Dernière mise à jour le 01/06/2026
TADIM 1 million d'unités internationales (UI) poudre pour solution pour inhalation par nébuliseur
Indications thérapeutiques
TADIM contient la substance active colistiméthate sodique et est administré par inhalation pour traiter les infections pulmonaires chroniques chez les patients atteints de mucoviscidose. Tadim est utilisé quand ces infections sont causées par une bactérie spécifique appelée Pseudomonas aeruginosa.
Cette bactérie très fréquente infecte les poumons de presque tous les patients atteints de mucoviscidose à un moment ou à un autre de leur vie. Si cette infection n’est pas correctement contrôlée, elle continuera à altérer vos poumons, en entraînant des difficultés supplémentaires.
Pour administrer TADIM, la poudre contenue dans le flacon doit être dissoute dans une solution diluante appropriée, du chlorure de sodium à 0,9 % (sérum physiologique) ou de l’eau stérile, puis directement inhalée (inspirée) dans les poumons à l’aide d’un inhalateur adéquat afin qu’une quantité plus importante d’antibiotique puisse atteindre la bactérie responsable de l’infection.
Présentations
> 30 flacon(s) en verre
Code CIP : 387 113-2 ou 34009 387 113 2 5
Déclaration de commercialisation : 27/01/2016
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 333,39 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 334,41 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 17/04/2019 | Renouvellement d'inscription (CT) | Le service médical rendu par TADIM est important chez l’adulte et l’enfant dans la prise en charge des infections pulmonaires chroniques dues à Pseudomonas aeruginosa chez les patients atteints de mucoviscidose. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 21/02/2018 | Réévaluation SMR | TADIM n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à COLIMYCINE 1 MUI, solution pour inhalation par nébuliseur. |
| V (Inexistant) | Avis du 18/12/2013 | Inscription (CT) | TADIM n’apporte pas d’amélioration du service médical rendu (ASMR V, inexistante) par rapport à COLIMYCINE 1 MUI, solution pour inhalation par nébuliseur. |
Autres informations
- Titulaire de l'autorisation : ZAMBON SPA
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière semestrielle
- renouvellement non restreint
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 419 918 0
ANSM - Mis à jour le : 26/03/2026
TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 1 million d’unités internationales (UI) (équivalent à environ 80 mg) de colistiméthate sodique.
Poudre pour solution pour inhalation par nébuliseur.
La poudre est blanche à blanc cassé.
4.1. Indications thérapeutiques
TADIM est indiqué chez adulte et l’enfant dans la prise en charge des infections pulmonaires chroniques dues à Pseudomonas aeruginosa chez les patients atteints de mucoviscidose (voir rubrique 5.1).
Il convient de tenir compte des recommandations officielles concernant l'utilisation appropriée des antibactériens.
4.2. Posologie et mode d'administration
Il est recommandé que le colistiméthate sodique (CMS) soit administré sous une surveillance médicale qui requiert une expérience appropriée pour son utilisation.
Posologie
La posologie peut être adaptée en fonction de la sévérité de l’infection et de la réponse clinique.
Posologies recommandées:
Administration par inhalation
Adultes, adolescents et enfants ≥ 2 ans
1-2 MUI deux à trois fois par jour (maximum 6 MUI / jour)
Enfants <2 ans
0,5-1 MUI deux fois par jour (max 2 MUI / jour)
Les recommandations cliniques en rapport avec les schémas thérapeutiques, y compris sur la durée de traitement, la périodicité et l’administration concomitante à d'autres antibiotiques doivent être respectées.
Sujet âgé
Aucune adaptation posologique n’est jugée nécessaire
Insuffisance rénale
Aucune adaptation posologique n’est jugée nécessaire, toutefois la prudence est recommandée chez les insuffisants rénaux (voir rubriques 4.4 et 5.2).
Insuffisance hépatique
Aucune adaptation posologique n’est jugée nécessaire
Mode d’administration
TADIM est destiné à être reconstitué avec une solution diluante et administré par nébulisation en utilisant un nébuliseur adapté.
Les caractéristiques de nébulisation du produit sur la base d’études in vitro réalisées avec différents systèmes de nébulisation, sont détaillées ci-dessous :
|
Caractéristique |
Système de nébulisation |
|||
|
Respironics I-neb AAD avec une chambre d’inhalation de 0,3 mL (grise) |
Pari eflow rapid |
Pari LC Sprint avec un compresseur Pari Boy SX |
||
|
Dose de TADIM placée dans le système de nébulisation |
||||
|
1 million UI dans 1 mL |
1 million UI dans 3mL |
1 million UI dans 3 mL |
||
|
(a) |
Distribution de la taille des gouttelettes; taille médiane des particules : d50 (µm) |
4,34 |
4,56 |
4,37 |
|
(b) |
Quantité totale de médicament libérée l’embout du nébuliseur # (millions UI) |
0,333 |
0,277 |
0,385 |
|
(c) |
Fraction de particules fines (% < 5 µm) |
59,55 |
58,19 |
57,73 |
|
(d) |
Dose de particules fines libérée à l’embout du nébuliseur # (millions UI < 5 µm) |
0,198 |
0,161 |
0,222 |
|
(e) |
Temps de distribution # |
3 minutes, 36 secondes |
5 minutes, 0 secondes |
6 minutes, 40 secondes |
|
(f) |
Taux de libération du médicament à l’embout du nébuliseur # (millions UI/minute) |
0,055 |
0,032 |
0,033 |
|
# Mesure effectuée au moyen d’une inhalation simulée : rapport d’exhalation (I/E) de 1:1, un volume courant de 500 mL et 15 respirations par minute. TADIM reconstitué avec un mélange 50:50 d’eau pour préparations injectables et de solution de chlorure de sodium à 0,9 % ajusté au volume recommandé pour chaque système de nébulisation. Pari Boy SX réglé à une pression de 1,6 bar et un débit de 5,1 L/min. (d) calculé à partir de (b) / 100 x (c) (f) = (d) / (e) |
||||
|
Caractéristique |
Système de nébulisation |
|||
|
Respironics I-neb AAD avec une chambre d’inhalation de 0,5 mL (lilas) |
Pari eflow rapid |
Pari LC Sprint avec un compresseur Pari Boy SX |
||
|
Dose de TADIM placée dans le système de nébulisation |
||||
|
1 million UI dans 1 mL |
2 millions UI dans 4 mL |
2 millions UI dans 4 mL |
||
|
(a) |
Distribution de la taille des gouttelettes ; taille médiane des particules : d50 (µm) |
4,81 |
4,31 |
4,35 |
|
(b) |
Quantité totale de médicament libérée à l’embout du nébuliseur # (millions UI) |
0,579 |
0,601 |
0,861 |
|
(c) |
Fraction de particules fines (% < 5 µm) |
53,01 |
63,11 |
57,73 |
|
(d) |
Dose de particules fines libérée à l’embout du nébuliseur # (millions UI < 5 µm) |
0,307 |
0,379 |
0,497 |
|
(e) |
Temps de distribution # |
8 minutes, 29 secondes |
6 minutes, 38 secondes |
11 minutes, 32 secondes |
|
(f) |
Taux de libération du médicament à l’embout du nébuliseur # (millions UI/minute) |
0,036 |
0,057 |
0,043 |
|
# Mesure effectuée au moyen d’une inhalation simulée : rapport d’exhalation (I/E) de 1:1, un volume courant de 500 mL et 15 respirations par minute. TADIM reconstitué avec un mélange 50:50 d’eau pour préparations injectables et de solution de chlorure de sodium à 0,9 % ajusté au volume recommandé pour chaque système de nébulisation. Pari Boy SX réglé à une pression de 1,6 bar et un débit de 5,1 L/min. (d) calculé à partir de (b) / 100 x (c) (f) = (d) / (e) |
||||
Dans une solution aqueuse, le colistiméthate sodique est hydrolysé en colistine, la substance active. Pour les précautions particulières d’élimination et de manipulation de la solution reconstituée, voir la rubrique 6.6.
Si d'autres traitements sont en cours, ils doivent être pris dans l'ordre recommandé par le médecin.
Tableau de conversion des posologies:
Dans l'Union Européenne (UE), la dose de colistiméthate sodique (CMS) doit être prescrite et administrée seulement en unités internationales (UI). L'étiquetage du produit indique le nombre d’UI par flacon.
Des confusions et des erreurs médicamenteuses ont eu lieu en raison des différentes expressions de la dose en termes d’activité. Aux Etats-Unis, et dans d’autres parties du monde, la dose exprimée est en milligrammes d’activité de colistine base (mg ACB).
Le tableau de conversion suivant est mentionné pour information et les valeurs doivent être seulement considérées comme indicatives et approximatives.
Tableau de conversion en CMS
|
Activité |
≈ masse de CMS (mg)* |
|
|
UI |
≈ mg ACB |
|
|
12 500 |
0,4 |
1 |
|
150 000 |
5 |
12 |
|
1 000 000 |
34 |
80 |
|
4 500 000 |
150 |
360 |
|
9 000 000 |
300 |
720 |
|
* Activité indicative de la substance active = 12 500 IU/mg |
||
TADIM est contre-indiqué chez les patients présentant une hypersensibilité connue au colistiméthate sodique ou à d’autres polymyxines.
4.4. Mises en garde spéciales et précautions d'emploi
L’inhalation de colistiméthate sodique peut être à l’origine d’une toux ou d’un bronchospasme. Une sensation d’asphyxie a été rapportée dans certains cas. Il est préférable d’administrer la première dose sous surveillance médicale. Un traitement préalable par un bronchodilatateur est recommandé et doit être systématique, notamment s’il entre dans le protocole thérapeutique habituel du patient. Il est nécessaire d’évaluer le VEMS avant et après administration.
La survenue d’une hyperréactivité bronchique induite par le colistiméthate sodique chez un patient sans traitement préalable par bronchodilatateurs nécessite de répéter le test à distance en utilisant un bronchodilatateur. Les signes d’une hyperréactivité bronchique en présence d’un bronchodilatateur peuvent indiquer une réponse allergique et le colistiméthate sodique doit être interrompu. Le bronchospasme doit alors faire l'objet d'un traitement médical approprié.
Il est possible que survienne une hyperréactivité bronchique en réponse au colistiméthate sodique lors d’administrations prolongées ; aussi il est recommandé de déterminer le VEMS avant et après administration de TADIM dans le cadre de visites médicales régulières.
Insuffisance rénale
Le colistiméthate sodique est excrété par voie rénale et peut être néphrotoxique à de fortes concentrations sériques. Même si cette toxicité est peu probable pendant le traitement par inhalation, il est recommandé de mesurer ses concentrations sériques surtout chez les patients insuffisants rénaux.
Toxicité rénale
Il a été rapporté une altération de la fonction rénale, habituellement après administration de doses intraveineuses ou intramusculaires plus élevées que les doses recommandées chez des patients présentant une fonction rénale normale, ou lorsque la posologie intraveineuse ou intramusculaire n’a pas été réduite chez des patients atteints d'insuffisance rénale, ou lors d'utilisation concomitante d'autres médicaments néphrotoxiques. Cet effet est généralement réversible à l'arrêt du traitement.
Neurotoxicité
Après administration intraveineuse ou intramusculaire, des concentrations sériques élevées de colistiméthate sodique peuvent être associées en cas de surdosage ou lorsque la posologie chez des patients atteints d'insuffisance rénale n’a pas été réduite ; cela peut induire une neurotoxicité. L'administration concomitante de myorelaxants non dépolarisants ou d'antibiotiques ayant des effets neurotoxiques similaires peut aussi induire une neurotoxicité. Une réduction de la dose de colistiméthate sodique peut soulager les symptômes. Les effets neurotoxiques rapportés comportent : vertige, paresthésie faciale transitoire, trouble de l'élocution, instabilité vasomotrice, troubles visuels, confusion, psychose et apnée (voir également rubrique 4.5).
Porphyrie
Ce médicament doit être utilisé avec la plus grande prudence chez les patients atteints de porphyrie.
Résistance microbiologique
Des cas de résistance acquise au colistiméthate sodique dans les infections à Pseudomonas aeruginosa mucoïde ont été rapportés lors de l’utilisation clinique. Il est nécessaire de tester la sensibilité chez les patients recevant un traitement au long cours lors de consultations régulières et en cas d’exacerbation de l'infection pulmonaire (voir rubrique 5.1).
Autre
Le colistiméthate sodique est connu pour réduire la libération présynaptique de l'acétylcholine à la jonction neuro-musculaire et doit être utilisé chez les patients atteints de myasthénie avec la plus grande prudence et seulement si cette prescription est absolument nécessaire.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Compte tenu des effets du colistiméthate sodique sur la libération d’acétylcholine, les myorelaxants non dépolarisants doivent être administrés avec la plus grande prudence chez les patients recevant du colistiméthate sodique, leurs effets pouvant être prolongés (voir rubrique 4.4).
Une extrême prudence s’impose en cas d’utilisation de colistiméthate sodique inhalé avec d’autres médicaments néphrotoxiques ou neurotoxiques (céfalotine sodique, aminosides et myorelaxants non dépolarisants, par exemple), y compris ceux administrés par voie intraveineuse ou voie intramusculaire (voir rubrique 4.4).
Un traitement concomitant de colistiméthate sodique avec les macrolides tels que l'azithromycine et la clarithromycine, ou les fluoroquinolones comme la norfloxacine et la ciprofloxacine, doit être envisagé avec prudence chez les patients présentant une myasthénie (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
La sécurité d’emploi chez la femme enceinte n’a pas été établie. Les études chez l’animal n’ont pas révélé d’effet tératogène potentiel. Il a cependant été montré que le colistiméthate sodique traverse la barrière placentaire et il existe par conséquent un risque de toxicité fœtale en cas d’administration pendant la grossesse. TADIM ne doit être prescrit pendant la grossesse que si les bénéfices sont supérieurs aux risques potentiels.
Allaitement
Le colistiméthate sodique étant excrété dans le lait maternel, l’allaitement est déconseillé pendant le traitement.
Fertilité
Il n'existe pas de données sur les effets du colistiméthate sodique sur la fertilité humaine. Les études effectuées chez l’animal avec le colistiméthate n'ont pas mis en évidence d'effets indésirables sur la fertilité (voir rubrique 5.3)..
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Une neurotoxicité caractérisée par des étourdissements, une confusion ou des troubles visuels, a été signalée suite à l’administration parentérale de colistiméthate sodique. Il est nécessaire de prévenir les patients de s’abstenir de conduire ou d’utiliser des machines si ces effets surviennent.
Les effets indésirables les plus fréquents après inhalation de colistiméthate sodique sont la toux et le bronchospasme (signalé par une sensation d’oppression thoracique qui peut être mise en évidence par une diminution du VEMS) chez environ 10 % des patients. (Voir également rubrique 4.4)
Les effets indésirables sont présentés ci-dessous par classe de système d'organes et par fréquence. La fréquence est définie comme suit : très fréquent (≥1/10), fréquent (≥1/100 à <1/10), peu fréquent (≥1/1000 à <1/100), rare (≥1/10000 à <1/1000), très rare (<1/10000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
classe de système d'organes |
fréquence |
effets indésirables rapportés |
|
Affections du système immunitaire |
fréquence indéterminée |
réactions d'hypersensibilité telles qu'éruption cutanée |
|
Affections respiratoires, thoraciques et médiastinales |
très fréquent |
toux, oppression thoracique, bronchoconstriction ou bronchospasme |
|
Troubles généraux et anomalies au site d’administration |
fréquence indéterminée |
mal de gorge et douleur dans la bouche |
Si des réactions d’hypersensibilité telles que des éruptions cutanées surviennent, le traitement par le colistiméthate sodique devra être interrompu.
Les cas de maux de gorge et de douleurs dans la bouche peuvent être dus à une hypersensibilité ou à une surinfection par Candida sp.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet :www.signalement-sante.gouv.fr.
Un surdosage peut entraîner une apnée, une faiblesse musculaire, un vertige, une paresthésie faciale transitoire, un trouble de l'élocution, une instabilité vasomotrice, des troubles visuels, de la confusion, une psychose et une insuffisance rénale.
Il n’existe pas d’antidote. La prise en charge d’un surdosage repose sur un traitement symptomatique et des mesures visant à augmenter la clairance du colistiméthate sodique, telles que la diurèse osmotique à l'aide de mannitol, une dialyse péritonéale ou une hémodialyse prolongée.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres antibactériens, polymyxines, code ATC : J01 XB01.
Mécanisme d’action
Le colistiméthate sodique est une prodrogue de la colistine, antibiotique de la famille des polymyxines (appartenant au groupe des polymyxines E). Il possède une structure polypeptidique et est dérivé de Bacillus polymyxa var. colistinus.
Les antibiotiques de la famille des polymyxines sont des agents surfactants qui agissent en se liant à la membrane cellulaire bactérienne et en modifiant sa perméabilité, ce qui provoque la mort des cellules bactériennes. Les polymyxines ont un effet bactéricide sur les bactéries à Gram négatif dont la membrane externe est hydrophobe.
Effets pharmacodynamiques
Un effet bactéricide concentration-dépendant des polymyxines sur les bactéries sensibles a été rapporté.
Mécanismes de résistance
La résistance apparaît en raison de modifications du lipopolysaccharide (LPS) ou d’autres composants de la membrane de la cellule bactérienne.
Sensibilité
La prévalence de la résistance acquise peut varier en fonction de la géographie et du temps pour certaines espèces. Il est donc utile de disposer d’informations sur la prévalence de la résistance locale, surtout pour le traitement d’infections sévères. Si nécessaire, il est souhaitable d’obtenir un avis spécialisé principalement lorsque l’intérêt du médicament dans certaines infections peut être mis en cause du fait du niveau de prévalence de la résistance locale.
|
Classes |
|
ESPÈCES HABITUELLEMENT SENSIBLES |
|
Aérobies à Gram négatif |
|
Acinetobacter sp. |
|
Haemophilus influenzae |
|
Klebsiella sp. |
|
Pseudomonas aeruginosa |
|
Espèces inconstamment sensibles (résistance acquise > 10%) |
|
Aérobies à Gram négatif |
|
Achromobacter xylosoxidans (Alcaligenes xylosoxidans) |
|
Stenotrophomonas maltophilia |
|
Classes |
|
ESPÈCES NATURELLEMENT RÉSISTANTES |
|
Aérobies à Gram négatif |
|
Burkholderia cepacia et espèces apparentées |
|
Proteus spp. |
|
Providencia spp. |
|
Serratia spp. |
Résistance
Le taux de résistance acquise au colistiméthate sodique dans les infections à Pseudomonas aeruginosa mucoïde est de 3 % environ. Toutefois, le taux de résistance peut varier localement et être plus élevé (voir rubrique 4.4).
Résistance croisée
La résistance aux polymyxines n’est pas croisée avec d’autres familles d’antibiotiques.
5.2. Propriétés pharmacocinétiques
L’absorption digestive est négligeable, aussi il est peu probable que si le colistiméthate sodique déposé dans le rhino-pharynx était avalé, cela augmente l’exposition systémique. L’absorption après administration dans les poumons est fonction du système de nébulisation, de la taille des gouttelettes de l’aérosol et de l’état des poumons.
Une étude chez des volontaires sains ayant reçu du colistiméthate sodique par voie inhalée a démontré que la Cmax de la polymyxine E1 (partie active) variait entre 40,0 et 69,9 ng/mL, et que l’ASC variait entre 350 et 668 ng/mL/h, selon le nébuliseur, le volume de remplissage et la concentration. Ces derniers paramètres faisaient varier la dose de 0,3 millions UI à 2 millions UI. La demi-vie était approximativement de 5,2h. Il a été calculé que la biodisponibilité absolue variait entre 5% et 18% selon le nébuliseur. L’ASC après administration intraveineuse d’une dose de 0,5 million UI était de 3352 ng/mL/h et la Cmax était de 1232 ng/mL.
Distribution
La liaison aux protéines est faible. Les antibiotiques de colistiméthate sodique persistent généralement dans les tissus musculaires, le foie, les reins, le cœur et le cerveau. Le volume de distribution a été calculé à 0,09 L/kg dans une seule étude chez des patients atteints de mucoviscidose.
Métabolisme
Le colistiméthate sodique est converti en base in vivo.
Élimination
Aucune information n’est disponible concernant l’élimination du colistiméthate sodique après inhalation.
Après administration par voie intraveineuse, l’excrétion s’effectue principalement par voie rénale, 62 % de la dose parentérale étant retrouvés inchangés dans l’urine dans un délai de 8 heures et environ 80 % dans un délai de 24 heures. Il n’y a pas d’excrétion biliaire.
5.3. Données de sécurité préclinique
Les données non cliniques issues des études de toxicité et de génotoxicité à doses répétées n'ont pas révélé de risque particulier pour l'homme.
Les études chez l’animal avec le colistiméthate n’indiquent pas d’effets délétères sur la fertilité ou le développement embryo-fœtal.
Il n’existe pas de données concernant la carcinogénicité éventuelle du colistiméthate sodique.
Ce médicament ne doit pas être mélangé avec d’autres médicaments à l’exception de ceux mentionnés dans la rubrique 6.6.
L’ajout d’autres antibiotiques à des solutions de TADIM peut entraîner une précipitation.
Avant ouverture : 3 ans
Après reconstitution :
La stabilité physico‑chimique de la solution reconstituée dans le flacon d’origine a été démontrée pendant 24 heures à une température comprise entre 2 et 8 ºC.
En cas d’auto-administration de l’antibiotique nébulisé, il doit être recommandé aux patients d’utiliser la solution immédiatement après sa préparation. Si cela n’est pas possible, la solution doit être conservée au réfrigérateur pendant 24 heures au maximum.
6.4. Précautions particulières de conservation
Pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Le produit est fourni dans un flacon ISO 10R (volume nominal 10 mL) en verre transparent de type I fermé hermétiquement par un bouchon en chlorobutyle siliconé de type I et protégé par une capsule déchirable en aluminium de 20 mm munie d’un opercule central en plastique rouge. Le produit est fourni en boîte de 30 flacons.
6.6. Précautions particulières d’élimination et de manipulation
TADIM peut être reconstitué pour obtenir une solution limpide, incolore à jaune pâle, soit avec de l’eau pour préparations injectables (eau ppi) afin d’obtenir une solution hypotonique limpide, incolore à jaune pâle, soit avec un mélange 50/50 d’eau pour préparations injectables et de solution de chlorure de sodium à 0,9 %, afin d’obtenir une solution isotonique limpide soit avec une solution de chlorure de sodium à 0,9 % afin d’obtenir une solution hypertonique. Le volume utilisé pour la reconstitution doit être celui indiqué dans le mode d’emploi du nébuliseur et n’est généralement pas supérieur à 4 mL. Pendant la reconstitution, faire tourner le flacon doucement pour éviter la formation de mousse.
Après reconstitution, TADIM peut être utilisé avec tout nébuliseur sous réserve qu'il soit adapté à l'inhalation de solution d’antibiotique.
Après reconstitution, les solutions doivent être utilisées immédiatement ; Si toutefois cela n’est pas possible, la solution doit être conservée au réfrigérateur et utilisée dans les 24 heures. Toute solution inutilisée restant dans le nébuliseur après le traitement doit être éliminée. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Les nébuliseurs habituellement utilisés fonctionnent sur la base d’un flux continu et il est vraisemblable qu’une partie du produit administré soit libéré dans l’environnement du patient. Avec un nébuliseur classique, il faut s’assurer que l’administration de TADIM s’effectue dans une pièce correctement ventilée, en particulier dans un contexte hospitalier où il est possible que plusieurs patients utilisent un nébuliseur en même temps. Un système de tuyaux ou des filtres peuvent permettre d’éviter la dispersion de l’aérosol dans l’environnement.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
ZAMBON S.p.A.
VIA LILLO DEL DUCA 10
20091 BRESSO (MI)
ITALIE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 387 113 2 5 : poudre en flacon (verre) : boîte de 30.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Prescription initiale hospitalière de 6 mois – Renouvellement non restreint.
ANSM - Mis à jour le : 26/03/2026
TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur
colistiméthate sodique
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien ou votre infirmier.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien ou votre infirmier. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur ?
3. Comment utiliser TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur ET DANS QUELS CAS EST-IL UTILISE ?
TADIM contient la substance active colistiméthate sodique et est administré par inhalation pour traiter les infections pulmonaires chroniques chez les patients atteints de mucoviscidose. Tadim est utilisé quand ces infections sont causées par une bactérie spécifique appelée Pseudomonas aeruginosa.
Cette bactérie très fréquente infecte les poumons de presque tous les patients atteints de mucoviscidose à un moment ou à un autre de leur vie. Si cette infection n’est pas correctement contrôlée, elle continuera à altérer vos poumons, en entraînant des difficultés supplémentaires.
Pour administrer TADIM, la poudre contenue dans le flacon doit être dissoute dans une solution diluante appropriée, du chlorure de sodium à 0,9 % (sérum physiologique) ou de l’eau stérile, puis directement inhalée (inspirée) dans les poumons à l’aide d’un inhalateur adéquat afin qu’une quantité plus importante d’antibiotique puisse atteindre la bactérie responsable de l’infection.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur ?
Dans certains cas, votre médecin peut décider de ne pas vous prescrire TADIM.
N’utilisez jamais TADIM:
· si vous êtes allergique (hypersensible) au colistiméthate sodique, à la colistine ou à d’autres polymyxines ;
Si cette situation vous concerne, consultez votre médecin avant de prendre TADIM.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou infirmier/ère avant d’utiliser TADIM :
· si vous avez ou avez eu des problèmes rénaux;
· si vous êtes atteint de myasthénie (une maladie rare qui provoque une faiblesse extrême des muscles et une fatigue très rapide);
· si vous souffrez de porphyrie (maladie héréditaire rare due à un trouble du métabolisme) ;
· si vous souffrez d’asthme.
Si l’une de ces situations vous concerne, parlez-en à votre médecin.
Chez les prématurés et les nouveau-nés, une attention particulière doit être prise lors de l'utilisation de TADIM car les reins ne sont pas encore totalement développés.
Autres médicaments et TADIM
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament. Ces autres médicaments pourraient perturber l’action de TADIM.
· médicaments qui peuvent affecter le fonctionnement de vos reins. Prendre ces médicaments en même temps que TADIM peut augmenter le risque de dysfonctionnement de vos reins.
· médicaments qui peuvent affecter votre système nerveux. Prendre ces médicaments en même temps que TADIM peut augmenter le risque d'effets indésirables neurologiques.
· médicaments appelés relaxants musculaires, souvent utilisés au cours d’une anesthésie générale. TADIM peut augmenter les effets de ces médicaments. Si vous devez recourir à une anesthésie générale, informez l’anesthésiste que vous prenez TADIM.
Si vous souffrez de myasthénie et que vous prenez également d'autres antibiotiques appelés macrolides (tels que l’azithromycine, la clarithromycine ou l’érythromycine) ou des antibiotiques appelés fluoroquinolones (comme l’ofloxacine, la norfloxacine et la ciprofloxacine), prendre TADIM augmente encore plus le risque de faiblesse musculaire et de difficultés respiratoires.
Recevoir du colistiméthate sodique par perfusion en même temps que TADIM par inhalation peut augmenter le risque d'effets indésirables.
TADIM avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou à votre pharmacien avant de prendre ce médicament.
Il n’existe pas de données sur la sécurité de TADIM chez la femme enceinte. Votre médecin évaluera le rapport bénéfice/risque avant que vous n’utilisiez TADIM.
Le colistiméthate sodique peut être sécrété dans le lait maternel. Demandez conseil à votre médecin avant d’utiliser TADIM.
Conduite de véhicules et utilisation de machines
Il est possible que l’utilisation de TADIM entraîne des vertiges, un état de confusion ou des problèmes de vue (vue trouble). Dans ce cas, vous ne devez pas conduire, ni utiliser d’outils ou de machines.
TADIM contient :
Sans objet.
3. COMMENT UTILISER TADIM, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin ou pharmacien.
Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Informez votre médecin si vous souffrez de problèmes rénaux car vous pourriez avoir besoin de prendre une dose plus faible de TADIM.
La première dose de TADIM sera toujours administrée en présence de votre médecin ou de votre infirmière.
Prenez TADIM après votre séance de kinésithérapie respiratoire (si vous avez des séances de kinésithérapie). Cela permettra de libérer vos poumons afin que TADIM puisse agir efficacement. Si vous prenez également d'autres médicaments par voie inhalée, votre médecin vous indiquera l'ordre dans lequel vous devez les prendre.
La dose recommandée chez les adultes, les adolescents et les enfants âgés de 2 ans ou plus est de 1-2 flacons (1-2 millions d'unités) deux à trois fois par jour (maximum 6 millions d'unités par jour).
La dose recommandée chez les enfants de moins de 2 ans est de un demi-flacon à un flacon entier (0,5 à 1 million d’unités) deux fois par jour (maximum de 2 millions d'unités par jour).
Votre médecin peut décider d’adapter la dose en fonction de votre situation.
TADIM est inhalé à l’aide d’un nébuliseur. TADIM peut être utilisé avec tout nébuliseur, sous réserve qu'il soit adapté pour délivrer l’antibiotique dans les poumons sous forme de brume. Assurez-vous que la pièce dans laquelle vous vous trouvez est bien ventilée.
Comment préparer une dose de TADIM.
Votre médecin ou votre infirmière vous montrera comment préparer et utiliser TADIM et vous servir du nébuliseur.
Avant de placer TADIM dans le nébuliseur pour l’inhalation, vous devez tout d’abord dissoudre le produit avec de l’eau pour préparations injectables, ou du chlorure de sodium à 0,9 % stérile (sérum physiologique) ou dans un mélange contenant 50 % d’eau pour préparations injectables et 50 % de chlorure de sodium stérile à 0,9 % (sérum physiologique), comme il est expliqué ci-dessous. Votre médecin ou infirmier/ère vous indiquera le volume correct de liquide à ajouter à chaque flacon de TADIM et le nombre de flacons de TADIM que vous devez préparer et utiliser avec votre nébuliseur pour chaque dose.
|
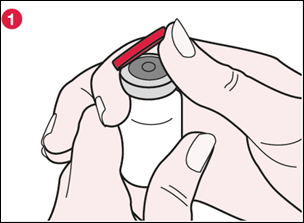
1) Repérez la languette sur l’opercule en plastique rouge près de la flèche marquée « FLIP UP » [TIRER VERS LE HAUT]. Puis en tenant le flacon d’une main et l’opercule en plastique de l’autre, tournez légèrement l’opercule dans le sens contraire des aiguilles d’une montre. Placez un pouce sous la languette et tirez l’opercule vers le haut à un angle d’environ 90° (voir illustration 1). |
|
|
|
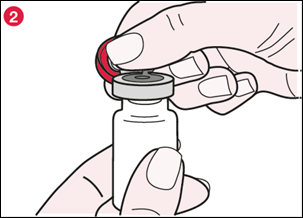
2) Tenez l’opercule en plastique comme le montre l’illustration 2 et tirez lentement pour ouvrir, comme une charnière, à presque 180°. |
|
|
|
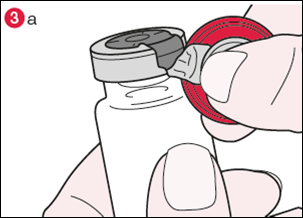
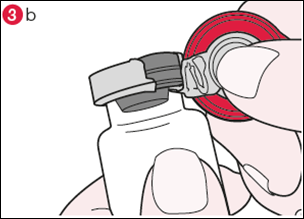
3) Tournez le flacon de façon à ce que l’opercule en plastique soit vers vous. En tenant le centre de l’opercule comme le montre l’illustration, tirez-le vers le bas et tournez légèrement vers la droite (illustration 3a) ou vers la gauche afin de briser la bague en aluminium d’un côté seulement (illustration 3b). |
|
|
|
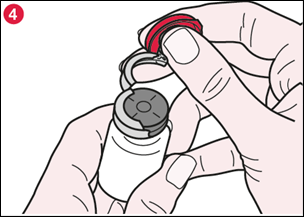
4) Une fois la bague ouverte, tenez fermement le flacon et retirez la bague en aluminium pour faire apparaître complètement le bouchon en caoutchouc (illustration 4). |
|
|
|
5) Retirez le bouchon en caoutchouc du flacon de TADIM en ne tenant que le bord extérieur du bouchon et placez-le à l’envers sur une surface propre. Ajoutez lentement l’eau pour préparations injectables, ou le chlorure de sodium à 0,9 % stérile ou le mélange eau pour préparations injectables + chlorure de sodium stérile à 0,9 % dans le flacon, en ajoutant le volume de liquide indiqué par votre médecin ou infirmier/ère. |
|
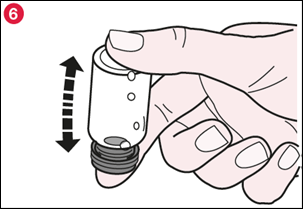
6) Replacez le bouchon en caoutchouc et retournez doucement le flacon deux fois (illustration 6). |
|
|
|
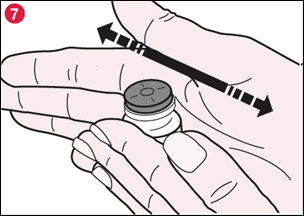
7) Faites rouler doucement le flacon entre vos mains pour dissoudre toute la poudre de TADIM visible au fond et sur le côté du flacon (illustration 7). N’agitez pas le flacon trop vigoureusement car cela pourrait entraîner la formation de mousse. |
|
|
|
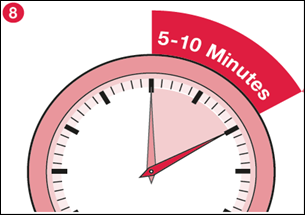
8) Lorsque presque toute la poudre est dissoute, laissez le flacon reposer pendant 5 à 10 minutes pour faire disparaître la mousse éventuelle et permettre à la poudre restante de se dissoudre. |
|
|
Versez la solution dans le nébuliseur et inhalez immédiatement. Si vous ne pouvez pas utiliser la solution immédiatement, replacez le bouchon sur le flacon et conservez le flacon au réfrigérateur pendant 24 heures au maximum. N’utilisez pas une solution ayant été préparée depuis plus de 24 heures. Reportez-vous à la rubrique 5 « Comment conserver TADIM » pour des instructions sur la conservation de TADIM ou l’élimination du médicament non utilisé.
Si vous avez pris plus de TADIM que vous n’auriez dû :
Si vous découvrez que vous avez pris une dose plus forte de TADIM que celle recommandée par votre médecin (ou si quelqu’un d’autre a pris ce produit), contactez immédiatement votre médecin.
Les symptômes liés à l'inhalation d'une quantité excessive de TADIM peuvent inclure :
· des picotements ou engourdissements sur le pourtour des lèvres et du visage
· des vertiges et étourdissements
· des troubles de l'élocution
· des troubles de la vue
· une confusion
· des troubles mentaux
· des bouffées de chaleur (rougeur du visage)
Si vous oubliez de prendre TADIM :
Prenez la dose oubliée dès que possible, sauf si l’heure de la dose suivante est proche.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre TADIM :
N’interrompez pas votre traitement prématurément. Votre médecin vous informera de la durée de votre traitement.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
TADIM peut parfois provoquer des réactions allergiques, notamment des éruptions sur la peau. Si vous constatez une telle réaction, interrompez votre traitement par TADIM et contactez immédiatement votre médecin.
Lors de l’inhalation de TADIM avec le nébuliseur, il arrive que certaines personnes ressentent une oppression dans la poitrine, sentent que leur respiration est sifflante, se mettent à tousser ou se sentent essoufflées (parfois décrit comme une sensation d’asphyxie). C’est pourquoi la première dose de TADIM doit être prise en présence de votre médecin ou de votre infirmier/ère. Votre médecin pourra également vous recommander de prendre un autre médicament pour éviter tout essoufflement. Votre médecin pourra contrôler votre respiration lors de vos prochaines consultations.
TADIM peut aussi affecter vos reins, généralement si la dose que vous avez prise est trop forte ou si vous prenez également d’autres médicaments susceptibles de causer des troubles rénaux.
TADIM peut parfois provoquer une irritation de la bouche ou de la gorge.
Déclaration des effets secondaires suspectés
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet :www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. Comment conserver Tadim, 1 million d’unités internationales (UI) poudre pour solution pour inhalation par nébuliseur ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon ou la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
Les flacons de TADIM non ouverts ne nécessitent pas de précautions particulières de conservation.
TADIM ne contient aucun conservateur. Une fois préparée, la solution reconstituée doit de préférence être utilisée immédiatement. Si cela n’est pas possible, la solution doit être conservée au réfrigérateur pendant 24 heures au maximum. Ne pas utiliser une solution ayant été préparée depuis plus de 24 heures.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Le colistiméthate sodique
Chaque flacon contient 1 million d’unités internationales (UI) (équivalent à environ 80 mg) de colistiméthate sodique.
· Les autres composants sont : le produit ne contient aucun autre composant.
TADIM est une poudre pour solution pour inhalation par nébuliseur qui se présente sous la forme d’une poudre blanche à blanc cassé contenue dans un flacon en verre.
TADIM est disponible en boîte de 30 flacons.
Titulaire de l’autorisation de mise sur le marché
ZAMBON S.p.A.
VIA LILLO DEL DUCA 10
20091 BRESSO (MI)
ITALIE
Exploitant de l’autorisation de mise sur le marché
ZAMBON FRANCE S.A.
13, RUE RENE JACQUES
92138 ISSY-LES-MOULINEAUX CEDEX
FRANCE
XELLIA PHARMACEUTICALS APS
DALSLANDSGADE 11
DK- 2300, COPENHAGEN S
DANEMARK
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen et au Royaume-Uni (Irlande du Nord) sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[À compléter ultérieurement par le titulaire]
CONSEILS / EDUCATION SANITAIRE
QUE SAVOIR SUR LES ANTIBIOTIQUES ?
Les antibiotiques sont efficaces pour combattre les infections dues aux bactéries. Ils ne sont pas efficaces contre les infections dues aux virus.
Aussi, votre médecin a choisi de vous prescrire cet antibiotique parce qu’il convient précisément à votre cas et à votre maladie actuelle.
Les bactéries ont la capacité de survivre ou de se reproduire malgré l’action d’un antibiotique. Ce phénomène est appelé résistance : il rend certains traitements antibiotiques inactifs.
La résistance s’accroît par l’usage abusif ou inapproprié des antibiotiques.
Vous risquez de favoriser l’apparition de bactéries résistantes et donc de retarder votre guérison ou même de rendre inactif ce médicament, si vous ne respectez pas :
· la dose à prendre,
· les moments de prise,
· et la durée de traitement.
En conséquence, pour préserver l’efficacité de ce médicament :
1- N’utilisez un antibiotique que lorsque votre médecin vous l’a prescrit.
2- Respectez strictement votre ordonnance.
3- Ne réutilisez pas un antibiotique sans prescription médicale même si vous pensez combattre une maladie apparemment semblable.
4- Ne donnez jamais votre antibiotique à une autre personne, il n’est peut-être pas adapté à sa maladie.
5- Une fois votre traitement terminé, rapportez à votre pharmacien toutes les boîtes entamées pour une destruction correcte et appropriée de ce médicament.