Dernière mise à jour le 28/04/2026
OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée
Indications thérapeutiques
Classe pharmacothérapeutique : Somatostatine et analogues, Code ATC : H01CB02.
OCTREOTIDE TEVA LP est un dérivé synthétique de la somatostatine, substance normalement présente dans l’organisme, et qui diminue l’effet de certaines hormones, comme l’hormone de croissance. L’avantage d’OCTREOTIDE TEVA LP par rapport à la somatostatine est que ce produit est plus puissant et que son effet se maintient plus longtemps.
OCTREOTIDE TEVA LP est utilisé :
· pour traiter l’acromégalie,
L’acromégalie est une affection dans laquelle l’organisme fabrique trop d’hormone de croissance. Normalement, l’hormone de croissance contrôle la croissance des tissus, des organes et des os. Un excès d’hormone de croissance entraîne une augmentation de la taille des os et des tissus, essentiellement au niveau des mains et des pieds. OCTREOTIDE TEVA LP diminue de façon marquée les symptômes de l’acromégalie tels que maux de tête, transpiration excessive, sensation d’engourdissement des mains et des pieds, fatigue et douleurs articulaires. Dans la plupart des cas, la production excessive d'hormone de croissance est due à une augmentation de la taille de l'hypophyse (adénome hypophysaire) ; le traitement par OCTREOTIDE TEVA LP peut réduire la taille de l'adénome.
OCTREOTIDE TEVA LP est utilisé pour traiter les patients atteints d’acromégalie :
o lorsque les autres traitements de l'acromégalie (chirurgie ou radiothérapie) ne sont pas appropriés ou sont inefficaces ;
o après une radiothérapie, pour couvrir la période intermédiaire jusqu’à ce que la radiothérapie devienne pleinement efficace.
· pour soulager les symptômes associés à une production excessive par l'estomac, l'intestin ou le pancréas de certaines hormones et autres substances,
Certaines maladies rares de l'estomac, de l'intestin ou du pancréas sont responsables d’une production excessive de certaines hormones et d’autres substances naturelles. Cela perturbe l'équilibre hormonal naturel de l'organisme, ce qui entraîne divers symptômes tels que bouffées de chaleur avec rougeurs cutanées, diarrhée, baisse de la pression artérielle, éruption cutanée et perte de poids. Le traitement par OCTREOTIDE TEVA LP aide à contrôler ces symptômes.
· pour le traitement des tumeurs neuroendocrines situées dans l'intestin (par ex. : dans l'appendice, l'intestin grêle ou le côlon),
Les tumeurs neuroendocrines sont des tumeurs rares qui peuvent être situées dans différentes parties du corps. OCTREOTIDE TEVA LP est également utilisé pour contrôler la croissance de ces tumeurs lorsqu'elles sont situées dans l'intestin (par ex. : appendice, intestin grêle ou colon).
· pour traiter les tumeurs hypophysaires qui produisent trop d’hormone de stimulation de la thyroïde (TSH).
Trop d’hormone de stimulation de la thyroïde (TSH) entraine une hyperthyroïdie. OCTREOTIDE TEVA LP est utilisé pour traiter les personnes atteintes de tumeurs hypophysaires qui produisent trop d’hormone de stimulation de la thyroïde (TSH) :
o lorsque d’autres types de traitement (chirurgie ou radiothérapie) ne conviennent pas ou n’ont pas fonctionné ;
o après radiothérapie, pour couvrir la période transitoire jusqu’à ce que la radiothérapie soit pleinement efficace.
Présentations
> 1 flacon(s) en verre - 1 seringue(s) préremplie(s) en verre de 2 ml avec adaptateur avec aiguille(s) sécurisée(s)
Code CIP : 34009 301 805 3 2
Déclaration de commercialisation : 15/01/2020
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 471,00 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 472,02 €
- Taux de remboursement :100 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : TEVA BV
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière annuelle
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 436 908 8
ANSM - Mis à jour le : 16/07/2024
OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Octréotide (sous forme d’acétate d’octréotide)........................................................................ 30 mg
Pour un flacon
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour suspension injectable à libération prolongée
Poudre : poudre blanche à blanc cassé, exempte de particules étrangères.
Solvant : solution limpide, incolore, pratiquement exempte de particules.
4.1. Indications thérapeutiques
Traitement de l’acromégalie chez les patients pour lesquels la chirurgie est inadaptée ou inefficace, ou pendant la période transitoire précédant la complète efficacité de la radiothérapie (voir rubrique 4.2).
Traitement des patients avec des symptômes associés aux tumeurs endocrines gastro-entéro-pancréatiques fonctionnelles, par exemple les tumeurs carcinoïdes avec signe(s) clinique(s) d'un syndrome carcinoïde (voir rubrique 5.1).
Traitement des patients atteints de tumeurs neuroendocrines avancées de l’intestin moyen ou de localisation primitive inconnue lorsque les sites primitifs ne correspondant pas à l’intestin moyen ont été exclus.
Traitement des adénomes thyréotropes :
· lorsque la sécrétion n’est pas normalisée après chirurgie et/ou radiothérapie ;
· chez les patients ne relevant pas d’un traitement chirurgical ;
· chez les patients irradiés, en attente de l’efficacité de la radiothérapie.
4.2. Posologie et mode d'administration
Acromégalie
Il est recommandé de débuter le traitement par OCTREOTIDE TEVA LP à la posologie de 20 mg toutes les 4 semaines pendant 3 mois. Chez les patients sous traitement par octréotide par voie sous-cutanée (s.c.), le traitement avec OCTREOTIDE TEVA LP peut être initié le lendemain de la dernière administration d’octréotide s.c. L'ajustement de la dose sera basé sur les taux sériques de l’hormone de croissance (GH) et de l’insulin-like growth factor IGF-1, encore appelée Somatomédine C, ainsi que sur les symptômes cliniques.
Si après 3 mois de traitement, les symptômes cliniques et les taux hormonaux (GH ; IGF-1) ne sont pas complètement contrôlés (concentration de GH toujours > 2,5 microgrammes/L), la dose d’OCTREOTIDE TEVA LP peut être augmentée à 30 mg administrée toutes les 4 semaines. Si trois mois plus tard, les concentrations de GH, IGF-1 et/ou les symptômes cliniques sont insuffisamment contrôlés par l'administration de 30 mg d’OCTREOTIDE TEVA LP, la dose pourra être augmentée à 40 mg toutes les 4 semaines.
Chez les patients dont le taux de GH se maintient, toujours, au-dessous de 1 microgramme/L, dont le taux d’IGF-1 est normalisé et chez qui les principaux signes/symptômes réversibles de l'acromégalie ont disparu après 3 mois de traitement à la dose de 20 mg, il est possible de réduire la dose à 10 mg d’OCTREOTIDE TEVA LP toutes les 4 semaines. Néanmoins, chez ces patients qui reçoivent de faibles doses d’OCTREOTIDE TEVA LP, une surveillance stricte des valeurs sériques de GH et d’IGF-1 et des signes/symptômes cliniques est recommandée.
Pour les patients qui sont traités par une dose stable d’OCTREOTIDE TEVA LP, un dosage des taux de GH et d'IGF-1 doit être réalisé tous les 6 mois.
Tumeurs endocrines gastro-entéro-pancréatiques
Traitement des patients présentant des symptômes associés aux tumeurs neuroendocrines gastro-entéro-pancréatiques fonctionnelles
Il est recommandé de débuter le traitement par OCTREOTIDE TEVA LP à la posologie de 20 mg toutes les 4 semaines. Chez les patients sous traitement par octréotide par voie sous-cutanée (s.c.), l’administration d’octréotide (s.c.) doit être poursuivie, à la même posologie efficace que précédemment, pendant les 2 semaines qui suivent la première injection d’OCTREOTIDE TEVA LP.
Chez les patients dont les symptômes et les marqueurs biologiques sont bien contrôlés après 3 mois de traitement, la posologie peut être réduite à 10 mg d’OCTREOTIDE TEVA LP toutes les 4 semaines.
Chez les patients dont les symptômes ne sont que partiellement contrôlés après 3 mois de traitement, la posologie peut être augmentée à 30 mg d’OCTREOTIDE TEVA LP toutes les 4 semaines.
Durant les périodes où les symptômes liés aux tumeurs neuroendocrines gastro-entéro-pancréatiques pourraient s’aggraver pendant le traitement par OCTREOTIDE TEVA LP, il est recommandé d’administrer simultanément l’octréotide par voie sous-cutanée à la dose qui était pratiquée avant l’instauration du traitement par OCTREOTIDE TEVA LP. Cela peut se produire principalement au cours des 2 premiers mois de traitement jusqu’à l’atteinte des concentrations thérapeutiques efficaces d’octréotide.
Traitement des patients atteints de tumeurs neuroendocrines avancées de l’intestin moyen ou de localisation primitive inconnue lorsque les sites primitifs ne correspondant pas à l’intestin moyen ont été exclus.
La dose recommandée d’OCTREOTIDE TEVA LP est de 30 mg toutes les 4 semaines (voir rubrique 5.1). Le traitement par OCTREOTIDE TEVA LP dans le but de contrôler la tumeur doit être poursuivi en l’absence de progression tumorale.
Adénomes thyréotropes
Le traitement par OCTREOTIDE TEVA LP doit débuter à la posologie de 20 mg toutes les 4 semaines pendant 3 mois avant d'envisager une adaptation posologique. La dose sera adaptée en fonction de la réponse de la TSH et des hormones thyroïdiennes.
Insuffisance rénale
L’insuffisance rénale n’a pas modifié l’aire sous la courbe (ASC) de l’octréotide quand l’octréotide est administré par voie sous-cutanée. Il n’est donc pas nécessaire d'ajuster la dose d’OCTREOTIDE TEVA LP.
Insuffisance hépatique
Lors d’une étude réalisée avec l’octréotide administré par voie sous-cutanée et par voie intraveineuse, il a été montré que la capacité d’élimination pouvait être réduite chez les patients atteints de cirrhose, mais pas chez les patients atteints de stéatose hépatique. Dans certains cas, un ajustement de la dose pourrait être nécessaire chez des patients présentant une insuffisance hépatique.
Population âgée
Dans une étude réalisée avec l’octréotide administré par voie sous-cutanée, aucun ajustement de la dose n’a été nécessaire chez des sujets âgés de 65 ans et plus. Ainsi, il n’est pas nécessaire d’adapter la dose d’OCTREOTIDE TEVA LP chez ces patients.
Population pédiatrique
Les données relatives à l'utilisation d’OCTREOTIDE TEVA LP chez l’enfant sont limitées.
Mode d’administration
OCTREOTIDE TEVA LP doit être exclusivement administrée par injection intramusculaire profonde. En cas d'administrations intramusculaires répétées, les injections doivent être effectuées alternativement dans le muscle fessier droit et gauche (voir rubrique 6.6).
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Générales
Les adénomes hypophysaires somatotropes peuvent parfois augmenter de volume, entraînant des complications sévères (par exemple, une altération du champ visuel). Il est donc important de surveiller attentivement tous les patients. En cas d’augmentation de volume de l’adénome, des alternatives thérapeutiques devraient être envisagées.
Les bénéfices thérapeutiques d’une diminution du taux de l’hormone de croissance (GH) et de la normalisation des taux d’IGF-1 chez les patientes acromégales sont susceptibles de restaurer la fertilité. Il est donc conseillé aux femmes en âge de procréer d’utiliser un moyen de contraception adéquat durant un traitement par octréotide (voir rubrique 4.6).
Un suivi de la fonction thyroïdienne doit être réalisé chez les patients traités au long cours par octréotide.
Un suivi de la fonction hépatique doit être réalisé au cours du traitement par octréotide.
Effets cardiovasculaires
Des cas fréquents de bradycardie ont été rapportés. Une adaptation posologique de médicaments tels que les bêta-bloquants, les inhibiteurs calciques ou les substances agissant sur l’équilibre hydro-électrolytique peut être nécessaire (voir rubrique 4.5).
Vésicule biliaire et réactions associées
La cholélithiase est un événement très fréquent au cours du traitement par octréotide et peut être associée à une cholécystite et à une dilatation des voies biliaires (voir rubrique 4.8). De plus, après commercialisation, des cas de cholangites ont été rapportés comme une complication de la cholélithiase chez les patients prenant OCTREOTIDE TEVA LP.Il est recommandé d’effectuer une échographie de la vésicule biliaire avant l’initiation du traitement par injection d’octréotide à libération prolongée puis tous les 6 mois environ pendant le traitement.
Métabolisme du glucose
OCTREOTIDE TEVA LP peut altérer la glycorégulation en raison de son action inhibitrice sur les sécrétions de GH, glucagon et insuline. La tolérance au glucose en post-prandial peut être perturbée. Comme cela a été rapporté avec l’octréotide s.c. dans certains cas, une hyperglycémie persistante peut résulter d’une administration chronique. Des cas d'hypoglycémie ont également été rapportés.
OCTREOTIDE TEVA LP peut interférer avec le métabolisme du glucose et réduire les besoins en insuline chez le diabétique de type 1. Chez les patients non diabétiques et les patients présentant un diabète de type 2 avec des réserves en insuline partiellement intactes, l’administration d’octréotide s.c. pourrait augmenter la glycémie post-prandiale. Ainsi, il est recommandé de suivre régulièrement la glycémie et le traitement antidiabétique.
Chez les patients ayant un insulinome, l’octréotide peut augmenter l’intensité et la durée de l’hypoglycémie. Ceci s’explique par le fait que l’octréotide inhibe de manière plus importante la sécrétion de la GH et du glucagon que celle de l’insuline, et que la durée de son action inhibitrice est plus courte sur l’insuline. Ces patients doivent être étroitement surveillés.
Fonction pancréatique
Une insuffisance pancréatique exocrine (IPE) a été observée chez certains patients recevant un traitement par l'octréotide pour des tumeurs neuroendocrines gastroentéropancréatiques. Les symptômes de l’IPE peuvent inclure une stéatorrhée, des selles molles, des ballonnements abdominaux et une perte de poids. Un dépistage et un traitement approprié de l’IPE conformément aux directives cliniques doivent être envisagés chez les patients symptomatiques.
Nutrition
Chez certains patients, l’octréotide peut diminuer l’absorption des lipides alimentaires.
Chez certains patients traités avec l’octréotide, une diminution du taux de vitamine B12 et un test de Schilling anormal ont été observés. Il est recommandé de contrôler le taux de vitamine B12 pendant le traitement par OCTREOTIDE TEVA LP chez les patients ayant des antécédents de carence en vitamine B12.
Teneur en sodium
OCTREOTIDE TEVA LP contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Une adaptation posologique de médicaments tels que les bêta-bloquants, les inhibiteurs calciques ou les substances agissant sur l’équilibre hydro-électrolytique peut être nécessaire lorsqu’ils sont administrés en même temps qu’OCTREOTIDE TEVA LP (voir rubrique 4.4).
Des adaptations posologiques de l’insuline et des antidiabétiques peuvent être nécessaires en cas d'administration concomitante d’OCTREOTIDE TEVA LP (voir rubrique 4.4).
Il a été montré que l’octréotide réduit l’absorption intestinale de la ciclosporine et retarde celle de la cimétidine.
L’administration concomitante d’octréotide et de bromocriptine augmente la biodisponibilité de cette dernière.
Des données limitées de la littérature, indiquent que les analogues de la somatostatine pourraient diminuer la clairance métabolique des substances métabolisées par le cytochrome P450, ce qui pourrait être lié à l’inhibition de l’hormone de croissance. Comme on ne peut exclure que l’octréotide puisse avoir cet effet, les médicaments métabolisés principalement par le CYP3A4 et possédant un faible index thérapeutique (ex : quinidine, terfénadine) doivent être utilisés avec prudence.
Association avec des analogues de la somatostatine radioactifs
La somatostatine et ses analogues tels que l'octréotide se lient de manière compétitive aux récepteurs de la somatostatine et peuvent interférer avec l'efficacité des analogues de la somatostatine radioactifs. L'administration d’OCTREOTIDE TEVA LP doit être évitée pendant au moins 4 semaines avant l'administration de lutécium (177Lu) oxodotréotide, un produit radiopharmaceutique se liant aux récepteurs de la somatostatine. Si nécessaire, les patients peuvent être traités par des analogues de la somatostatine de courte durée d'action jusqu'à 24 heures avant l'administration de lutécium (177Lu) oxodotréotide.
Après l'administration de lutécium (177Lu) oxodotréotide, le traitement par d’OCTREOTIDE TEVA LP (peut être repris dans les 4 à 24 heures et doit être à nouveau interrompu 4 semaines avant la prochaine administration de lutécium (177Lu) oxodotréotide.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données sur l’exposition de la femme enceinte à l’octréotide sont limitées (moins de 300 grossesses), et dans environ un tiers de ces cas, les données sur l’issue des grossesses ne sont pas connues. La majorité des rapports a été reçue après la commercialisation de l’octréotide et plus de la moitié des expositions à l’octréotide pendant la grossesse a été rapportée chez des patientes acromégales. La plupart des patientes avaient été exposées à l’octréotide pendant le premier trimestre de la grossesse, à des doses comprises entre 100 et 1 200 microgrammes/jour d’octréotide s.c. ou entre 10 et 40 mg/mois d’injection d’octréotide à libération prolongée. Des anomalies congénitales ont été rapportées dans environ 4 % des cas de grossesse dont l’issue est connue sans qu’aucun lien de causalité n’ait été établi avec la prise d’octréotide.
Les études effectuées chez l’animal n’ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction (voir rubrique 5.3).
Par précaution, il est préférable de ne pas utiliser OCTREOTIDE TEVA LP au cours de la grossesse (voir rubrique 4.4).
On ne sait pas si l'octréotide est excrété dans le lait maternel. Des études chez l’animal ont montré que l’octréotide est excrété dans le lait maternel. Au cours du traitement par OCTREOTIDE TEVA LP, les patientes ne doivent pas allaiter.
Fertilité
On ne sait pas si l'octréotide a un effet sur la fertilité humaine. Une descente tardive des testicules a été observée chez les descendants mâles des femelles traitées durant la grossesse et l’allaitement. Cependant, l'octréotide n’a pas altéré la fertilité des rats mâle et femelle traités à des doses allant jusqu'à 1 mg/kg de poids corporel/jour (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Synthèse du profil de sécurité
Les effets indésirables les plus fréquemment rapportés avec l’octréotide sont des affections gastro-intestinales, des affections du système nerveux, des affections hépatobiliaires, et des troubles du métabolisme et de la nutrition.
Les effets indésirables les plus fréquemment rapportés dans les études cliniques avec l'octréotide étaient : diarrhées, douleurs abdominales, nausées, flatulences, céphalées, cholélithiase, hyperglycémie et constipation. D'autres effets indésirables ont été fréquemment rapportés comme des sensations vertigineuses, des douleurs localisées, des boues biliaires, des dysfonctionnements thyroïdiens (par ex. : diminution de la TSH, diminution de la T4 totale et diminution de la T4 libre), des selles molles, une intolérance au glucose, des vomissements, une asthénie et une hypoglycémie.
Liste tabulée des effets indésirables
Les effets indésirables listés dans le tableau 1, ci-dessous, ont été rapportés lors des études cliniques avec l’octréotide.
Les effets indésirables (tableau 1) sont classés par ordre décroissant de fréquence selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000) et très rare (< 1/10 000), incluant les cas isolés. Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1 : Effets indésirables rapportés au cours des essais cliniques
|
Affections gastro-intestinales |
|
|
Très fréquent : |
Diarrhée, douleurs abdominales, nausées, constipation, flatulences. |
|
Fréquent : |
Dyspepsie, vomissements, ballonnements, stéatorrhée, selles molles, décoloration des selles. |
|
Affections du système nerveux |
|
|
Très fréquent : |
Céphalées. |
|
Fréquent : |
Sensation vertigineuse. |
|
Affections endocriniennes |
|
|
Fréquent : |
Hypothyroïdie, dysthyroïdie (par ex : diminution de la TSH, diminution de la T4 totale et diminution de la T4 libre) |
|
Affections hépatobiliaires |
|
|
Très fréquent : |
Cholélithiase. |
|
Fréquent : |
Cholécystite, boue biliaire, hyperbilirubinémie. |
|
Troubles du métabolisme et de la nutrition |
|
|
Très fréquent : |
Hyperglycémie. |
|
Fréquent : |
Hypoglycémie, altération de la tolérance au glucose, anorexie. |
|
Peu fréquent : |
Déshydratation. |
|
Troubles généraux et anomalies au site d'administration |
|
|
Très fréquent : |
Réactions au site d'injection. |
|
Fréquent : |
Asthénie. |
|
Investigations |
|
|
Fréquent : |
Elévation du taux des transaminases. |
|
Affections de la peau et du tissu sous-cutané |
|
|
Fréquent : |
Prurit, rash, alopécie. |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Fréquent : |
Dyspnée. |
|
Affections cardiaques |
|
|
Fréquent : |
Bradycardie. |
|
Peu fréquent : |
Tachycardie. |
Post-commercialisation
Les effets indésirables listés dans le tableau 2 ont été rapportés spontanément et il n’est pas toujours possible d’évaluer leur fréquence ou la relation de cause à effet avec l’exposition au médicament.
Tableau 2 : Effets indésirables issus de la notification spontanée
|
Affections hématologiques et du système lymphatique |
|
Thrombocytopénie. |
|
Affections du système immunitaire |
|
Anaphylaxie, allergie/réactions d’hypersensibilité. |
|
Affections de la peau et du tissu sous-cutané |
|
Urticaire. |
|
Affections hépatobiliaires |
|
Pancréatite aiguë, hépatite aiguë sans cholestase, hépatite cholestatique, cholestase, ictère, ictère cholestatique. |
|
Affections cardiaques |
|
Arythmies. |
|
Investigations |
|
Élévation du taux de phosphatases alcalines, élévation du taux de gamma-glutamyltransférase. |
Description de certains effets indésirables
Vésicule biliaire et réactions associées
Il a été démontré que les analogues de la somatostatine inhibent la contractilité vésiculaire et diminuent la sécrétion biliaire, ce qui peut entraîner des anomalies vésiculaires ou la formation de boue biliaire (ou sludge). Le développement de calculs biliaires a été rapporté chez 15 à 30 % des patients traités à long terme par octréotide s.c.. L'incidence dans la population générale (âgée de 40 à 60 ans) est d'environ 5 à 20 %. L'exposition à long terme à l’injection d’octréotide à libération prolongée de patients atteints d'acromégalie ou de tumeurs gastro-entéro-pancréatiques suggère que le traitement par injection d’octréotide à libération prolongée n'augmente pas l'incidence de la formation de calculs biliaires, comparé au traitement par octréotide s.c..
Les calculs biliaires sont généralement asymptomatiques. S’ils deviennent symptomatiques, ils doivent être traités soit par une thérapie de dissolution avec des acides biliaires, soit par chirurgie.
Affections gastro-intestinales
Dans de rares cas, les effets indésirables gastro-intestinaux peuvent évoquer une occlusion intestinale aiguë avec distension abdominale progressive, douleur épigastrique sévère, sensibilité et défense abdominales.
En général, la fréquence des événements gastro-intestinaux décroît progressivement au cours du traitement.
Hypersensibilité et réactions anaphylactiques
Une hypersensibilité et des réactions allergiques ont été rapportées au cours de l'expérience post-commercialisation. Lorsque ceux-ci se produisent, ils affectent principalement la peau, rarement la bouche et les voies respiratoires. Des cas isolés de choc anaphylactique ont été signalés.
Réactions au site d'injection
Des réactions au site d'injection, notamment douleur, rougeur, hémorragie, prurit, œdème ou induration, ont été fréquemment rapportées chez les patients recevant un traitement par injection d’octréotide à libération prolongée. Cependant, ces événements n'ont pas nécessité d’intervention médicale dans la majorité des cas.
Troubles du métabolisme et de la nutrition
Bien que l’excrétion de graisses dans les selles puisse être augmentée, il n’y a pas de preuve à ce jour que le traitement au long cours par l’octréotide puisse conduire à une carence nutritionnelle par malabsorption.
Enzymes pancréatiques
Dans de très rares cas, des pancréatites aiguës ont été rapportées dans les premières heures ou les premiers jours du traitement par octréotide s.c. et se sont résolues à l’arrêt du traitement. Par ailleurs, des cas de pancréatites dues à une lithiase biliaire ont été rapportés chez des patients traités au long cours par octréotide s.c.
Affections cardiaques
La bradycardie est un effet indésirable fréquent avec les analogues de la somatostatine. Chez des patients atteints d’acromégalie et de syndromes carcinoïdes, des modifications de l’ECG, telles que : allongement de l’intervalle QT, déviation axiale, repolarisation précoce, microvoltage, transition R/S, onde R précoce et modifications non spécifiques du segment ST-T ont été observés. La relation entre ces événements et le traitement par acétate d’octréotide n’a cependant pas été établie car de nombreux patients présentaient des pathologies cardiaques associées (voir rubrique 4.4).
Thrombocytopénie
Une thrombocytopénie a été rapportée au cours de l’expérience post-commercialisation, en particulier pendant le traitement par injection d’octréotide (i.v.) chez des patients atteints de cirrhose du foie et pendant le traitement par injection d’octréotide à libération prolongée. La thrombocytopénie est réversible après l'arrêt du traitement.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Un nombre limité de surdosages accidentels avec l’injection d’octréotide à libération prolongée a été rapporté. La dose allait de 100 mg à 163 mg/mois d’injection d’octréotide à libération prolongée. Le seul effet indésirable rapporté a été des bouffées de chaleur.
Des cas de patients atteints de cancer recevant des doses d’injection d’octréotide à libération prolongée allant jusqu’à 60 mg/mois et jusqu’à 90 mg toutes les 2 semaines ont été rapportés. Ces doses ont été en général bien tolérées. Cependant, les effets indésirables suivants ont été rapportés : mictions fréquentes, fatigue, dépression, anxiété et altération de la concentration.
La prise en charge du surdosage est symptomatique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Somatostatine et analogues, Code ATC : H01CB02.
Mécanisme d’action
L’octréotide est un octapeptidique de synthèse, dérivant de la somatostatine naturelle, possédant les mêmes effets pharmacologiques et dont la durée d'action est nettement prolongée. Il inhibe l'augmentation pathologique de la sécrétion de l'hormone de croissance (GH) ainsi que de peptides et de la sérotonine produits par le système endocrinien gastro-entéro-pancréatique (GEP).
Chez l'animal, l’octréotide est, par rapport à la somatostatine, un inhibiteur plus puissant de la sécrétion de GH, de glucagon et d'insuline, avec une plus grande sélectivité pour l’inhibition de la GH et du glucagon.
Chez le sujet sain, il a été constaté que l’octréotide, comme la somatostatine inhibait :
· la libération de la GH stimulée par l'arginine, par l'exercice ou par l'hypoglycémie induite par l'insuline ;
· la libération post-prandiale d'insuline, de glucagon, de gastrine et d'autres peptides du système endocrinien GEP, de même que la libération d'insuline et de glucagon provoquée par l'arginine ;
· la libération d'hormone thyréotrope (TSH) induite par la TRH.
Contrairement à la somatostatine, l'octréotide inhibe la sécrétion de GH préférentiellement à celle de l'insuline et son administration n'est pas suivie d'un rebond de sécrétion hormonale (c-à-d. de GH chez les acromégales).
Chez les patients acromégales, OCTREOTIDE TEVA LP, une forme galénique de l'octréotide permettant l'administration répétée toutes les 4 semaines, permet d'obtenir des concentrations sériques thérapeutiques et stables d'octréotide entraînant une baisse constante de la GH et une normalisation des concentrations sériques d'IGF-1, chez la majorité des patients. Chez la plupart des patients, l’injection d’octréotide à libération prolongée réduit significativement les symptômes cliniques de la maladie tels que : céphalées, transpiration, paresthésie, fatigue, douleur ostéo-articulaire et syndrome du canal carpien. Chez les patients acromégales non traités auparavant ayant un adénome hypophysaire somatotrope, l’injection d’octréotide à libération prolongée permet une réduction du volume tumoral supérieure à 20 % pour une proportion significative de patients (50 %).
Chez certains patients présentant un adénome hypophysaire somatotrope, l’injection d’octréotide à libération prolongée peut entraîner une diminution du volume tumoral (avant l'intervention chirurgicale). Toutefois, l'intervention chirurgicale ne doit pas être retardée.
Chez les patients atteints de tumeurs endocrines gastro-entéro-pancréatiques fonctionnelles, OCTREOTIDE TEVA LP assure un contrôle continu des symptômes liés à l’affection sous-jacente. Les effets de l’octréotide dans les différents types de tumeurs gastro-entéro-pancréatiques sont les suivants :
Tumeurs carcinoïdes
L’administration d’octréotide peut entraîner une amélioration des symptômes, notamment des « flushs » et de la diarrhée. Dans de nombreux cas, cela s’accompagne d’une diminution des taux plasmatiques de sérotonine et de l’excrétion urinaire d’acide 5-hydroxy-indole acétique.
VIPomes
La caractéristique biologique de ces tumeurs est une surproduction de peptide intestinal vasoactif (VIP). Dans la plupart des cas, l’administration d’octréotide permet un soulagement de la diarrhée sécrétoire sévère qui caractérise cette affection, ce qui contribue à améliorer de façon importante la qualité de vie des patients. Cela s’accompagne d’une amélioration des troubles électrolytiques associés (notamment de l’hypokaliémie), ce qui permet de suspendre les apports hydro-électrolytiques par voies entérale et parentérale. Chez certains patients, l'examen par tomodensitométrie suggère que l'évolution tumorale a été ralentie ou stoppée, ou même une réduction de la masse tumorale, notamment de métastases hépatiques, a pu être observée. L'amélioration clinique s'accompagne généralement d'une réduction du taux plasmatique de VIP, qui peut même se normaliser.
Glucagonomes
L'administration d’octréotide entraîne dans la plupart des cas une amélioration notable de l'érythème migratoire nécrolytique qui caractérise ces tumeurs. L’octréotide a souvent un effet sur le diabète léger, mais cet effet n'est pas prononcé et n'est généralement pas suffisant pour entraîner une diminution des besoins en insuline ou en antidiabétiques oraux. L’octréotide permet une amélioration des diarrhées, ainsi qu'une prise de poids. Bien que l'administration d’octréotide provoque souvent une baisse immédiate du taux plasmatique de glucagon, elle ne se maintient généralement pas au cours d'une administration prolongée, bien que l'amélioration des symptômes se maintienne.
Gastrinomes/syndrome de Zollinger-Ellison
Le traitement par inhibiteurs de la pompe à protons et antagonistes des récepteurs H2 permet généralement de contrôler l'hypersécrétion d'acide gastrique. Cependant, il est possible que la diarrhée, qui est aussi un symptôme majeur, ne soit pas soulagée de manière adéquate par les inhibiteurs de la pompe à protons et les antagonistes des récepteurs H2. OCTREOTIDE TEVA LP peut aider à réduire davantage l'hypersécrétion d'acide gastrique et à soulager les symptômes, y compris la diarrhée, dans la mesure où elle permet de réduire les hypergastrinémies de certains patients.
Insulinomes
L'administration d’octréotide entraîne une chute de l'insuline immunoréactive circulante. Chez les patients porteurs de tumeurs opérables, l’octréotide peut contribuer à rétablir et à maintenir une glycémie normale avant l'intervention. Chez les patients porteurs de tumeurs bénignes inopérables ou malignes, le contrôle de la glycémie peut être amélioré même en l’absence d’une réduction concomitante et durable des taux circulants d'insuline.
Traitement des patients atteints de tumeurs neuroendocrines avancées de l’intestin moyen ou de localisation primitive inconnue lorsque les sites primitifs ne correspondant pas à l’intestin moyen ont été exclus.
Une étude de phase III randomisée, en double aveugle, contrôlée versus placebo (PROMID) a démontré que l’injection d’octréotide à libération prolongée inhibe la croissance tumorale chez les patients atteints de tumeurs neuroendocrines avancées de l’intestin moyen. 85 patients ont été randomisés entre une administration d’injection d’octréotide à libération prolongée 30 mg toutes les 4 semaines (n = 42) ou de placebo (n = 43) pendant 18 mois ou jusqu’à progression de la tumeur ou décès.
Les principaux critères d’inclusion étaient : naïf de traitement médical, confirmation histologique, tumeur localisée inopérable ou métastatique bien différenciée, tumeurs/carcinomes neuroendocrines fonctionnels ou non, tumeurs primitives de l’intestin moyen ou d’origine inconnue présumées provenir de l’intestin moyen après exclusion d’une origine pancréatique, thoracique ou autre.
Le critère principal d’évaluation était le temps jusqu’à progression de la tumeur ou décès lié à la tumeur (TTP : Time To Progression).
Dans la population en intention de traiter (ITT) (tous les patients randomisés), 26 et 41 cas de progression de la tumeur ou décès lié à la tumeur ont été rapportés dans les groupes injection d’octréotide à libération prolongée et placebo, respectivement (HR = 0,32 ; IC à 95 %, 0,19 à 0,55 ; p = 0,000015).
Dans la population en intention de traiter « conservatrice » (ITTc), dans laquelle, 3 patients ont été censurés à la date de la randomisation, 26 et 40 cas de progression de la tumeur ou décès lié à la tumeur ont été rapportés dans les groupes injection d’octréotide à libération prolongée et placebo, respectivement (HR = 0,34 ; IC à 95 %, 0,20 à 0,59 ; p = 0,000072 ; fig. 1). Le temps médian jusqu’à progression de la tumeur était de 14,3 mois (IC à 95 %, 11,0 à 28,8 mois) dans le groupe injection d’octréotide à libération prolongée et 6,0 mois (IC à 95 %, 3,7 à 9,4 mois) dans le groupe placebo.
Dans la population per protocole (PP), dans laquelle des patients additionnels ont été censurés à la fin du traitement, 19 et 38 cas de progression de la tumeur ou décès lié à la tumeur ont été rapportés dans les groupes injection d’octréotide à libération prolongée et placebo, respectivement (HR = 0,24 ; IC à 95 %, 0,13 à 0,45 ; p = 0,0000036).
Figure 1 : Estimations de Kaplan-Meier du TTP, comparant l’injection d’octréotide à libération prolongée à un placebo (population ITT conservatrice)
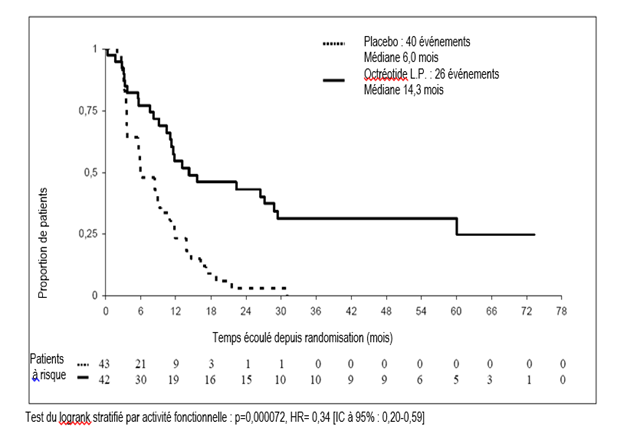
Tableau 3 : Temps jusqu’à progression (TTP) Résultats selon la population analysée
|
|
Événements TTP |
TTP médian en mois [IC à 95 %] |
HR [IC à 95 %] Valeur de p* |
||
|
|
Injection d’octréotide à libération prolongée |
Placebo |
Injection d’octréotide à libération prolongée |
Placebo |
|
|
ITT |
26 |
41 |
NM |
NM |
0,32 [IC à 95 % : 0,19 à 0,55] ; P = 0,000015 |
|
ITTc |
26 |
40 |
14,3 [IC à 95 % : 11,0 à 28,8] |
6,0 [IC à 95 % : 3,7 à 9,4] |
0,34 [IC à 95 %, 0,20 à 0,59] ; P = 0,000072 |
|
PP |
19 |
38 |
NM |
NM |
0,24 [IC à 95 % : 0,13 à 0,45] ; P = 0,0000036 |
|
NM = non mentionné ; HR = hazard ratio ; TTP = délai jusqu’à progression de la tumeur ; ITT = intention de traiter ; ITTc = ITT conservatrice ; PP = per protocole * Test du log-rank stratifié par activité fonctionnelle |
|||||
L’efficacité du traitement est similaire chez les patients atteints de tumeurs neuroendocrines fonctionnelles (HR = 0,23 ; IC à 95 %, 0,09 à 0,57) ou non fonctionnelles (HR = 0,25 ; IC à 95 %, 0,10 à 0,59).
Après 6 mois de traitement, une stabilisation de la maladie est observée chez 67 % des patients du groupe injection d’octréotide à libération prolongée contre 37 % dans le groupe placebo.
Du fait du bénéfice clinique significatif de l’injection d’octréotide à libération prolongée au moment de l’analyse intermédiaire planifiée, le recrutement dans l’étude a été arrêté.
La tolérance de l’injection d’octréotide à libération prolongée dans cette étude était en accord avec son profil de tolérance connu.
Adénomes hypophysaires thyréotropes
Il a été montré qu'une injection intramusculaire d’octréotide à libération prolongée toutes les 4 semaines permet de réguler les taux d'hormones thyroïdiennes élevés, de normaliser le taux de TSH et d'améliorer les signes et symptômes cliniques d'hyperthyroïdie chez les patients présentant des adénomes thyréotropes. Un effet statistiquement significatif par rapport aux données initiales a été atteint après 28 jours du traitement par injection d’octréotide à libération prolongée et les bénéfices du traitement ont perduré jusqu'à 6 mois.
5.2. Propriétés pharmacocinétiques
Absorption
Après une injection intramusculaire unique d’octréotide à libération prolongée, la concentration sérique en octréotide atteint un pic initial transitoire dans l’heure qui suit l'administration, suivie par une diminution progressive jusqu’à parvenir dans les 24 heures à un taux bas, non détectable d’octréotide. Après ce pic initial le premier jour, la concentration en octréotide se maintient à des taux infra-thérapeutiques chez la majorité des patients pendant les 7 jours suivants. Par la suite, la concentration d’octréotide s'élève à nouveau, atteint un plateau vers le 14e jour et se maintient relativement constante au cours des 3 à 4 semaines suivantes. Le niveau du pic au jour 1 est plus bas que les niveaux atteints lors de la phase de plateau et pas plus de 0,5 % de la quantité totale de médicament n’est libéré au cours du jour 1. Aux environs du 42e jour, la concentration d'octréotide diminue lentement, parallèlement à la dégradation finale de la matrice de polymère de cette forme galénique.
Chez les acromégales, les concentrations plateau d’équilibre après des doses uniques de 10 mg, 20 mg et 30 mg d’injection d’octréotide à libération prolongée s’élèvent respectivement à 358 ng/L, 926 ng/L et 1 710 ng/L. À l’état d’équilibre, les concentrations sériques d'octréotide obtenues après 3 injections à 4 semaines d’intervalle, sont 1,6 à 1,8 fois plus élevées et atteignent 1 557 ng/L et 2 384 ng/L respectivement après des injections répétées de 20 mg et 30 mg d’octréotide à libération prolongée.
Chez les patients porteurs de tumeurs carcinoïdes, les taux sériques moyens (et médians) d’octréotide à l’état d’équilibre après injections répétées de 10 mg, 20 mg et 30 mg d’octréotide à libération prolongée à 4 semaines d’intervalle augmentent également proportionnellement à la dose et atteignent respectivement 1 231 (894) ng/L, 2 620 (2 270) ng/L et 3 928 (3 010) ng/L.
Sur une période incluant jusqu'à 28 injections mensuelles d’octréotide à libération prolongée, l’octréotide ne s’est pas accumulé au-delà de ce qui était attendu à partir du chevauchement des courbes de libération.
Distribution et biotransformation
Le profil pharmacocinétique de l’octréotide après injection d’octréotide à libération prolongée reflète le profil de libération à partir de la matrice en polymère et sa biodégradation. Une fois l’octréotide libéré dans la circulation systémique, sa distribution se fait selon ses propriétés pharmacocinétiques connues, décrites lors de l’administration sous-cutanée. Le volume de distribution de l'octréotide à l'état d'équilibre est de 0,27 L/kg, la clairance corporelle totale est de 160 mL/min. La liaison aux protéines plasmatiques est de l'ordre de 65 %. La quantité d'octréotide liée aux hématies est négligeable.
Les données pharmacocinétiques issues des prélèvements sanguins limités réalisés chez des patients pédiatriques présentant une obésité d'origine hypothalamique, âgés de 7 à 17 ans et traités par injection d’octréotide à libération prolongée 40 mg une fois par mois, ont montré des concentrations plasmatiques résiduelles moyennes d'octréotide de 1 395 ng/L après la première injection et de 2 973 ng/L à l'état d'équilibre. Une variabilité interindividuelle importante a été observée.
Les concentrations résiduelles d'octréotide à l'état d'équilibre n'étaient pas corrélées avec l'âge et l'IMC, mais étaient modérément corrélées avec le poids corporel (52,3-133 kg) et les différences entre hommes et femmes étaient significatives, avec des valeurs plus élevées de 17 % environ chez les femmes.
5.3. Données de sécurité préclinique
Les études sur la reproduction menées chez l'animal n'ont mis en évidence aucun effet tératogène, embryo/fœtotoxique ou délétère sur la reproduction après administration de l'octréotide aux parents à des doses allant jusqu'à 1 mg/kg/jour. Un léger ralentissement de la croissance physiologique a été noté dans la descendance du rat, mais ce ralentissement était transitoire et imputable à l'inhibition de la GH due à une activité pharmacodynamique excessive (voir rubrique 4.6).
Aucune étude spécifique n'a été menée chez le rat jeune. Lors des études de développement pré- et post-natal, un retard de croissance et de maturation a été observé chez les sujets de la génération F1 après administration d'octréotide à la mère pendant toute la durée de la grossesse et de la période de lactation. Une descente tardive des testicules a été observée chez les descendants mâles de la génération F1, mais aucun effet délétère n’a été observé sur la fertilité des mâles de la génération F1 touchés. Ainsi, les effets mentionnés ci-dessus étaient temporaires et considérés comme consécutifs à l'inhibition de la GH.
Poly (DL-lactide-co-glycolide), mannitol (E421).
Solvant (en seringue préremplie) :
Carmellose sodique, mannitol (E421), poloxamère, eau pour préparations injectables.
3 ans.
Le produit ne doit pas être conservé après reconstitution (il doit être utilisé immédiatement).
6.4. Précautions particulières de conservation
A conserver dans l’emballage d’origine, à l'abri de la lumière.
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
OCTREOTIDE TEVA LP peut être conservé à une température ne dépassant pas 25 °C le jour de l’injection.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Les conditionnements unitaires contiennent un flacon de verre, fermé par un bouchon en caoutchouc (caoutchouc chlorobutyle) scellé par une capsule d’aluminium avec joint d’étanchéité rouge foncé, contenant la poudre pour suspension injectable et une seringue préremplie en verre incolore avec un bouchon à l’avant et un autre côté piston (caoutchouc bromobutyle) avec 2 mL de solvant, conditionnés ensemble dans une barquette scellée avec un adaptateur pour flacon et une aiguille d'injection sécurisée.
Des conditionnements unitaires et triples sont disponibles
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Instructions relatives à la préparation et à l'administration intramusculaire d’OCTREOTIDE TEVA LP
POUR INJECTION INTRAMUSCULAIRE PROFONDE SEULEMENT
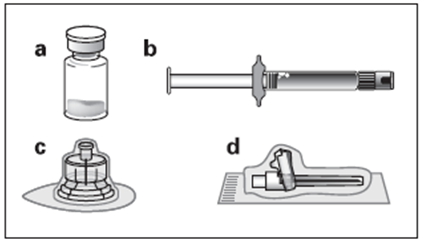
|
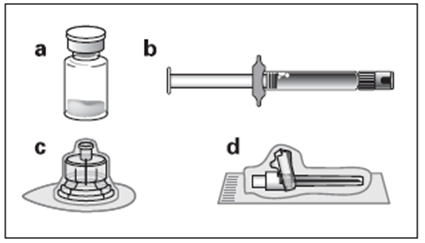
Le kit d’injection comprend : |
|
|
|
a. Un flacon contenant la poudre d’OCTREOTIDE TEVA LP b. Une seringue préremplie contenant le solvant de reconstitution c. Un adaptateur pour flacon pour la reconstitution du produit d. Une aiguille d’injection sécurisée |
Les instructions ci-dessous doivent être suivies avec attention afin de garantir la bonne reconstitution d’OCTREOTIDE TEVA LP avant son administration intramusculaire profonde.
Il y a 3 étapes critiques lors de la reconstitution de la suspension d’OCTREOTIDE TEVA LP. Ne pas les respecter peut empêcher la bonne administration du produit.
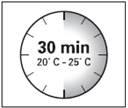
· Le kit d'injection doit être à température ambiante. Sortir le kit d'injection du réfrigérateur et le laisser à température ambiante pendant au-moins 30 minutes avant la reconstitution, mais sans dépasser 24 heures.
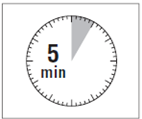
· Après avoir ajouté le solvant dans le flacon, laisser reposer ce dernier pendant 5 minutes pour laisser la poudre s’humidifier totalement.
· Après humidification, secouer modérément le flacon à l’horizontale pendant 30 secondes minimum jusqu'à la formation d'une suspension homogène. La suspension d’OCTREOTIDE TEVA LP doit être préparée immédiatement avant l'administration.
OCTREOTIDE TEVA LP doit être administrée exclusivement par un personnel de santé entraîné.
|
Etape 1 |
|
|
· Sortir le kit d’injection OCTREOTIDE TEVA LP du réfrigérateur. |
|
|
ATTENTION : il est essentiel de commencer la reconstitution une fois que le kit d’injection est revenu à température ambiante. Pour cela, laisser le kit à température ambiante pendant un minimum de 30 minutes avant la reconstitution, mais sans dépasser 24 heures. |
|
|
Remarque : le kit d’injection peut être remis au réfrigérateur si nécessaire. |
|
|
Etape 2 |
|
|
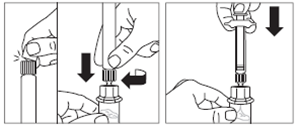
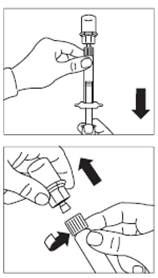
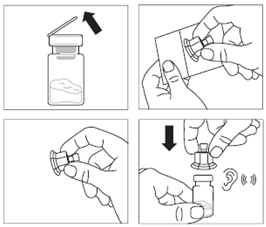
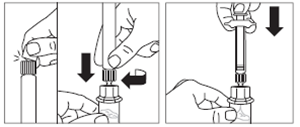
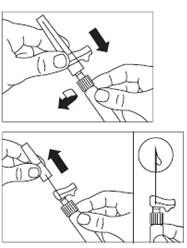
· Retirer l’opercule de plastique du flacon et désinfecter le bouchon de caoutchouc du flacon à l’aide d’un tampon alcoolisé. · Retirer le film de protection et retirer l’adaptateur pour flacon de son emballage en le tenant entre le bouchon Luer blanc et la collerette. NE PAS toucher l’extrémité du dispositif d’accès, à quelque endroit que ce soit. · Placer le flacon sur une surface plane. Positionner l’adaptateur pour flacon sur le dessus du flacon et pousser l’adaptateur complètement vers le bas de telle sorte qu’il se fixe en laissant entendre un « clic ». · Nettoyer l’extrémité de l'adaptateur pour flacon à l’aide d’un tampon alcoolisé. |
|
|
Etape 3 |
|
|
· Retirer le capuchon blanc et lisse de la seringue préremplie de solvant et visser la seringue sur l'adaptateur pour flacon. · Pousser lentement le piston jusqu’en bas pour transférer tout le solvant dans le flacon. |
|
|
Etape 4 |
|
|
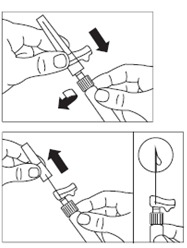
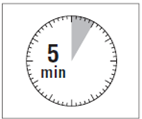
ATTENTION : il est essentiel de laisser le flacon reposer pendant 5 minutes afin de s’assurer que le solvant a complètement humidifié la poudre. Remarque : il est normal que le piston remonte car il peut y avoir une légère surpression dans le flacon. · A ce stade, préparer le patient pour l'injection. |
|
|
Etape 5 |
|
|
· Après la période d’humidification, s’assurer de bien repousser le piston jusqu’en bas de la seringue. |
|
|
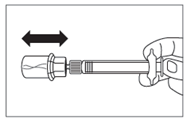
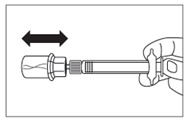
ATTENTION : Maintenir le piston appuyé et secouer le flacon modérément à l’horizontale pendant minimum 30 secondes de telle sorte que la poudre soit complètement mise en suspension (suspension laiteuse homogène). Secouer de nouveau modérément pendant 30 secondes si la poudre n’est pas complètement mise en suspension. |
|
|
Etape 6 |
|
|
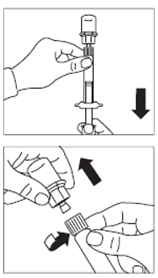
· Retourner la seringue et le flacon tête en bas, tirer doucement le piston et aspirer entièrement le contenu du flacon dans la seringue. · Dévisser la seringue de l’adaptateur pour flacon. |
|
|
Etape 7 |
|
|
· Préparer le site d’injection à l’aide d’un tampon alcoolisé. · Visser l’aiguille d’injection sécurisée sur la seringue. · Si l'administration immédiate est retardée, agiter de nouveau la seringue doucement pour s’assurer d’une suspension laiteuse homogène. · Retirer le capuchon protecteur de l'aiguille d’injection. · Tapoter doucement la seringue pour éliminer les bulles visibles et les chasser hors de la seringue. · Procéder immédiatement à l'étape 8 pour l'administration au patient. Tout retard peut entraîner une sédimentation. |
|
|
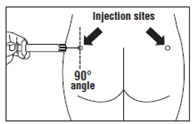
Etape 8 |
|
|
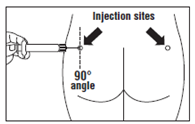
· OCTREOTIDE TEVA LP doit être injecté exclusivement par voie intramusculaire profonde ; JAMAIS par voie intraveineuse. · Piquer l’aiguille profondément dans le muscle fessier gauche ou droit avec un angle de 90º par rapport à la peau. · Tirer lentement le piston pour vérifier qu’aucun vaisseau n’a été touché (repiquer si un vaisseau a été touché). · Pousser lentement le piston jusqu’à ce que la seringue soit vide. Retirer l’aiguille du point d’injection et utiliser le dispositif de sécurité (comme expliqué à l’étape 9). |
|
|
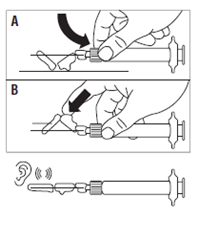
Etape 9 |
|
|
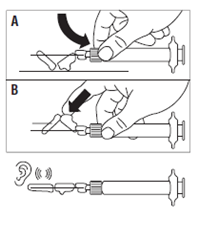
· Placer le dispositif de sécurité sur l’aiguille selon l’une des 2 méthodes suivantes : o soit presser la charnière du volet de sécurité à plat sur une surface rigide (figure A) o soit pousser la charnière en avant avec le doigt (figure B). · Un « clic » sonore confirme la mise en place correcte. · Remarque : inscrire le site d'injection au dossier du patient et changer de site tous les mois. · Jeter immédiatement la seringue (dans un conteneur adapté). |
|
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
SWENSWEG 5
2031 GA HAARLEM
PAYS-BAS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 805 3 2 : Poudre en flacon (verre) et 2 mL de solvant en seringue préremplie (verre).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Prescription initiale hospitalière annuelle.
ANSM - Mis à jour le : 16/07/2024
OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée ?
3. Comment utiliser OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Somatostatine et analogues, Code ATC : H01CB02.
OCTREOTIDE TEVA LP est un dérivé synthétique de la somatostatine, substance normalement présente dans l’organisme, et qui diminue l’effet de certaines hormones, comme l’hormone de croissance. L’avantage d’OCTREOTIDE TEVA LP par rapport à la somatostatine est que ce produit est plus puissant et que son effet se maintient plus longtemps.
OCTREOTIDE TEVA LP est utilisé :
· pour traiter l’acromégalie,
L’acromégalie est une affection dans laquelle l’organisme fabrique trop d’hormone de croissance. Normalement, l’hormone de croissance contrôle la croissance des tissus, des organes et des os. Un excès d’hormone de croissance entraîne une augmentation de la taille des os et des tissus, essentiellement au niveau des mains et des pieds. OCTREOTIDE TEVA LP diminue de façon marquée les symptômes de l’acromégalie tels que maux de tête, transpiration excessive, sensation d’engourdissement des mains et des pieds, fatigue et douleurs articulaires. Dans la plupart des cas, la production excessive d'hormone de croissance est due à une augmentation de la taille de l'hypophyse (adénome hypophysaire) ; le traitement par OCTREOTIDE TEVA LP peut réduire la taille de l'adénome.
OCTREOTIDE TEVA LP est utilisé pour traiter les patients atteints d’acromégalie :
o lorsque les autres traitements de l'acromégalie (chirurgie ou radiothérapie) ne sont pas appropriés ou sont inefficaces ;
o après une radiothérapie, pour couvrir la période intermédiaire jusqu’à ce que la radiothérapie devienne pleinement efficace.
· pour soulager les symptômes associés à une production excessive par l'estomac, l'intestin ou le pancréas de certaines hormones et autres substances,
Certaines maladies rares de l'estomac, de l'intestin ou du pancréas sont responsables d’une production excessive de certaines hormones et d’autres substances naturelles. Cela perturbe l'équilibre hormonal naturel de l'organisme, ce qui entraîne divers symptômes tels que bouffées de chaleur avec rougeurs cutanées, diarrhée, baisse de la pression artérielle, éruption cutanée et perte de poids. Le traitement par OCTREOTIDE TEVA LP aide à contrôler ces symptômes.
· pour le traitement des tumeurs neuroendocrines situées dans l'intestin (par ex. : dans l'appendice, l'intestin grêle ou le côlon),
Les tumeurs neuroendocrines sont des tumeurs rares qui peuvent être situées dans différentes parties du corps. OCTREOTIDE TEVA LP est également utilisé pour contrôler la croissance de ces tumeurs lorsqu'elles sont situées dans l'intestin (par ex. : appendice, intestin grêle ou colon).
· pour traiter les tumeurs hypophysaires qui produisent trop d’hormone de stimulation de la thyroïde (TSH).
Trop d’hormone de stimulation de la thyroïde (TSH) entraine une hyperthyroïdie. OCTREOTIDE TEVA LP est utilisé pour traiter les personnes atteintes de tumeurs hypophysaires qui produisent trop d’hormone de stimulation de la thyroïde (TSH) :
o lorsque d’autres types de traitement (chirurgie ou radiothérapie) ne conviennent pas ou n’ont pas fonctionné ;
o après radiothérapie, pour couvrir la période transitoire jusqu’à ce que la radiothérapie soit pleinement efficace.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée ?
Suivez attentivement toutes les instructions données par votre médecin. Elles peuvent différer des informations mentionnées dans cette notice.
Veuillez lire les explications suivantes avant d'utiliser OCTREOTIDE TEVA LP.
· si vous êtes allergique à l’octréotide ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin avant d’utiliser OCTREOTIDE TEVA LP :
· si vous savez que vous avez actuellement ou avez eu par le passé des calculs biliaires ou que vous avez des complications comme de la fièvre, des frissons, des douleurs abdominales ou un jaunissement de la peau ou des yeux, parlez-en à votre médecin, car une utilisation prolongée d’OCTREOTIDE TEVA LP peut favoriser la formation de calculs biliaires. Votre médecin pourra décider de contrôler votre vésicule biliaire régulièrement ;
· si vous savez que vous êtes diabétique, car OCTREOTIDE TEVA LP peut modifier vos taux de sucre dans le sang. Si vous êtes diabétique, vos taux de sucre doivent être contrôlés régulièrement ;
· si vous avez eu dans le passé une carence en vitamine B12, votre médecin pourra décider de contrôler votre taux de vitamine B12 régulièrement.
Bilans et contrôles
Si vous êtes traité(e) par OCTREOTIDE TEVA LP pendant une période prolongée, votre médecin pourra décider de contrôler le fonctionnement de votre thyroïde régulièrement.
Votre médecin contrôlera le fonctionnement de votre foie.
Votre médecin peut souhaiter vérifier votre fonction enzymatique pancréatique.
Enfants
Les données relatives à l'utilisation d’OCTREOTIDE TEVA LP chez l’enfant sont limitées.
Autres médicaments et OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Vous pourrez généralement continuer à prendre vos autres médicaments pendant votre traitement par OCTREOTIDE TEVA LP. Cependant, des modifications de l'effet de certains médicaments, tels que la cimétidine, la ciclosporine, la bromocriptine, la quinidine et la terfénadine ont été rapportées au cours du traitement par OCTREOTIDE TEVA LP
Si vous prenez un médicament pour contrôler votre tension artérielle (par ex. un bêta-bloquant ou un inhibiteur calcique) ou un médicament destiné à contrôler l’équilibre hydro-électrolytique, votre médecin pourra être amené à adapter la dose que vous devez prendre.
Si vous êtes diabétique, votre médecin pourra être amené à adapter votre dose d'insuline.
Si vous êtes sur le point de recevoir du lutécium (177Lu) oxodotréotide, un traitement radio-pharmaceutique, votre médecin pourra arrêter et/ou adapter votre traitement par OCTREOTIDE TEVA LP. pendant une courte période.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
OCTREOTIDE TEVA LP ne doit être utilisé pendant la grossesse que si ce traitement est vraiment indispensable.
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement.
N’allaitez pas pendant le traitement par OCTREOTIDE TEVA LP. On ne sait pas si OCTREOTIDE TEVA LP passe dans le lait maternel.
Conduite de véhicules et utilisation de machines
OCTREOTIDE TEVA LP n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Cependant, certains des effets indésirables que vous pourriez ressentir au cours du traitement par OCTREOTIDE TEVA LP, tels que des maux de tête et de la fatigue, pourraient diminuer votre aptitude à conduire des véhicules et à utiliser des machines.
OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée contient du sodium
OCTREOTIDE TEVA LP contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée ?
Si vous avez utilisé plus d’OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée que vous n’auriez dû
Aucune réaction pouvant mettre en jeu la vie du patient n'a été déclarée après un surdosage d’OCTREOTIDE TEVA LP.
Les symptômes d'un surdosage sont les suivants : bouffées de chaleur, besoin fréquent d'uriner, fatigue, dépression, anxiété et manque de concentration.
Si vous pensez subir les effets d'un surdosage et ressentez ces symptômes, consultez immédiatement votre médecin.
Si vous oubliez d’utiliser OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée
Si vous oubliez une injection, il est recommandé de la faire faire dès que vous vous en rendez compte, puis de continuer le traitement comme prévu. Un retard de quelques jours pour l'injection d'une dose ne sera pas néfaste, mais certains symptômes pourraient réapparaître temporairement le temps que vous repreniez les injections.
Si vous arrêtez d’utiliser OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée
Si vous interrompez votre traitement par OCTREOTIDE TEVA LP, les symptômes peuvent réapparaître. Par conséquent, vous ne devez pas arrêter d’utiliser OCTREOTIDE TEVA LP à moins que votre médecin ne vous l’ait demandé.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Certains effets indésirables peuvent être graves. Prévenez immédiatement votre médecin si vous présentez l'un des effets indésirables suivants :
Très fréquent (pouvant affecter plus de 1 personne sur 10) :
· Calculs biliaires, entraînant une douleur brutale dans le dos.
· Taux de sucre dans le sang trop élevé.
Fréquent (pouvant affecter jusqu’à 1 personne sur 10) :
· Thyroïde insuffisamment active (hypothyroïdie) entraînant une modification du rythme cardiaque, de l'appétit ou du poids ; fatigue, sensation de froid, ou gonflement de la gorge.
· Résultats anormaux lors des contrôles du fonctionnement de la thyroïde.
· Inflammation de la vésicule biliaire (cholécystite) ; les symptômes peuvent inclure une douleur abdominale en haut à droite, de la fièvre, des nausées, un jaunissement de la peau et des yeux (jaunisse).
· Taux de sucre dans le sang trop faible.
· Altération de la tolérance au glucose.
· Rythme cardiaque lent.
Peu fréquent (pouvant affecter jusqu’à 1 personne sur 100) :
· Soif, faible quantité d’urines, urines foncées, sécheresse et rougeur de la peau.
· Rythme cardiaque rapide.
Autres effets indésirables graves
· Réactions d'hypersensibilité (allergie), notamment éruption cutanée.
· Anaphylaxie : type de réaction allergique pouvant entraîner des difficultés à avaler ou à respirer, un gonflement et des picotements, possiblement avec une chute de la pression artérielle et des vertiges ou une perte de conscience.
· Inflammation du pancréas (pancréatite) ; les symptômes peuvent inclure une douleur soudaine en haut de l’abdomen, des nausées, des vomissements et des diarrhées.
· Inflammation du foie (hépatite) ; les symptômes sont notamment un jaunissement de la peau et des yeux (jaunisse), des nausées, des vomissements, une perte d'appétit, un sentiment général de malaise, des démangeaisons et des urines claires.
· Rythme cardiaque irrégulier.
· Faible taux de plaquettes dans le sang ; cela pourrait entraîner une augmentation des saignements ou des ecchymoses.
Prévenez immédiatement votre médecin si vous remarquez l'un des effets indésirables mentionnés ci-dessus.
Autres effets indésirables
Prévenez votre médecin, votre pharmacien ou votre infirmier/ère si vous remarquez l'un des effets indésirables mentionnés ci-dessous. Ils sont généralement sans gravité et ont tendance à disparaître au cours du traitement.
Très fréquent (pouvant affecter plus de 1 personne sur 10) :
· Diarrhée.
· Douleurs abdominales.
· Nausées.
· Constipation.
· Flatulences (gaz).
· Céphalées.
· Douleur localisée au site d’injection.
Fréquent (pouvant affecter jusqu’à 1 personne sur 10) :
· Inconfort au niveau de l’estomac après les repas (dyspepsie).
· Vomissements.
· Sensation de plénitude de l’estomac.
· Selles grasses.
· Selles molles.
· Selles décolorées.
· Sensation vertigineuse.
· Perte d’appétit.
· Résultats anormaux lors des contrôles du fonctionnement du foie.
· Chute de cheveux.
· Essoufflement.
· Faiblesse.
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre infirmier/ère ou votre pharmacien.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à libération prolongée ?
Tenir ce médicament hors de la vue et de la portée des enfants.
A conserver dans l’emballage d’origine, à l'abri de la lumière.
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
OCTREOTIDE TEVA LP peut être conservé à une température ne dépassant pas 25 °C le jour de l’injection.
Ne pas conserver OCTREOTIDE TEVA LP après reconstitution (il doit être utilisé immédiatement).
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
N’utilisez pas ce médicament si vous remarquez des particules ou un changement de couleur.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Octréotide....................................................................................................................... 30 mg
sous forme d’acétate d’octréotide
Pour un flacon.
· Les autres composants sont :
Dans la poudre (en flacon) : poly (DL-lactide-co-glycolide) et mannitol (E421).
Dans le solvant (en seringue préremplie) : carmellose sodique, mannitol (E421), poloxamère et eau pour préparations injectables.
Chaque conditionnement unitaire contient 1 flacon de verre de 30 mg d’octréotide, fermé par un bouchon en caoutchouc scellé par une capsule d’aluminium avec joint d’étanchéité rouge foncé, 1 seringue préremplie en verre avec 2 mL de solvant, 1 aiguille d’injection sécurisée et 1 adaptateur pour flacon et chaque conditionnement triple contient 3 flacons de 30 mg d’octréotide, 3 seringues préremplies avec 2 mL de solvant, 3 aiguilles d’injection sécurisées et 3 adaptateurs pour flacon.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
SWENSWEG 5
2031 GA HAARLEM
PAYS-BAS
Exploitant de l’autorisation de mise sur le marché
TEVA SANTE
100-110 ESPLANADE DU GENERAL DE GAULLE
92931 LA DEFENSE CEDEX
MERCKLE GMBH
LUDWIG-MERCKLE-STR. 3,
BLAUBEUREN
89143
ALLEMAGNE
Ou
PLIVA HRVATSKA D.O.O. (PLIVA CROATIA LTD.)
PRILAZ BARUNA FILIPOVICA 25,
ZAGREB
10000
REPUBLIQUE DE CROATIE
Ou
PHARMATHEN INTERNATIONAL S.A
INDUSTRIAL PARK SAPES,
RODOPI PREFECTURE, BLOCK No 5,
RODOPI 69300,
GRECE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Dose d’OCTREOTIDE TEVA LP 30 mg, poudre et solvant pour suspension injectable à utiliser
Acromégalie
Il est recommandé de débuter le traitement par OCTREOTIDE TEVA LP à la posologie de 20 mg toutes les 4 semaines pendant 3 mois. Chez les patients sous traitement par octréotide par voie sous-cutanée (s.c.), le traitement avec OCTREOTIDE TEVA LP peut être initié le lendemain de la dernière administration d’octréotide s.c. L'ajustement de la dose sera basé sur les taux sériques de l’hormone de croissance (GH) et de l’insulin-like growth factor IGF-1, encore appelée Somatomédine C, ainsi que sur les symptômes cliniques.
Si après 3 mois de traitement, les symptômes cliniques et les taux hormonaux (GH ; IGF-1) ne sont pas complètement contrôlés (concentration de GH toujours > 2,5 microgrammes/L), la dose d’OCTREOTIDE TEVA LP peut être augmentée à 30 mg administrée toutes les 4 semaines. Si trois mois plus tard, les concentrations de GH, IGF-1 et/ou les symptômes cliniques sont insuffisamment contrôlés par l'administration de 30 mg d’OCTREOTIDE TEVA LP, la dose pourra être augmentée à 40 mg toutes les 4 semaines.
Chez les patients dont le taux de GH se maintient, toujours, au-dessous de 1 microgramme/L, dont le taux d’IGF-1 est normalisé et chez qui les principaux signes/symptômes réversibles de l'acromégalie ont disparu après 3 mois de traitement à la dose de 20 mg, il est possible de réduire la dose à 10 mg d’OCTREOTIDE TEVA LP toutes les 4 semaines. Néanmoins, chez ces patients qui reçoivent de faibles doses d’OCTREOTIDE TEVA LP, une surveillance stricte des valeurs sériques de GH et d’IGF-1 et des signes/symptômes cliniques est recommandée.
Pour les patients qui sont traités par une dose stable d’OCTREOTIDE TEVA LP, un dosage des taux de GH et d'IGF-1 doit être réalisé tous les 6 mois.
Tumeurs endocrines gastro-entéro-pancréatiques
Traitement des patients présentant des symptômes associés aux tumeurs neuroendocrines gastro-entéro-pancréatiques fonctionnelles
Il est recommandé de débuter le traitement par OCTREOTIDE TEVA LP à la posologie de 20 mg toutes les 4 semaines. Chez les patients sous traitement par octréotide par voie sous-cutanée (s.c.), l’administration d’octréotide (s.c.) doit être poursuivie, à la même posologie efficace que précédemment, pendant les 2 semaines qui suivent la première injection d’OCTREOTIDE TEVA LP.
Chez les patients dont les symptômes et les marqueurs biologiques sont bien contrôlés après 3 mois de traitement, la posologie peut être réduite à 10 mg d’OCTREOTIDE TEVA LP toutes les 4 semaines.
Chez les patients dont les symptômes ne sont que partiellement contrôlés après 3 mois de traitement, la posologie peut être augmentée à 30 mg d’OCTREOTIDE TEVA LP toutes les 4 semaines.
Durant les périodes où les symptômes liés aux tumeurs neuroendocrines gastro-entéro-pancréatiques pourraient s’aggraver pendant le traitement par OCTREOTIDE TEVA LP, il est recommandé d’administrer simultanément l’octréotide par voie sous-cutanée à la dose qui était pratiquée avant l’instauration du traitement par OCTREOTIDE TEVA LP. Cela peut se produire principalement au cours des 2 premiers mois de traitement jusqu’à l’atteinte des concentrations thérapeutiques efficaces d’octréotide.
Traitement des patients atteints de tumeurs neuroendocrines avancées de l’intestin moyen ou de localisation primitive inconnue lorsque les sites primitifs ne correspondant pas à l’intestin moyen ont été exclus.
La dose recommandée d’OCTREOTIDE TEVA LP est de 30 mg toutes les 4 semaines (voir rubrique 5.1). Le traitement par OCTREOTIDE TEVA LP dans le but de contrôler la tumeur doit être poursuivi en l’absence de progression tumorale.
Adénomes thyréotropes
Le traitement par OCTREOTIDE TEVA LP doit débuter à la posologie de 20 mg toutes les 4 semaines pendant 3 mois avant d'envisager une adaptation posologique. La dose sera adaptée en fonction de la réponse de la TSH et des hormones thyroïdiennes.
Instructions relatives à la préparation et à l'administration intramusculaire d’OCTREOTIDE TEVA LP
POUR INJECTION INTRAMUSCULAIRE PROFONDE SEULEMENT
|
Le kit d’injection comprend : |
|
|
|
a. Un flacon contenant la poudre d’OCTREOTIDE TEVA LP b. Une seringue préremplie contenant le solvant de reconstitution c. Un adaptateur pour flacon pour la reconstitution du produit d. Une aiguille d’injection sécurisée |
Les instructions ci-dessous doivent être suivies avec attention afin de garantir la bonne reconstitution d’OCTREOTIDE TEVA LP avant son administration intramusculaire profonde.
Il y a 3 étapes critiques lors de la reconstitution de la suspension d’OCTREOTIDE TEVA LP. Ne pas les respecter peut empêcher la bonne administration du produit.
· Le kit d'injection doit être à température ambiante. Sortir le kit d'injection du réfrigérateur et le laisser à température ambiante pendant au-moins 30 minutes avant la reconstitution, mais sans dépasser 24 heures.
· Après avoir ajouté le solvant dans le flacon, laisser reposer ce dernier pendant 5 minutes pour laisser la poudre s’humidifier totalement.
· Après humidification, secouer modérément le flacon à l’horizontale pendant 30 secondes minimum jusqu'à la formation d'une suspension homogène. La suspension d’OCTREOTIDE TEVA LP doit être préparée immédiatement avant l'administration.
OCTREOTIDE TEVA LP doit être administrée exclusivement par un personnel de santé entraîné.
|
Etape 1 |
|
|
· Sortir le kit d’injection OCTREOTIDE TEVA LP du réfrigérateur. |
|
|
ATTENTION : il est essentiel de commencer la reconstitution une fois que le kit d’injection est revenu à température ambiante. Pour cela, laisser le kit à température ambiante pendant un minimum de 30 minutes avant la reconstitution, mais sans dépasser 24 heures. |
|
|
Remarque : le kit d’injection peut être remis au réfrigérateur si nécessaire. |
|
|
Etape 2 |
|
|
· Retirer l’opercule de plastique du flacon et désinfecter le bouchon de caoutchouc du flacon à l’aide d’un tampon alcoolisé. · Retirer le film de protection et retirer l’adaptateur pour flacon de son emballage en le tenant entre le bouchon Luer blanc et la collerette. NE PAS toucher l’extrémité du dispositif d’accès, à quelque endroit que ce soit. · Placer le flacon sur une surface plane. Positionner l’adaptateur pour flacon sur le dessus du flacon et pousser l’adaptateur complètement vers le bas de telle sorte qu’il se fixe en laissant entendre un « clic ». · Nettoyer l’extrémité de l'adaptateur pour flacon à l’aide d’un tampon alcoolisé. |
|
|
Etape 3 |
|
|
· Retirer le capuchon blanc et lisse de la seringue préremplie de solvant et visser la seringue sur l'adaptateur pour flacon. · Pousser lentement le piston jusqu’en bas pour transférer tout le solvant dans le flacon. |
|
|
Etape 4 |
|
|
ATTENTION : il est essentiel de laisser le flacon reposer pendant 5 minutes afin de s’assurer que le solvant a complètement humidifié la poudre. Remarque : il est normal que le piston remonte car il peut y avoir une légère surpression dans le flacon. · A ce stade, préparer le patient pour l'injection. |
|
|
Etape 5 |
|
|
· Après la période d’humidification, s’assurer de bien repousser le piston jusqu’en bas de la seringue. |
|
|
ATTENTION : Maintenir le piston appuyé et secouer le flacon modérément à l’horizontale pendant minimum 30 secondes de telle sorte que la poudre soit complètement mise en suspension (suspension laiteuse homogène). Secouer de nouveau modérément pendant 30 secondes si la poudre n’est pas complètement mise en suspension. |
|
|
Etape 6 |
|
|
· Retourner la seringue et le flacon tête en bas, tirer doucement le piston et aspirer entièrement le contenu du flacon dans la seringue. · Dévisser la seringue de l’adaptateur pour flacon. |
|
|
Etape 7 |
|
|
· Préparer le site d’injection à l’aide d’un tampon alcoolisé. · Visser l’aiguille d’injection sécurisée sur la seringue. · Si l'administration immédiate est retardée, agiter de nouveau la seringue doucement pour s’assurer d’une suspension laiteuse homogène. · Retirer le capuchon protecteur de l'aiguille d’injection. · Tapoter doucement la seringue pour éliminer les bulles visibles et les chasser hors de la seringue. · Procéder immédiatement à l'étape 8 pour l'administration au patient. Tout retard peut entraîner une sédimentation. |
|
|
Etape 8 |
|
|
· OCTREOTIDE TEVA LP doit être injecté exclusivement par voie intramusculaire profonde ; JAMAIS par voie intraveineuse. · Piquer l’aiguille profondément dans le muscle fessier gauche ou droit avec un angle de 90º par rapport à la peau. · Tirer lentement le piston pour vérifier qu’aucun vaisseau n’a été touché (repiquer si un vaisseau a été touché). · Pousser lentement le piston jusqu’à ce que la seringue soit vide. Retirer l’aiguille du point d’injection et utiliser le dispositif de sécurité (comme expliqué à l’étape 9). |
|
|
Etape 9 |
|
|
· Placer le dispositif de sécurité sur l’aiguille selon l’une des 2 méthodes suivantes : o soit presser la charnière du volet de sécurité à plat sur une surface rigide (figure A) o soit pousser la charnière en avant avec le doigt (figure B). · Un « clic » sonore confirme la mise en place correcte. · Remarque : inscrire le site d'injection au dossier du patient et changer de site tous les mois. · Jeter immédiatement la seringue (dans un conteneur adapté). |
|