Dernière mise à jour le 01/06/2026
VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable
Indications thérapeutiques
VANCOMYCINE SANDOZ 500 mg poudre pour solution à diluer pour perfusion ou pour solution buvable est destinée à être utilisée en une solution pour perfusion ou une solution orale.
Pour usage intraveineux
La vancomycine en perfusion est utilisée dans toutes les tranches d’âges pour le traitement des infections sévères suivantes :
· infections de la peau et des tissus situés sous la peau,
· infections des os et des articulations,
· infection des poumons appelée « pneumonie »,
· infection de la membrane entourant l’intérieur du cœur (endocardite), et pour prévenir une endocardite chez des patients à risque lors d’opérations chirurgicales majeures,
· méningite,
· infection dans le sang pouvant être associée avec l’une des infections listées ci-dessus.
Pour usage intra-péritonéal
Chez les patients bénéficiant d’une dialyse péritonéale, la vancomycine est utilisée chez les adultes et les enfants pour le traitement d’infections liées à la dialyse péritonéale.
Pour usage oral
La vancomycine peut être utilisée par voie orale chez les adultes et les enfants pour le traitement des infections de la muqueuse de l’intestin grêle et du colon avec atteinte de la muqueuse (colite pseudo-membraneuse), causée par la bactérie Clostridium difficile.
Présentations
> 10 flacon(s) en verre de 13,5 ml
Code CIP : 566 133-8 ou 34009 566 133 8 0
Déclaration de commercialisation : 06/10/2005
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
Autres informations
- Titulaire de l'autorisation : SANDOZ
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 402 245 4
ANSM - Mis à jour le : 23/02/2026
VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 500 mg de chlorhydrate de vancomycine équivalant à 500 000 UI de vancomycine.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution à diluer pour perfusion ou pour solution buvable.
4.1. Indications thérapeutiques
Administration intraveineuse
La vancomycine est indiquée pour tous les groupes d'âges pour le traitement des infections suivantes (voir rubriques 4.2, 4.4 et 5.1) :
· infections compliquées de la peau et des tissus mous (ICPTM),
· infections des os et des articulations,
· pneumonies communautaires (PC),
· pneumonies nosocomiales (PN), y compris pneumonies acquises sous ventilation mécanique (PAVM),
· endocardites infectieuses,
· méningites bactériennes aiguës,
· bactériémies associées ou suspectées d’être associées à l’une des infections listées ci-dessus.
La vancomycine est également indiquée dans tous les groupes d'âges en prophylaxie péri-opératoire antibactérienne chez les patients présentant un risque élevé de développer une endocardite bactérienne lors d'interventions chirurgicales majeures.
Administration intra-péritonéale
La vancomycine est indiquée dans tous les groupes d'âges pour le traitement par voie intra-péritonéale de péritonite associée à une dialyse (voir rubriques 4.2, 4.4 et 5.1).
Administration orale
La vancomycine est indiquée dans tous les groupes d'âges pour le traitement des infections à Clostridium difficile (CDI) (voir rubriques 4.2, 4.4 et 5.1).
Il convient de tenir compte des recommandations officielles concernant l'utilisation appropriée des antibactériens.
4.2. Posologie et mode d'administration
Si nécessaire, la vancomycine doit être administrée en association à d'autres antibiotiques.
Administration intraveineuse
La dose initiale est à adapter au poids corporel total. Il est attendu que les adaptations des doses suivantes soient basées sur les concentrations sériques afin d'atteindre les concentrations thérapeutiques cibles. La fonction rénale doit être prise en considération pour les doses ultérieures et l'intervalle d'administration.
Patients âgés de 12 ans et plus
La dose recommandée est de 15 à 20 mg/kg de poids corporel toutes les 8 à 12 heures (ne pas dépasser 2 g par dose).
Chez les patients avec présentation péjorative de la maladie, une dose charge de 25– 30 mg/kg de poids corporel peut être envisagée afin d’obtenir rapidement les concentrations plasmatiques résiduelles cibles de vancomycine.
Nourrissons et enfants âgés entre un mois et moins de 12 ans :
La dose recommandée est de 10 à 15 mg/kg de poids corporel toutes les 6 heures (voir rubrique 4.4).
Nouveau-nés à terme (depuis la naissance jusqu'à 27 jours d'âge post-natal) et nouveau-nés prématurés (depuis la naissance jusqu'à 27 jours après la date de naissance attendue)
Pour établir le schéma d’administration chez les nouveau-nés, l'avis d'un médecin expérimenté dans la prise en charge des nouveau-nés devrait être requis. Des schémas d’administration de la vancomycine chez les nouveau-nés sont proposés dans le tableau suivant : (voir rubrique 4.4).
|
APM (semaines) |
Dose (mg/kg) |
Intervalle d'administration (h) |
|
< 29 |
15 |
24 |
|
29-35 |
15 |
12 |
|
> 35 |
15 |
8 |
APM : âge post-menstruel [(temps écoulé entre le premier jour des dernières menstruations et la naissance (âge gestationnel) plus le temps écoulé après la naissance (âge post-natal)].
Prophylaxie péri-opératoire contre l'endocardite bactérienne dans tous les groupes d'âges
La dose recommandée est une dose initiale de 15 mg/kg avant l'induction anesthésique. En fonction de la durée de l'intervention chirurgicale, une deuxième dose de vancomycine peut être nécessaire.
Durée du traitement
Des durées de traitement sont proposées dans le tableau ci-après. Dans tous les cas, la durée du traitement devrait être adaptée au type et à la sévérité de l'infection et à la réponse clinique du patient.
|
Indication |
Durée de traitement |
||||||
|
7 à 14 jours 4 à 6 semaines *
|
||||||
|
Infections des os et des articulations |
4 à 6 semaines ** |
||||||
|
Pneumonies communautaires |
7 à 14 jours |
||||||
|
Pneumonies nosocomiales, y compris pneumonies acquises sous ventilation mécanique |
7 à 14 jours |
||||||
|
Endocardites infectieuses |
4 à 6 semaines *** |
||||||
|
Méningites bactériennes aiguës |
10 à 21 jours |
||||||
* Continuer jusqu'à ce que le débridement ne soit plus nécessaire, que le patient soit amélioré au plan clinique et qu’il ne soit plus fébrile depuis 48 à 72 heures.
** Des traitements d’antibiothérapie suppressive par voie orale de plus longue durée seraient à considérer pour des infections sur prothèses articulaires.
*** La durée et la nécessité d’une association thérapeutique sont basées sur le type de valve et le type de bactérie.
Populations particulières
Personnes âgées
Des doses d’entretien plus basses pourraient être requises en raison de la diminution de la fonction rénale liée à l'âge.
Insuffisance rénale
Chez les patients adultes et pédiatriques avec insuffisance rénale, il convient de considérer d'administrer une dose initiale suivie d’une surveillance des taux plasmatiques résiduels de vancomycine, plutôt que de recourir à un schéma d’administration planifié, en particulier chez les patients avec insuffisance rénale sévère ou chez ceux qui bénéficient d’un traitement de substitution rénale (RRT), en raison de plusieurs facteurs variables qui peuvent affecter leurs taux de vancomycine.
Chez les patients avec insuffisance rénale légère ou modérée, la dose initiale ne doit pas être diminuée. Chez les patients avec insuffisance rénale sévère, il est préférable de prolonger l'intervalle d'administration plutôt que d'administrer des doses journalières plus faibles.
L'administration concomitante de médicaments qui pourraient réduire la clairance de la vancomycine et/ou potentialiser ses effets indésirables doit être prise en considération (voir rubrique 4.4).
La vancomycine est peu dialysable par hémodialyse intermittente. Cependant, l'utilisation de membranes à haute perméabilité et d’un traitement continu de substitution rénale (CRRT) augmente la clairance de la vancomycine et généralement demande une dose de remplacement (habituellement après la séance d'hémodialyse en cas d'hémodialyse intermittente).
Adultes
Les adaptations de dose chez les patients adultes peuvent être basées sur le débit de filtration glomérulaire estimé (eGFR) selon la formule suivante :
Hommes : [poids (kg) x 140 - âge (ans)]/ 72 x créatinine plasmatique (mg/dL).
Femmes : 0,85 x valeur calculée selon la formule ci-dessus.
La dose initiale habituelle pour les patients adultes est de 15 à 20 mg/kg pouvant être administrée toutes les 24 heures chez les patients avec une clairance de la créatinine entre 20 et 49 mL/min. Chez les patients avec insuffisance rénale sévère (clairance de la créatinine en dessous de 20 mL/min) ou chez ceux en traitement de substitution rénale, les temps d’administration et la quantité de vancomycine pour les doses ultérieures dépendent principalement de la modalité du RRT et il est attendu qu’ils soient basés sur les taux sériques résiduels de vancomycine et sur la fonction rénale résiduelle (voir rubrique 4.4).
Selon le contexte clinique, il peut être approprié d'attendre les résultats des taux de vancomycine avant d'administrer la dose suivante.
Chez les patients avec présentation péjorative de la maladie et en insuffisance rénale, il est attendu que la dose de charge initiale (25 à 30 mg/kg) ne soit pas diminuée.
Population pédiatrique
Les adaptations de dose chez les patients pédiatriques âgés d'1 an et plus, peuvent être basées sur le débit de filtration glomérulaire estimé (eGFR) selon la formule de Schwartz révisée :
eGFR (mL/min/1,73m2) = (taille cm x 0,413)/ créatinine plasmatique (mg/dL),
eGFR (mL/min/1,73m2) = (taille cm x 36,2/ créatinine plasmatique (μmol/L).
Pour les nouveau-nés et nourrissons âgés de moins de 1 an, l'avis d'un expert devrait être sollicité car la formule révisée de Schwartz ne peut pas leur être appliquée.
Des recommandations orientant sur les doses pour la population pédiatrique sont présentées dans le tableau ci-après en suivant les mêmes principes que pour les patients adultes.
|
GFR (mL/min/1.73 m2) |
Dose IV |
Fréquence |
|
50-30 |
15 mg/kg |
toutes les 12 heures |
|
29-10 |
15 mg/kg |
toutes les 24 heures |
|
< 10 |
10-15 mg/kg |
Nouvelle dose basée sur les taux* |
|
Hémodialyse intermittente |
||
|
Dialyse péritonéale |
||
|
Traitement continu de substitution rénale |
15 mg/kg |
Nouvelle dose basée sur les taux* |
* Les temps d’administration et la quantité de vancomycine pour les doses ultérieures dépendent principalement de la modalité du RRT et il est attendu qu’ils soient basés sur les taux sériques de vancomycine obtenus avant l'administration d'une nouvelle dose et sur la fonction rénale résiduelle. Selon le contexte clinique, il peut être approprié d'attendre les résultats des taux de vancomycine avant d’administrer la dose suivante.
Insuffisance hépatique
Il n'est pas nécessaire d'adapter la dose chez les patients insuffisants hépatiques.
Grossesse
Des doses significativement plus élevées peuvent être requises pour atteindre des concentrations sériques thérapeutiques chez les femmes enceintes (voir rubrique 4.6).
Patients obèses
Chez les patients obèses, la dose initiale devrait être adaptée individuellement selon le poids corporel total, comme chez les patients non-obèses.
Administration intra-péritonéale
Péritonite associée à la dialyse péritonéale
Adultes
Administration intermittente : la dose recommandée est de 15-30 mg/kg dans l’échange long, tous les 5-7 jours.
Perfusion continue : dose de charge de 30 mg/kg suivie d'une dose d’entretien de 1,5 mg/kg/poche dans tous les échanges.
Population pédiatrique
Administration intermittente : dose initiale de 30 mg/kg dans l’échange long, suivie de 15 mg/kg tous les 3-5 jours pendant l’échange long (l’administration de la deuxième dose devrait être basée sur les taux plasmatiques obtenus 2-4 jours après la dose initiale, voir rubrique 4.4).
Perfusion continue : dose de charge de 1000 mg/L par litre de dialysat, suivie de 25 mg/L (après 3-6 heures de la dose de charge) dans tous les échanges.
Des doses additionnelles pourraient être nécessaires chez les patients en dialyse péritonéale automatisée (APD) car les échanges rapides lors de l'APD peuvent conduire à un intervalle inadapté pour atteindre les taux thérapeutiques de vancomycine qui est administrée par voie intra-péritonéale intermittente.
Administration orale
Patients âgés de 12 ans et plus
Traitement des infections à Clostridium difficile (ICD) :
La dose de vancomycine recommandée est de 125 mg toutes les 6 heures pendant 10 jours pour un premier épisode d’ICD non sévère. Cette dose peut être augmentée à 500 mg toutes les 6 heures pendant 10 jours en cas d’ICD sévère ou compliquée. La dose maximale journalière ne devrait pas dépasser 2g.
Chez les patients avec récurrences multiples, l'épisode d’ICD en cours peut être traité avec la vancomycine, 125 mg 4 fois par jour pendant 10 jours, suivi soit d’une diminution progressive de la dose, par exemple en diminuant progressivement jusqu'à 125 mg par jour, ou soit suivi d’un traitement itératif, par exemple, 125-500 mg/jour tous les 2-3 jours pendant au moins 3 semaines.
Nouveau-nés, nourrissons et enfants âgés de moins de 12 ans
La dose de vancomycine recommandée est de 10 mg/kg par voie orale toutes les 6 heures pendant 10 jours. La dose journalière maximale ne devrait pas dépasser 2 g.
La durée du traitement par vancomycine peut nécessiter d’être adaptée à l'évolution clinique du patient. Dès que possible, l'antibiotique suspecté d’être à l’origine de l’ICD doit être arrêté. Un apport adéquat en liquides et électrolytes devra être effectué.
Surveillance des concentrations sériques de vancomycine
La fréquence du suivi thérapeutique des médicaments (Therapeutic Drug monitoring, TDM) doit être individualisée selon la situation clinique et la réponse au traitement, variant entre un prélèvement par jour qui pourrait être requis chez certains patients instables au plan hémodynamique à au moins une fois par semaine chez les patients stables répondant au traitement. Chez les patients à fonction rénale normale, il est attendu que la concentration sérique de vancomycine soit mesurée le deuxième jour de traitement, immédiatement avant la dose suivante.
Chez les patients en hémodialyse intermittente, les taux de vancomycine devraient être généralement obtenus avant le début de la séance d'hémodialyse.
Après administration orale, il est attendu que la surveillance des concentrations sériques de vancomycine soit effectuée chez les patients présentant des maladies inflammatoires intestinales (voir rubrique 4.4).
Il est attendu que les taux sanguins thérapeutiques résiduels de vancomycine soient de 10-20 mg/L, dépendant du site de l'infection et de la sensibilité du pathogène. Des taux résiduels de 15-20 mg/L sont généralement recommandés par les laboratoires cliniques pour mieux couvrir les pathogènes classés comme sensibles avec CMI ≥1 mg/L (voir rubriques 4.4 et 5.1).
Des méthodes basées sur des modèles peuvent être utiles dans la prédiction des doses individuelles requises pour obtenir une ASC appropriée. L'approche modélisée peut être utilisée pour calculer la dose initiale individuelle ainsi que pour les adaptations de doses basées sur les résultats du TDM (voir rubrique 5.1).
Mode d’administration
|
Le mode d’administration et les modalités de préparation de la solution diffèrent selon la voie d’administration. |
Administration intraveineuse
La vancomycine par voie intraveineuse est généralement administrée en perfusion intermittente et les recommandations de doses présentées dans cette rubrique correspondent à ce type d'administration.
La vancomycine doit être administrée uniquement en perfusion intraveineuse lente d'une durée d'au moins une heure ou à une vitesse maximale de 10 mg/min (choisir la modalité la plus longue) et suffisamment diluée (au moins 100 mL par 500 mg ou au moins 200 mL par 1000 mg) (voir rubrique 4.4).
Les patients nécessitant une restriction hydrique peuvent aussi recevoir une solution de 500 mg/50 mL ou 1000 mg/100 mL, malgré le risque accru de survenue d'effets indésirables liés à la perfusion avec ces concentrations plus élevées.
Pour des informations sur la préparation de la solution, voir la rubrique 6.6.
La perfusion continue de vancomycine peut être envisagée, par exemple, chez les patients ayant une clairance de vancomycine instable.
Administration intra-péritonéale
Il est attendu que les antibiotiques administrés par voie intra-péritonéale soient ajoutés au dialysat en utilisant une méthode stérile.
Administration orale
Une préparation spécifique de la solution est requise pour prendre le médicament par voie orale.
Une seringue graduée en mL est nécessaire à la préparation de la solution de vancomycine administrée par voie orale.
Le principe de cette préparation est le suivant : la solution buvable de vancomycine doit être reconstituée avec de l’eau potable puis peut être mélangée à de l’eau sucrée, du jus d’orange ou du sirop de fraise pour masquer le goût. La solution doit être administrée immédiatement.
Ne jamais injecter la solution reconstituée destinée à l’administration orale.
Pour des informations sur la préparation de la solution compte tenu des différents modes d’administration, voir la rubrique 6.6.
La vancomycine ne doit pas être administrée par voie intramusculaire en raison du risque de nécrose au niveau du site d'administration.
4.4. Mises en garde spéciales et précautions d'emploi
Des réactions d'hypersensibilité sévères et parfois d’issue fatale peuvent survenir (voir rubriques 4.3 et 4.8). En cas de réactions d'hypersensibilité, le traitement par vancomycine doit être arrêté immédiatement et les mesures d'urgence appropriées doivent être instaurées.
Chez les patients traités par vancomycine sur une longue période ou avec d'autres médicaments qui peuvent causer une neutropénie ou une agranulocytose, le taux des leucocytes doit être contrôlé à intervalles réguliers. Il est attendu que tous les patients traités par la vancomycine fassent l’objet périodiquement d’un bilan hématologique, d’analyses d'urines, de tests de la fonction rénale et hépatique.
La vancomycine doit être utilisée avec précaution chez les patients présentant des réactions allergiques à la teicoplanine, car des réactions d'hypersensibilité croisée, incluant des chocs anaphylactiques d’issue fatale, peuvent survenir.
Effets cardiovasculaires et cérébrovasculaires
Des cas de syndrome de Kounis ont été rapportés chez des patients traités par vancomycine. Le syndrome de Kounis a été défini comme des symptômes cardiovasculaires secondaires à une réaction allergique ou hypersensible associée à une constriction des artères coronaires pouvant entraîner un infarctus du myocarde.
Spectre d'activité antibactérienne
La vancomycine a un spectre d'activité antibactérienne limitée à des bactéries à Gram positif. L’utilisation de la vancomycine en monothérapie n'est pas adaptée au traitement de certains types d'infections sauf si le pathogène identifié est déjà connu comme étant sensible, ou s'il y a une forte suspicion que le(s) pathogène(s) le(s) plus suspecté(s) soi(en)t sensible(s) à la vancomycine.
L'utilisation appropriée de la vancomycine doit tenir compte du spectre d'activité antibactérienne, du profil de sécurité d’emploi et de la pertinence du traitement antibiotique avec la situation du patient.
Ototoxicité
L'ototoxicité, qui peut être transitoire ou permanente (voir rubrique 4.8) a été rapportée chez des patients avec antécédents de surdité, qui ont reçu des doses intraveineuses excessives, ou qui reçoivent un traitement concomitant avec une autre substance active ototoxique comme un aminoglycoside. La vancomycine doit être également évitée chez les patients avec antécédents de perte de l'audition. La surdité peut être précédée par des acouphènes. L'expérience avec d'autres antibiotiques suggère que la surdité puisse être évolutive malgré l'arrêt du traitement. Pour réduire le risque d'ototoxicité, les taux sanguins doivent être contrôlés régulièrement et un contrôle régulier de la fonction auditive est recommandé.
Les personnes âgées sont particulièrement exposées aux lésions auditives. Il est attendu que les fonctions auditive et vestibulaire soient surveillées pendant et après le traitement, et que l'usage concomitant ou séquentiel avec d'autres substances ototoxiques soit évité.
Réactions liées à la perfusion
L’administration rapide en bolus (par exemple en quelques minutes) peut être associée à une hypotension importante (y compris avec un choc et rarement un arrêt cardiaque), des réactions de type histaminique et une éruption maculo-papuleuse ou érythémateuse (« syndrome de l’homme rouge » ou « syndrome du cou rouge »). La vancomycine doit être perfusée lentement sous forme d’une solution diluée (2,5 à 5,0 mg/mL), à une vitesse ne dépassant pas 10 mg/min et sur une période d’au moins 60 minutes, afin d’éviter des réactions liées aux perfusions rapides. L’arrêt de la perfusion entraine généralement la disparition rapide de ces réactions.
La fréquence des réactions liées à la perfusion (hypotension, éruptions érythémateuses brusques, érythème, urticaire et prurit) augmente en cas d’administration concomitante d’anesthésiques (voir rubrique 4.5). Ces réactions peuvent être réduites en administrant la vancomycine en perfusion sur au moins 60 minutes, avant l’induction anesthésique.
Réactions cutanées sévères (SCAR)
Des réactions indésirables cutanées sévères (SCAR), y compris syndrome de Stevens-Johnson (SJS), nécrolyse épidermique toxique (NET), réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) et pustulose exanthématique aiguë généralisée (PEAG), qui peuvent engager le pronostic vital ou être mortelles, ont été rapportées en association avec un traitement avec la vancomycine (voir rubrique 4.8). La plupart de ces réactions sont survenues en quelques jours et jusqu'à huit semaines après le début du traitement par la vancomycine.
Au moment de la prescription, les patients doivent être informés des signes et des symptômes et doivent être étroitement surveillés en cas de réactions cutanées. Si des signes et symptômes évocateurs de ces réactions apparaissent, la vancomycine doit être arrêtée immédiatement et un traitement alternatif doit être envisagé. Si le patient a développé un SCAR avec l'utilisation de la vancomycine, le traitement par la vancomycine ne doit à aucun moment être redémarré.
Réactions liées au site d’administration
Une douleur et une thrombophlébite peuvent survenir chez de nombreux patients recevant de la vancomycine administrée par voie intraveineuse et elles sont parfois sévères. La fréquence et la sévérité de la thrombophlébite peuvent être réduites en administrant lentement le médicament sous forme de solution diluée (voir rubrique 4.2) et en variant régulièrement les sites de perfusion.
L’efficacité et la sécurité de la vancomycine n’a pas été établie pour les voies d’administration intrathécale, intra-lombaire et intra-ventriculaire.
L’administration de vancomycine par injection intra-péritonéale pendant une dialyse péritonéale continue ambulatoire a été associée à un syndrome de péritonite chimique.
Néphrotoxicité
La vancomycine doit être utilisée avec précaution chez les patients insuffisants rénaux, incluant les patients anuriques, car le risque de développer des effets toxiques est beaucoup plus élevé en présence de concentrations sanguines élevées et prolongées. Le risque de toxicité est plus élevé en cas de fortes concentrations sanguines ou de traitement prolongé.
Un contrôle régulier des taux sanguins de vancomycine est indiqué en cas de traitement à doses élevées et d’utilisation prolongée, en particulier chez les patients présentant un dysfonctionnement rénal ou une altération de l’audition, ainsi qu’en cas d’administration concomitante de substances néphrotoxiques ou ototoxiques, respectivement (voir rubriques 4.2 et 4.5).
Affections oculaires
La vancomycine n'est pas autorisée pour une utilisation intracamérulaire ou intravitréenne, y compris en prophylaxie de l'endophtalmie.
Des vasculites rétiniennes occlusives hémorragiques (HORV), incluant une perte permanente de la vision, ont été observées dans des cas individuels à la suite de l'utilisation intracamérulaire ou intravitréenne de vancomycine pendant ou après une chirurgie de la cataracte.
Population pédiatrique
Les recommandations actuelles de doses pour l’administration intraveineuse dans la population pédiatrique, en particulier pour les enfants âgés de moins de 12 ans, peuvent conduire à des taux sub-thérapeutiques de vancomycine chez un nombre important d’enfants. Cependant, la sécurité de doses élevées de vancomycine n’a pas été complètement évaluée et des doses dépassant 60 mg/kg/jour ne peuvent généralement pas être recommandées.
La vancomycine doit être utilisée avec une prudence particulière chez les nouveau-nés prématurés et les jeunes nourrissons, à cause de leur immaturité rénale et de l’augmentation possible des concentrations sériques de vancomycine. Les concentrations sanguines de vancomycine doivent par conséquent faire l’objet d’un suivi minutieux chez ces enfants. L’administration concomitante de vancomycine et d’anesthésiques a été associée à un érythème et des bouffées congestives de type histaminique chez les enfants. De manière similaire, l’usage concomitant de médicaments néphrotoxiques comme les antibiotiques aminoglycosides, les AINS (par exemple ibuprofène pour la fermeture du canal artériel persistant) ou l’amphotéricine B est associé à un risque accru de néphrotoxicité (voir rubrique 4.5) et par conséquent un suivi plus fréquent des taux sériques de vancomycine et de la fonction rénale est indiqué.
Pour le traitement intra-péritonéal d’une péritonite associée à une dialyse péritonéale (PDP) chez les enfants avec fonction rénale résiduelle, l’administration intermittente est indiquée uniquement quand les taux sériques de vancomycine peuvent être contrôlés de manière appropriée.
Utilisation chez les personnes âgées
La diminution naturelle de la filtration glomérulaire liée à l’âge peut entrainer une augmentation des concentrations sériques de vancomycine si la posologie n’est pas adaptée (voir rubrique 4.2).
Interactions médicamenteuses avec les anesthésiques
La dépression myocardique induite par les anesthésiques peut être majorée par la vancomycine. Pendant l’anesthésie, les doses doivent être bien diluées et administrées lentement sous surveillance cardiaque étroite. Il convient de ne pas procéder à des changements de position jusqu’à la fin de la perfusion pour permettre ensuite une adaptation posturale (voir rubrique 4.5).
Entérocolite pseudo-membraneuse
En cas de diarrhée sévère persistante, la possibilité d’une entérocolite pseudo-membraneuse pouvant menacer le pronostic vital doit être prise en compte (voir rubrique 4.8). Les anti-diarrhéiques ne doivent pas être administrés.
Surinfection
L’utilisation prolongée de la vancomycine peut induire la prolifération d’organismes non sensibles. Les patients doivent être attentivement surveillés. Des mesures appropriées doivent être prises en cas de surinfection au cours du traitement.
Administration orale
L’administration intraveineuse de vancomycine n’est pas efficace pour le traitement des infections à Clostridium difficile. Pour cette indication, la vancomycine doit être administrée par voie orale.
La recherche de Clostridium difficile ou de sa toxine n’est pas recommandée chez l’enfant âgé de moins 1 an en raison du taux élevé de colonisation asymptomatique, sauf en cas de diarrhée sévère avec des facteurs de risque de stase telle que la maladie de Hirschsprung, une atrésie anale opérée ou d’autres troubles sévères de la motilité. D’autres étiologies devraient toujours être recherchées et l’entérocolite à Clostridium difficile doit être documentée.
Potentiel d'absorption systémique
L'absorption peut être augmentée chez les patients atteints de maladies inflammatoires de la muqueuse intestinale ou de colite pseudo-membraneuse due à Clostridium difficile. Ces patients sont à risque de présenter des effets indésirables, surtout en cas d'insuffisance rénale concomitante. Plus l'insuffisance rénale est sévère, plus grand est le risque que surviennent des effets indésirables comme ceux décrits avec l'administration parentérale de vancomycine. Il est attendu que la surveillance des concentrations plasmatiques de vancomycine soit effectuée chez les patients présentant des maladies inflammatoires de la muqueuse intestinale.
Néphrotoxicité
Il est attendu que la surveillance régulière de la fonction rénale soit effectuée chez les patients présentant un dysfonctionnement rénal ou chez les patients recevant en association un traitement avec un aminoglycoside ou avec un autre médicament néphrotoxique.
Ototoxicité
Il est attendu que la surveillance régulière de la fonction auditive soit effectuée pour minimiser le risque d'ototoxicité chez les patients atteints d'une perte de l'audition, ou qui reçoivent en association un traitement avec un médicament ototoxique tel qu'un aminoglycoside.
Interactions médicamenteuses avec les médicaments inhibant la motilité gastro-intestinale et les inhibiteurs de la pompe à protons
Les médicaments inhibant la motilité gastro-intestinale doivent être évités et l'utilisation des inhibiteurs de la pompe à protons devrait être réévaluée.
Développement des bactéries résistantes au médicament
La vancomycine orale augmente le risque de sélection d’Entérocoques résistants à la vancomycine dans le tractus gastro-intestinal. Par conséquent, un usage prudent de la vancomycine orale est recommandé.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Autres médicaments potentiellement néphrotoxiques ou ototoxiques
L’administration concomitante ou séquentielle de vancomycine et d’autres médicaments potentiellement ototoxiques ou néphrotoxiques peut accroître l’ototoxicité ou la néphrotoxicité (voir rubrique 4.4). Les médicaments néphrotoxiques peuvent être des produits de contraste iodés, des aminosides, des organoplatines, l’association d’antibactériens pipéracilline/tazobactam, du méthotrexate à doses élevées, et certains médicaments antirétroviraux comme la pentamidine, le foscarnet, l’aciclovir, le ganciclovir, le famciclovir, le valaciclovir, le valganciclovir, la ciclosporine, ou le tacrolimus. Les médicaments ototoxiques peuvent être des aminosides, des organoplatines, certains diurétiques. Une surveillance étroite du patient est nécessaire, particulièrement en cas d’administration concomitante d’aminosides. La dose maximale de vancomycine sera alors limitée à 500 mg toutes les 8 heures.
Anesthésiques
Il a été signalé que l’incidence des effets indésirables éventuels (par exemple hypotension, érubescence de la peau, érythème, urticaire, dépression myocardique ou prurit) augmentait lorsque la vancomycine était administrée conjointement à des anesthésiques. Pour éviter ces effets indésirables, la vancomycine doit être administrée au moins 60 minutes avant l’induction de l’anesthésie (voir rubrique 4.4).
Relaxants musculaires
Si le chlorhydrate de vancomycine est administré pendant ou immédiatement après l’intervention chirurgicale, les effets des relaxants musculaires conjointement administrés (notamment la succinylcholine), comme un blocage neuromusculaire, peuvent être renforcés ou prolongés.
Anticoagulants oraux
L’administration concomitante de vancomycine et de warfarine peut augmenter les effets de ces anticoagulants. De nombreux cas d’augmentation de l’activité des anticoagulants oraux ont été rapportés chez des patients recevant des antibiotiques. Le contexte infectieux ou inflammatoire marqué, l’âge et l’état général du patient apparaissent comme des facteurs de risque. Dans ces circonstances, il apparaît difficile de faire la part entre la pathologie infectieuse et son traitement dans la survenue du déséquilibre de l’INR. Il est recommandé de suivre l’INR fréquemment pendant et rapidement après l’administration concomitante de vancomycine avec des anticoagulants oraux.
4.6. Fertilité, grossesse et allaitement
Grossesse
Le bénéfice thérapeutique élevé de cette molécule justifie que son utilisation puisse être envisagée si besoin au cours de la grossesse, quel qu'en soit le terme. Les données animales n'ont pas mis en évidence d'effet tératogène, cependant les données cliniques sont encore insuffisantes. Compte tenu de l'ototoxicité de la vancomycine, une évaluation de la fonction auditive (oto émissions) du nouveau-né peut être réalisée en cas d'utilisation pendant la grossesse.
Compte tenu du très faible passage de la vancomycine dans le lait maternel et de sa faible absorption digestive, l’utilisation de la vancomycine administrée par voie injectable ou par voie orale est envisageable en cours d’allaitement si nécessaire.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Des réactions cutanées sévères (SCAR), y compris le syndrome de Stevens-Johnson (SJS), une nécrolyse épidermique toxique (NET), une réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) et une pustulose exanthématique aiguë généralisée (PEAG) ont été rapportées en association avec un traitement par la vancomycine (voir rubrique 4.4).
Les effets indésirables les plus fréquents sont la phlébite, les réactions pseudo-allergiques et une éruption érythémateuse brusque de la partie supérieure du corps (« syndrome du cou rouge ») en relation avec des perfusions intraveineuses de vancomycine trop rapides.
L'absorption de la vancomycine au niveau du tractus gastro-intestinal est négligeable. Cependant, en cas d'inflammation sévère de la muqueuse intestinale, surtout en présence d'une insuffisance rénale concomitante, des effets indésirables comme ceux décrits avec l'administration parentérale de vancomycine peuvent survenir.
Tableau des effets indésirables
Dans chaque groupe de fréquence, les effets indésirables sont présentés en ordre décroissant de gravité.
Les effets indésirables listés ci-après sont définis en utilisant la convention suivante du dictionnaire MedDRA et des classes de système d'organes :
Très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur base des données disponibles).
|
Classe de système d'organes |
|
|
Fréquence |
Effet indésirable |
|
Affections hématologiques et du système lymphatique |
|
|
Rare |
Neutropénie réversible1, agranulocytose, éosinophilie, thrombocytopénie, pancytopénie. |
|
Fréquence indéterminée |
Anémie hémolytique. |
|
Affections du système immunitaire |
|
|
Rare |
Réactions d'hypersensibilité, réactions anaphylactiques2. |
|
Affections de l'oreille et du labyrinthe |
|
|
Peu fréquent |
Perte transitoire ou permanente de l'audition4. |
|
Rare |
Vertiges, acouphènes3, étourdissements. |
|
Affections cardiaques |
|
|
Très rare |
Arrêt cardiaque. |
|
Fréquence indéterminée |
Syndrome de Kounis. |
|
Affections vasculaires |
|
|
Fréquent |
Baisse de la tension artérielle. |
|
Rare |
Vascularite. |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Fréquent |
Dyspnée, stridor. |
|
Affections gastro-intestinales |
|
|
Rare |
Nausées. |
|
Très rare |
Entérocolite pseudomembraneuse. |
|
Fréquence indéterminée |
Vomissements, diarrhée. |
|
Affections hépatobiliaires |
|
|
Fréquent |
Alanine aminotransférase augmentée, aspartate aminotransférase augmentée. |
|
Affections de la peau et du tissu sous-cutané |
|
|
Fréquent |
Eruption érythémateuse brusque de la partie supérieure du corps (« syndrome de l'homme rouge »), exanthème et inflammation des muqueuses, prurit, urticaire. |
|
Très rare |
Dermatite exfoliatrice, syndrome de Stevens-Johnson, nécrolyse épidermique toxique (NET), dermatose bulleuse à IgA linéaire. |
|
Fréquence indéterminée |
Eosinophilie et symptômes systémiques (syndrome DRESS (syndrome d’hypersensibilité médicamenteuse)). Pustulose Exanthématique Aiguë Généralisée (PEAG). |
|
Affections du rein et des voies urinaires |
|
|
Fréquent |
Insuffisance rénale se manifestant principalement par une augmentation des taux plasmatiques de la créatinine et de l'urée. |
|
Rare |
Néphrite interstitielle, insuffisance rénale aiguë. |
|
Fréquence indéterminée |
Nécrose tubulaire aiguë. |
|
Troubles généraux et anomalies du site d'administration |
|
|
Fréquent |
Phlébite, rougeur de la partie supérieure du corps et du visage. |
|
Rare |
Fièvre médicamenteuse, frissons, douleurs et spasmes musculaires des muscles pectoraux et dorsaux. |
Description des effets indésirables sélectionnés
1 Une neutropénie réversible débute habituellement une semaine ou plus après le début du traitement intraveineux ou après une dose totale de plus de 25 g.
2 Pendant ou peu de temps après une perfusion rapide, des réactions anaphylactiques/anaphylactoïdes y compris un wheezing peuvent survenir. Les réactions disparaissent à l’arrêt de l’administration, généralement entre 20 minutes et 2 heures. La vancomycine doit être perfusée lentement (voir rubriques 4.2 et 4.4). Une nécrose peut survenir après une injection intramusculaire.
3 Des acouphènes, qui peuvent précéder l'apparition d’une surdité, devraient être considérés comme une indication à arrêter le traitement.
4 L'ototoxicité a été principalement rapportée chez des patients ayant reçu des doses élevées, ou chez ceux recevant en association un traitement avec d'autres médicaments ototoxiques comme un aminoglycoside, ou chez ceux présentant une altération préexistante de la fonction rénale ou de l'audition.
Population pédiatrique
Le profil de sécurité est généralement cohérent entre les patients pédiatriques et les patients adultes. La néphrotoxicité a été décrite chez les enfants, généralement en association avec d’autres médicaments néphrotoxiques tels que les aminoglycosides.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Traitement symptomatique avec maintien de la filtration glomérulaire. La vancomycine est difficilement éliminée par dialyse. L'hémoperfusion sur résine Amberlite XAD-4 est d'efficacité limitée.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d'action
La vancomycine est un antibiotique glycopeptide tricyclique qui inhibe la synthèse de la paroi cellulaire des bactéries sensibles en se liant avec une forte affinité à la terminaison D-alanyl-D-alanine des précurseurs de la paroi cellulaire. Le médicament est bactéricide sur les micro-organismes en division. De surcroît, il bloque la perméabilité de la membrane de la cellule bactérienne et la synthèse d'ARN.
Relation Pharmacocinétique/ Pharmacodynamique
La vancomycine présente une activité non dépendante de la concentration, avec le rapport de l’aire sous la courbe (ASC) de concentration divisée par la concentration minimale inhibitrice (CMI) de la bactérie cible comme principal paramètre prédictif d’efficacité. Sur la base de données in vitro, de données chez l’animal et de données limitées chez l’homme, un rapport ASC/CMI de 400 a été établi comme étant la valeur cible PK/PD de l’efficacité clinique de la vancomycine. Pour atteindre cette cible lorsque les CMI sont ≥ 1,0 mg/L, un schéma posologique se situant dans la fourchette supérieure et des concentrations sériques résiduelles élevées (15-20 mg/L) sont nécessaires (voir rubrique 4.2).
Mécanisme de résistance
La résistance acquise aux glycopeptides est plus fréquente chez les entérocoques et est basée sur l'acquisition de plusieurs complexes de gènes van qui modifient la cible D-alanyl-D-alanine en D-alanyl-D-lactate ou D-alanyl-D-sérine qui se lie faiblement à la vancomycine. Dans certains pays, une augmentation des cas de résistance est observée en particulier chez les entérocoques ; des souches multi-résistantes d'Enterococcus faecium sont particulièrement préoccupantes.
Les gènes van ont été rarement retrouvés chez Staphylococcus aureus, où les modifications de la paroi cellulaire induisent une sensibilité « intermédiaire », qui est le plus souvent hétérogène. Aussi, des souches de Staphylococcus résistantes à la méticilline (SARM) avec sensibilité diminuée à la vancomycine ont été rapportées. La sensibilité diminuée ou la résistance à la vancomycine pour Staphylococcus n'est pas encore bien élucidée. Plusieurs éléments génétiques et de nombreuses mutations sont nécessaires.
Il n'y a pas de résistance croisée entre la vancomycine et les autres classes d'antibiotiques. Une résistance croisée avec d'autres antibiotiques glycopeptides, comme la teicoplanine, peut apparaitre. Le développement de résistance secondaire en cours de traitement est rare.
Synergie
L’association de la vancomycine à un antibiotique aminoglycoside a un effet synergique sur de nombreuses souches de Staphylococcus aureus, streptocoques de groupe D non entérocoques, entérocoques et streptocoques du groupe Viridans. L’association de la vancomycine à une céphalosporine a un effet synergique sur certaines souches de Staphylococcus epidermidis résistantes à l’oxacilline, et l’association de la vancomycine à la rifampicine a un effet synergique sur Staphylococcus epidermidis et un effet synergique partiel sur certaines souches de Staphylococcus aureus. Comme la vancomycine en association avec une céphalosporine pourrait également avoir un effet antagoniste contre certaines souches de Staphylococcus epidermidis et en association avec la rifampicine un effet antagoniste contre certaines souches de Staphylococcus aureus, il serait utile de réaliser préalablement un test de synergie.
Il devrait être procédé à des prélèvements pour une culture bactérienne afin d’isoler et d’identifier la bactérie causale et déterminer la sensibilité à la vancomycine.
Concentrations critiques
La vancomycine est active contre des bactéries à Gram positif, tels que staphylocoques, streptocoques, entérocoques, pneumocoques et clostridies. Les bactéries à Gram négatif sont résistantes.
La prévalence de la résistance acquise peut varier en fonction de la zone géographique et du temps pour certaines espèces. Il est donc utile de disposer d’'information sur la résistance locale, notamment pour le traitement d'infections sévères.
Si nécessaire, il est souhaitable de disposer d’un avis spécialisé principalement lorsque l’intérêt du médicament dans certaines infections peut être mis en cause du fait du niveau de prévalence de la résistance locale. Cette information donne une orientation sur la probabilité qu'un micro-organisme soit sensible à la vancomycine.
Les valeurs seuils des concentrations minimales inhibitrices établies par l’EUCAST (European Committee on Antimicrobial Susceptibility Testing) sont comme suit :
|
|
Sensible |
Résistant |
|
Staphylococcus aureus 1 |
≤ 2 mg/L |
> 2 mg/L |
|
Staphylocoques coagulase négative 1 |
≤ 4 mg/L |
> 4 mg/L |
|
Enterococcus spp. |
≤ 4 mg/L |
> 4 mg/L |
|
Streptocoques des groupes A, B, C et G |
≤ 2 mg/L |
> 2 mg/L |
|
Streptococcus pneumoniae |
≤ 2 mg/L |
> 2 mg/L |
|
Anaérobies à Gram positif |
≤ 2 mg/L |
> 2 mg/L |
1 Les S. aureus avec valeurs de CMI de 2 mg/L pour la vancomycine sont à la limite de la distribution du type sauvage et il pourrait y avoir une moindre réponse clinique.
|
|||||||||
|
|||||||||
|
|||||||||
|
L’émergence de la résistance à la vancomycine diffère d’un hôpital à l’autre et un laboratoire microbiologique local devrait être consulté pour obtenir des informations locales pertinentes. |
5.2. Propriétés pharmacocinétiques
La vancomycine est administrée par voie intraveineuse pour le traitement d’infections systémiques.
Chez les patients à fonction rénale normale, les concentrations plasmatiques moyennes après perfusion intraveineuse de plusieurs doses de 1 g de vancomycine (15 mg/kg) pendant 60 minutes, sont environ de 50-60 mg/L, 20-25 mg/L et 5-10 mg/L, immédiatement, 2 heures et 11 heures après la fin de la perfusion, respectivement. Les taux plasmatiques obtenus après plusieurs doses sont similaires à ceux atteints après une dose unique.
Si la vancomycine est administrée par voie intra-péritonéale lors d’une dialyse péritonéale, environ 30-65 % atteint le cycle systémique au cours des 6 premières heures. Après administration intra-péritonéale de 30 mg/kg, des taux plasmatiques d’environ 10 mg/L sont atteints.
La vancomycine n’est généralement pas absorbée dans le sang après administration orale. Cependant, une absorption peut se produire chez les patients présentant une colite (pseudo-membraneuse). Ceci peut conduire à l’accumulation de vancomycine chez les patients présentant également une insuffisance rénale.
Distribution
Le volume de distribution est environ de 60 L/1,73 m2de surface corporelle. A des concentrations sériques de vancomycine de 10 mg/L à 100 mg/L, la liaison de la vancomycine aux protéines plasmatiques est de l’ordre de 30-55 %, mesurée par ultrafiltration.
La vancomycine diffuse facilement au travers du placenta et est distribuée dans le sang ombilical. Dans le cas de méninges non inflammées, seules de petites quantités de vancomycine traversent la barrière hémato-encéphalique.
Biotransformation
Le médicament est faiblement métabolisé. Après administration parentérale, la vancomycine est excrétée par voie rénale par filtration glomérulaire presqu’entièrement sous forme de substance microbiologiquement active (approximativement 75-90 % dans les 24 heures).
Elimination
La demi-vie d’élimination de la vancomycine est de 4 à 6 heures chez les patients avec fonction rénale normale, et 2,2 à 3 heures chez les enfants. La clairance plasmatique est de l’ordre de 0,058 L/kg/h et la clairance rénale de l’ordre de 0,048 L/kg/h. Dans les premières 24 heures, approximativement 80 % d’une dose administrée de vancomycine est excrétée dans l’urine par filtration glomérulaire. Le dysfonctionnement rénal retarde l’élimination de la vancomycine. Chez les patients anéphriques, la demi-vie moyenne est de 7,5 jours. A cause de l’ototoxicité de la vancomycine, la surveillance des concentrations plasmatiques est indiquée pendant le traitement.
L’excrétion biliaire est peu significative (moins de 5 % d’une dose).
Même si la vancomycine n’est pas éliminée efficacement par hémodialyse ou dialyse péritonéale, des cas d’augmentation de la clairance de la vancomycine avec l’hémoperfusion et l’hémofiltration ont été rapportés.
Après administration orale, seulement une fraction de la dose administrée est retrouvée dans l’urine. En revanche, des concentrations élevées de vancomycine sont retrouvées dans les fèces (> 3100 mg/kg avec des doses de 2 g/jour).
Linéarité/non-linéarité
La concentration de vancomycine augmente en règle générale de manière proportionnelle à l’augmentation de la dose. Les concentrations plasmatiques obtenues après l’administration de plusieurs doses sont similaires à celles qui sont atteintes après administration d’une dose unique.
Caractéristiques dans des populations particulières
Insuffisance rénale
La vancomycine est principalement éliminée par filtration glomérulaire.
Chez les patients avec une défaillance de la fonction rénale, la demi-vie d’élimination terminale de la vancomycine est prolongée et la clairance corporelle totale est diminuée. En conséquence, il est attendu que la dose optimale soit calculée selon les recommandations posologiques mentionnées dans la rubrique 4.2. Posologie et mode d’administration.
Insuffisance hépatique
La pharmacocinétique de la vancomycine n’est pas modifiée chez les patients avec insuffisance hépatique.
Femmes enceintes
Des doses significativement plus élevées peuvent être requises pour atteindre des concentrations sériques thérapeutiques chez les femmes enceintes (voir rubrique 4.6).
Patients avec excès pondéral
La distribution de la vancomycine peut être modifiée chez les patients présentant un excès pondéral en raison de l’augmentation du volume de distribution, de la clairance rénale et des possibles changements dans la liaison aux protéines plasmatiques. Chez ces sous-populations, les concentrations sériques de vancomycine ont été plus élevées que celles attendues chez les adultes sains de sexe masculin (voir rubrique 4.2).
Population pédiatrique
La pharmacocinétique de la vancomycine a montré une large variabilité inter-individuelle chez les nouveau-nés prématurés et les nouveau-nés à terme. Chez les nouveau-nés, après administration intraveineuse, le volume de distribution de la vancomycine varie entre 0,38 et 0,97 L/kg, similaire aux valeurs de chez l’adulte, tandis que la clairance varie entre 0,63 et 1,4 mL/kg/min. La demi-vie varie entre 3,5 et 10 heures et est plus longue que chez l’adulte, ce qui reflète les valeurs habituellement plus basses chez le nouveau-né.
Chez les nourrissons et enfants plus âgés, le volume de distribution varie entre 0,26-1,05 L/kg tandis que la clairance varie entre 0,33-1,87 mL/kg/min.
5.3. Données de sécurité préclinique
Il n'a pas été mis en évidence d'effet génotoxique lors de la réalisation des tests standards.
Les études à long terme pour évaluer le potentiel carcinogène de la vancomycine n'ont pas été effectuées.
Mannitol, hydroxyde de sodium, acide chlorhydrique.
Traitement en association
En cas de traitement associant la vancomycine à d’autres antibiotiques/agents chimiothérapiques, les préparations doivent être administrées séparément.
La vancomycine n’est pas compatible avec les bêta-lactamines. Le risque de précipitation augmente avec l’augmentation des concentrations de vancomycine. Il est recommandé de rincer les tubulures de perfusion entre l’administration de vancomycine et ces antibiotiques. Il est également recommandé de diluer les solutions de vancomycine jusqu’à une concentration égale ou inférieure à 5 mg/mL (sauf cas particulier, voir rubrique 4.2).
L’administration de vancomycine par injection intravitréenne n’est pas autorisée. Une précipitation a été observée après une injection intravitréenne de vancomycine et de ceftazidime à l’aide de seringues et d’aiguilles séparées utilisées pour traiter une endophtalmie.
Le précipité dans le corps vitré s’est dissous complètement, mais lentement, sur une période de 2 mois au cours desquels l’acuité visuelle s’est également améliorée.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique 6.6.
Avant ouverture
Flacon de poudre : 18 mois.
Après ouverture
Solution pour perfusion après dilution : la stabilité physico-chimique a été démontrée avec du chlorure de sodium (NaCl 0,9 %) ou du glucose à 5 % pendant 24 heures à une température ne dépassant pas 25°C, ou 96 heures à une température comprise entre 2 et 8°C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation en cours d’utilisation ne devraient pas dépasser 24 heures à une température comprise entre 2 et 8°C, sauf en cas de reconstitution/dilution réalisées en conditions d’asepsie dument contrôlées et validées.
Solution buvable : la stabilité physico-chimique de la solution buvable reconstituée avec de l’eau potable est de 24 heures à une température ne dépassant pas 25°C.
6.4. Précautions particulières de conservation
Avant ouverture : à conserver à une température ne dépassant pas 25°C.
Après ouverture : pour les conditions de conservation du médicament après reconstitution/dilution, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
500 mg de poudre en flacon (verre type I) de 13,5 mL. Boîte de 1 ou 10.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les modalités de préparation de la solution de vancomycine diffèrent selon la voie d’administration (voir rubrique 4.2).
Administration intraveineuse
La poudre doit être reconstituée avec de l’eau pour préparations injectables et la solution ainsi obtenue doit ensuite être diluée avec du chlorure de sodium (NaCl 0,9 %) ou du glucose à 5 % avant d’être utilisée.
Avant leur administration, les solutions reconstituées et diluées doivent être inspectées visuellement afin de déceler la présence éventuelle de particules ou de décoloration. Les solutions ne doivent être utilisées que si elles sont limpides et exemptes de particules.
A- Préparation de la solution reconstituée
Dissoudre le contenu d'un flacon de 500 mg de vancomycine avec 10 mL d'eau pour préparations injectables.
NE JAMAIS INJECTER LA SOLUTION TELLE QUELLE MAIS LA DILUER COMME SUIT :
B- Préparation de la solution pour perfusion
La solution reconstituée doit être diluée avec du chlorure de sodium (NaCl 0,9 %) ou du glucose à 5 %.
Perfusion discontinue : la solution reconstituée contenant 500 mg de vancomycine doit être diluée avec au moins 100 mL de solvant.
La dose souhaitée doit être administrée en perfusion intraveineuse lente d’une durée d’au moins une heure, à une vitesse maximale de 10 mg/min et suffisamment diluée (au moins 100 mL par 500 mg ou au moins 200 mL par 1000 mg).
Les patients nécessitant une restriction hydrique peuvent aussi recevoir une solution de 500 mg/50 mL ou 1000 mg/100 mL, malgré le risque accru de survenue d’effets indésirables liés à la perfusion avec ces concentrations plus élevées.
Perfusion continue : la solution reconstituée préparée en fonction de la dose à administrer au patient doit être ajoutée à un volume suffisamment important de solvant, afin que la dose souhaitée puisse être administrée lentement sur une période de 24 heures.
Les flacons sont à usage unique pour la voie intraveineuse. Le produit non utilisé doit être éliminé.
Administration orale
A- Préparation de la solution reconstituée dans le flacon de vancomycine
1- Remplir un verre d’eau potable.
2- Prélever 5 mL d’eau potable à l’aide d’une seringue graduée en mL (seringue graduée jusqu’à 10 mL).
3- Ajouter les 5 mL d’eau potable dans le flacon de vancomycine puis agiter énergiquement.
4- Renouveler la manipulation en ajoutant de nouveau dans le flacon, 5 mL d’eau potable prélevés dans le verre à l’aide de la seringue graduée et agiter énergiquement jusqu’à l’obtention d’une dissolution complète de la poudre.
|
Le flacon contient ainsi 10 mL de solution buvable reconstituée correspondant à 500 mg de vancomycine (500 mg/10 mL). |
5- Jeter l’eau potable qui resterait dans le verre. Garder la seringue graduée pour les étapes suivantes.
B- Préparation de la solution pour l’administration orale
6- Selon la posologie de vancomycine prescrite, calculer le volume en mL de solution reconstituée à administrer pour une prise à l’aide du tableau de correspondance ci-dessous :
|
Dose (mg) à administrer par prise |
Volume de solution reconstituée à administrer par prise (mL) |
|
50 mg 100 mg 200 mg 250 mg 300 mg 350 mg 400 mg 450 mg 500 mg |
1 mL 2 mL 4 mL 5 mL 6 mL 7 mL 8 mL 9 mL 10 mL |
Ce volume correspond à la dose requise par prise.
Par exemple : chez l’enfant, la posologie recommandée est de 40 mg/kg/jour répartis en 4 prises journalières. Pour un enfant de 20 kg, la dose par prise calculée est de 40 mg x 20 kg / 4, soit 200 mg par prise. Le volume calculé de solution reconstituée à administrer par prise est donc de 200 mg x 10 mL/ 500 mg, soit 4 mL par prise.
7- Le volume ainsi calculé est à prélever dans le flacon de vancomycine contenant la solution reconstituée. Prélever ce volume à l’aide de la même seringue graduée puis transférer ce volume dans un verre ou un biberon.
8- Le goût de la solution reconstituée peut être masqué en y ajoutant de l’eau sucrée, du jus d’orange ou du sirop de fraise. Le volume d’eau sucrée, de jus de fruit ou de sirop ajouté devra être adapté à la quantité de liquide que le patient est capable d’avaler, en particulier pour une administration à un enfant.
9- Mélanger toujours la solution prête à être administrée avant la prise du médicament.
|
Une fois mélangée à l’eau sucrée, au jus d’orange ou au sirop de fraise, la solution buvable ne peut pas être conservée et doit être ingérée immédiatement. |
10- Après chaque utilisation, rincer la seringue graduée pour administration orale et la ranger dans la boite de VANCOMYCINE SANDOZ 500 mg.
|
Avertissement La solution reconstituée de 500 mg de vancomycine avec 10 mL d’eau potable peut être conservée maximum 24 heures dans le flacon à une température ne dépassant pas 25°C. Dans la limite d’une période de 24 heures et selon le volume nécessaire pour une prise, plusieurs prélèvements de la solution reconstituée peuvent être effectués dans un même flacon de 500 mg. Dans ce cas, il est impératif d’identifier ce flacon dans son lieu de conservation afin de s’assurer que son utilisation est réservée à l’usage oral. NE JAMAIS INJECTER LA SOLUTION RECONSTITUEE DESTINEE A L’ADMINISTRATION ORALE. |
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
49 AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 556 527 3 1 : 500 mg de poudre en flacon en verre de 13,5 mL ; boîte de 1.
· 34009 566 133 8 0 : 500 mg de poudre en flacon en verre de 13,5 mL ; boîte de 10.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 23/02/2026
VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable
Chlorhydrate de vancomycine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable ?
3. Comment utiliser VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable ET DANS QUELS CAS EST-IL UTILISE ?
VANCOMYCINE SANDOZ 500 mg poudre pour solution à diluer pour perfusion ou pour solution buvable est destinée à être utilisée en une solution pour perfusion ou une solution orale.
Pour usage intraveineux
La vancomycine en perfusion est utilisée dans toutes les tranches d’âges pour le traitement des infections sévères suivantes :
· infections de la peau et des tissus situés sous la peau,
· infections des os et des articulations,
· infection des poumons appelée « pneumonie »,
· infection de la membrane entourant l’intérieur du cœur (endocardite), et pour prévenir une endocardite chez des patients à risque lors d’opérations chirurgicales majeures,
· méningite,
· infection dans le sang pouvant être associée avec l’une des infections listées ci-dessus.
Pour usage intra-péritonéal
Chez les patients bénéficiant d’une dialyse péritonéale, la vancomycine est utilisée chez les adultes et les enfants pour le traitement d’infections liées à la dialyse péritonéale.
Pour usage oral
La vancomycine peut être utilisée par voie orale chez les adultes et les enfants pour le traitement des infections de la muqueuse de l’intestin grêle et du colon avec atteinte de la muqueuse (colite pseudo-membraneuse), causée par la bactérie Clostridium difficile.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable ?
· si vous êtes allergique à la vancomycine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Des signes de réaction allergique à ce médicament, y compris des problèmes respiratoires et des douleurs thoraciques, ont été signalés avec VANCOMYCINE SANDOZ. Arrêtez immédiatement VANCOMYCINE SANDOZ et contactez immédiatement votre médecin ou un service d’urgence médicale si vous remarquez l’un de ces signes.
Des effets indésirables graves pouvant entraîner une perte de vision ont été rapportés après l'injection de vancomycine dans les yeux.
Adressez-vous à votre médecin, pharmacien ou infirmier/ère avant d’utiliser VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable si :
· vous avez déjà présenté une réaction allergique à la teicoplanine, parce que cela pourrait signifier que vous êtes également allergique à la vancomycine,
· vous avez des troubles de l’audition, en particulier si vous êtes âgé (vous pouvez avoir des tests auditifs pendant le traitement),
· vous avez des problèmes au niveau du rein (vous aurez des examens de sang et d’urines pendant le traitement),
· vous recevez la vancomycine administrée en perfusion au lieu d’une administration par voie orale pour le traitement de diarrhée associée à une infection due à Clostridium difficile,
· vous avez déjà développé une éruption cutanée sévère ou une desquamation de la peau, des cloques et/ou des plaies dans la bouche après avoir pris de la vancomycine.
Adressez-vous à votre médecin ou pharmacien ou infirmier/ère pendant le traitement par la vancomycine si :
· vous recevez la vancomycine depuis longtemps (vous pouvez avoir des examens sanguins, des examens pour mesurer le fonctionnement du foie et des reins pendant le traitement),
· vous présentez une réaction cutanée pendant le traitement. Des réactions cutanées graves, y compris le syndrome de Stevens-Johnson, une nécrolyse épidermique toxique, une réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) et une pustulose exanthématique aiguë généralisée (PEAG) ont été rapportées en association avec un traitement à la vancomycine. Arrêtez d'utiliser la vancomycine et consultez immédiatement un médecin si vous remarquez l'un des symptômes décrits dans la rubrique 4,
· vous présentez une diarrhée sévère ou prolongée pendant ou après l’utilisation de vancomycine, consultez votre médecin immédiatement. Ceci pourrait être un signe d’inflammation des intestins (colite pseudo-membraneuse) qui peut survenir lors des traitements avec des antibiotiques.
Enfants
La vancomycine sera utilisée avec prudence chez les nouveau-nés prématurés et chez les jeunes nourrissons, car leurs reins ne sont pas complètement développés et ils pourraient accumuler la vancomycine dans le sang. Des examens sanguins peuvent être nécessaires chez ces enfants pour contrôler les niveaux de vancomycine dans le sang.
L’administration concomitante de vancomycine et de médicaments anesthésiques a été associée à des rougeurs de la peau (érythème) et des réactions allergiques chez les enfants. De manière similaire, l’utilisation concomitante de médicaments comme les antibiotiques aminoglycosides, des anti-inflammatoires non-stéroïdiens (AINS, par exemple ibuprofène) ou l’amphotéricine B (médicament pour traiter des infections à champignons) peuvent augmenter le risque d’atteinte du rein et par conséquent des examens sanguins et pour mesurer le fonctionnement des reins plus fréquents peuvent être nécessaires.
Autres médicaments et VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Une attention particulière est nécessaire si vous prenez / utilisez d'autres médicaments car certains pourraient interagir avec la vancomycine, par exemple :
Des médicaments potentiellement nocifs pour les reins (par exemple : des aminosides, des produits de contraste iodés, des produits de chimiothérapie à base de platine, l’association d’antibactériens pipéracilline/tazobactam, du méthotrexate à doses élevées et certains antirétroviraux comme la pentamidine, le foscarnet, l’aciclovir, le ganciclovir, le famciclovir, le valaciclovir, le valganciclovir, la ciclosporine, ou du tacrolimus). Si vous recevez simultanément de la vancomycine et d’autres médicaments potentiellement nocifs pour les reins, cet effet nocif peut être accru. En pareil cas, un contrôle minutieux et régulier des reins est nécessaire.
Des médicaments potentiellement nocifs pour l’audition (par exemple : aminosides, les produits de chimiothérapie à base de platine, certains diurétiques). Si vous recevez simultanément ces médicaments et de la vancomycine, cet effet nocif peut être accru. Dans ce cas, un contrôle minutieux et régulier de l’audition est nécessaire.
Anesthésiques : l’utilisation d’anesthésiques augmente le risque de développer certains effets indésirables de la vancomycine, comme une chute de la tension artérielle, des rougeurs de la peau, une urticaire, une diminution de la fonction cardiaque et des démangeaisons.
Relaxants musculaires : si vous prenez simultanément des relaxants musculaires (par exemple : succinylcholine), leur effet myorelaxant peut être intensifié ou prolongé.
Anticoagulants oraux : si vous prenez simultanément de la warfarine, l’effet de cet anticoagulant peut être augmenté.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Durant la grossesse l'utilisation de ce médicament peut être envisagée si besoin. Si vous découvrez que vous êtes enceinte pendant le traitement, consultez votre médecin, car lui seul peut juger de la nécessité de la poursuivre.
L'administration de ce médicament est envisageable en cours d’allaitement si nécessaire.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
La vancomycine n’a aucun effet ou qu’un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable contient du sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par flacon, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable ?
Posologie
La dose que vous recevrez dépend de :
· votre âge,
· votre poids,
· l’infection que vous présentez,
· l’état de fonctionnement de vos reins,
· votre capacité à entendre,
· tout autre médicament que vous prenez.
Administration intraveineuse
Adultes et adolescents (à partir de l’âge de 12 ans et plus)
La dose est calculée en fonction de votre poids. La dose habituelle de vancomycine pour perfusion est de 15 à 20 mg par kilogramme de poids. Elle est habituellement administrée toutes les 8 à 12 heures. Dans certains cas, votre médecin peut décider d’administrer une dose initiale jusqu’à 30 mg par kilogramme de poids. La dose maximale ne devrait pas dépasser 2 g par dose.
Enfants
Enfants âgés de 1 mois à moins de 12 ans
La dose est calculée en fonction de votre poids. La dose habituelle de vancomycine pour perfusion est de 10 à 15 mg par kilogramme de poids. Elle est habituellement administrée toutes les 6 heures.
Nouveau-nés prématurés et nouveau-nés à terme (de 0 à 27 jours)
La dose sera calculée selon l’âge post-menstruel (temps écoulé depuis le premier jour des dernières règles et la naissance (âge gestationnel) plus le temps écoulé après la naissance (âge post-natal).
Les personnes âgées, les femmes enceintes et les patients présentant une défaillance des reins peuvent nécessiter une dose différente.
Administration intra-péritonéale
Adultes et enfants
Lors du traitement d’infections liées à la dialyse péritonéale, votre médecin décidera la dose exacte de vancomycine dont vous avez besoin.
Administration orale
Adultes et adolescents (de l’âge de 12 à 18 ans)
La dose recommandée est de 125 mg toutes les 6 heures. Dans certains cas, votre médecin peut décider l’administration d’une dose journalière plus élevée jusqu'à 500 mg toutes les 6 heures. La dose maximale journalière ne devrait pas dépasser 2 g.
Si vous avez présenté auparavant d'autres épisodes (d’infection de la muqueuse), il est probable que vous nécessitiez d'une dose et d'une durée de traitement différentes.
Enfants
Nouveau-nés, nourrissons et enfants âgés de moins de 12 ans
La dose recommandée est de 10 mg par chaque kg de poids corporel. Elle est habituellement administrée toutes les 6 heures. La dose maximale journalière ne devrait pas dépasser 2 g.
Mode d'administration
Administration par voie intraveineuse
Une administration par perfusion intraveineuse signifie que le médicament s’écoule d'un flacon ou d’une poche de perfusion par une tubulure dans vos vaisseaux sanguins et dans votre corps. Votre médecin ou infirmier/ère vont toujours administrer la vancomycine dans le sang et non dans le muscle.
L’administration de la vancomycine dans vos veines dure au moins 60 minutes.
Administration intra-péritonéale
Pour le traitement d’infections liées à la dialyse péritonéale, la vancomycine sera ajoutée au dialysat pendant l'échange long.
Administration par voie orale
Pour le traitement des infections de l’intestin (appelées colites pseudo-membraneuses), le médicament doit être administré comme une solution à usage oral (vous prendrez le médicament par la bouche).
Modalités de préparation de la solution
|
Les modalités de préparation de la solution de vancomycine diffèrent selon la voie d’administration. |
Administration intraveineuse
Voir la rubrique « Information destinée exclusivement aux professionnels de santé ».
Administration par voie orale
Une préparation spécifique de la solution est requise pour prendre le médicament par voie orale.
Une seringue graduée en mL est nécessaire à la préparation de la solution de vancomycine administrée par voie orale.
Les différentes étapes de préparation de la solution pour une administration orale sont décrites ci-dessous. Il convient de respecter scrupuleusement chaque étape.
Procéder de la manière suivante pour un flacon de poudre de 500 mg :
A- Préparation de la solution reconstituée dans le flacon de vancomycine
|
|
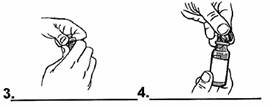
1. Retirer un flacon de VANCOMYCINE SANDOZ 500 mg de la boîte.
2. Retirer l’opercule rouge du flacon. |
|
|
3. Enlever la capsule en aluminium.
4. Retirer le bouchon en chlorobutyle pour ouvrir le flacon. |
|
|
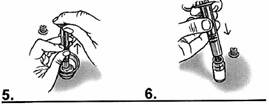
5. Remplir un verre d’eau potable. A l’aide de la seringue graduée, prélever 5 millilitres d’eau.
6. Ajouter l’eau prélevée dans le flacon de VANCOMYCINE SANDOZ 500 mg. |
|
|
7. Refermer le flacon avec le bouchon.
8. Prendre le flacon et agiter vigoureusement (pendant au moins 30 secondes) jusqu’à dissolution complète. |
|
|
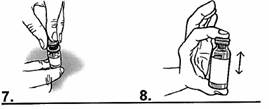
9. Renouveler la manipulation : à l’aide de la seringue graduée, prélever de nouveau dans le verre 5 millilitres d’eau potable.
10. Retirer le bouchon pour y introduire l’eau prélevée. |
|
|
11. Refermer le flacon avec le bouchon.
12. Agiter jusqu’à dissolution complète de la poudre. |


|
Le flacon contient ainsi 10 mL de solution buvable reconstituée correspondant à 500 mg de vancomycine (500 mg/10 mL). |
Jeter l’eau potable qui resterait dans le verre.
Garder la seringue graduée pour les étapes suivantes.
NE JAMAIS INJECTER LA SOLUTION RECONSTITUEE DESTINEE A L’ADMINISTRATION ORALE.
B- Préparation de la prise du médicament pour l’administration orale
En fonction de la dose de vancomycine prescrite, le volume en mL de solution reconstituée à administrer pour une prise est calculé à l’aide du tableau de correspondance ci-dessous :
|
Dose (mg) à administrer par prise |
Volume de la solution reconstituée à administrer par prise (mL) |
|
50 mg 100 mg 200 mg 250 mg 300 mg 350 mg 400 mg 450 mg 500 mg |
1 mL 2 mL 4 mL 5 mL 6 mL 7 mL 8 mL 9 mL 10 mL |
Le volume à administrer par prise est prélevé dans le flacon de vancomycine contenant la solution reconstituée, à l’aide de la même seringue graduée.
|
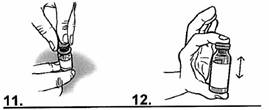
|
1. Retirer le bouchon pour ouvrir le flacon de vancomycine contenant la solution reconstituée. 2. A l’aide de la même seringue graduée, prélever le volume à administrer pour une prise dans le flacon. Par exemple : si la dose est de 200 mg pour une prise, prélever 4 mL de solution reconstituée dans le flacon. Le volume en mL se lit directement sur les graduations de la seringue graduée en mL. |
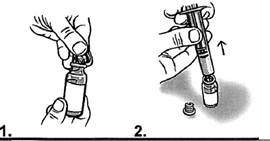
3- Puis, transférer le volume ainsi prélevé dans un verre ou un biberon.
4- Le goût de la solution reconstituée peut-être masqué en y ajoutant de l’eau sucrée, du jus d’orange ou du sirop de fraise. Le volume d’eau sucrée, de jus de fruit ou de sirop ajouté devra être adapté à la quantité de liquide que le patient est capable d’avaler, en particulier pour une administration à un enfant.
5- Mélanger toujours la solution prête à être administrée avant la prise du médicament.
|
Une fois mélangée à l’eau sucrée, au jus d’orange ou au sirop de fraise, la solution buvable ne peut pas être conservée. Par conséquent effectuer cette préparation juste avant la prise orale du médicament et PRENDRE LE MEDICAMENT IMMEDIATEMENT. |
6- Après chaque utilisation, rincer la seringue graduée pour administration orale et la ranger dans la boite de VANCOMYCINE SANDOZ 500 mg.
|
Avertissement La solution reconstituée de 500 mg de vancomycine avec 10 mL d’eau potable peut être conservée maximum 24 heures dans le flacon à une température ne dépassant pas 25°C. Dans la limite d’une période de 24 heures et selon le volume nécessaire pour une prise, plusieurs prélèvements de la solution reconstituée peuvent être effectués dans un même flacon de 500 mg. Dans ce cas, il est impératif d’identifier ce flacon dans son lieu de conservation afin de s’assurer que son utilisation est réservée à l’usage oral. |
Voir la rubrique « Information destinée exclusivement aux professionnels de santé ».
Durée du traitement
La durée du traitement dépend de l'infection que vous avez et peut durer quelques semaines.
La durée du traitement peut être différente en fonction de la réponse au traitement de chaque patient.
Pendant le traitement, vous pouvez avoir des examens sanguins, urinaires à partir d’un recueil d'urines que vous aurez donné, et peut-être des tests auditifs afin de détecter des signes d'effets indésirables éventuels.
Si vous avez utilisé plus de VANCOMYCINE SANDOZ 500 mg, poudre pour solution pour perfusion que vous n’auriez dû
Sans objet.
Si vous oubliez d’utiliser VANCOMYCINE SANDOZ 500 mg, poudre pour solution pour perfusion
Si vous arrêtez d’utiliser VANCOMYCINE SANDOZ 500 mg, poudre pour solution pour perfusion
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Arrêtez d'utiliser la vancomycine et consultez immédiatement un médecin si vous remarquez l'un des symptômes suivants :
· plaques rougeâtres non surélevées, en forme de cible ou circulaires sur le tronc, souvent accompagnées de cloques centrales, desquamation de la peau, ulcères de la bouche, de la gorge, du nez, des organes génitaux et des yeux. Ces éruptions cutanées graves peuvent être précédées de fièvre et de symptômes pseudo-grippaux (syndrome de Stevens-Johnson et nécrolyse épidermique toxique),
· éruption cutanée étendue, température corporelle élevée et ganglions lymphatiques hypertrophiés (syndrome DRESS ou syndrome d'hypersensibilité médicamenteuse),
· éruption cutanée rouge et squameuse avec des bosses sous la peau et des cloques accompagnée de fièvre au début du traitement (pustulose exanthématique aiguë généralisée),
· douleur thoracique pouvant être le signe d’une réaction allergique potentiellement grave appelée syndrome de Kounis.
La vancomycine peut provoquer des réactions allergiques, bien que les réactions allergiques graves (choc anaphylactique) soient rares. Prévenez immédiatement votre médecin si vous avez brusquement une respiration sifflante, des difficultés à respirer, une rougeur sur la partie supérieure du corps, une éruption ou des démangeaisons.
L'absorption de vancomycine au niveau de l’appareil digestif est négligeable. Cependant, si vous présentez une maladie inflammatoire de l’intestin, et en particulier si vous avez également des problèmes au niveau du rein, des effets indésirables comme ceux décrits avec l’administration de la vancomycine en perfusion, peuvent survenir.
Effets indésirables fréquents (peuvent affecter jusqu'à 1 personne sur 10) :
· augmentation des enzymes hépatiques,
· baisse de la tension artérielle,
· essoufflement, respiration bruyante (un son aigu résultant d'une obstruction des voies respiratoires supérieures),
· éruption et inflammation de la muqueuse de la bouche, démangeaisons, éruption accompagnée de démangeaisons, urticaire,
· problèmes rénaux qui peuvent être détectés principalement par des tests sanguins,
· rougeur de la partie supérieure du corps et du visage, inflammation d'une veine.
Effets indésirables peu fréquents (peuvent affecter jusqu'à 1 personne sur 100) :
· perte transitoire ou permanente de l'audition.
Effets indésirables rares (peuvent affecter jusqu'à 1 personne sur 1000) :
· baisse des globules blancs, globules rouges et plaquettes (cellules sanguines responsables de la coagulation sanguine),
· augmentation de certains globules blancs dans le sang,
· perte d'équilibre, bruits dans les oreilles, vertiges,
· inflammation des vaisseaux sanguins,
· nausées (envie de vomir),
· inflammation des reins et défaillance rénale,
· douleurs dans les muscles de la cage thoracique et du dos,
· fièvre, frissons.
Effets indésirables très rares (peuvent affecter jusqu'à 1 personne sur 10000) :
· apparition soudaine de réaction cutanée allergique sévère avec desquamation de la peau ou avec apparition de cloques. Ceci peut être associé avec une fièvre élevée et des douleurs articulaires,
· arrêt cardiaque,
· inflammation de l'intestin provoquant des douleurs abdominales et une diarrhée, pouvant contenir du sang.
Fréquence indéterminée (la fréquence ne peut pas être estimée sur base des données disponibles) :
· vomissements, diarrhée,
· confusion, somnolence, manque d'énergie, gonflement, rétention d’eau, diminution de la quantité d'urines émises,
· éruption avec gonflement ou douleur derrière les oreilles, dans le cou, dans l'aine, sous le menton et au niveau des aisselles (ganglions gonflés), résultats de laboratoire anormaux du sang et du fonctionnement du foie,
· éruption avec des cloques et de la fièvre,
· dégradation excessive des globules rouges entraînant une fatigue et une peau pâle (anémie hémolytique).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER VANCOMYCINE SANDOZ 500 mg, poudre pour solution à diluer pour perfusion ou pour solution buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon après {EXP}. La date de péremption fait référence au dernier jour de ce mois.
Avant ouverture
A conserver à une température ne dépassant pas 25°C.
Après ouverture
Solution pour perfusion après dilution : la stabilité physico-chimique a été démontrée avec du chlorure de sodium (NaCl 0,9 %) ou du glucose à 5 % pendant 24 heures à une température ne dépassant pas 25°C, ou 96 heures à une température comprise entre 2 et 8°C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation en cours d’utilisation ne devraient pas dépasser 24 heures à une température comprise entre 2 et 8°C, sauf en cas de reconstitution/dilution réalisées en conditions d’asepsie dument contrôlées et validées.
Solution buvable : la stabilité physico-chimique de la solution buvable reconstituée avec de l’eau potable est de 24 heures à une température ne dépassant pas 25°C.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Chaque flacon contient 500 mg de chlorhydrate de vancomycine équivalant à 500 000 UI de vancomycine.
· Les autres composants sont : mannitol, hydroxyde de sodium, acide chlorhydrique.
Ce médicament se présente sous forme de poudre pour solution à diluer pour perfusion ou pour solution buvable. Boîte de 1 ou 10 flacon(s).
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
49, AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
Exploitant de l’autorisation de mise sur le marché
9 PLACE MARIE-JEANNE BASSOT
92300 LEVALLOIS-PERRET
BIOLOGICI ITALIA LABORATORIES S.R.L.
VIA FILIPPO SERPERO, 2
20060 MASATE (MI)
ITALIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Incompatibilités
La valeur du pH de la vancomycine est faible. Ceci peut entraîner une instabilité chimique ou physique en cas de mélange avec d’autres substances. Par conséquent, chaque solution parentérale doit, avant utilisation, être inspectée visuellement afin de déceler la présence d’une précipitation ou d’une décoloration.
Traitement en association
En cas de traitement associant la vancomycine à d’autres antibiotiques/agents chimiothérapiques, les préparations doivent être administrées séparément.
La vancomycine n’est pas compatible avec les bêta-lactamines. Le risque de précipitation augmente avec l’augmentation des concentrations de vancomycine. Il est recommandé de rincer les tubulures de perfusion entre l’administration de vancomycine et ces antibiotiques. Il est également recommandé de diluer les solutions de vancomycine jusqu’à une concentration égale ou inférieure à 5 mg/mL (sauf cas particulier, voir rubrique 4.2 du résumé des caractéristiques du produit (RCP)).
Modalités de préparation
Les modalités de préparation de la solution de vancomycine diffèrent selon la voie d’administration (voir rubrique 4.2 du RCP).
Ø Administration intraveineuse
La poudre doit être reconstituée et la solution reconstituée ainsi obtenue doit ensuite être diluée avant d’être utilisée.
Avant leur administration, les solutions reconstituées et diluées doivent être inspectées visuellement afin de déceler la présence éventuelle de particules ou de décoloration. Les solutions ne doivent être utilisées que si elles sont limpides et exemptes de particules.
A- Préparation de la solution reconstituée
Dissoudre le contenu d'un flacon de 500 mg de vancomycine avec 10 mL d'eau pour préparations injectables.
NE JAMAIS INJECTER LA SOLUTION TELLE QUELLE MAIS LA DILUER COMME SUIT :
B- Préparation de la solution pour perfusion
La solution reconstituée doit ensuite être diluée avec du chlorure de sodium (NaCl 0,9 %) ou du glucose à 5 %.
Perfusion discontinue : la solution reconstituée contenant 500 mg de vancomycine doit être diluée avec au moins 100 mL de solvant.
La dose souhaitée doit être administrée en perfusion intraveineuse lente d’une durée d’au moins une heure, à une vitesse maximale de 10 mg/min et suffisamment diluée (au moins 100 mL par 500 mg ou au moins 200 mL par 1000 mg).
Les patients nécessitant une restriction hydrique peuvent aussi recevoir une solution de 500 mg/50 mL ou 1000 mg/100 mL, malgré le risque accru de survenue d’effets indésirables liés à la perfusion avec ces concentrations plus élevées.
Perfusion continue : la solution reconstituée préparée en fonction de la dose à administrer au patient doit être ajoutée à un volume suffisamment important de solvant, afin que la dose souhaitée puisse être administrée lentement sur une période de 24 heures.
Les flacons sont à usage unique pour la voie intraveineuse. Le produit non utilisé doit être éliminé.
Ø Administration orale
Voir la rubrique 3 de la notice.
Le flacon de poudre contient 500 mg de vancomycine. Après reconstitution, 10 mL de solution buvable reconstituée correspondent à 500 mg de vancomycine (500 mg/10 mL).
Le volume (mL) de solution reconstituée à administrer pour une prise est calculé en fonction de la dose de vancomycine prescrite. Ce volume est prélevé dans le flacon de vancomycine contenant la solution reconstituée à l’aide d’une seringue graduée en mL.
Le goût de la solution reconstituée peut être masqué en y ajoutant de l’eau sucrée, du jus d’orange ou du sirop de fraise. Une fois mélangée à l’eau sucrée, au jus d’orange ou au sirop de fraise, la solution buvable ne peut pas être conservée et doit être administrée immédiatement.
La solution reconstituée de 500 mg de vancomycine avec 10 mL d’eau potable peut être conservée maximum 24 heures dans le flacon à une température ne dépassant pas 25°C. Dans la limite d’une période de 24 heures et selon le volume nécessaire pour une prise, plusieurs prélèvements de la solution reconstituée peuvent être effectués dans un même flacon de 500 mg. Dans ce cas, il est impératif d’identifier ce flacon dans son lieu de conservation afin de s’assurer que son utilisation est réservée à l’usage oral.
Conseils d’éducation sanitaire
Les antibiotiques sont utilisés pour combattre les infections dues aux bactéries. Ils ne sont pas efficaces contre les infections dues aux virus.
Quand votre médecin choisit de vous prescrire un antibiotique c’est parce qu’il convient précisément à votre maladie actuelle. Malgré l’action d’un antibiotique, certaines bactéries ont la capacité de survivre et de se reproduire. Ce phénomène est appelé résistance : il rend certains traitements antibiotiques inactifs.
La résistance s’accroit par l’usage inapproprié des antibiotiques. Vous risquez de favoriser l’apparition de bactéries résistantes et donc de retarder votre guérison ou rendre inactif l'antibiotique, si vous ne respectez pas de manière appropriée :
· la dose à prendre,
· les moments de prise,
· la durée du traitement.
Par conséquent, pour préserver l'efficacité de ce médicament :
1 – N’utilisez un antibiotique que lorsqu’un médecin vous l’a prescrit.
2 – Respectez strictement votre ordonnance.
3 – Ne réutilisez pas un antibiotique sans prescription médicale, même si vous pensez combattre une maladie apparemment semblable.