Dernière mise à jour le 01/06/2026
PROLASTIN 1000 mg, poudre et solvant pour solution pour perfusion
Indications thérapeutiques
Classe pharmacothérapeutique : inhibiteur de protéase, alpha-1 antitrypsine humaine, code ATC : B02AB02.
PROLASTIN appartient à la famille des médicaments appelés alpha-1 antitrypsine.
L’alpha-1 antitrypsine est une substance qui est fabriquée par notre organisme pour freiner les élastases, des substances qui endommagent les poumons. En cas de déficit héréditaire en alpha-1 antitrypsine, l’équilibre entre l’alpha-1 antitrypsine et les élastases est rompu, ce qui peut entraîner une destruction progressive du tissu pulmonaire et l’apparition d’un emphysème pulmonaire.
L’emphysème pulmonaire consiste en un agrandissement anormal des poumons, s’accompagnant d’une destruction du tissu pulmonaire. PROLASTIN est utilisé pour rétablir l’équilibre entre l’alpha-1 antitrypsine et les élastases pulmonaires afin d’éviter que l’emphysème pulmonaire ne continue à s’aggraver.
PROLASTIN est utilisé en traitement à long terme chez des patients présentant une forme particulière de carence en alpha-1 antitrypsine, comme celle qui a été diagnostiquée par votre médecin.
Présentations
> 1 flacon(s) de poudre en verre de 1000 mg - 1 flacon(s) de solvant en verre de 40 ml avec dispositif(s) de transfert
Code CIP : 34009 301 883 1 6
Déclaration de commercialisation : 13/01/2021
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Faible | Avis du 08/07/2020 | Inscription (CT) | Le service médical rendu par PROLASTIN (alpha-1 antitrypsine humaine) est faible dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 08/07/2020 | Inscription (CT) | Compte tenu : • des données cliniques disponibles notamment une étude randomisée, en double aveugle versus placebo déjà évaluées par la commission dans le cadre de l’évaluation des autres spécialités à base d’alpha-1 antitrypsine humaine, • de l’absence de définition d’un seuil de pertinence clinique permettant d’estimer la pertinence clinique de l’effet observé sur le ralentissement de la perte de densité parenchymateuse pulmonaire avec l’alpha-1 antitrypsine par rapport au placebo, • du besoin médical actuellement partiellement couvert par 2 spécialités à base d’alpha-1 antitrypsine humaine, mais de la nécessité de disposer d’alternatives supplémentaires pour pallier les situations de tension d’approvisionnement ou de pénurie qui ont été antérieurement rapportées avec les 2 spécialités disponibles, la Commission considère que PROLASTIN (alpha-1 antitrypsine humaine) n’apporte pas d’amélioration du service médical rendu (ASMR V) dans la prise en charge actuelle des patients déficitaires en alpha-1 antitrypsine. |
Autres informations
- Titulaire de l'autorisation : GRIFOLS DEUTSCHLAND GMBH
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 419 755 3
ANSM - Mis à jour le : 25/02/2026
PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un flacon de poudre contient environ 1 000 mg d’alpha-1 antitrypsine humaine*.
Après reconstitution avec 40 mL de solvant, la solution contient environ 25 mg/ml d’alpha-1 antitrypsine humaine.
*Produite à partir de plasma de donneurs humains.
Excipient(s) à effet notoire :
PROLASTIN contient 2,76 mg de sodium par mL de solution reconstituée (120 mmol/L).
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution pour perfusion.
Poudre ou masse friable : de couleur blanche, jaune clair ou marron clair.
Solvant : solution limpide et incolore.
La solution reconstituée apparaît comme une solution transparente à légèrement opalescente, incolore ou d’une couleur vert clair, jaune clair ou marron clair.
4.1. Indications thérapeutiques
Les patients doivent recevoir un traitement pharmacologique et non pharmacologique optimal et montrer des signes de maladie pulmonaire évolutive (ex : diminution du volume expiratoire maximal par seconde [VEMS] attendu, réduction de la capacité de marche ou augmentation du nombre d'exacerbations) évalués par un professionnel de santé expérimenté dans le traitement du déficit en alpha-1 antitrypsine.
4.2. Posologie et mode d'administration
Le traitement doit être instauré par un médecin expérimenté dans le traitement des bronchopneumopathies chroniques obstructives et surveillé lors des premières perfusions. Les perfusions suivantes peuvent être administrées par un professionnel de santé, voir rubrique 4.4.
La durée du traitement est laissée à l’appréciation du médecin traitant. Aucune limite de durée de traitement spécifique n’a été fixée.
Posologie
Adultes, y compris personnes âgées
Sauf prescription contraire, la dose hebdomadaire est de 60 mg de substance active par kg de poids corporel (équivalent à 180 mL de solution reconstituée pour perfusion, qui contient 25 mg/mL d’alpha-1 antitrypsine humaine pour un patient pesant 75 kg), administrée sous la forme d’une perfusion de courte durée qui suffit habituellement pour maintenir un taux d'alpha-1 antitrypsine sérique constant supérieur à 80 mg/dL correspondant à un taux de 1,3 μM au niveau des poumons. Ces taux dans le sang et dans le liquide recouvrant l’épithélium pulmonaire sont, en théorie, censés protéger contre l’aggravation de l'emphysème pulmonaire.
Population pédiatrique
La sécurité et l’efficacité de PROLASTIN dans la population pédiatrique (<18 ans) n’ont pas encore été établies. Aucune donnée n’est disponible.
Mode d’administration
PROLASTIN ne doit être administré qu’en perfusion intraveineuse après reconstitution.
La poudre doit être dissoute avec le solvant fourni dans la boîte (eau pour préparations injectables) comme décrit dans la rubrique 6.6 et administrée en utilisant un set pour perfusion approprié (non fourni).
La solution reconstituée doit être administrée dans les 3 heures qui suivent sa préparation.
La vitesse de perfusion ne doit pas dépasser 0,08 mL/kg de poids par minute (correspond à 6 mL par minute pour un patient de 75 kg). Cette vitesse de perfusion peut être ajustée en fonction de la tolérance par le patient. Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
PROLASTIN ne doit pas être utilisé chez les patients :
· souffrant d'un déficit en IgA sélectif, chez qui la présence d'anticorps anti-IgA a été démontrée, en raison du risque de réactions allergiques voire de choc anaphylactique ;
· ayant une hypersensibilité à l’alpha-1 antitrypsine ou à l’un des excipients mentionnés à la rubrique 6.1 (voir également rubrique 4.4).
4.4. Mises en garde spéciales et précautions d'emploi
PROLASTIN pouvant provoquer une augmentation transitoire du volume sanguin, la prudence s’impose plus particulièrement chez les patients présentant une insuffisance cardiaque sévère et chez les patients présentant un risque de surcharge volémique.
Hypersensibilité
Dans de rares cas, des réactions d’hypersensibilité peuvent survenir, y compris chez les patients ayant toléré un traitement précédent par alpha-1 antitrypsine humaine. En cas de réaction d’hypersensibilité sévère (entraînant une baisse de la tension artérielle à moins de 90 mmHg, une dyspnée ou même un choc anaphylactique), le traitement par PROLASTIN doit être immédiatement interrompu et si nécessaire, un traitement adapté au choc doit être instauré.
Traitement à domicile
Les données sur l’utilisation de PROLASTIN à domicile sont limitées.
Les risques potentiels associés au traitement à domicile sont liés à la manipulation et à l’administration du médicament ainsi qu’à la prise en charge des réactions indésirables. Les patients doivent dans tous les cas être informés des signes de réaction d’hypersensibilité.
Le bien-fondé du traitement à domicile pour le patient est laissé à l’appréciation du médecin traitant, qui doit s’assurer qu’une formation appropriée est dispensée (concernant par exemple la reconstitution, l’utilisation du dispositif de transfert pour la reconstitution, le branchement de la tubulure intraveineuse, les techniques de perfusion, la tenue à jour d’un carnet de traitement, l’identification des effets indésirables et la conduite à tenir en cas de survenue de tels effets) et que l’utilisation du médicament est réévaluée à intervalles réguliers.
Agents infectieux transmissibles
Les mesures standard destinées à prévenir les infections provoquées par l’utilisation des médicaments préparés à partir de sang ou de plasma humains, comprennent la sélection des donneurs, l’analyse des dons de sang individuels et des pools plasmatiques au niveau de certains marqueurs infectieux spécifiques ainsi que la mise en œuvre dans le procédé de fabrication, d’étapes efficaces d’inactivation/élimination des virus. Malgré ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humains sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents et à d’autres agents pathogènes.
Les mesures prises sont considérées comme efficaces contre les virus enveloppés, notamment le virus de l’immunodéficience humaine (VIH), les virus de l’hépatite B (VHB) et de l’hépatite C (VHC). Les mesures prises peuvent avoir un effet limité contre les virus non enveloppés tels que le virus de l’hépatite A et le parvovirus B19.
L’infection par le parvovirus B19 peut être grave chez la femme enceinte (infection fœtale) ainsi que chez les patients immunodéprimés ou ayant une augmentation de l’érythropoïèse (par exemple en cas d’anémie hémolytique).
Une vaccination appropriée (hépatite A et B) des patients recevant régulièrement ou de façon répétée de l’alpha-1 antitrypsine préparée à partir de plasma humain est recommandée.
Traçabilité
Chaque fois que PROLASTIN est administré à un patient, le nom et le numéro de lot du produit doivent être clairement enregistrés afin de conserver un lien entre le patient et le lot du produit.
Tabagisme
Le traitement de PROLASTIN ne peut pas être refusé aux fumeurs. Mais étant donné que l’efficacité de PROLASTIN peut être compromise par la présence de fumée dans les poumons, il est fortement recommandé à ces patients d’arrêter de fumer.
Teneur en sodium
Ce médicament contient environ 110,4 mg (4,8 mmol) de sodium par flacon de 1 000 mg.
Dans le cas d’un patient pesant 75 kg, la teneur en sodium de la dose recommandée équivaut à 24,84 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
A prendre en compte chez les patients contrôlant leur apport alimentaire en sodium.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée.
4.6. Fertilité, grossesse et allaitement
Grossesse
On ne dispose d’aucune donnée clinique sur l’exposition à PROLASTIN pendant la grossesse. Des études chez l’animal n’ont pas été réalisées. Lors de la prescription de PROLASTIN à des femmes enceintes, la prudence s’impose.
On ne sait pas si l’alpha-1 antitrypsine est excrétée dans le lait maternel. Le passage de l’alpha-1 antitrypsine dans le lait n’a pas été étudié chez l’animal. La décision de poursuivre/d’interrompre l’allaitement ou de poursuivre/d’arrêter le traitement avec PROLASTIN doit prendre en considération le bénéfice de l’allaitement pour l’enfant et au regard du bénéfice du traitement par PROLASTIN pour la femme.
Fertilité
Aucune étude sur la fertilité chez l’animal n’a été réalisée avec PROLASTIN. L’inhibiteur alpha1-protéinase étant une protéine endogène humaine, aucun effet indésirable sur la fertilité n’est attendu lorsqu’il est administré aux doses recommandées.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
PROLASTIN n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
Résumé du profil de sécurité
Le traitement par PROLASTIN peut provoquer des réactions connues telles que fièvre, symptômes pseudo-grippaux, dyspnée, urticaire, nausées, etc.
Comme avec tout traitement protéique, des réactions immunologiques peu fréquentes ou rares peuvent survenir, même lorsque le patient n’a pas présenté d’hypersensibilité ou de réaction allergique lors d’une administration antérieure. Il peut s’agir de réactions allergiques telles qu’urticaire ou dyspnée, et très rarement choc anaphylactique (voir rubrique 4.4).
Les symptômes pouvant être de nature immunologique doivent être évalués avant que le patient ne reprenne son traitement.
Liste tabulée des effets indésirables
Les effets indésirables sont présentés dans le tableau ci-dessous selon la classification de systèmes d’organes MedDRA (SOC et niveau de terme préférentiel).
Les fréquences ont été définies selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Les effets indésirables suivants ont été observés dans le cadre de l’utilisation de PROLASTIN :
|
Classe de systèmes d’organes |
Peu fréquent |
Rare |
Très rare |
|
Affections du système immunitaire |
Urticaire |
Réactions d’hypersensibilité |
Choc anaphylactique |
|
Affections du système nerveux |
Vertiges/confusion/ céphalées |
|
|
|
Affections cardiaques |
|
Tachycardie |
|
|
Affections vasculaires |
|
Hypotension/ |
|
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée |
|
|
|
Affections de la peau et du tissu sous-cutané |
Eruption cutanée |
|
|
|
Affections gastro-intestinales |
Nausées |
|
|
|
Affections musculo-squelettiques et du tissu conjonctif |
Douleurs articulaires/ arthralgies |
Dorsalgies |
|
|
Troubles généraux et anomalies au niveau du site d’administration |
Frissons, fièvre, symptômes pseudo-grippaux, douleur thoracique |
|
|
En ce qui concerne la sécurité virale, voir rubrique 4.4.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Les conséquences du surdosage ne sont pas connues.
En cas de surdosage, le patient doit être étroitement surveillé afin de détecter d’éventuels effets indésirables et des mesures de soutien doivent être disponibles.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
L'alpha-1 antitrypsine est un composant normal du sang humain qui inhibe l’activité de l’élastase du neutrophile et d’autres enzymes. L’alpha-1 antitrypsine a un poids moléculaire de 51 kDa et appartient à la famille des inhibiteurs de la sérine-protéase.
Mécanisme d’action :
Actuellement, on suppose que la pathogenèse de l’emphysème lié à un déficit en alpha-1 antitrypsine est attribuable à la perturbation biochimique chronique de l’équilibre entre l’élastase et l’alpha-1 antitrypsine. L’élastase, qui est synthétisée par les cellules pro-inflammatoires dans les voies respiratoires inférieures, est capable de dégrader les tissus élastiques. Un des principaux inhibiteurs de l’élastase est l’alpha-1 antitrypsine, absent en cas de déficit héréditaire en alpha-1 antitrypsine. Dans ce cas, les structures alvéolaires ne sont pas protégées contre l’élastase qui est libérée par les neutrophiles dans les voies respiratoires inférieures, à laquelle elles sont donc exposées de manière chronique.
Cette situation entraîne une dégradation progressive des tissus élastiques et lorsque les taux d’alpha-1 antitrypsine baissent en dessous de 80 mg/dL (correspondant à un taux de 1,3 μM au niveau des poumons), le risque d’emphysème augmente.
Effets pharmacodynamiques
L’administration de PROLASTIN augmente et maintient les taux dans le sang et dans le liquide recouvrant l’épithélium pulmonaire de l’inhibiteur alpha1-protéinase, menant à une diminution de l’aggravation de l’emphysème pulmonaire.
Deux études observationnelles contrôlées ont montré que le ralentissement le plus significatif de la réduction du VEMS était celui observé chez les patients ayant un VEMS de 35 à 60 % de la valeur attendue.
5.2. Propriétés pharmacocinétiques
Les paramètres pharmacocinétiques qui ont été étudiés pour PROLASTIN dans l’étude ChAMP, une étude multicentrique, randomisée, en double-aveugle, croisée, pour évaluer la comparabilité pharmacocinétique de l’alpha-1 MP (processus modifié) à celles de PROLASTIN chez 24 sujets adultes au déficit en alpha-1 antitrypsine (DAAT), sont présentés dans le tableau 1 ci-dessous :
Tableau 1 : Critère d’évaluation principale pharmacocinétique et autres paramètres pharmacocinétiques importants dérivés des résultats des tests sur l’efficacité
|
AUC0-7 jours Moyenne (%CV)
|
Cmax Moyen (%CV)
|
tmax Médian (Fourchette)
|
t1/2 Moyenne (%CV)
|
Ctrough moyen Moyenne (%CV) |
|
|
|
(mg*h/ml) |
(mg/ml) |
(h) |
(h) |
(mg/ml)
|
|
Prolastin |
152,4 (16%) |
1,848 (15%) |
0,820 (0,25-2,90) |
139,3 (18%) |
0,574 (20%) |
5.3. Données de sécurité préclinique
La substance active de PROLASTIN, l’alpha-1 antitrypsine, est obtenue à partir de plasma humain et se comporte comme un composant plasmatique endogène. L’administration d’une dose unique de PROLASTIN à différentes espèces animales aussi bien que l’administration de doses quotidiennes pendant 5 jours consécutifs à des lapins n'ont montré aucun effet toxique. On ne dispose pas d’études précliniques complémentaires sur l’administration de doses répétées (toxicité chronique, carcinogénicité, reprotoxicité). Il n’y aurait aucun intérêt à mener ces études sur les modèles animaux traditionnels étant donné qu’il est fort probable que les animaux développeraient des anticorps contre les protéines hétérologues humaines administrées. Etant donné que l’inhibiteur alpha1-protéinase humain est une protéine et un composant physiologique du sang humain, on ne s’attend pas à ce qu’il présente des effets carcinogénétiques, génotoxiques ou tératogènes.
Poudre : chlorure de sodium, phosphate monosodique.
Solvant : eau pour préparations injectables.
3 ans.
La solution reconstituée doit être utilisée dans les 3 heures suivant sa préparation.
Une fois préparée, la solution pour perfusion ne doit pas être conservée au réfrigérateur. Toute solution non utilisée doit être éliminée conformément à la réglementation en vigueur.
6.4. Précautions particulières de conservation
À conserver à une température ne dépassant pas 25 °C.
Ne pas congeler.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre : flacon en verre de type I avec bouchon en caoutchouc chlorobutyle et opercule en aluminium.
Solvant : flacon en verre de type I avec bouchon en caoutchouc et opercule en aluminium.
Présentations :
Emballage unitaire
Chaque boite de PROLASTIN 1 000 mg, poudre et solvant pour solution pour perfusion contient :
· un flacon de poudre (1 000 mg d'alpha-1 antitrypsine humaine) ;
· un flacon de solvant (40 mL d’eau pour préparations injectables) ;
· un dispositif de transfert pour la reconstitution.
Emballage multiple
Chaque emballage multiple contient :
4 boites de PROLASTIN 1 000 mg, poudre et solvant pour solution pour perfusion
Toutes les présentations peuvent ne pas être commercialisés.
6.6. Précautions particulières d’élimination et de manipulation
La poudre doit être mélangée et dissoute dans le contenu du flacon d’eau pour préparations injectables fourni dans la boîte, comme décrit ci-dessous. La solution reconstituée apparaît comme une solution transparente à légèrement opalescente, incolore ou d’une couleur vert clair, jaune clair ou marron clair. La reconstitution totale doit être obtenue en 5 minutes.
Préparation de la solution reconstituée pour perfusion
1. Utiliser une technique aseptique (matériel propre et stérilisé) afin de maintenir la stérilité. La reconstitution doit être effectuée sur une surface de travail plane.
2. S’assurer que les flacons de PROLASTIN et de solvant (eau pour préparations injectables) de PROLASTIN sont à température ambiante (20 °C à 25 ºC).
3. Retirer l’opercule du flacon de PROLASTIN et nettoyer les bords et les bouchons avec un coton imprégné d’alcool. Laisser les bouchons en caoutchouc sécher.
|
4. |
5. |
6. Retirer l’emballage extérieur |
|
7. |
8. |
9. |
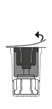
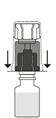
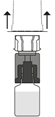
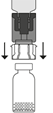
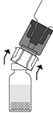
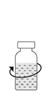
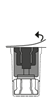
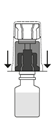
10. Si plusieurs flacons sont nécessaires pour obtenir la dose requise, répéter les étapes ci-dessus en utilisant le nouveau dispositif de transfert contenu dans chacun des flacons pour flacon neuf. Ne pas réutiliser un dispositif de transfert déjà utilisé.
Seules les solutions transparentes à légèrement opalescentes, incolore ou de couleur vert clair, jaune clair ou marron clair et exemptes de particules visibles peuvent être utilisées. La solution reconstituée doit toujours être utilisée dans les 3 heures qui suivent sa reconstitution. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
COLMARER STRASSE 22
60528 FRANKFURT
ALLEMAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 883 1 6 : Poudre en flacon (verre) et solvant en flacon (verre) avec dispositif de transfert.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM.
Liste I
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 25/02/2026
PROLASTIN 1 000 mg poudre et solvant pour solution pour perfusion
Alpha-1 antitrypsine humaine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion ?
3. Comment utiliser PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion ?
6. Contenu de l’emballage et autres informations
1. QU’EST-CE QUE PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : inhibiteur de protéase, alpha-1 antitrypsine humaine, code ATC : B02AB02.
PROLASTIN appartient à la famille des médicaments appelés alpha-1 antitrypsine.
L’alpha-1 antitrypsine est une substance qui est fabriquée par notre organisme pour freiner les élastases, des substances qui endommagent les poumons. En cas de déficit héréditaire en alpha-1 antitrypsine, l’équilibre entre l’alpha-1 antitrypsine et les élastases est rompu, ce qui peut entraîner une destruction progressive du tissu pulmonaire et l’apparition d’un emphysème pulmonaire.
L’emphysème pulmonaire consiste en un agrandissement anormal des poumons, s’accompagnant d’une destruction du tissu pulmonaire. PROLASTIN est utilisé pour rétablir l’équilibre entre l’alpha-1 antitrypsine et les élastases pulmonaires afin d’éviter que l’emphysème pulmonaire ne continue à s’aggraver.
PROLASTIN est utilisé en traitement à long terme chez des patients présentant une forme particulière de carence en alpha-1 antitrypsine, comme celle qui a été diagnostiquée par votre médecin.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion ?
N’utilisez jamais PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion
· si vous êtes allergique ou hypersensible à la substance active ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6 ;
· si vous souffrez d’un déficit connu en certaines immunoglobulines (IgA) car dans ce cas des réactions allergiques ou même un choc anaphylactique pourraient se produire
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou infirmier/ère avant d’utiliser PROLASTIN.
· Informez votre médecin si votre cœur est gravement affaibli (insuffisance cardiaque). Une prudence particulière est recommandée car PROLASTIN peut entraîner une augmentation passagère du volume sanguin.
Réactions allergiques (hypersensibilité)
Dans de rares cas, des réactions d’hypersensibilité à PROLASTIN peuvent survenir, même si vous avez bien toléré l’alpha-1 antitrypsine lors d’administrations précédentes.
Votre médecin vous expliquera quels sont les signes de réaction allergique et ce que vous devez faire en cas de survenue d’une telle réaction (voir également rubrique 4.4).
Si vous développez des signes de réaction allergique sévère pendant la perfusion du médicament, informez immédiatement votre médecin ou infirmier/ère.
Informations sur la sécurité concernant le risque d’infections
Lorsque des médicaments sont préparés à partir de sang ou de plasma humain, certaines mesures sont prises pour éviter la transmission d’infections au patient. Ces mesures comprennent notamment :
· une sélection soigneuse des donneurs de sang et de plasma (pour garantir l’exclusion des personnes pouvant être porteuses d’infections) ;
· l’analyse de chaque don et de chaque pool de plasma à la recherche de traces de virus/de signes d’infection ;
· l’inclusion dans le procédé de traitement du sang ou du plasma, d’étapes pouvant inactiver ou éliminer les virus.
Malgré ces mesures, une transmission éventuelle d’une infection ne peut être complètement exclue en cas d’utilisation de médicaments préparés à partir de sang ou de plasma. Il en va de même pour des virus qui sont encore inconnus ou pour des nouveaux virus, ou pour d’autres types d’infections.
On pense que les mesures prises sont efficaces contre les virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), les virus de l’hépatite B et de l’hépatite C. Il se peut qu’elles soient insuffisantes vis-à-vis des virus non enveloppés, tels que le virus de l’hépatite A et le parvovirus B19. L’infection à parvovirus B19 peut être grave chez la femme enceinte (infection fœtale) et pour les patients dont le système immunitaire est déprimé ou qui présentent certains types d’anémie (par exemple drépanocytose ou anémie hémolytique).
Il se peut que votre médecin vous conseille d’envisager une vaccination anti-hépatite A et hépatite B, si l’on vous administre régulièrement/de manière répétitive de l’alpha-1 antitrypsine fabriquée à partir de plasma humain.
Il est fortement recommandé de noter le nom et le numéro de lot du produit lors de chaque administration de PROLASTIN afin de maintenir la traçabilité des lots utilisés.
Tabagisme
L’arrêt du tabagisme est fortement recommandé car l’efficacité de PROLASTIN est compromise par la présence de fumée de tabac dans les poumons.
Enfants et adolescents
Il n’y a aucune donnée disponible sur l’utilisation de PROLASTIN chez les enfants et les adolescents âgés de moins de 18 ans.
Autres médicaments et PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion
Aucune interaction médicamenteuse avec PROLASTIN n’est connue à ce jour.
Informez néanmoins votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament, y compris un médicament obtenu sans ordonnance.
PROLASTIN avec des aliments et boissons et de l’alcool
Sans objet
Grossesse et allaitement
Demandez conseil à votre médecin ou pharmacien avant de prendre tout médicament.
Il n’y a pas de donnée concernant l’utilisation de PROLASTIN au cours de la grossesse. Informez votre médecin si vous êtes enceinte ou planifiez une grossesse. On ignore si PROLASTIN passe dans le lait maternel. Demandez conseil à votre médecin si vous allaitez.
Conduite de véhicules et utilisation de machines
Rien n’indique que PROLASTIN altère l’aptitude à conduire des véhicules ou à utiliser des machines.
PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion contient du sodium
PROLASTIN 1 000 mg contient environ 110,4 mg de sodium (composant principal du sel de cuisine/table).
Dans le cas d’un patient pesant 75 kg, la dose recommandée équivaut à 24,84 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte. Parlez-en à votre médecin ou pharmacien si vous devez suivre un régime à faible teneur en sel (sodium).
3. COMMENT UTILISER PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion ?
Traitement à domicile
Après les premières perfusions, un professionnel de santé pourrait également administrer PROLASTIN, mais uniquement après avoir reçu une formation appropriée. Votre médecin déterminera si le traitement à domicile vous convient et s’assurera que le professionnel de santé a reçu des instructions concernant :
· la façon de préparer et d’administrer la solution reconstituée pour perfusion (voir les instructions illustrées à la fin de cette notice ;
· la façon de maintenir la stérilité du produit (techniques de perfusion aseptiques) ;
· la façon de tenir à jour un carnet de traitement ;
· la façon d’identifier des effets indésirables, y compris les signes de réaction allergique, et les mesures à prendre si de tels effets surviennent (voir également rubrique 4).
Posologie
La dose de PROLASTIN que vous recevez est déterminée en fonction de votre poids corporel. L’administration d’une dose de 60 mg de substance active par kg de poids corporel (équivalant à 180 mL de solution reconstituée pour perfusion contenant 25 mg/mL d’alpha-1 antitrypsine (humaine) dans le cas d’un patient pesant 75 kg) est habituellement suffisante pour maintenir un taux sérique d'alpha-1 antitrypsine protecteur empêchant que l’emphysème pulmonaire continue à s’aggraver.
La durée du traitement est fixée par votre médecin. À ce jour, rien n'indique qu’il soit nécessaire de limiter la durée du traitement.
Si vous avez l’impression que l’effet de PROLASTIN est trop fort ou trop faible, parlez-en à votre médecin ou à votre pharmacien.
Si vous avez utilisé plus de PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion que vous n’auriez dû
A ce jour, on ignore les conséquences d’un surdosage.
· Informez votre médecin ou le professionnel de santé si vous pensez que vous avez utilisé plus de PROLASTIN que vous n’auriez dû. Il/elle prendra les mesures appropriées.
Si vous oubliez d’utiliser PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion
· Adressez-vous à votre médecin qui déterminera si la dose oubliée doit être administrée.
· Ne prenez pas de dose double pour compenser une perfusion oubliée.
Si vous arrêtez d’utiliser PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion
Votre état peut s’aggraver à l’arrêt de PROLASTIN. Veuillez consulter votre médecin traitant si vous souhaitez arrêter prématurément votre traitement par PROLASTIN.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Si des effets indésirables surviennent pendant la perfusion de PROLASTIN, la perfusion doit être interrompue ou arrêtée, selon la nature et la sévérité de l’effet indésirable.
Effets indésirables graves éventuels
Dans de rares cas (peuvent affecter jusqu’à 1 patient sur 1 000), des réactions d’hypersensibilité peuvent survenir ; dans de très rares cas (peuvent affecter jusqu’à 1 patient sur 10 000) ces réactions peuvent se manifester sous forme de réactions anaphylactiques de tout type, même si vous n’aviez pas présenté de signes d’allergie lors des perfusions précédentes.
Informez immédiatement votre médecin ou infirmier/ère si vous remarquez l’un des signes suivants :
· éruption cutanée, urticaire, démangeaisons ;
· difficultés pour avaler ;
· gonflement du visage ou de la bouche ;
· bouffées congestives ;
· difficultés pour respirer (dyspnée) ;
· chute de la tension artérielle ;
· modification du rythme cardiaque ;
· frissons.
Votre médecin ou le professionnel de santé décidera de ralentir ou d’arrêter la perfusion et d’instaurer un traitement approprié si nécessaire.
En cas de traitement à domicile, arrêtez immédiatement la perfusion et contactez votre médecin ou le professionnel de santé.
Les effets indésirables suivants ont été observés pendant le traitement par PROLASTIN.
Peu fréquents (peuvent affecter jusqu’à 1 patient sur 100) :
· frissons, fièvre, symptômes pseudo-grippaux, douleur à la poitrine ;
· urticaire ;
· vertiges, hébétude, maux de tête ;
· difficultés respiratoires (dyspnée) ;
· éruption cutanée ;
· nausées ;
· douleurs articulaires (arthralgies).
Rares (peuvent affecter jusqu’à 1 patient sur 1 000) :
· réactions d’hypersensibilité ;
· pouls rapide (tachycardie) ;
· diminution de la tension artérielle (hypotension) ;
· élévation de la tension artérielle (hypertension) ;
· maux de dos.
Très rares (peuvent affecter jusqu’à 1 patient sur 10 000) :
· choc allergique.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion ?
A conserver à une température ne dépassant pas 25 °C.
Ne pas congeler.
La solution reconstituée ne doit pas être conservée au réfrigérateur et doit toujours être utilisée dans les 3 heures suivant la préparation. Tout médicament non utilisé doit être éliminé.
Ne jetez aucun médicament au tout-à-l’égout ni avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette du flacon et sur la boîte. La date de péremption fait référence au dernier jour de ce mois.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient PROLASTIN 1000 mg poudre et solvant pour solution pour perfusion ?
· La substance active est l’alpha-1 antitrypsine humaine (provenant de sang ou de plasma humain).
· Les autres composants sont : chlorure de sodium, phosphate monosodique et eau pour préparations injectables (solvant/diluant).
L’alpha-1 antitrypsine est une poudre ou une masse friable de couleur blanche, jaune clair ou marron clair.
Après reconstitution avec l’eau pour préparation injectables, la solution doit être transparente à légèrement opalescente, incolore ou de couleur vert clair, jaune clair ou marron clair, et exempte de particules visibles.
1 mL de la solution reconstituée contient 25 mg d’alpha-1 antitrypsine.
Une boîte unitaire de PROLASTIN 1 000 mg, poudre et solvant pour solution pour perfusion contient :
· un flacon de poudre contenant 1 000 mg d’alpha-1 antitrypsine humaine ;
· un flacon contenant 40 mL de solvant (eau pour préparations injectables) ;
· un dispositif de transfert pour la reconstitution.
Les emballages multiples de PROLASTIN 1 000 mg contiennent :
· 4 boites (emballage unitaire) de PROLASTIN 1000 mg
Titulaire de l’autorisation de mise sur le marché
COLMARER STRASSE 22
60528 FRANCFORT
ALLEMAGNE
Exploitant de l’autorisation de mise sur le marché
24 RUE DE PRONY
75017 PARIS
POLIGNO LEVANTE
C/CAN GUASCH 2
08150 PARETS DEL VALLES, BARCELONE
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[À compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
------------------------------------------------------------------------------------------------------------------------
Informations destinées aux professionnels de la santé et aux patients aptes au traitement à domicile
Préparation de la solution reconstituée pour perfusion :
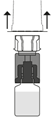
1. Utiliser une technique aseptique (matériel propre et stérilisé) afin de maintenir la stérilité. La reconstitution doit être effectuée sur une surface de travail plane.
2. S’assurer que les flacons de PROLASTIN et de solvant (eau stérile pour préparations injectables) de PROLASTIN sont à température ambiante (20 °C à 25 ºC).
3. Retirer l’opercule du flacon de PROLASTIN et du flacon de solvant et nettoyer les bords et les bouchons avec un coton imprégné d’alcool. Laisser les bouchons en caoutchouc sécher.
|
4. Ouvrir l’emballage du dispositif de transfert stérile en retirant complètement le film protecteur. Ne pas sortir le dispositif de l’emballage. |
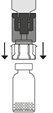
5. |
6. Retirer l’emballage extérieur |
|
7. |
8. |
9. |
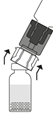 Grâce au
Grâce au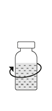
10. Si plusieurs flacons sont nécessaires pour obtenir la dose requise, répéter les étapes ci-dessus en utilisant le nouveau dispositif de transfert contenu dans chacun des flacons pour flacon neuf. Ne pas réutiliser un dispositif de transfert déjà utilisé.
La dissolution complète doit être obtenue en 5 minutes environ pour la présentation 1 000 mg.
N’utiliser que des solutions transparentes à légèrement opalescentes, incolores ou de couleur vert clair, jaune clair ou marron clair, et exemptes de particules visibles. PROLASTIN ne doit pas être mélangé avec d’autres solutions pour perfusion. La solution reconstituée doit toujours être utilisée dans les 3 heures suivant sa préparation.
La solution reconstituée doit être administrée en perfusion intraveineuse lente, en utilisant un set de perfusion approprié (non fourni). La vitesse de perfusion ne doit pas dépasser 0,08 mL/kg de poids corporel par minute (ce qui correspond à 6 mL pour un patient pesant 75 kg).