une protéine naturellement présente dans l’organisme. Le rôle de cette protéine est de participer à une coagulation normale du sang et d’empêcher les saignements trop longs.
CLOTTAFACT est utilisé dans les situations suivantes :
· Comme traitement dans la prise en charge de saignements importants (hémorragies) spontanés, provoqués par un traumatisme ou en cas de chirurgie, chez les adultes, les adolescents et les enfants ayant une maladie héréditaire entraînant un taux de fibrinogène dans le sang en dessous de la normale (hypofibrinogénémie constitutionnelle), un mauvais fonctionnement du fibrinogène (dysfibrinogénémie constitutionnelle) ou une absence de fibrinogène (afibrinogénémie constitutionnelle).
· Comme traitement complémentaire dans la prise en charge de saignements très importants ne pouvant être arrêtés (hémorragie sévère incontrôlée) chez les patients ayant un taux de fibrinogène dans le sang inférieur à la normale (hypofibrinogénémie acquise) causé par une :
o Augmentation de la consommation du fibrinogène en cas de saignements incontrôlés menaçant la vie du patient lors de complications d’accouchement, de chirurgie ou de traumatisme.
o Diminution de la production du fibrinogène par le foie en cas de maladie grave du foie (insuffisance hépatique sévère ou secondaire à un traitement par la L-Asparaginase).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER CLOTTAFACT 1,5 g, poudre et solvant pour solution injectable ?
N’utilisez jamais CLOTTAFACT :
· si vous êtes allergique (hypersensible) à la substance active (le fibrinogène) ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser CLOTTAFACT.
La surveillance clinique et biologique du patient sera faite de manière adaptée en fonction de la situation et des traitements utilisés.
L'état clinique du patient et les autres thérapies de substitution utilisées feront l'objet d'une surveillance continue.
Dans le traitement des hémorragies aiguës sévères, la prescription de CLOTTAFACT doit s’effectuer en association avec les mesures de réanimation adaptées à la situation clinique et biologique du patient.
Traçabilité
A chaque administration de CLOTTAFACT à un patient, le nom et le numéro de lot du produit doivent être enregistrés afin de maintenir un lien entre le patient et le numéro de lot du médicament.
Risque de caillots dans les vaisseaux sanguins
Il existe un risque de thrombose (formation de caillots dans le sang), chez les patients traités pour un manque de fibrinogène, particulièrement à forte dose ou à doses répétées.
Les patients traités par du fibrinogène humain devront être surveillés étroitement pour détecter les signes ou symptômes de thrombose.
Le bénéfice potentiel du traitement par CLOTTAFACT sera évalué par votre médecin en fonction du risque de thrombose dans les situations suivantes :
· chez les patients ayant déjà eu une maladie grave du cœur (maladie coronarienne) ou une crise cardiaque (infarctus du myocarde),
· chez les patients ayant une maladie du foie,
· chez les nouveau-nés,
· chez les patients à risque de formation de caillots dans le sang (thrombose) ou de formation de multiples caillots dans la circulation sanguine (coagulation intra vasculaire disséminée),
· avant, pendant et/ou après une intervention chirurgicale.
Dans ces cas, une étroite surveillance devra être réalisée.
Risque d’allergies
Votre médecin vous informera des signes annonciateurs d’une réaction allergique/réaction allergique grave (réaction anaphylactique) (voir rubrique 4. « Quels sont les effets indésirables éventuels »). Si l’un de ces effets survient, ce médicament doit être arrêté immédiatement.
Sécurité virale
Lorsque des médicaments sont préparés à partir de sang ou de plasma humain, des mesures de prévention du risque de transmission d’agents infectieux sont mises en place. Celles-ci comprennent :
· une sélection soigneuse des donneurs de sang et de plasma pour assurer que les donneurs susceptibles d’être porteurs d’infections sont exclus,
· la recherche de virus/marqueurs d’infection sur chaque don et sur les mélanges de plasma,
· l’inclusion d’étapes d’inactivation et d’élimination des virus dans le traitement du sang et du plasma.
Malgré ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission de maladies infectieuses ne peut être totalement exclu. Ceci s’applique également à tous les virus inconnus ou émergents ou autres types d’agents infectieux.
Les mesures prises sont considérées comme efficaces pour lutter contre le risque d’infection par le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B et le virus de l’hépatite C.
Les mesures prises peuvent être d’efficacité limitée vis-à-vis des virus non-enveloppés tels que le virus de l’hépatite A et le parvovirus B 19.
L’infection par le parvovirus B 19 peut être grave pour les femmes enceintes (infection foetale) et les personnes dont le système immunitaire est affaibli ou qui sont atteintes de certains types d'anémies (par exemple, une drépanocytose ou une anémie hémolytique).
Votre médecin peut vous recommander d’envisager une vaccination contre les hépatites A et B si vous recevez de manière régulière/répétée du fibrinogène dérivé de plasma humain.
Risque d’anticorps
En cas de traitement substitutif d’autres déficits constitutionnels (héréditaires) avec des facteurs de coagulation, l’apparition d’anticorps peut être observée mais aucun cas n’a été rapporté à ce jour avec le fibrinogène.
Enfants et adolescents
Les mêmes mises en garde et précautions d’emploi s’appliquent à la population pédiatrique.
Autres médicaments et CLOTTAFACT
Aucune interaction médicamenteuse avec CLOTTAFACT n'est connue à ce jour. Néanmoins le mélange préalable avec d’autres produits et/ou médicaments est formellement déconseillé.
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
CLOTTAFACT avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Ce médicament ne sera utilisé pendant la grossesse et l’allaitement que sur les conseils de votre médecin.
Si vous découvrez que vous êtes enceinte pendant le traitement, consultez votre médecin car lui seul peut juger de la nécessité de le poursuivre.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Sportifs
Sans objet.
Conduite de véhicules et utilisation de machines
Aucun effet sur l’aptitude à conduire des véhicules et à utiliser des machines n’a été observé.
CLOTTAFACT contient du sodium
Ce médicament contient au maximum 3 mmol (ou 69 mg) de sodium (composant principal du sel de cuisine/table) dans chaque flacon. Cela équivaut à 3,45% de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte. Vous devez en tenir compte si vous suivez un régime sans sel ou pauvre en sel.
3. COMMENT UTILISER CLOTTAFACT 1,5 g, poudre et solvant pour solution injectable ?
Le traitement doit être initié sous la surveillance d’un médecin spécialiste.
Posologie
La dose et la durée de la substitution dépendent de votre poids, de la sévérité de votre maladie, de la localisation du saignement et de son étendue ainsi que de votre état de santé.
Votre médecin mesurera également votre concentration de fibrinogène dans le plasma (c’est la partie liquide du sang) avant de commencer le traitement et régulièrement au cours du traitement, pour adapter la dose et la fréquence d’administration.
Mode d’administration
Ce médicament sera préparé par un professionnel de santé avant l’injection. Ce médicament sera ensuite injecté dans une veine par perfusion. L’utilisation d’un nécessaire de perfusion muni d’un filtre de 15 µm, tel que celui fourni dans l’emballage, est obligatoire.
Si vous avez utilisé plus de CLOTTAFACT que vous n’auriez dû
Pour éviter tout risque de surdosage, votre médecin effectuera régulièrement des analyses de sang pour contrôler votre taux de fibrinogène.
En cas de surdosage, un risque de formation anormale de caillots dans le sang ne peut être exclu.
Si vous oubliez d’utiliser CLOTTAFACT
Si vous arrêtez d’utiliser CLOTTAFACT
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
· Réactions allergiques : comme avec tout médicament contenant des protéines et administré dans une veine, des réactions allergiques peuvent survenir. Dans certains cas, ces réactions ont évolué vers une réaction allergique grave, éventuellement avec chute de la pression artérielle (choc anaphylactique).
Les signes annonciateurs d’une réaction allergique sont :
· des sensations de brûlure et picotement au site d’injection,
· des fourmillements,
· des rougeurs, des démangeaisons et des éruptions de la peau,
· des démangeaisons (urticaire),
· une inflammation de la peau,
· une pâleur,
· un gonflement du visage ou de la gorge,
· une toux,
· une respiration sifflante (de type asthmatique)
· une oppression au niveau de la poitrine et du thorax,
· une accélération du rythme cardiaque,
· une diminution de la pression artérielle,
· une fatigue extrême (léthargie),
· une agitation,
· des frissons,
· des nausées et vomissements.
|
Si l'un de ces effets survient, contactez immédiatement votre médecin qui, selon le type et la sévérité de la réaction, décidera d’arrêter le traitement et/ou d’initier un traitement adapté |
· Caillots sanguins : la formation de caillots sanguins obstruant les vaisseaux sanguins peut survenir.
Ils peuvent alors entrainer :
· une crise cardiaque. Les signes annonciateurs sont : une douleur soudaine dans la poitrine ou un essoufflement.
· un accident vasculaire cérébral (AVC). Les signes annonciateurs sont : l’apparition soudaine d’une faiblesse musculaire, une perte de sensation et/ou de l’équilibre, une baisse de la vigilance ou une difficulté à parler.
· une embolie pulmonaire (caillot sanguin obstruant une artère irrigant le poumon qui est une maladie grave). Les signes annonciateurs sont : des douleurs dans la poitrine ou le thorax, une difficulté à respirer ou une toux avec sang.
· un caillot de sang dans les veines (thrombose veineuse). Les signes annonciateurs sont : des rougeurs, une sensation de chaleur, une douleur, une sensibilité ou un gonflement d’une ou des deux jambes.
|
Si l'un de ces effets survient, contactez immédiatement votre médecin qui, selon le type et la sévérité de la réaction, décidera d’arrêter le traitement et/ou d’initier un traitement adapté |
Les effets indésirables suivants sont peu fréquents (entre 1 injection sur 100 et 1 injection sur 1 000) :
· des maux de tête,
· des sueurs nocturnes.
Les effets indésirables suivants sont rares (entre 1 injection sur 1 000 et 1 injection sur 10 000) :
· des étourdissements,
· des vomissements
· un bourdonnement d’oreilles,
· des difficultés pour respirer (asthme),
· des rougeurs, des irritations cutanées, des éruptions de la peau,
· une transpiration excessive,
· une sensation de chaleur.
Effets indésirables chez les enfants et les adolescents
La fréquence, le type et la sévérité des effets secondaires chez les enfants et les adolescents, sont les mêmes que chez l’adulte, à l’exception des réactions allergiques/anaphylactiques qui sont plus fréquentes chez les enfants.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER CLOTTAFACT 1,5 g, poudre et solvant pour solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon et la boîte. La date de péremption fait référence au dernier jour de ce mois.
A conserver dans l’emballage extérieur d’origine à une température ne dépassant pas 25°C, à l’abri de la lumière. Ne pas congeler.
Après reconstitution, le médicament doit être administré immédiatement.
N’utilisez pas ce médicament si vous remarquez que la solution est trouble ou contient un dépôt.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient CLOTTAFACT 1,5 g, poudre et solvant pour solution injectable
· La substance active est :
Fibrinogène humain (1,5 g de fibrinogène humain pour 100 mL de solution reconstituée).
· Les autres composants sont :
Poudre : chlorhydrate d’arginine, isoleucine, chlorhydrate de lysine, glycine, citrate de sodium.
Solvant : eau pour préparations injectables.
Titulaire de l’autorisation de mise sur le marché
3, AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
Exploitant de l’autorisation de mise sur le marché
3, AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
3, AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Faites attention avec CLOTTAFACT
L’état clinique du patient et les autres thérapies de substitution utilisées feront l’objet d’une surveillance continue.
Dans le traitement des hémorragies aiguës sévères, la prescription de CLOTTAFACT doit s’effectuer en association avec les mesures de réanimation adaptées à la situation clinique et biologique du patient.
Il existe un risque de thrombose, chez les patients traités pour un déficit acquis ou constitutionnel avec du fibrinogène humain particulièrement à forte dose ou à doses répétées. Les patients traités par du fibrinogène humain devront être surveillés étroitement pour détecter les signes ou symptômes de thrombose.
Le bénéfice potentiel du traitement par du fibrinogène humain devra être évalué en fonction du risque thromboembolique dans les situations suivantes : patients avec antécédents de maladie coronarienne ou infarctus du myocarde, avec une maladie hépatique, en péri ou post opératoire, chez les nouveau-nés ou les patients à risque d’événements thromboemboliques ou de coagulation intra vasculaire disséminée. Une étroite surveillance devra être observée. L’hypofibrinogénémie acquise est associée à des concentrations plasmatiques basses de tous les facteurs (pas uniquement le fibrinogène) et inhibiteurs de coagulation. Par conséquent, l’utilisation de produits sanguins contenant des facteurs de coagulation devra être prise en considération. Une surveillance stricte de la coagulation est nécessaire.
En cas d’allergie, d’allergie généralisée ou de réaction anaphylactique, l’administration devra être interrompue immédiatement et un traitement symptomatique adapté devra être instauré.
Posologie
· Traitement dans les déficits constitutionnels
L’objectif de la première injection est d’atteindre un taux de fibrinogène circulant supérieur à 1 g/L. Ce taux est à maintenir jusqu’au contrôle et à la stabilisation de l’hémostase, puis un taux au-dessus de 0,5 g/L jusqu’à cicatrisation complète.
En cas d’intervention chirurgicale majeure, une surveillance étroite du traitement par des tests de coagulation est nécessaire.
En cas de procédure chirurgicale ou de traitement d’un épisode de saignement non-chirurgical, la dose doit être calculée comme suit :
Quantité à injecter (g) = (taux à obtenir (g/L) - taux basal (g/L)) ´ 1/ taux de récupération (g/L)/(g/kg)´ poids corporel (kg).
Le ratio « 1/taux de récupération » est déterminé à partir du taux de récupération du patient* (voir section 5.2 du RCP), ou si le taux de récupération est inconnu :
· 0,053 (g/kg)/(g/L) pour les enfants et adolescents <40 kg
· 0,043 (g/kg)/(g/L) pour les adultes et adolescents ³40 kg.
* Exemple pour le calcul du taux de récupération du patient et de la dose
Dans le cas d’un patient de 60 kg avec un taux de fibrinogène indétectable en basal et à 1,20 g/L 1 heure après l’injection de 0,060 g/kg de CLOTTAFACT :
· Le calcul du taux de récupération du patient est :
1,20 (g/L) / 0,060 (g/kg) = 20,0 (g/L)/(g/kg)
· Le calcul de la dose pour une augmentation du taux à 1,0 g/L est :
1,0 g/L x 1 / 20,0 (g/L)/(g/kg) [ou 0,050 (g/kg)/(g/L)] x 60 kg = 3 g.
En cas d’urgence si le taux basal de fibrinogène n’est pas disponible, la dose initiale recommandée est de 0,05 g/kg administré en intraveineux chez les adultes et adolescents ³40 kg, et doit être majoré de 20-25% chez les enfants et adolescents <40 kg.
La posologie ultérieure (doses et fréquence des injections) sera adaptée à l'état clinique et au suivi biologique du patient.
La demi-vie biologique du fibrinogène est de 3-4 jours. Ainsi, en l’absence de dégradation, un traitement répété avec le fibrinogène humain n’est habituellement pas nécessaire. Compte tenu de l’accumulation survenant en cas d’administration répétée dans un but prophylactique, la dose et la fréquence doivent être déterminées en fonction des objectifs thérapeutiques du médecin pour chaque patient.
Population pédiatrique
Les données montrent que le taux de récupération et la demi-vie in vivo chez les enfants et les adolescents ayant un poids corporel <40 kg sont inférieurs à ceux des adultes et des adolescents ayant un poids corporel ³40 kg (voir la rubrique 5.2). Par conséquent, lorsqu’il est inconnu, il convient d’utiliser des taux de récupération adaptés pour calculer la dose de CLOTTAFACT dans les groupes de poids corporel respectif, en considérant qu’un poids corporel inférieur à 40 kg couvre la tranche d’âge allant de la naissance à environ 12 ans. La posologie (doses et fréquence des injections) doit être adaptée en fonction de la réponse clinique individuelle.
Traitement dans les déficits acquis
· Adultes : généralement, une dose initiale de 1 à 2 g est administrée, et éventuellement répétée. Dans les hémorragies aiguës sévères obstétricales, des quantités plus importantes de fibrinogène (4 à 8 g) peuvent être nécessaires.
· En cas d’urgence dans les hémorragies aiguës, si la concentration de fibrinogène n’est pas disponible, une dose initiale sera administrée et les doses suivantes seront adaptées aux concentrations disponibles dans l’intervalle.
· Enfants : la posologie déterminée en fonction du poids corporel et de la situation clinique est généralement de 0,02 à 0,03 g/kg.
Mode et voies d’administration
Reconstitution :
Respecter les règles d’asepsie habituelles.
|
|
· Si nécessaire, amener les deux flacons (poudre et solvant) à température ambiante.
|
|
|
· Retirer la capsule protectrice du flacon de solvant (eau pour préparations injectables) et du flacon de poudre. · Désinfecter la surface de chaque bouchon. |
|
|
· Retirer le capuchon protecteur translucide du système de transfert et insérer à fond le biseau, ainsi dégagé, au centre du bouchon du flacon de solvant en opérant simultanément un mouvement de rotation.
|
|
|
· Retirer le deuxième capuchon protecteur de l'autre extrémité du système de transfert. · Retourner le flacon de solvant et enfoncer rapidement l’extrémité libre du biseau au centre du bouchon du flacon de poudre afin de permettre le transfert du solvant vers la poudre. · Veiller à ce que le biseau soit toujours immergé dans le solvant pour éviter un cassage précoce du vide.
|
|
|
· Pendant le transfert, diriger le jet de solvant sur toute la surface de la poudre et sur la paroi du flacon par un mouvement de rotation horizontale. Veiller à ce que la totalité du solvant soit transférée. · A la fin du transfert, le vide est automatiquement cassé (air stérile). |
|
|
· Retirer le flacon vide (solvant) avec le système de transfert. · Agiter modérément par un mouvement de rotation doux pour éviter la formation de mousse, jusqu'à dissolution complète de la poudre. |



Le produit reconstitué doit être examiné visuellement avant administration, afin de s’assurer qu’il ne contient pas de particules. La solution reconstituée présente une opalescence plus ou moins prononcée. Ne pas utiliser de solution trouble ou contenant un dépôt.
Après reconstitution, le produit doit être administré immédiatement.
Administration :
CLOTTAFACT doit être exclusivement administré par voie intraveineuse, en une seule fois et immédiatement après reconstitution sans dépasser 4 mL/min. En situations d’hémorragies sévères, le débit d’administration peut atteindre 20 mL/min.
CLOTTAFACT ne doit pas être mélangé avec d’autres médicaments et doit être administré avec une voie d’injection/perfusion séparée.
L’utilisation d’un nécessaire de perfusion muni d’un filtre de 15 µm, tel que celui fourni dans l’emballage, est obligatoire.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.