Dernière mise à jour le 03/08/2026
FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (S.C.) en seringue pré-remplie
Indications thérapeutiques
ANTI-THROMBOTIQUES – Code ATC : B01AB06.
Ce médicament est un anticoagulant de la famille des héparines dites de « bas poids moléculaire ». Il vous est prescrit en traitement curatif, dans le cas d’une thrombose déjà existante.
Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien.
Présentations
> 10 seringue(s) préremplie(s) en verre de 1 ml avec système de sécurité
Code CIP : 347 336-0 ou 34009 347 336 0 4
Déclaration de commercialisation : 01/09/1998
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 129,50 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 130,52 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 22/06/2016 | Renouvellement d'inscription (CT) | Le service médical rendu par les spécialités FRAXODI et FRAXIPARINE reste important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 05/04/2023
FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (S.C.) en seringue pré-remplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Nadroparine calcique................................................................................................. 19 000 U.I. Axa
Pour une seringue pré-remplie.
1 ml de solution injectable contient 19 000 UI Axa de nadroparine calcique.
Excipient à effet notoire : latex (composant du protège aiguille)
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable.
4.1. Indications thérapeutiques
Cette héparine est une héparine de bas poids moléculaire (HBPM).
Son indication est la suivante:
· traitement curatif des thromboses veineuses profondes constituées.
4.2. Posologie et mode d'administration
Posologie
VOlE SOUS-CUTANEE.
Cette présentation est adaptée à l'adulte.
Ne pas injecter par voie I.M.
1 ml de FRAXODI correspond environ à 19 000 UI anti-Xa de nadroparine.
Mode d’administration
Technique de l'injection sous-cutanée
Ne pas purger la bulle d'air.
L'injection sous-cutanée de la nadroparine doit être réalisée de préférence chez le patient en décubitus, dans le tissu cellulaire sous-cutané de la ceinture abdominale antérolatérale et postérolatérale, alternativement du côté droit et du côté gauche.
L'aiguille doit être introduite perpendiculairement et non tangentiellement, sur toute sa longueur, dans l'épaisseur d'un pli cutané réalisé entre le pouce et l'index de l’opérateur. Ce pli cutané doit être maintenu pendant toute la durée de l’injection.
Recommandations générales
La surveillance régulière de la numération plaquettaire est impérative pendant toute la durée du traitement en raison du risque de thrombopénie induite par l'héparine (TIH) (voir rubrique 4.4).
Pour les techniques de rachianesthésie et d’anesthésie péridurale, l’intérêt de l’injection préopératoire doit être évalué en raison du risque théorique accru d’hématome intrarachidien (voir rubrique 4.4).
Des recommandations spécifiques relatives au délai d’injection de la nadroparine encadrant une rachianesthésie, une anesthésie péridurale ou une ponction lombaire doivent être respectées (voir rubrique 4.4).
· Traitement curatif des thromboses veineuses profondes (TVP)
Toute suspicion de thrombose veineuse profonde doit être confirmée rapidement par des examens adaptés.
Fréquence d'administration
1 injection par jour.
Dose administrée
La dose par injection est de 171 UI anti-Xa/kg.
La posologie des HBPM n'a pas été évaluée en fonction du poids corporel chez les patients d'un poids supérieur à 100 kg ou inférieur à 40 kg. II peut apparaître une moindre efficacité des HBPM pour les patients de poids supérieurs à 100 kg, ou un risque hémorragique accru pour les patients de poids inférieur à 40 kg. Une surveillance clinique particulière s'impose.
A titre indicatif, les posologies à administrer en fonction du poids des patients sont de 0,1 ml/10 kg 1 fois par jour, comme indiqué dans le tableau ci-dessous :
|
Poids corporel |
Volume de FRAXODI par injection |
|
40-49 kg 50-59.kg 60-69 kg 70-79 kg 80-89 kg 90-99 kg ≥100 kg |
0,4 ml 0,5 ml 0,6 ml 0,7 ml 0,8 ml 0,9 ml 1,0 ml |
Lorsque la posologie est adaptée au poids des patients, ajuster, si nécessaire, le volume à administrer en amenant le piston à la graduation désirée en tenant la seringue verticalement.
Durée de traitement des TVP
Le traitement par HBPM doit être relayé rapidement par les anticoagulants oraux, sauf contre-indication. La durée du traitement par HBPM ne doit pas excéder 10 jours, délai d'équilibration par les AVK inclus, sauf en cas de difficultés d'équilibration (voir rubrique 4.4 Précautions d'emploi : surveillance biologique). Le traitement anticoagulant oral doit donc être débuté le plus tôt possible.
Populations particulières
Insuffisance rénale
Traitement curatif de la maladie thromboembolique
Une réduction de dose n'est pas nécessaire chez les patients ayant une insuffisance rénale légère (clairance de la créatinine ≥ 50 ml/min).
Une insuffisance rénale modérée ou sévère entraine une augmentation de l’exposition à la nadroparine. Ces patients ont un risque majoré d'évènements thromboemboliques et de saignements.
Si le prescripteur estime qu'une réduction de dose est appropriée, en considérant les facteurs de risque individuels de saignements et d'évènements thromboemboliques chez les patients ayant une insuffisance rénale modérée (clairance de la créatinine ≥ 30 ml/min et < 50 ml/min), la dose doit être réduite de 25 à 33% (voir rubriques 4.4 et 5.2).
La nadroparine est contre-indiquée chez des patients ayant une insuffisance rénale sévère (voir rubriques 4.4 et 5.2).
lnsuffisance hépatique
Aucune étude clinique n'a été conduite chez les patients ayant une insuffisance hépatique.
La nadroparine est contre-indiquée dans les situations suivantes :
· hypersensibilité à la nadroparine, à l'héparine ou à ses dérivés, y compris les autres héparines de bas poids moléculaire, ou à l'un des excipients mentionnés à la rubrique 6.1 ;
· antécédents de thrombopénie induite par l'héparine (ou TIH) grave de type II induite sous héparine non fractionnée ou sous héparine de bas poids moléculaire ou toute autre antécédent de thrombopénie avec la nadroparine (voir rubrique 4.4 Précautions d'emploi);
· manifestations ou tendances hémorragiques liées à des troubles de l'hémostase (les coagulations intravasculaires disséminées peuvent être une exception à cette règle lorsqu'elles ne sont pas liées à un traitement par l'héparine - voir rubrique 4.4 Précautions d'emploi);
· lésion organique susceptible de saigner (telle qu'un ulcère gastroduodénal évolutif) ;
· accident vasculaire cérébral hémorragique,
· en l'absence de données, en cas d'insuffisance rénale sévère (définie par une clairance de la créatinine < 30 ml/min selon l'estimation de la formule de Cockroft), à doses curatives dans le traitement des thromboses veineuses profondes, des évènements thromboemboliques, de l'angor instable et de l'infarctus du myocarde sans onde Q en dehors de l'indication au cours de l'hémodialyse.
· de plus, une anesthésie péridurale, ou une rachianesthésie ne doivent jamais être effectuées lors d'un traitement curatif par HBPM,
· les formes multidoses contiennent de l'alcool benzylique, et ne devraient donc pas être utilisées chez les enfants âgés de moins de 3 ans,
· endocardite infectieuse aiguë (en dehors de certaines cardiopathies emboligènes).
A dose curative, ce médicament est GÉNÉRALEMENT DECONSEILLE dans les cas suivants :
· accident vasculaire cérébral ischémique étendu à la phase aiguë, avec ou sans troubles de la conscience. Lorsque l'accident vasculaire cérébral est d'origine embolique, le délai à respecter est de 72 heures. La preuve de l'efficacité des HBPM à dose curative n'a cependant pas été établie à ce jour, quelles que soient la cause, l'étendue et la sévérité clinique de l'infarctus cérébral ;
· l'utilisation de la nadroparine est généralement déconseillée en cas d’insuffisance rénale légère à modérée. Cependant, si son utilisation est jugée nécessaire dans ces situations, il convient de :
o si le prescripteur estime qu'une réduction de dose est appropriée, en considérant les facteurs de risque individuels de saignements et d'évènements thromboemboliques chez les patients ayant une insuffisance rénale modérée (clairance de la créatinine >30 et <50 ml/min), la dose doit être réduite de 25 à 33% (voir rubriques 4.2, 4.4 et 5.2),
o la réduction de dose n'est pas nécessaire en cas d'insuffisance rénale légère (clairance de la créatinine supérieure ou égale à 50 ml/mn) (voir rubrique 4.2).
De plus, ce médicament à doses curatives est GÉNÉRALEMENT DECONSEILLE, chez tous les sujets quel que soit l'âge, en association avec (voir rubrique 4.5) :
· l'acide acétylsalicylique aux doses antalgiques, anti-pyrétiques et anti-inflammatoires,
· les AlNS (voie générale),
· le dextran 40 (voie parentérale).
4.4. Mises en garde spéciales et précautions d'emploi
Bien que les différentes spécialités d'héparines de bas poids moléculaire aient toutes des concentrations exprimées en unités internationales anti-Xa, leur efficacité ne se limite pas qu'à cette activité anti-Xa. II serait dangereux de substituer le schéma posologique d'une HBPM par celui d'une autre HBPM ou par celui d'un autre polysaccharide de synthèse, chaque schéma ayant été validé par des études cliniques spécifiques. II y a donc lieu d'être particulièrement vigilant et de respecter le mode d'emploi spécifique de chacune des spécialités.
Mises en garde spéciales
Risque hémorragique
II est impératif de respecter les schémas thérapeutiques recommandés (posologies et durées de traitement). Dans le cas contraire, des accidents hémorragiques peuvent s'observer, surtout chez les sujets à risque (sujets âgés, insuffisants rénaux ).
Les accidents hémorragiques graves ont notamment été observés :
· chez le sujet âgé, notamment du fait de la détérioration de la fonction rénale liée à l'âge,
· en cas d'insuffisance rénale,
· en cas de poids inférieur à 40 kg,
· en cas de traitement prolongé au-delà de la durée moyenne préconisée de 10 jours,
· en cas de non-respect des modalités thérapeutiques conseillées (notamment durées de traitement et adaptation de la dose en fonction du poids pour les traitements curatifs),
· en cas d'association à des médicaments majorant le risque hémorragique (voir rubrique 4.5).
Dans tous les cas, une surveillance particulière est indispensable chez les patients âgés et/ou insuffisants rénaux, ainsi qu'en cas de traitement prolongé au-delà de 10 jours.
Pour détecter une accumulation, une mesure de l'activité anti-Xa peut-être utile dans certains cas (voir Précautions d'emploi/Surveillance biologique).
Risque de thrombopénie induite par l'héparine (TIH)
La surveillance régulière de la numération plaquettaire est impérative pendant toute la durée du traitement en raison du risque de thrombopénie induite par l’héparine (TIH).
De rares cas de TIH, parfois sévères, ont été rapportés, pouvant être associées à une thrombose artérielle ou veineuse.
Un tel diagnostic doit être envisagé dans les situations suivantes :
· une thrombopénie,
· une diminution significative du nombre de plaquettes (de 30 à 50% par rapport à la valeur de base) et/ou un nombre de plaquettes < 150.000/mm3 (ou 150 Giga/L),
· une thrombose survenant pendant le traitement (phlébite, embolie pulmonaire, ischémie aigue des membres inférieurs, voire un infarctus du myocarde ou un accident vasculaire cérébral ischémique),
· une aggravation de la thrombose initiale pendant le traitement,
· une coagulation intravasculaire disséminée.
Dans ce cas, il faut systématiquement penser à une thrombopénie induite par l'héparine (TIH) et faire pratiquer en urgence une numération des plaquettes (voir Précautions d'emploi). Le traitement par nadroparine doit être arrêté.
Population pédiatrique
En l'absence de données, l'utilisation des HBPM chez l'enfant n'est pas recommandée.
Précautions d'emploi
Fonction rénale
La nadroparine est principalement éliminée par le rein, ce qui se traduit par une augmentation de l’imprégnation en nadroparine chez les patients ayant une insuffisance rénale (voir rubrique 5.2 – insuffisance rénale). Les patients présentant une insuffisance rénale ont un risque augmenté de saignements et doivent être traités avec précaution.
Chez les patients ayant une clairance de la créatinine comprise entre 30 et 50 ml/mn, la décision du prescripteur de réduire la dose de nadroparine doit reposer sur l'évaluation par le prescripteur du risque individuel de saignement par rapport au risque d'évènement thromboembolique du patient (voir rubrique 4.2).
Avant d'instaurer un traitement par HBPM, il est indispensable d'évaluer la fonction rénale, et plus particulièrement chez le sujet âgé à partir de 75 ans, en calculant la clairance de la créatinine (Clcr) à l'aide de la formule de Cockroft, en disposant d'un poids récent du patient :
Chez l'homme, Clcr = (140-âge) x poids / (0,814 x créatininémie) avec l'âge exprimé en années, le poids en kg, la créatininémie en µmol/l.
Cette formule doit être corrigée pour les femmes en multipliant le résultat par 0,85. Lorsque la créatinine est exprimée en mg/ml, multiplier par un facteur 8,8.
La mise en évidence d'une insuffisance rénale sévère (Clcr de l'ordre de 30 ml/min) contre-indique la prescription d'HBPM dans les indications curatives (voir rubrique 4.3).
Surveillance biologique
Surveillance plaquettaire des patients sous HBPM et risque de Thrombopénie Induite par l’Héparine (ou TIH de type II)
Afin de pouvoir détecter les TIH de manière optimale, il est nécessaire de surveiller les patients de la manière suivante :
· Dans un contexte chirurgical ou traumatique récent (dans les 3 mois) : Une surveillance biologique systématique est nécessaire, chez tous les patients, compte tenu de l'incidence des TlH > 0,1%, voire >1 %, en chirurgie et en traumatologie. Elle consiste à pratiquer une numération plaquettaire :
o avant le traitement par HBPM ou au plus tard dans les 24 heures après l'instauration du traitement,
o puis 2 fois par semaine pendant un mois (période de risque maximal),
o puis une fois par semaine jusqu'à l'arrêt du traitement en cas de traitement prolongé.
· En dehors d’un contexte chirurgical ou traumatique récent (dans les 3 mois) : Une surveillance biologique systématique est nécessaire, selon les mêmes modalités qu'en chirurgie et en traumatologie (voir paragraphe ci-dessus) chez les patients:
o ayant des antécédents d'exposition à l'HNF ou aux HBPM dans les 6 derniers mois, compte tenu de l'incidence des TIH > 0,1%, voire >1%,
o atteints de comorbidités importantes, compte tenu de la gravité potentielle des TIH chez ces patients.
Dans les autres cas, compte tenu de l'incidence des TIH plus faible (< 0,1%), la surveillance de la numération plaquettaire peut être réduite à :
· une seule numération plaquettaire en début de traitement ou au plus tard dans les 24 heures après l'instauration du traitement,
· une numération plaquettaire en cas de manifestation clinique évocatrice de TIH (tout nouvel épisode thromboembolique artériel et/ou veineux, toute lésion cutanée douloureuse au site d'injection, toute manifestation allergique ou anaphylactoïde sous traitement). Le patient doit être informé de la possibilité de survenue de ces manifestations et de la nécessité de prévenir son médecin référent le cas échéant.
· Une TIH doit être suspectée devant un nombre de plaquettes < 150.000/m3 (ou 150 Giga/L) et/ou une chute relative des plaquettes de l’ordre de 50%, voire 30%, par rapport à la numération plaquettaire avant traitement.
Les effets d'une TIH sont probablement de nature immunoallergique, et apparaissent généralement entre le 5ème et le 21ème jour suivant l'instauration du traitement héparinique (avec un pic de fréquence aux environs du 10ème jour). Mais elle peut survenir beaucoup plus précocément, lorsque des antécédents de thrombopénie induite par l'héparine existent, et des cas isolés ont été rapportés au-delà de 21 jours.
En cas d'antécédent de thrombopénie (à l'exception d'une TIH de type II, voir rubrique 4.3) sous héparine (soit non fractionnée, soit de bas poids moléculaire), un traitement par nadroparine peut être envisagé si nécessaire. Dans ce cas, le traitement doit faire l'objet d'une surveillance clinique étroite et d'un contrôle de la numération plaquettaire au moins quotidiennement. Si une thrombopénie survient, le traitement doit être arrêté immédiatement.
En cas de survenue de thrombocytopénie sous héparine (standard ou héparine de bas poids moléculaire), la substitution par un autre agent anti-thrombotique peut être envisagée. Si cet autre agent anti-thrombotique n'est pas disponible, un relais par une autre héparine de bas poids moléculaire peut être envisagé si l'administration d'héparine est nécessaire. Dans ce cas, la surveillance de la numération plaquettaire doit être réalisée au moins quotidiennement et le traitement devra être arrêté dès que possible, considérant que des cas de thrombopénies initiales se poursuivant après substitution ont été rapportés (voir rubrique 4.3).
De tels antécédents seront donc systématiquement recherchés au cours d'un interrogatoire approfondi avant le début du traitement.
Dans tous les cas, l'apparition d'une TIH constitue une situation d'urgence et nécessite un avis spécialisé.
Toute baisse significative (30% à 50% de la valeur initiale) de la numération plaquettaire doit donner l'alerte, avant même que cette valeur n'atteigne un seuil critique. La constatation d'une diminution du nombre de plaquettes impose dans tous les cas :
1). un contrôle immédiat de la numération ;
2). la suspension du traitement héparinique, si la baisse est confirmée, voire accentuée, lors de ce contrôle, en l'absence d'une autre étiologie évidente.
Un prélèvement doit être réalisé sur tube citraté pour réaliser des tests d'agrégation plaquettaire in vitro et des tests immunologiques. Mais, dans ces conditions, la conduite à tenir immédiate ne repose pas sur le résultat de ces tests d'agrégation plaquettaire in vitro ou immunologiques, car seuls quelques laboratoires spécialisés les pratiquent en routine et le résultat n'est obtenu, dans le meilleur des cas, qu'au bout de plusieurs heures. Ces tests doivent cependant être réalisés pour aider au diagnostic de cette complication, car en cas de poursuite du traitement héparinique, le risque de thrombose est majeur.
3). la prévention ou le traitement des complications thrombotiques de la TIH.
Si la poursuite de l'anticoagulation semble indispensable, l'héparine doit être relayée par une autre classe d'antithrombotiques : danaparoïde sodique ou lepirudine, prescrits suivant les cas à dose préventive ou curative.
Le relais par les AVK ne sera pris qu'après normalisation de la numération plaquettaire, en raison du risque d'aggravation du phénomène thrombotique par les AVK.
Relais de l'héparine par les AVK
· Renforcer alors la surveillance clinique et biologique (temps de Quick exprimé en INR) pour contrôler l'effet des AVK.
· En raison du temps de latence précédant le plein effet de l'antivitamine K utilisé, l'héparine doit être maintenue à dose équivalente pendant toute la durée nécessaire pour que I'INR soit dans la zone thérapeutique souhaitable de l'indication lors de deux contrôles successifs.
Contrôle de l’activité anti-Xa
· La majorité des études cliniques qui ont démontré l'efficacité des HBPM ayant été conduites avec une dose adaptée au poids et sans surveillance biologique particulière, l'utilité d'une surveillance biologique n'a pas été établie pour apprécier l'efficacité d'un traitement par HBPM. Toutefois, la surveillance biologique par détermination de l'activité anti-Xa peut être utile pour gérer le risque hémorragique, dans certaines situations cliniques fréquemment associées à un risque de surdosage.
· Ces situations concernent essentiellement les indications curatives des HBPM, en raison des doses administrées, quand existe :
o une insuffisance rénale légère à modérée (clairance estimée selon la formule de Cockroft de l'ordre de 30 ml/min à 60 ml/min) : en effet, contrairement à l'héparine standard non fractionnée, les HBPM s'éliminent en grande partie par le rein et toute insuffisance rénale peut conduire à un surdosage relatif. L'insuffisance rénale sévère constitue quant à elle, une contre-indication à l'utilisation des HBPM aux doses curatives (voir rubrique 4.3);
o un poids extrême (maigreur voire cachexie, obésité) ;
o une hémorragie inexpliquée.
A l'inverse, la surveillance biologique n'est pas recommandée aux doses prophylactiques si le traitement par HBPM est conforme aux modalités thérapeutiques conseillées (en particulier pour la durée du traitement), ainsi qu'au cours de l'hémodialyse.
Afin de détecter une possible accumulation après plusieurs administrations, il est le cas échéant recommandé de prélever le sang du patient au pic maximal d'activité (selon les données disponibles), c'est à dire :
· environ 4 heures après la 3ème administration, lorsque le médicament est délivré en 2 injections SC par jour,
· environ 4 heures après la 2ème administration, lorsque le médicament est délivré en 1 injection SC par jour.
La répétition du dosage de l'activité anti-Xa pour mesurer l'héparinémie, par exemple tous les 2 à 3 jours, sera discutée au cas par cas, en fonction des résultats du dosage précédent, et une éventuelle modification de la dose d'HBPM sera envisagée.
Pour chaque HBPM et chaque schéma thérapeutique, l'activité anti-Xa générée est différente.
A titre indicatif, d'après les données disponibles, la moyenne observée (± écart- type) à la 4ème heure pour la nadroparine, délivrée :
· à la dose de 83 UI/kg par injection, en 2 injections par 24h, a été de 1,01±0,18 UI.
· à la dose de 166 UI/kg en 1 injection par 24h a été de 1,34±0,15 UI.
Ces valeurs moyennes ont été observées au cours des essais cliniques pour les dosages d'activité anti-Xa effectués par méthode chromogénique (amidolytique).
Temps de céphaline avec activateur (TCA)
Certaines HBPM allongent modérément le TCA. En l'absence de pertinence clinique établie, toute surveillance du traitement fondée sur ce test est inutile.
Réalisation d’une rachianesthésie/anesthésie péridurale ou d’une ponction lombaire en cas de traitement par HBPM
· Comme avec les autres anticoagulants, de rares cas d'hématomes intra-rachidiens entraînant une paralysie prolongée ou permanente ont été rapportés lors de l'administration d'HBPM au cours d'une rachianesthésie ou d'une anesthésie péridurale.
· Le risque d’hématome intra-rachidien ou épidural est augmenté par la présence de cathéters périduraux à demeure et en cas d’association à des traitements interférant avec l’hémostase comme les anti-inflammatoires non-stéroïdiens (AINS), les antiagrégants plaquettaires ou tout autre anticoagulant. Le risque semble également accru dans le cas d’une ponction lombaire traumatique ou répétée.
· Si un traitement préopératoire par HBPM est nécessaire (alitement prolongé, traumatisme) et que le bénéfice d'une anesthésie locorégionale rachidienne ou épidurale ou d'une ponction lombaire a été soigneusement évalué, un délai minimum de 24 heures pour les doses curatives doit être respecté entre la dernière injection de nadroparine et l'insertion ou le retrait du cathéter ou de l'aiguille utilisés pour l'anesthésie rachidienne ou épidurale, en tenant compte des caractéristiques du produit et du profil du patient.
· Pour les patients présentant une insuffisance rénale, des délais plus longs seront nécessaires.
· Dans la quasi-totalité des cas, le traitement prophylactique par HBPM pourra être débuté dans les 6 à 8 heures qui suivent la réalisation de la technique ou l'ablation du cathéter, sous couvert d'une surveillance neurologique.
· La réintroduction de la nadroparine devra être décalée jusqu'à ce que la procédure chirurgicale soit terminée.
· Les patients devront être suivis fréquemment pour rechercher des signes ou symptômes d'une atteinte neurologique, tels que des douleurs dorsales, une déficience sensorielle ou motrice (engourdissement et faiblesse des membres inférieurs), dysfonctionnement des intestins et/ou de la vessie. Si une atteinte neurologique est détectée, un traitement d'urgence est nécessaire.
· L'équipe médicale doit être formée à la détection de ces signes ou symptômes. Les patients doivent être sensibilisés à l'importance de prévenir immédiatement leur médecin s'ils ressentent l'un de ces signes ou symptômes.
· Si un hématome intrarachidien est suspecté, le diagnostic doit être posé en urgence et un traitement incluant une décompression de la moelle épinière doit être initié.
· Si un saignement significatif ou manifeste a été constaté lors de la pose du cathéter, une évaluation minutieuse du rapport bénéfice I risque devrait être faite avant d'initier ou de reprendre le traitement héparinique.
· Une attention particulière sera portée en cas d'association avec d'autres médicaments interférant avec l'hémostase (notamment anti-inflammatoires non stéroïdiens, aspirine).
Situations à risque
La surveillance du traitement sera renforcée dans les cas suivants en raison d’un risque accru de saignements:
· insuffisance hépatique,
· hypertension artérielle sévère
· antécédents d'ulcères digestifs ou de toute autre lésion organique susceptible de saigner,
· maladies vasculaires de la choriorétine,
· en période post-opératoire après chirurgie du cerveau et de la moelle épinière ou des yeux,
· Hyperkaliémie,
· la réalisation d'une ponction lombaire devra être discutée en tenant compte du risque de saignement intrarachidien. Elle devra être différée chaque fois que possible,
L'héparine peut freiner la sécrétion d'aldostérone et entraîner une hyperkaliémie.
Ceci a été observé particulièrement chez les patients ayant une kaliémie élevée et chez les patients à risque (diabétiques, insuffisants rénaux chroniques, acidose métabolique préexistante ou traitement par des médicaments susceptibles d'augmenter la kaliémie tels que les inhibiteurs de l’enzyme de conversion de l’angiotensine et les anti-inflammatoires non stéroïdiens).
Le risque d'hyperkaliémie augmente avec la durée du traitement et est habituellement réversible. En cas de traitement prolongé, une surveillance de la kaliémie peut être effectuée chez les patients à risque.
Traitements salicylés, anti-inflammatoires non-stéroïdiens et antiagrégants plaquettaires
Dans le traitement curatif de la maladie thromboembolique veineuse et en prévention de la coagulation durant l'hémodialyse, l'association à l'aspirine, aux autres traitements salicylés, aux anti- inflammatoires non-stéroïdiens et aux antiagrégants plaquettaires, n'est pas recommandée en raison du risque accru de saignement. Dans le cas où cette association ne peut être évitée, une surveillance clinique et biologique étroite devra être réalisée. Dans les études cliniques portant sur le traitement de l'angor instable et de l'infarctus du myocarde sans onde Q, la nadroparine a été associée à l'aspirine à des doses ne dépassant pas 325 mg par jour (voir rubriques 4.2 et 4.5).
Nécrose cutanée
Des cas de nécrose cutanée ont été très rarement rapportés. Ces réactions sont précédées de plaques érythémateuses, infiltrées ou douloureuses, ou d'un purpura, avec ou sans signes généraux. Si l'un de ces cas se présente, le traitement doit être arrêté immédiatement.
Allergie au latex
Le protège aiguille de la seringue pré-remplie contient du latex, qui peut causer, chez les personnes allergiques au latex, des réactions allergiques graves.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Certains médicaments ou classes thérapeutiques sont susceptibles de favoriser la survenue d'une hyperkaliémie : les sels de potassium, les diurétiques hyperkaliémants, les inhibiteurs de l'enzyme de conversion, les inhibiteurs de l'angiotensine ll, les anti-inflammatoires non stéroïdiens, les héparines (de bas poids moléculaire ou non fractionnées), la ciclosporine et le tacrolimus, le triméthoprime.
La survenue d'une hyperkaliémie peut dépendre de l'existence de facteurs de risque associés.
Ce risque est majoré en cas d'association des médicaments sus-cités.
Associations déconseillées
Chez le sujet de moins de 65 ans aux doses curatives d’HBPM, et chez le sujet âgé (> 65 ans) quelle que soit la dose d’HBPM.
+ Acide acétylsalicylique aux doses antalgiques (et autres salicylés), anti-inflammatoires (A.l.N.S et glucocorticoïdes par voie systémique) et antiagrégants plaquettaires (abciximab, acide acétylsalicylique aux doses antiagrégants dans les indications cardiologiques et neurologiques, beraprost, clopidogrel, eptifibatide, iloprost, ticlopidine, tirofiban) :
Augmentation du risque hémorragique (inhibition de la fonction plaquettaire et agression de la muqueuse gastro-duodénale par les salicylés et les A.l.N.S).
Utiliser un analgésique antipyrétique non salicylé (type paracétamol).
Dans les études cliniques, la nadroparine a été associée à l’aspirine à des doses ne dépassant pas 325 mg par jour dans le traitement de l’angor instable ou de l’infarctus du myocarde sans onde Q (voir rubrique 4.2 et 4.4).
Si l’association avec les A.l.N.S ne peut être évitée, une surveillance clinique étroite est recommandée.
+ Dextran 40 (voie parentérale)
Augmentation du risque hémorragique (inhibition de la fonction plaquettaire par le Dextran 40).
Associations faisant l'objet de précautions d’emploi
+ Anticoagulants oraux
La nadroparine doit être administrée avec précaution chez les patients recevant des anticoagulants oraux, du fait de la potentialisation de l'action anticoagulante par cette association.
Lors du relais de l'héparine par un anticoagulant oral, renforcer la surveillance clinique et poursuivre le traitement par la nadroparine jusqu'à stabilisation de l'lNR (Rapport lnternational Normalise) a la valeur cible.
Associations à prendre en compte
L'utilisation conjointe de médicaments agissant à divers niveaux de l'hémostase majore le risque de saignement. Ainsi, quel que soit l'âge, l'association des HBPM aux anticoagulants oraux, aux antiagrégants plaquettaires (abciximab, AlNS, acide acétylsalicylique quelle que soit la dose, clopidogrel, eptifibatide, iloprost, ticlopidine, tirofiban) et aux thrombolytiques doit être prise en compte en maintenant une surveillance clinique et éventuellement biologique.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études effectuées chez l'animal n'ont pas mis en évidence d'effet tératogène ou fœtotoxique de la nadroparine.
Traitement curatif
En clinique, il n'existe pas actuellement de données suffisamment pertinentes pour évaluer un éventuel effet malformatif ou fœtotoxique de la nadroparine lorsqu'elle est administrée à dose curative pendant toute la grossesse.
En conséquence, par mesure de précaution, il est préférable de ne pas utiliser la nadroparine à dose curative pendant toute la grossesse.
Allaitement
Les informations disponibles sur l'excrétion de la nadroparine dans le lait maternel sont limitées. Cependant, la résorption digestive chez le nouveau-né est à priori improbable. Le traitement par nadroparine est donc compatible avec l'allaitement.
Fertilité
ll n'existe aucune étude clinique sur l'effet de la nadroparine sur la fécondité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Aucune donnée de l'effet de la nadroparine sur l'aptitude à conduire des véhicules et à utiliser des machines n'est disponible.
Les fréquences des effets indésirables (de très fréquent à très rare) ont été déterminées par les données de très nombreux essais cliniques. Les fréquences assignées aux autres effets indésirables (comme ceux < 1/10000) ont été déterminées par des données post-marketing et font plutôt référence à des taux observés qu’à des taux réels.
La classification des effets indésirables utilisée est la suivante :
· très fréquent : ≥ 1/10
· fréquent : ≥ 1/100 ; < 1/10
· peu fréquent : ≥1/1000 ; < 1/100
· rare : ≥ 1/10000 ; < 1/1000
· très rare : < 1/10000
· fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
Affections hématologiques et du système lymphatique
Très fréquent : manifestations hémorragiques pouvant toucher différents sites, qui surviennent essentiellement en présence :
· De facteurs de risques associés : lésions organiques susceptibles de saigner, certaines associations médicamenteuses (voir Rubrique 4.3 Contre-indications et 4.5 Interactions avec d’autres médicaments et autres formes d’interactions), âge, insuffisance rénale, faible poids.
· De non-respect des modalités thérapeutiques, notamment durée de traitement et adaptation de la dose en fonction du poids (voir 4.4 Mises en garde/risque hémorragique).
Rare :
· Hématomes intrarachidiens qui peuvent survenir lors de l’administration d’héparine de bas poids moléculaire au cours d’une rachianesthésie, d’une analgésie ou d’une anesthésie péridurale.
· Thrombopénies (voir rubrique 4.4 Mises en garde et précautions d’emploi). Elles sont de deux types :
o Les plus fréquentes, de type l, sont habituellement modérées (> 100 000/mm3) précoces (avant le 5ème jour) et ne nécessitant pas l’arrêt du traitement,
o Rarement des thrombopénies immuno-allergiques graves de type II (TIH) parfois compliquées de thromboses artérielles ou veineuses. Leur prévalence est encore mal évaluée (voir Rubrique 4.4 Mises en garde et précautions d'emploi).
· Thrombocytose (élévation asymptomatique et réversible des plaquettes).
Très rare : hyperéosinophilie, isolée ou associée à des effets cutanés, réversible à l’arrêt du traitement.
Affections du système immunitaire
Très rare : réactions d’hypersensibilité immédiate (incluant des réactions cutanées, des angio-œdème, des bronchospasmes voire des chocs de type anaphylactique) qui sont susceptibles, dans certains cas, de conduire à l'arrêt du traitement.
Affections du système nerveux
Fréquence indéterminée : maux de tête, migraine.
Troubles du métabolisme et de la nutrition
Très rare : hyperkaliémie réversible due au freinage de la sécrétion d’aldostérone par l’héparine, particulièrement chez les patients à risque (voir rubrique 4.4).
Affections hépatobiliaires
Fréquent : augmentation des transaminases généralement transitoire
Affections des organes de reproduction et du sein
Très rare : Priapisme
Affections de la peau et du tissu sous-cutané
Rare : Rash, urticaire, érythème, prurit
Très rare : Nécrose cutanée, le plus souvent au point d'injection (voir rubrique 4.4).
Affections musculo-squelettiques et systémiques
Le risque d’ostéoporose ne peut être exclu, comme avec les héparines non fractionnées, lors de traitement prolongé.
Troubles généraux et anomalies au site d’administration
Très fréquent : hématomes au point d’injection
Ils sont majorés par le non-respect de la technique d’injection ou l’utilisation d’un matériel d’injection inadéquat.
Dans certains cas, l'apparition de nodules fermes traduisant un processus inflammatoire et non liée à un enkystement de l'héparine peut être observée. Ces nodules disparaissent généralement en quelques jours et ne sont pas un motif d'arrêt du traitement.
Fréquent : réactions au point d’injection (incluant inflammation, prurit, érythème).
Plus rarement, des réactions de type IV ou hypersensibilité retardée, se présentant comme un eczéma de contact ont également été rapportées.
Rare: calcinoses au point d'injection
La calcinose est plus fréquente chez les patients présentant un produit phosphocalcique anormal, tels que dans certains cas d'insuffisance rénale chronique.
Très rare: nécrose cutanée au point d'injection
Ces réactions peuvent être précédées d'un purpura ou de placards érythémateux, infiltrés et douloureux. La suspension du traitement doit être immédiate.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Le surdosage accidentel après administration sous-cutanée de doses massives d'héparine de bas poids moléculaire pourrait entraîner des complications hémorragiques. La numération des plaquettes et des autres paramètres de la coagulation doit être réalisée. Un saignement mineur nécessite rarement un traitement spécifique : réduire ou retarder l'administration des doses suivantes de nadroparine est généralement suffisant.
En cas d'hémorragie sévère, un traitement par sulfate de protamine peut être indiqué dans certains cas, en tenant compte des faits suivants :
· la protamine neutralise largement l'effet anticoagulant de la nadroparine mais une partie de l'activité anti Xa sera maintenue ;
· l'efficacité de la nadroparine est nettement inférieure à celle rapportée lors d'un surdosage par l'héparine non fractionnée ;
· en raison de ses effets indésirables (notamment choc anaphylactique), le rapport bénéfice/risque du sulfate de protamine sera soigneusement évalué avant prescription.
La neutralisation est dans ce cas effectuée par l'injection intraveineuse lente de protamine (sulfate ou chlorhydrate).
La dose de protamine utile est fonction :
· de la dose d'héparine injectée (on peut utiliser 100 UAH de protamine pour neutraliser l'activité de 100 Ul anti-Xa d'héparine de bas poids moléculaire),
· du temps écoulé depuis l'injection de l'héparine, avec éventuellement une réduction des doses de l'antidote.
Néanmoins, il n'est pas possible de neutraliser totalement l'activité anti-Xa.
Par ailleurs, la cinétique de résorption de l'héparine de bas poids moléculaire peut rendre cette neutralisation transitoire et nécessiter de fragmenter la dose totale calculée de protamine en plusieurs injections (2 à 4), réparties sur 24 heures.
En cas d'ingestion, même massive, d'héparine de bas poids moléculaire (aucun cas rapporté), aucune conséquence grave n'est, a priori, à redouter, compte tenu de la très faible résorption du produit aux niveaux gastrique et intestinal.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : ANTI-THROMBOTIQUES, code ATC : B01AB06.
Mécanisme d’action
· La nadroparine est une héparine de bas poids moléculaire dans laquelle les activités antithrombotique et anticoagulantes de l'héparine standard ont été dissociées.
· Elle est caractérisée par une activité anti-Xa plus élevée que l'activité anti-Ila ou anti-thrombotique.
· Pour la nadroparine, le rapport entre ces deux activités est compris entre 2,5 et 4.
Effets pharmacodynamiques
· Aux doses curatives, au pic maximum d'activité, le TCA peut être allongé de 1,4 fois le temps du témoin. Cet allongement est le reflet de l'activité antithrombinique résiduelle de la nadroparine.
5.2. Propriétés pharmacocinétiques
Les paramètres pharmacocinétiques sont étudiés à partir de l'évolution des activités anti-Xa plasmatiques.
Biodisponibilité
Après injection par voie sous-cutanée, la résorption du produit est rapide et proche de 100%; l'activité plasmatique maximale est observée entre la 3ème et la 4ème heure si la nadroparine est administrée en 2 injections par jour. Ce pic est décalé entre la 4ème et la 6ème heure si la nadroparine est administrée en 1 injection par jour.
Métabolisme
Il s'effectue essentiellement au niveau hépatique (désulfatation, dépolymérisation).
Distribution
Après injection par voie sous-cutanée, la demi-vie de l'activité anti-Xa est supérieure pour les héparines de bas poids moléculaire, comparativement aux héparines non fractionnées.
Cette demi-vie est de l'ordre de 3 à 4 heures.
Quant à l'activité anti-Ila, elle disparaît plus rapidement du plasma que l'activité anti-Xa avec les héparines de bas poids moléculaire.
Elimination
L'élimination s'effectue principalement par voie rénale sous forme peu ou pas métabolisée.
Populations à risque
Sujet âgé
Chez le sujet âgé, la fonction rénale étant physiologiquement diminuée, l'élimination est ralentie.
Il est indispensable d'évaluer systématiquement la fonction rénale des sujets âgés de plus de 75 ans par la formule de Cockroft, avant l'instauration d'un traitement par HBPM (voir rubrique 4.4).
lnsuffisance rénale
Dans une étude pharmacocinétique réalisée en 1988, chez 5 patients présentant une insuffisance rénale modérée, 7 patients présentant une insuffisance rénale sévère et 7 patients dialysés ayant reçu une dose unique de nadroparine par voie intraveineuse, une corrélation a été démontrée entre la clairance de la nadroparine et celle de la créatinine.
Chez les patients présentant une insuffisance rénale modérée (clairance de la créatinine 36-43 ml/min), l’AUC moyenne et la demi-vie étaient augmentées respectivement de 52% et 39% par rapport aux volontaires sains. Chez ces patients, la clairance plasmatique moyenne de nadroparine était diminuée de 63% par rapport à la normale. Une large variabilité interindividuelle a été observée dans cette étude.
Chez les sujets présentant une insuffisance rénale sévère (clairance de la créatinine 10-20 ml/min), l’AUC moyenne et la demi-vie étaient augmentées respectivement de 95% et 112% par rapport aux volontaires sains. Leur clairance plasmatique était diminuée de 50% par rapport à celle observée chez les patients ayant une fonction rénale normale.
Hémodialyse
L'héparine de bas poids moléculaire est injectée dans la ligne artérielle du circuit de dialyse, à des doses suffisantes pour éviter la coagulation du circuit.
Entre deux séances de dialyse, chez les patients présentant une insuffisance rénale sévère, hémodialysés (clairance de la créatinine 3-6 ml/min), l’AUC moyenne et la demi-vie étaient augmentées respectivement de 62% et 65% par rapport aux volontaires sains. La clairance plasmatique des patients présentant une insuffisance rénale sévère, hémodialysés était diminuée de 67% par rapport à celle observée chez les patients ayant une fonction rénale normale (voir rubriques 4.2 et 4.4).
En cas de surdosage, le passage de nadroparine dans la circulation générale peut donner lieu à une activité anti-Xa élevée, en rapport avec l'insuffisance rénale terminale.
5.3. Données de sécurité préclinique
Sans objet.
Solution d’acide chlorhydrique ou soluté d’hydroxyde de calcium officinal, eau pour préparations injectables.
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
3 ans.
6.4. Précautions particulières de conservation
A conserver à une température inférieure à 25°C.
A conserver dans son emballage jusqu’à l’utilisation.
Après ouverture : le produit doit être utilisé immédiatement.
6.5. Nature et contenu de l'emballage extérieur
1 ml de solution injectable en seringue pré-remplie (verre) avec système de sécurité : manchon plastique transparent. Boite de 2, 6 ou 10.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Après injection, le système de protection doit être glissé sur l’aiguille utilisée, de manière à ce qu’elle soit complètement protégée. Tenir d’une main la seringue par le manchon et tirer fermement sur la bague pour déverrouiller le manchon et l’amener jusqu’au clic de verrouillage. La seringue peut alors être mise dans le conteneur à déchets.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Viatris SANTE
1 Rue de Turin
69007 Lyon
France
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 347 334 8 2 : 1 ml de solution injectable en seringue pré-remplie (verre) avec système de sécurité manchon plastique transparent; boîte de 2.
· 34009 347 335 4 3 : 1 ml de solution injectable en seringue pré-remplie (verre) avec système de sécurité manchon plastique transparent; boîte de 6.
· 34009 347 336 0 4 : 1 ml de solution injectable en seringue pré-remplie (verre) avec système de sécurité manchon plastique transparent; boîte de 10.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
A compléter ultérieurement par le titulaire
10. DATE DE MISE A JOUR DU TEXTE
A compléter ultérieurement par le titulaire
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I.
ANSM - Mis à jour le : 05/04/2023
FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie
Nadroparine calcique
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous
· Gardez cette notice, vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien ou votre infirmier/ère.
· Ce traitement doit être suivi avec une très grande rigueur et une très grande vigilance, car mal équilibré, il peut entraîner des complications hémorragiques ou une rechute de votre maladie. Il nécessite une surveillance biologique particulière, effectuée à l’aide d’un test appelé INR (International Normalized Ratio).
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie et dans quels cas est-il utilisé?
2. Quelles sont les informations à connaître avant d'utiliser FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie?
3. Comment utiliser FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie?
4. Quels sont les effets indésirables éventuels?
5. Comment conserver FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie ET DANS QUELS CAS EST-IL UTILISE ?
ANTI-THROMBOTIQUES – Code ATC : B01AB06.
Ce médicament est un anticoagulant de la famille des héparines dites de « bas poids moléculaire ». Il vous est prescrit en traitement curatif, dans le cas d’une thrombose déjà existante.
Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie ?
Si votre médecin vous a informé(e) que vous étiez allergique au latex, contactez-le avant de prendre ce médicament.
N'utilisez jamais FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie dans les cas suivants :
|
|
CONTRE-INDIQUEE |
DECONSEILLEE |
|
DANS TOUS LES CAS
|
· vous êtes allergique à la nadroparine, à l'héparine ou à un produit similaire (tel que l'énoxaparine, la bémiparine ou la daltéparine), ou à l'un des composants contenus dans ce médicament (mentionnés dans la rubrique 6) · si vous avez déjà eu dans le passé un épisode grave de baisse des plaquettes due à l’héparine (les plaquettes sont des éléments du sang importants pour la coagulation sanguine) · si vous avez une maladie connue de la coagulation · en cas de lésion (interne ou externe) risquant de saigner. · lors de la plupart des endocardites (infections du cœur) · en cas d’hémorragie cérébrale · si vous avez une insuffisance rénale sévère (sauf en cas de dialyse) · une anesthésie péridurale ou une rachianesthésie est contre-indiquée lors d’un traitement curatif. |
· quel que soit l’âge, en cas de traitement concomitant par l’aspirine (aux doses utilisées pour la douleur et la fièvre et les inflammations), par les anti-inflammatoires non stéroïdiens (AINS) ou par le dextran (médicament utilisé en réanimation) · dans les premiers jours qui suivent un accident vasculaire cérébral non hémorragique · en cas d’insuffisance rénale légère ou modérée. |
Avertissements et précautions
FRAXODI ne doit pas être remplacée par d’autres médicaments appartenant au groupe HBPM. Cela est dû au fait que les HBPM ne sont pas exactement identiques et n’ont pas la même activité, ni les mêmes instructions d’utilisation.
FRAXODI est réservé au traitement des thromboses veineuses profondes.
FRAXODI s'administre en une seule injection quotidienne.
Pour éviter la survenue de saignement, il est impératif de ne pas dépasser la dose et la durée de traitement que votre médecin vous a indiquées.
Ce traitement nécessite des prises de sang répétées pour un contrôle régulier du nombre de vos plaquettes (en général deux fois par semaine).
En effet, très rarement, il peut survenir au cours du traitement par héparine une baisse importante du nombre de plaquettes. Ceci impose un arrêt de l'héparine et une surveillance accrue car des complications graves peuvent survenir, notamment des thromboses de manière paradoxale.
NE PAS INJECTER PAR VOIE INTRAMUSCULAIRE. Les modalités d'injection doivent être très précisément respectées.
Dans certains cas, notamment lors d'un traitement curatif, il peut exister un risque de saignement :
· pour les patients âgés,
· en cas de poids inférieur à 40 kg,
· en cas d'insuffisance rénale (voir Dans quels cas ne pas utiliser ce médicament et Posologie),
· en cas de traitement prolongé au-delà de la durée habituelle de 10 jours,
· en cas de non-respect des modalités thérapeutiques conseillées
· ,en cas d'association à des médicaments majorant le risque hémorragique (voir Prise ou utilisation d'autres médicaments).
Ces situations peuvent nécessiter une surveillance particulière: examens médicaux et prises de sang éventuelles.
Si vous avez ou si vous avez eu une maladie hépatique ou rénale, un ulcère ou une autre lésion susceptible de saigner, prévenez votre médecin.
FRAXODI peut entraîner une élévation du potassium dans le sang.
Si vous avez du diabète, une insuffisance rénale chronique, une acidose métabolique préexistante ou si vous prenez d'autres médicaments susceptibles d'augmenter le potassium dans le sang, prévenez votre médecin, il pourra être amené à vous prescrire des prises de sang pour surveiller cet effet secondaire.
Le protège aiguille de la seringue pré-remplie contient du latex de caoutchouc naturel et peut provoquer des réactions allergiques graves.
Enfants et adolescents
Ce médicament n'est habituellement pas recommandé chez l'enfant.
Autres médicaments et FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie
En raison de la survenue possible de saignement, prévenez systématiquement votre médecin si vous prenez l'un des médicaments suivants:
· de l'aspirine, aux doses antalgiques (et autres salicylés),
· des anti-inflammatoires non stéroïdiens (AINS), et glucocorticoïdes par voie systémique,
· des anti-agrégants plaquettaires (abciximab, acide acétylsalicylique aux doses anti-agrégants berapost, clopidrogel, eptifibatide, iloprost, ticlopidine, tirofiban),
· du dextran (médicament utilisé en réanimation),
· des anticoagulants oraux (anti-vitamines K).
Informez votre médecin ou pharmacien si vous prenez, avez pris récemment ou pourriez prendre un autre médicament, y compris un médicament obtenu sans ordonnance.
Votre médecin pourra adapter les modalités de votre traitement en conséquence.
Grossesse et allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Par mesure de précaution il est préférable de ne pas utiliser ce médicament pendant toute la grossesse dans le traitement curatif des thromboses.
A l'accouchement avec anesthésie péridurale, la prudence doit être accrue.
Ce traitement n'est pas contre-indiqué chez la femme qui allaite.
FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie contient du latex.
Le protège aiguille de la seringue pré-remplie contient du latex et peut provoquer des réactions allergiques graves.
3. COMMENT UTILISER FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie ?
Veillez à toujours utiliser ce médicament en suivant exactement les instructions de cette notice ou les indications de votre médecin, pharmacien ou infirmier/ère. Vérifiez auprès de votre médecin, pharmacien ou infirmier/ère en cas de doute.
La dose et la durée du traitement sont déterminées par votre médecin en fonction de votre poids. Ce médicament est réservé au traitement curatif.
1 ml de FRAXODI correspond environ à 19 000 Ul anti-Xa de nadroparine.
Si ce médicament doit être remplacé par un anticoagulant pris par voie orale, les injections ne seront arrêtées qu'après quelques jours pendant lesquels vous prendrez les 2 traitements en même temps. Il s'agit du temps nécessaire pour que le second traitement soit actif et que les examens sanguins de la coagulation soient au niveau souhaité par votre médecin.
Mode et voie d’administration
VOIE SOUS-CUTANEE.
Ne pas injecter par voie intramusculaire.
Fréquence d’administration
1 seule injection par jour.
Durée du traitement
La durée du traitement ne dépasse habituellement pas 10 jours.
Si vous avez pris plus de FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie que vous n'auriez dû :
Prévenir rapidement un médecin, en raison d'un risque de saignement.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Les effets les plus fréquemment observés:
· Saignements qui peuvent être graves. Si cela se produit, il faut avertir immédiatement votre médecin ou l'infirmière ; ils peuvent être favorisés par une lésion risquant de saigner, par une insuffisance rénale ou par certains médicaments pris pendant la même période.
· Il peut se former des hématomes ou des nodules (« boules ») sous la peau au point d'injection, ce qui peut être plus ou moins douloureux. Ceux-ci disparaîtront spontanément et ne doivent pas faire interrompre le traitement.
Les effets fréquemment observés:
· Réactions cutanées au point d'injection.
· Elévation du taux sanguin de certaines enzymes du foie.
Les effets rarement observés:
· Diminution du nombre de plaquettes (cellules sanguines nécessaire à la coagulation), qui peut dans certains cas être grave et qu'il importe de signaler immédiatement au médecin traitant (voir 3. b Mises en garde spéciales). C'est pourquoi le nombre de plaquettes sera contrôlé régulièrement.
· Augmentation du nombre de plaquettes sans symptômes associés.
· Dépôts de calcium sous la peau au point d'injection.
· Lésions neurologiques (hématomes intrarachidiens) de gravité variable survenant lors de l'administration de ce type de médicament au cours de certaines anesthésies.
Rash, urticaire, érythème, pruritLes effets très rarement observés:
· Manifestations allergiques avec rash et gonflement du visage, de la bouche, des lèvres et de la gorge entraînant des troubles de la respiration et pouvant mettre en danger la vie du malade (anaphylaxie).
· Douleurs, rougeurs ou nécrose de la peau autour du point d'injection
· Elévation du nombre de certains globules blancs appelés polynucléaires éosinophiles.
· Elévation du potassium dans le sang.
· Erection anormale du pénis (priapisme).
·
Les effets à fréquence indéterminée:
· maux de tête, migraine.
Autres effets:
Risque d'ostéoporose (déminéralisation du squelette entraînant une fragilité osseuse) lors de traitement prolongé.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte.
Après ouverture : le produit doit être utilisé immédiatement.
A conserver à une température inférieure à 25°C.
A conserver dans son emballage jusqu'à l'utilisation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient FRAXODI 19 000 U.I. Axa/1 ml, solution injectable (SC) en seringue pré-remplie
· La substance active est :
Nadroparine calcique .. 19 000 UI Axa
Pour une seringue pré-remplie.
· Les autres composants sont :
Soluté d'hydroxyde de calcium officinal ou solution d'acide chlorhydrique, eau pour préparations injectables.
Ce médicament se présente sous forme d'une solution injectable en seringue pré-remplie. Boîte de 2, 6 ou 10.
Titulaire de l’autorisation de mise sur le marché
Viatris Sante
1 Rue de Turin
69007 Lyon
France
Exploitant de l’autorisation de mise sur le marché
Viatris Sante
1 Rue de Turin
69007 Lyon
France
ASPEN NOTRE DAME DE BONDEVILLE
1, RUE DE L'ABBAYE
76960 NOTRE-DAME-DE-BONDEVILLE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
INSTRUCTIONS DESTINEES A LA PERSONNE QUI ADMINISTRE CE MEDICAMENT
Utiliser la présentation adaptée à la dose prescrite.
1 ml de FRAXODI correspond environ à 19 000 UI anti-Xa de nadroparine.
Mode et voie d’administration
VOIE SOUS CUTANEE (en dehors de l’indication dans les circuits de dialyse)
Ne pas injecter par voie intramusculaire.
Préparation et technique de l’injection sous cutanée
· Ne pas purger la bulle d’air.
· L’utilisation d’aiguilles de très fin calibre (au maximum 0,5 mm de diamètre) est recommandée.
· L’injection doit être réalisée, de préférence sur un patient allongé, dans le tissu cellulaire sous-cutané de la ceinture abdominale antéro-latérale et postéro-latérale, tantôt à droite, tantôt à gauche.
|
· |
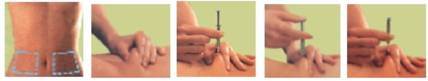
L'aiguille doit être introduite perpendiculairement et non tangentiellement, dans l'épaisseur d'un pli cutané réalisé entre le pouce et l'index de l'opérateur. Le pli doit être maintenu durant toute la durée de l'injection.
Utilisation du système de protection de l'aiguille
Cet étui contient une seringue pour FRAXODI avec un système de protection de l'aiguille après injection.
|
|
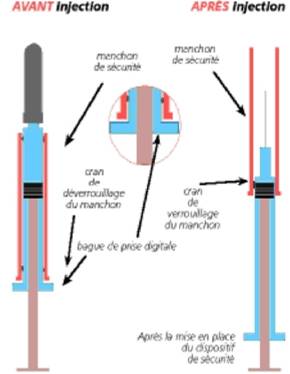
Après injection, mettre en place le système de sécurité de FRAXODI:
Tenir d'une main la seringue par le manchon et tirer fermement de l'autre sur la bague pour déverrouiller le manchon et l'amener jusqu'au clic de verrouillage (schéma).
Pour une meilleure sécurité, le dispositif oppose une résistance normale lors du verrouillage ou du déverrouillage.
|
|
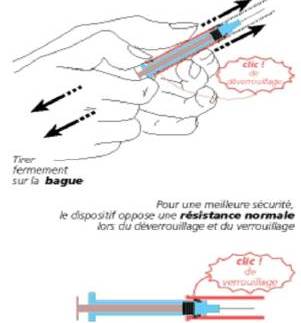
L'aiguille souillée est maintenant entièrement protégée. La seringue peut être éliminée selon la procédure normale prévue pour les déchets.