Dernière mise à jour le 01/06/2026
ACULAR 0,5 %, collyre en solution
Indications thérapeutiques
Classe pharmacothérapeutique : ANTI-INFLAMMATOIRES - code ATC : S01BC05
ACULAR appartient à un groupe de médicaments appelés anti-inflammatoires non stéroïdiens (AINS).
ACULAR est utilisé dans la prévention et le traitement de l'inflammation oculaire post-opératoire après chirurgie de la cataracte.
Présentations
> 1 flacon(s) compte-gouttes polyéthylène de 5 ml
Code CIP : 334 295-9 ou 34009 334 295 9 1
Déclaration de commercialisation : 07/06/1999
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 2,60 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 3,62 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 03/10/2018 | Renouvellement d'inscription (CT) | le service médical rendu par ACULAR reste important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 10/02/2026
ACULAR 0,5%, collyre en solution
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Kétorolac trométamol ........................................................................................................ 5 mg/ml
Excipient(s) à effet notoire : Chlorure de benzalkonium 0,1 mg/ml.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution aqueuse limpide, incolore à jaune pâle.
4.1. Indications thérapeutiques
ACULAR est indiqué chez l'adulte.
4.2. Posologie et mode d'administration
La posologie recommandée est de 1 ou 2 gouttes dans l’œil atteint toutes les 6 à 8 heures pendant 21 jours, en commençant les instillations 24 heures avant l'intervention.
Population pédiatrique
Il n’existe pas d’utilisation justifiée d’ACULAR dans la population pédiatrique dans cette indication.
Patients âgés
Aucune différence en termes de sécurité et d’efficacité n’a été observée entre les patients âgés et les patients plus jeunes.
Mode d’administration
Voie oculaire.
Instiller une goutte de collyre dans le cul-de-sac conjonctival inférieur de l'œil à traiter, en tirant la paupière inférieure légèrement vers le bas et en regardant vers le haut.
En cas d'utilisation concomitante d'ACULAR 0,5%, collyre en solution, avec d'autres produits ophtalmiques topiques, un intervalle d'au moins 5 minutes doit être respecté entre les administrations des deux médicaments.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Grossesse, à partir du début du 6ème mois (au-delà de 24 semaines d’aménorrhée) (voir rubrique 4.6)
Il existe une possibilité de réaction croisée avec l’acide acétylsalicylique ou d'autres anti-inflammatoires non stéroïdiens (AINS). C’est la raison pour laquelle, ACULAR 0,5%, collyre en solution est contre-indiqué chez les patients ayant développé des hypersensibilités à ces produits.
4.4. Mises en garde spéciales et précautions d'emploi
Comme les autres AINS, ACULAR 0,5%, collyre en solution peut masquer les signes habituels d'une infection. S’il existe un risque d’infection, un traitement adéquat doit être prescrit.
Tous les AINS sont susceptibles de ralentir ou de retarder la cicatrisation des plaies. L'utilisation concomitante d'AINS et de corticoïdes topiques peut augmenter le risque d’apparition de problèmes de cicatrisation. L'utilisation concomitante d'ACULAR 0,5%, collyre en solution et des corticoïdes topiques doit être effectuée avec précaution chez les patients présentant une prédisposition à la dégradation épithéliale cornéenne.
L'utilisation des AINS topiques peut entraîner une kératite. Chez certains patients, un traitement continu avec des AINS topiques peut entraîner une dégradation de l’épithélium, un amincissement cornéen, une ulcération cornéenne ou une perforation de la cornée. Ces événements peuvent engager le pronostic visuel. Les patients présentant des signes de dégradation épithéliale cornéenne doivent cesser immédiatement d'utiliser les AINS topiques, et surveiller étroitement l'état de leur cornée.
Les AINS topiques doivent être utilisés avec précaution chez les patients ayant subi des chirurgies oculaires lourdes, ou présentant une dénervation cornéenne, des anomalies de l'épithélium cornéen, un diabète, des maladies de la surface oculaire (telles qu’un syndrome de l'œil sec), une polyarthrite rhumatoïde, ou des chirurgies oculaires répétées sur une courte période de temps, car ils pourraient encourir un risque accru d'événements indésirables de la cornée pouvant engager le pronostic visuel.
L’expérience post-commercialisation avec les AINS topiques suggère également qu'une utilisation des AINS topiques plus de 24 heures avant la chirurgie ou plus de 14 jours après la chirurgie pourrait augmenter le risque de survenue et la sévérité des événements indésirables cornéens.
Des cas de bronchospasme ou d’exacerbation d'un asthme ont été rapportés au cours de la surveillance post-commercialisation chez des patients ayant soit une hypersensibilité connue à l'aspirine / aux AINS, soit des antécédents médicaux d'asthme, en association à l'utilisation d'ACULAR 0,5%, collyre en solution, lequel pourrait avoir contribué à la survenue de ces événements. Des précautions sont recommandées pour l'utilisation d'ACULAR 0,5%, collyre en solution chez ces patients (voir rubrique 4.8).
Refermer soigneusement le flacon après utilisation.
Les patients doivent être informés qu’ils doivent éviter tout contact entre l’embout du flacon et l’œil ou les structures avoisinantes afin d’éviter de se blesser ou de contaminer le collyre.
Les effets indésirables peuvent être réduits en utilisant la dose minimale efficace pendant la durée la plus courte nécessaire au soulagement des symptômes.
Informations destinées aux porteurs de lentilles de contact
Le conservateur présent dans ACULAR 0,5%, collyre en solution, le chlorure de benzalkonium, peut causer une irritation oculaire et une coloration des lentilles de contact souples. Éviter tout contact avec des lentilles souples. Retirer les lentilles de contact avant l’instillation et attendre au moins 15 minutes avant de les remettre.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée.
ACULAR 0,5%, collyre en solution a été administré, sans mise en évidence de risque particulier de tolérance, avec des médicaments systémiques et ophtalmiques tels que les antibiotiques, les sédatifs, les bétabloquants, les inhibiteurs de l'anhydrase carbonique, les myotiques, les mydriatiques, les anesthésiques locaux et les cycloplégiques.
ACULAR 0,5%, collyre en solution peut ralentir ou retarder la cicatrisation. Les corticoïdes topiques sont également connus pour ralentir ou retarder la cicatrisation. L'utilisation concomitante d'AINS topiques et de corticoïdes topiques peut augmenter le risque de problèmes de cicatrisation (voir rubrique 4.4).
En cas d'utilisation concomitante d'ACULAR 0,5%, collyre en solution avec d'autres produits ophtalmiques topiques, un intervalle d'au moins 5 minutes doit être respecté entre les administrations des deux médicaments.
4.6. Fertilité, grossesse et allaitement
Il convient de tenir compte des informations et recommandations relatives aux AINS administrés par voie générale présentées dans cette rubrique, compte tenu des risques liés à ces produits, bien que les quantités de kétorolac trométamol passant dans la circulation systémique soient potentiellement faibles après administration oculaire.
Grossesse
L’inhibition de la synthèse des prostaglandines par les AINS peut affecter le déroulement de la grossesse et/ou le développement de l’embryon ou du fœtus.
Risques associés à l’utilisation au cours du 1er trimestre
Les données des études épidémiologiques suggèrent une augmentation du risque de fausse-couche, de malformations cardiaques et de gastroschisis, après traitement par un inhibiteur de la synthèse des prostaglandines en début de grossesse. Le risque absolu de malformation cardiovasculaire est passé de moins de 1% dans la population générale, à approximativement 1,5 % chez les personnes exposées aux AINS. Le risque paraît augmenter en fonction de la dose et de la durée du traitement. Chez l’animal, il a été montré que l’administration d’un inhibiteur de la synthèse des prostaglandines provoquait une perte pré et post-implantatoire accrue et une augmentation de la létalité embryo-foetale. De plus, une incidence supérieure de certaines malformations, y compris cardiovasculaires, a été rapportée chez des animaux ayant reçu un inhibiteur de la synthèse des prostaglandines au cours de la phase d’organogénèse de la gestation.
Risques associés à l’utilisation à partir de la 12ème semaine d’aménorrhée et jusqu’à la naissance :
A partir de la 12ème semaine d’aménorrhée et jusqu’à la naissance, tous les AINS, par l’inhibition de la synthèse des prostaglandines, peuvent exposer le fœtus à une atteinte fonctionnelle rénale :
· in utero pouvant s'observer dès 12 semaines d'aménorrhée (mise en route de la diurèse fœtale) : oligoamnios (le plus souvent réversible à l'arrêt du traitement), voire anamnios en particulier lors d'une exposition prolongée.
· à la naissance, une insuffisance rénale (réversible ou non) peut persister en particulier en cas d'exposition tardive et prolongée (avec un risque d'hyperkaliémie sévère retardée).
Risques associés à l’utilisation à partir de la 24ème semaine d’aménorrhée :
A partir de la 24ème semaine d’aménorrhée, les AINS peuvent exposer le fœtus à une toxicité cardio-pulmonaire (fermeture prématurée du canal artériel et hypertension artérielle pulmonaire). La constriction du canal artériel peut survenir à partir du début du 6ème mois (au-delà de la 24ème semaine d’aménorrhée) et peut conduire à une insuffisance cardiaque droite fœtale ou néonatale voire à une mort fœtale in utero. Ce risque est d'autant plus important que la prise est proche du terme (moindre réversibilité). Cet effet existe même pour une prise ponctuelle.
En fin de grossesse, la mère et le nouveau-né peuvent présenter :
· un allongement du temps de saignement du fait d’une action anti-agrégante pouvant survenir même après administration de très faibles doses de médicament ;
· une inhibition des contractions utérines entraînant un retard de terme ou un accouchement prolongé.
En conséquence :
Sauf nécessité absolue, ce médicament ne doit pas être prescrit chez une femme qui envisage une grossesse ou au cours des 5 premiers mois de grossesse (24 premières semaines d’aménorrhée). Si ce médicament est administré chez une femme souhaitant être enceinte ou enceinte de moins de 6 mois, la dose devra être la plus faible possible et la durée du traitement la plus courte possible. Une prise prolongée est fortement déconseillée.
A partir du début du 6ème mois (au-delà de 24 semaines d'aménorrhée): toute prise de ce médicament, même ponctuelle, est contre-indiquée. Une prise par mégarde à partir de cette date justifie une surveillance cardiaque et rénale, fœtale et/ou néonatale selon le terme d'exposition. La durée de cette surveillance sera adaptée à la demi-vie d'élimination de la molécule
Allaitement
Les A.I.N.S. passant dans le lait maternel, ce médicament est déconseillé chez la femme qui allaite.
Fertilité
Comme tous les AINS, l'utilisation de ce médicament peut temporairement altérer la fertilité féminine en agissant sur l’ovulation ; il est donc déconseillé chez les femmes souhaitant concevoir un enfant. Chez les femmes rencontrant des difficultés pour concevoir ou réalisant des tests de fertilité, l'arrêt du traitement doit être envisagé.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables les plus fréquemment rapportés avec l'utilisation d'ACULAR 0,5%, collyre en solution sont une sensation transitoire de picotement et de brûlure lors de l'instillation.
La fréquence des effets indésirables documentée durant les essais cliniques est présentée plus loin et définie comme suit : Très fréquent (≥ 1/10) ; Fréquent (≥1/100 à <1/10) ; Peu fréquent (≥1/1000 à <1/100) ; Rare (≥1/10000 à <1/1000) ; Très rare <1/10000) ; Fréquence indéterminée (ne peut être estimée à partir des données disponibles).
Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Affections du système immunitaire
Fréquents : Hypersensibilité, y compris réactions allergiques locales
Affections du système nerveux
Fréquents : Céphalées
Affections oculaires
Très fréquents : Irritation oculaire (y compris sensation de brûlure)
Douleur oculaire (y compris picotement)
Fréquents : Kératite superficielle (punctiforme)
Œdème oculaire et/ou palpébral
Prurit oculaire
Hyperémie conjonctivale
Infection oculaire
Inflammation oculaire
Iritis
Précipités kératiques
Hémorragies rétiniennes
Œdème maculaire cystoïde
Traumatisme oculaire
Augmentation de la pression intraoculaire
Vision floue et/ou diminuée
Peu fréquents : Kératite ulcérative
Infiltrats cornéens
Sécheresse oculaire
Épiphora
Fréquence indéterminée : Lésions de la cornée, telles qu’amincissement, érosion, dégradation et perforation épithéliales*
Gonflement oculaire
Hyperhémie oculaire
Affections respiratoires, thoraciques et médiastinales
Fréquence indéterminée : Bronchospasme ou exacerbation d'un asthme**
*Il y a eu un nombre limité de notifications spontanées de pharmacovigilance de lésions de la cornée comprenant un amincissement cornéen, une érosion cornéenne, des lésions épithéliales et une perforation de la cornée. Ces événements sont survenus principalement chez des patients ayant utilisé concomitamment des corticoïdes topiques et/ou chez ceux présentant une prédisposition (voir rubrique 4.4 « Mises en garde spéciales et précautions d’emploi »).
**Des cas de bronchospasme ou d'exacerbation d'un asthme ont été rapportés au cours de la surveillance post-commercialisation chez des patients ayant soit une hypersensibilité connue à l'aspirine / aux AINS, soit des antécédents médicaux d'asthme, en association à l'utilisation d'ACULAR 0,5%, collyre en solution, lequel pourrait avoir contribué à la survenue de ces événements.
Aux doses usuelles utilisées en ophtalmologie, aucun des effets indésirables rapportés lors d’une utilisation systémique des AINS (y compris le kétorolac trométamol) n'a été observé.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Aucun cas de surdosage n'a été rapporté. La survenue d'un surdosage est peu probable avec le mode d'administration recommandé.
En cas d'ingestion accidentelle, il faut boire des liquides afin de diluer le produit.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : anti-inflammatoires non-stéroïdiens, code ATC : S01BC05.
ACULAR 0,5 %, collyre en solution (kétorolac trométamol) est un anti-inflammatoire non stéroïdien possédant une activité anti-inflammatoire et antalgique. Il agirait en inhibant la cyclo-oxygénase, enzyme essentielle pour la biosynthèse des prostaglandines. ACULAR 0,5 %, collyre en solution a montré qu’il réduisait les taux de prostaglandines dans l’humeur aqueuse après administration ophtalmique.
Le kétorolac trométamol administré par voie systémique n'entraîne pas de myosis. Les résultats des études cliniques montrent qu’ACULAR 0,5 %, collyre en solution n’a pas d’effet significatif sur la pression intraoculaire.
5.2. Propriétés pharmacocinétiques
Dans une étude de tolérance de 21 jours à dose répétée (x3/j) menée auprès de volontaires sains, seul 1 des 13 sujets présentait un taux plasmatique détectable (0,021 µg/ml) de kétorolac avant la prise suivante. Chez un autre groupe de 13 sujets, seuls 4 sujets ont présenté de très faibles taux plasmatiques de kétorolac (0,011 à 0,023 µg/ml) 15 minutes après l’administration oculaire.
Ainsi, la détection de taux plus élevés de kétorolac dans l'humeur aqueuse et de taux plasmatiques très faibles ou indétectables après administration ophtalmique suggère que l'utilisation du kétorolac trométamol par voie ophtalmique pour le traitement de troubles oculaires entraîne une absorption systémique plutôt faible chez les patients.
5.3. Données de sécurité préclinique
Des études expérimentales de toxicité aiguë, subaiguë et chronique d'ACULAR 0,5 %, collyre en solution chez l'animal ont établi la sécurité d'emploi du médicament. En outre, l'octoxinol 40 a été évalué séparément en termes de tolérance oculaire. ACULAR s'est avéré non-irritant, n'a pas démontré d'effet anesthésique local, n'a pas influencé la cicatrisation de plaies de la cornée dans des conditions expérimentales chez le lapin, n'a pas favorisé la propagation d'infections oculaires expérimentales de Candida albicans, du virus Herpès simplex de type 1, ou de Pseudomonas aeruginosa chez le lapin, et n'a pas augmenté la pression intraoculaire des yeux sains du lapin.
Chlorure de benzalkonium.
Edétate disodique.
Octoxinol 40.
Hydroxyde de sodium ou acide chlorhydrique (dilué) pour ajuster le pH.
Eau purifiée.
Avant ouverture : 2 ans.
Après ouverture : à utiliser dans les 15 jours après ouverture du flacon.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
6.5. Nature et contenu de l'emballage extérieur
Flacon compte-gouttes en polyéthylène basse densité (avec embout compte-gouttes en PEBD) contenant 5 ml de solution. La taille de la goutte est de 35 microlitres. Chaque flacon dispose d'un capuchon vissé en polystyrène MIPS.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 334 295 9 1 : 5 ml en flacon compte-gouttes (PE).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 10/02/2026
ACULAR 0,5%, collyre en solution
Kétorolac trométamol
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ACULAR 0,5%, collyre en solution et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser ACULAR 0,5%, collyre en solution ?
3. Comment utiliser ACULAR 0,5%, collyre en solution ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ACULAR 0,5%, collyre en solution ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ACULAR 0,5%, collyre en solution ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : ANTI-INFLAMMATOIRES - code ATC : S01BC05
ACULAR appartient à un groupe de médicaments appelés anti-inflammatoires non stéroïdiens (AINS).
ACULAR est utilisé dans la prévention et le traitement de l'inflammation oculaire post-opératoire après chirurgie de la cataracte.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ACULAR 0,5%, collyre en solution ?
N’utilisez jamais ACULAR 0,5%, collyre en solution :
· si vous êtes allergique au kétorolac trométamol ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous êtes allergique à l'aspirine ou à tout autre produit similaire tel que les autres agents anti-inflammatoires non stéroïdiens.
· si vous êtes enceinte, à partir du début du 6ème mois de grossesse (au-delà de 24 semaines d’aménorrhée)
Avertissements et précautions
Adressez-vous à votre médecin avant d’utiliser ACULAR 0,5%, collyre en solution.
Si vous souffrez, ou avez souffert :
· d'infections virales ou bactériennes de l'œil,
· de tendances hémorragiques (par exemple anémie) ou d’ulcères gastriques,
· de diabète,
· de polyarthrite rhumatoïde,
· de syndrome de l'œil sec,
· d’asthme après utilisation d'anti-inflammatoires non stéroïdiens,
· ou si vous avez subi récemment une chirurgie oculaire,
· de perte de sensibilité de la cornée (la surface transparente couvrant la pupille et l'iris), ou de lésion de surface, normalement lisse, de la cornée.
En cas de réaction d'hypersensibilité ou de signes indiquant une allergie à ce médicament, en particulier une crise d'asthme ou un gonflement brutal du visage ou du cou, stoppez le traitement et contactez immédiatement un médecin ou un service médical d'urgence.
Enfants
ACULAR 0,5%, collyre en solution ne doit pas être prescrit chez l'enfant.
Autres médicaments et ACULAR 0,5%, collyre en solution
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Si vous utilisez ACULAR en même temps qu’un autre produit oculaire, attendre au moins 5 minutes entre l'instillation d'ACULAR et l'instillation de l'autre produit.
ACULAR 0,5%, collyre en solution avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre tout médicament.
Grossesse
Avant le début du 6ème mois de grossesse (jusqu’à la 24ème semaine d’aménorrhée), vous ne devez pas prendre ce médicament, sauf en cas d’absolue nécessité déterminée par votre médecin, en raison du risque potentiel de fausses couches ou de malformations. Dans ce cas, la dose devra être la plus faible possible et la durée du traitement la plus courte possible.
A partir du début du 6ème mois jusqu’à la fin de la grossesse (au-delà de la 24ème semaine d’aménorrhée), ce médicament est contre-indiqué, vous ne devez EN AUCUN CAS prendre ce médicament, car ses effets sur votre enfant peuvent avoir des conséquences graves voire fatales, notamment sur le cœur, les poumons et/ou les reins, et cela même avec une seule prise.
Si vous avez pris ce médicament alors que vous étiez enceinte, parlez-en immédiatement à votre gynécologue obstétricien, afin qu’une surveillance adaptée vous soit proposée si nécessaire.
Allaitement
Ce médicament passant dans le lait maternel, il est déconseillé de l'utiliser pendant l'allaitement.
Fertilité
Ce médicament, comme tous les anti-inflammatoires non stéroïdiens (AINS), peut altérer la fertilité des femmes et entraîner des difficultés pour devenir enceinte, de façon réversible à l’arrêt du traitement. Informez votre médecin si vous planifiez une grossesse ou si vous avez des difficultés à concevoir.
Conduite de véhicules et utilisation de machines
ACULAR 0,5%, collyre en solution peut provoquer une vision trouble chez certains patients. Ne pas conduire de véhicule ni utiliser de machine tant que les symptômes n'ont pas disparu.
ACULAR 0,5%, collyre en solution contient du chlorure de benzalkonium.
· Ce médicament contient 0,1 mg de chlorure de benzalkonium par millilitre, soit 0,1 mg/ml.
· Le chlorure de benzalkonium peut être absorbé par les lentilles de contact souples et peut changer la couleur des lentilles de contact. Si vous portez des lentilles de contact, retirez-les avant l'application de ce médicament et attendez au moins 15 minutes avant de les remettre.
· Le chlorure de benzalkonium peut également causer une irritation oculaire, notamment si vous avez les yeux secs ou des troubles de la cornée (la couche claire située à l’avant de l’œil). Si vous ressentez des sensations anormales dans les yeux, des picotements ou des douleurs dans l’œil après avoir utilisé ce médicament, parlez-en à votre médecin.
3. COMMENT UTILISER ACULAR 0,5%, collyre en solution ?
La dose recommandée est de 1 ou 2 gouttes toutes les 6 à 8 heures dans l’œil atteint pendant 3 semaines après l’intervention ; les instillations débuteront 24 heures avant l'intervention.
Mode et voie d'administration
Voie oculaire.
Appliquez le collyre de la manière suivante :
|
|
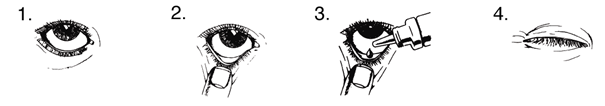
1. Lavez-vous les mains. Penchez la tête en arrière et regardez le plafond.
2. Tirez doucement la paupière inférieure vers le bas jusqu'à formation d'une petite poche.
3. Retournez le flacon vers le bas et pressez-le pour libérer une goutte dans chaque œil nécessitant le traitement.
4. Lâchez la paupière inférieure et fermez l'œil durant 30 secondes.
Si la goutte n’atteint pas votre œil, recommencez.
Pour éviter toute contamination ou blessure, ne laissez pas l'extrémité du compte-gouttes toucher votre œil ou autre chose.
Remettez le capuchon en place et vissez-le fermement après usage.
Essuyez tout excès de liquide sur votre joue à l'aide d'un mouchoir propre.
L'application correcte de votre collyre est très importante.
Pour toute question, adressez-vous à votre médecin ou à votre pharmacien.
Si vous avez utilisé plus de ACULAR 0,5%, collyre en solution que vous n’auriez dû
La prise d'un nombre trop important de gouttes ne devrait entraîner aucun effet indésirable. Appliquez la dose suivante à l'horaire habituel. Si, par accident, ce médicament était ingéré, buvez ou faites boire des liquides pour le diluer et contactez votre médecin.
Si vous oubliez d’utiliser ACULAR 0,5%, collyre en solution
Si vous oubliez une dose, appliquez-la dès que vous vous en rendrez compte, à moins qu'il ne soit presque l'heure de la dose suivante, auquel cas ne prenez pas la dose oubliée. Puis instillez la dose suivante comme d'habitude et poursuivez votre traitement normalement.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez d’utiliser ACULAR 0,5%, collyre en solution
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Effets indésirables très fréquents (pouvant affecter plus d’1 patient sur 10) : picotement et/ou brûlure oculaire, douleur oculaire, irritation oculaire.
Effets indésirables fréquents (pouvant affecter jusqu’à 1 patient sur 10) : réaction allergique, œdème/gonflement de l’œil ou de la paupière, démangeaisons de l’œil, œil rouge, infection de l’œil, inflammation de l’œil (en surface ou à l’intérieur), saignement de la rétine, gonflement de la partie centrale de la rétine (couche de l’œil sensible à la lumière), maux de tête, blessure accidentelle causée par l’embout du flacon touchant l’œil, augmentation de la pression dans l’œil, vision floue et/ou diminuée.
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 patient sur 1 00) : lésion ulcéreuse de l’œil ou inflammation de la couche transparente de la surface de l’œil, sécheresse de l’œil ou larmoiements.
Effets indésirables de fréquence indéterminée (ne pouvant être estimée à partir des données disponibles) : lésion de la surface de l'œil telle qu'un amincissement, une érosion, une perforation, une dégradation cellulaire, une difficulté ou un sifflement respiratoire, l’aggravation d'un asthme.
Les effets indésirables liés à la cornée (la surface de l'œil) peuvent survenir davantage en cas d'utilisation d’ACULAR 0,5%, collyre en solution pour une durée excédant deux semaines, ou si vous utilisez au même moment des gouttes de corticoïdes topiques, ou si vous avez une affection oculaire sous-jacente. Consultez immédiatement votre médecin en cas de douleur, d'irritation oculaire accrue, ou de modification de votre vision.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ACULAR 0,5%, collyre en solution ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Jetez le flacon 15 jours après ouverture, même s’il reste de la solution.
Ce médicament doit être conservé à une température ne dépassant pas 25°C.
N’utilisez pas ce médicament si l’opercule d’inviolabilité fixé sur le bouchon est endommagé.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ACULAR 0,5%, collyre en solution
· La substance active est :
Kétorolac trométamol .................................................................................................. 5 mg/ml
· Les autres composants sont :
Chlorure de benzalkonium, chlorure de sodium, édétate disodique, octoxinol 40, hydroxyde de sodium ou acide chlorhydrique (pour ajuster le pH), eau purifiée.
Qu’est-ce que ACULAR 0,5%, collyre en solution et contenu de l’emballage extérieur ?
ACULAR 0,5%, collyre en solution est un collyre en solution limpide, incolore à jaune pâle, dans un flacon en plastique.
Chaque boîte contient 1 flacon en plastique de 10ml doté d’un capuchon vissé. Chaque flacon est rempli à moitié et contient 5 ml de collyre.
Titulaire de l’autorisation de mise sur le marché
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
Exploitant de l’autorisation de mise sur le marché
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
ALLERGAN PHARMACEUTICALS IRELAND
WESTPORT
CO. MAYO
IRLANDE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).