Dernière mise à jour le 29/06/2026
ACTILYSE, poudre et solvant pour solution injectable et perfusion
Indications thérapeutiques
Classe pharmacothérapeutique : Thrombolytiques - code ATC : B01AD02
La substance active présente dans ACTILYSE est l’altéplase.
Elle fait partie du groupe de médicaments nommés agents thrombolytiques. Ces médicaments agissent en dissolvant les caillots de sang qui se sont formés dans les vaisseaux sanguins.
ACTILYSE 10, 20 ou 50 mg sont utilisés dans le traitement d’un certain nombre d’affections provoquées par les caillots de sang qui se forment dans les vaisseaux sanguins, notamment :
· la crise cardiaque provoquée par des caillots sanguins présents dans les artères du cœur (infarctus du myocarde à la phase aiguë),
· les caillots sanguins présents dans les artères des poumons (embolie pulmonaire massive à la phase aiguë),
· l’accident vasculaire cérébral provoqué par un caillot sanguin présent dans une artère du cerveau (accident vasculaire cérébral ischémique aigu) s’il s’est écoulé moins de 4 heures 30 depuis la dernière fois où vous avez été vu sans symptômes de l’AVC en cours.
Présentations
> 1 flacon(s) en verre - 1 flacon(s) en verre de 10 ml
Code CIP : 557 184-2 ou 34009 557 184 2 0
Déclaration de commercialisation : 19/01/1993
Cette présentation est agréée aux collectivités
> 1 flacon(s) en verre - 1 flacon(s) en verre de 20 ml avec canule(s) de transfert
Code CIP : 558 529-3 ou 34009 558 529 3 3
Déclaration de commercialisation : 19/05/1996
Cette présentation est agréée aux collectivités
> 1 flacon(s) en verre - 1 flacon(s) en verre de 50 ml avec canule(s) de transfert
Code CIP : 558 530-1 ou 34009 558 530 1 5
Déclaration de commercialisation : 19/05/1996
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 04/07/2012 | Extension d'indication | Le service médical rendu par ACTILYSE est important dans la nouvelle indication : traitement fibrinolytique de l’accident vasculaire cérébral ischémique à la phase aiguë dans le délai de 4h30 suivant l’apparition des symptômes, au lieu du délai de 3 heures dans la précédente indication. |
| Important | Avis du 02/07/2003 | Extension d'indication | Le service médical rendu par cette spécialité est important, dans les strictes conditions d’utilisation (en particulier délai inférieur à 3 heures, respect des contre-indications et des critères cliniques, admission en UNV) dans la nouvelle indication Traitement fibrinolytique de l’accident vasculaire cérébral ischémique à la phase aiguë. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| III (Modéré) | Avis du 04/07/2012 | Extension d'indication | Compte tenu de l'absence de donnée d'efficacité à long terme, de la faible quantité d'effet de l'altéplase versus placebo et du risque d'hémorragie intracrânienne, ACTILYSE apporte une amélioration du service médical rendu modérée (ASMR III) dans la prise en charge de l'AVC ischémique dans l'extension de la fenêtre thérapeutique (0 - 4h30 au lieu de 0 - 3h) suivant l'apparition des symptômes de l'AVC. |
| I (Majeur) | Avis du 02/07/2003 | Extension d'indication | Dans les conditions d'utilisation strictes indiquées par l'AMM et dans le seul cadre de son usage dans les UNV, cette spécialité représente une avancée thérapeutique majeure. |
Autres informations
- Titulaire de l'autorisation : BOEHRINGER INGELHEIM FRANCE
- Conditions de prescription et de délivrance :
- liste I
- réservé à l'usage en situation d'urgence selon l'article R5121-96 du code de la santé publique
- réservé à l'usage HOSPITALIER
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 361 787 6
ANSM - Mis à jour le : 16/04/2026
ACTILYSE, poudre et solvant pour solution injectable et perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un flacon de poudre contient :
Altéplase................................................................................. 10 mg (correspondant à 5 800 000 UI)
ou
Altéplase............................................................................... 20 mg (correspondant à 11 600 000 UI)
ou
Altéplase.............................................................................. 50 mg (correspondant à 29 000 000 UI),
respectivement.
L’altéplase est produite par la technique de l’ADN recombinant dans une lignée cellulaire d’ovaire de hamster chinois.
L'activité spécifique de la substance de référence interne est de 580 000 UI/mg, cette valeur étant confirmée par comparaison avec le deuxième standard international de l'OMS pour le tPA. La spécification pour l'activité spécifique de l’altéplase est de 522 000 à 696 000 UI/mg.
Excipient(s) à effet notoire
Chaque flacon de 10 mg contient 1,0 mg de polysorbate 80 (E433).
Chaque flacon de 20 mg contient 2,0 mg de polysorbate 80 (E433).
Chaque flacon de 50 mg contient 5,0 mg de polysorbate 80 (E433).
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable et perfusion.
La poudre se présente sous la forme d’un gâteau de lyophilisation blanc à jaune pâle. La préparation reconstituée est une solution limpide, incolore à jaune pâle.
4.1. Indications thérapeutiques
Traitement thrombolytique de l'infarctus du myocarde (IDM) aigu.
· Schéma thérapeutique dit "accéléré" (90 minutes) (voir rubrique 4.2) : destiné aux patients chez qui le traitement peut être débuté dans les 6 heures suivant l'apparition des symptômes.
· Schéma thérapeutique dit "des 3 heures" (voir rubrique 4.2) : destiné aux patients chez qui le traitement peut être débuté entre 6 et 12 heures après l'apparition des symptômes, à condition que l'indication soit évidente.
L'altéplase permet de réduire le taux de mortalité à 30 jours après infarctus du myocarde.
Traitement thrombolytique après embolie pulmonaire (EP) massive à la phase aiguë avec instabilité hémodynamique.
Le diagnostic devra être confirmé dans la mesure du possible par des méthodes objectives (angiographie, scanner).
Il n'existe pas de preuve d'un bénéfice en termes de morbi-mortalité dans cette indication.
Traitement thrilbolytique de l’accident vasculaire cérébral (AVC) ischémique à la phase aiguë dans les 4 heures 30 suivant le dernier moment où le patient a été vu en bonne santé, et après exclusion d’une hémorragie intracrânienne.
4.2. Posologie et mode d'administration
Le traitement par ACTILYSE devra être instauré aussitôt que possible après l'apparition des symptômes. Les recommandations suivantes concernant la posologie doivent être appliquées :
Infarctus du myocarde à la phase aiguë
Posologie
a) Schéma posologique dit "accéléré" (90 minutes) adapté aux patients à la phase aiguë de l’infarctus du myocarde pouvant être traités dans les 6 heures suivant l'apparition des symptômes.
Chez les patients de poids corporel ≥ 65 kg :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 15 mg, suivi immédiatement par |
15 ml |
7,5 ml |
|
Perfusion intraveineuse à débit constant de 50 mg sur les 30 premières minutes, immédiatement suivi par |
50 ml |
25 ml |
|
Perfusion intraveineuse à débit constant de 35 mg sur 60 minutes, sans dépasser la dose maximale totale de 100 mg |
35 ml |
17,5 ml |
Chez les patients de poids corporel < 65 kg, la dose totale doit être adaptée en fonction du poids selon le schéma d’administration suivant :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 15 mg, suivi immédiatement par |
15 ml |
7,5 ml |
|
Perfusion intraveineuse à débit constant, de 0,75 mg/kg de poids corporel (pc) sur les 30 premières minutes, suivi immédiatement par |
0,75 ml/kg |
0,375 ml/kg |
|
Perfusion intraveineuse à débit constant, de 0,5 mg/kg de poids corporel (pc) sur 60 minutes |
0,5 ml/kg |
0,25 ml/kg |
b) Schéma posologique dit "des 3 heures" adapté aux patients souffrant d’un infarctus du myocarde en phase aiguë, chez qui le traitement est mis en œuvre entre la 6e et la 12e heure suivant l'apparition des symptômes.
Chez les patients de poids corporel ≥ 65 kg :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 10 mg, suivi immédiatement par |
10 ml |
5 ml |
|
Perfusion intraveineuse à débit constant de 50 mg sur les 60 premières minutes, suivi immédiatement par |
50 ml |
25 ml |
|
Perfusion intraveineuse à débit constant de 40 mg sur 2 heures jusqu’à une dose maximale totale de 100 mg |
40 ml |
20 ml |
Chez les patients de poids corporel < 65 kg :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 10 mg, suivi immédiatement par |
10 ml |
5 ml |
|
Perfusion intraveineuse à débit constant sur 3 heures, jusqu’à la dose maximale totale de 1,5 mg/kg pc |
1,5 ml/kg pc |
0,75 ml/kg |
Traitements associés
Un traitement adjuvant anti-thrombotique est recommandé conformément aux recommandations internationales actuelles concernant la prise en charge des patients présentant un infarctus du myocarde avec sus-décalage du segment ST.
Mode d’administration
La solution reconstituée doit être administrée par voie intraveineuse et doit être utilisée immédiatement.
Le flacon de 2 mg d’altéplase n’est pas adapté pour une utilisation dans cette indication. Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Embolie pulmonaire massive à la phase aiguë
Posologie
Chez les patients de poids corporel ≥ 65 kg :
Une dose totale de 100 mg d’altéplase doit être administrée en 2 heures. L'expérience acquise porte essentiellement sur le schéma posologique suivant :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 10 mg sur 1 à 2 minutes, suivi immédiatement par |
10 ml |
5 ml |
|
Perfusion intraveineuse à débit constant de 90 mg sur 2 heures jusqu’à une dose maximale de 100 mg |
90 ml |
45 ml |
Chez les patients de poids corporel < 65 kg :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 10 mg sur 1 à 2 minutes, suivi immédiatement par |
10 ml |
5 ml |
|
Perfusion intraveineuse à débit constant sur 2 heures jusqu’à la dose maximale totale de 1,5 mg/kg pc |
1,5 ml/kg |
0,75 ml/kg |
Traitement associé
Après le traitement par ACTILYSE, une héparinothérapie doit être instaurée (ou reprise) si la valeur du TCA est inférieure à deux fois la limite supérieure de la normale. La perfusion doit être ajustée afin d'obtenir un TCA de 50 à 70 secondes (1,5 à 2,5 fois la valeur de référence).
Mode d’administration
La solution reconstituée doit être administrée par voie intraveineuse et doit être utilisée immédiatement.
Le flacon de 2 mg d’altéplase n’est pas adapté pour une utilisation dans cette indication. Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Accident vasculaire cérébral ischémique à la phase aiguë
L’instauration et le suivi du traitement doivent être réalisés sous la responsabilité d’un médecin formé et expérimenté en pathologie neurovasculaire (voir rubriques 4.3 et 4.4).
Le traitement par ACTILYSE doit être initié, chez les adultes et les adolescents âgés de 16 ans ou plus (voir rubriques 4.3 et 4.4), le plus tôt possible dans les 4 heures 30 suivant le dernier moment où le patient a été vu en bonne santé, et après exclusion d’une hémorragie intracrânienne par des techniques d’imagerie adaptées. L’effet du traitement étant temps-dépendant, l’instauration précoce du traitement augmente les chances d’évolution favorable. Au-delà de 4h30 après l’apparition des symptômes, l’administration d’ACTILYSE est associée à un rapport bénéfice/risque défavorable, ACTILYSE ne doit donc pas être administré (voir rubrique 5.1).
Posologie
La posologie totale recommandée est de 0,9 mg d’altéplase/kg de poids corporel (dose maximale de 90 mg), en commençant par 10% de la dose totale devant être administrée comme bolus intraveineux initial, suivi immédiatement par une perfusion intraveineuse sur 60 minutes de la dose totale restante.
|
TABLE DES DOSES POUR L’AVC ISCHEMIQUE A LA PHASE AIGUË |
|||
|
En utilisant la concentration standard recommandée de 1 mg/ml, le volume (ml) à administrer est égal à la valeur de la dose recommandée (mg) |
|||
|
Poids (kg) |
Dose totale (mg) |
Dose en bolus (mg) |
Dose en perfusion* (mg) |
|
40 |
36,0 |
3,6 |
32,4 |
|
42 |
37,8 |
3,8 |
34,0 |
|
44 |
39,6 |
4,0 |
35,6 |
|
46 |
41,4 |
4,1 |
37,3 |
|
48 |
43,2 |
4,3 |
38,9 |
|
50 |
45,0 |
4,5 |
40,5 |
|
52 |
46,8 |
4,7 |
42,1 |
|
54 |
48,6 |
4,9 |
43,7 |
|
56 |
50,4 |
5,0 |
45,4 |
|
58 |
52,2 |
5,2 |
47,0 |
|
60 |
54,0 |
5,4 |
48,6 |
|
62 |
55,8 |
5,6 |
50,2 |
|
64 |
57,6 |
5,8 |
51,8 |
|
66 |
59,4 |
5,9 |
53,5 |
|
68 |
61,2 |
6,1 |
55,1 |
|
70 |
63,0 |
6,3 |
56,7 |
|
72 |
64,8 |
6,5 |
58,3 |
|
74 |
66,6 |
6,7 |
59,9 |
|
76 |
68,4 |
6,8 |
61,6 |
|
78 |
70,2 |
7,0 |
63,2 |
|
80 |
72,0 |
7,2 |
64,8 |
|
82 |
73,8 |
7,4 |
66,4 |
|
84 |
75,6 |
7,6 |
68,0 |
|
86 |
77,4 |
7,7 |
69,7 |
|
88 |
79,2 |
7,9 |
71,3 |
|
90 |
81,0 |
8,1 |
72,9 |
|
92 |
82,8 |
8,3 |
74,5 |
|
94 |
84,6 |
8,5 |
76,1 |
|
96 |
86,4 |
8,6 |
77,8 |
|
98 |
88,2 |
8,8 |
79,4 |
|
100+ |
90,0 |
9,0 |
81,0 |
*administré à une concentration de 1 mg/ml sur 60 minutes, en perfusion à débit constant.
Traitement associé
Médicaments affectant la coagulation / fonction plaquettaire
La tolérance et l’efficacité de ce protocole d’administration en association avec l’héparine ou un antiagrégant plaquettaire comme l’acide acétylsalicylique au cours des 24 premières heures suivant l’administration d’ACTILYSE n’ont pas été suffisamment étudiées. Par conséquent, l’administration d’héparine par voie intraveineuse ou d’un antiagrégant plaquettaire comme l’acide acétylsalicylique doit être évité au cours des premières 24 heures suivant l’administration d’ACTILYSE en raison d’un risque d’hémorragie augmenté. Si l’administration d’héparine est rendue nécessaire pour d’autres indications (par exemple en prévention de thrombose veineuse profonde), la posologie ne doit pas dépasser 10 000 UI par jour, par voie sous-cutanée.
Mode d’administration
La solution reconstituée doit être administrée immédiatement, par voie intraveineuse.
Le flacon de 2 mg d’altéplase n’est pas adapté pour une utilisation dans cette indication. Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Population pédiatrique
L’expérience de l’utilisation d’ACTILYSE chez les enfants et les adolescents est limitée. ACTILYSE est contre-indiqué chez les enfants et les adolescents âgés de moins de 16 ans pour le traitement de l’accident vasculaire cérébral ischémique à la phase aiguë (voir rubrique 4.3). La dose pour les adolescents âgés de 16 ans ou plus est la même que celle pour les adultes (voir rubrique 4.4 pour les recommandations sur les techniques d’imageries à utiliser en amont).
Hypersensibilité connue à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Contre-indications dans l’infarctus du myocarde à la phase aiguë, dans l’embolie pulmonaire massive à la phase aiguë et dans l’accident vasculaire cérébral ischémique à la phase aiguë
ACTILYSE est contre-indiqué dans toutes les situations associées à un risque élevé de saignement :
· trouble hémorragique significatif actuel ou au cours des six derniers mois,
· diathèse hémorragique connue,
· hémorragie sévère ou potentiellement dangereuse, manifeste ou récente,
· antécédents de lésion du système nerveux central (par exemple néoplasie, anévrisme, intervention chirurgicale intracérébrale ou intra-rachidienne),
· accouchement récent (moins de 10 jours), ponction récente d'un vaisseau non accessible à la compression (par exemple, ponction de la veine sous-clavière ou jugulaire),
· hypertension artérielle sévère non contrôlée (voir rubrique 4.4),
· endocardite bactérienne, péricardite,
· pancréatite aiguë,
· ulcères gastro-intestinaux actifs , varices œsophagiennes,anévrisme artériel connu et/ou de malformations artérielles ou veineuses,
· néoplasie majorant le risque hémorragique,
· hépatopathie sévère, y compris insuffisance hépatique, cirrhose, hypertension portale (varices œsophagiennes) et hépatite évolutive,
· intervention chirurgicale ou traumatismes importants au cours des 3 derniers mois.
Contre-indications complémentaires dans l’indication d’infarctus du myocarde à la phase aiguë et dans l’embolie pulmonaire massive à la phase aiguë
· tout antécédent connu d’accident vasculaire cérébral hémorragique ou d’origine inconnue,
· antécédents connus d’accident vasculaire cérébral ischémique ou d’accident ischémique transitoire (AIT) au cours des six mois précédents, sauf si l’accident vasculaire cérébral ischémique à la phase aiguë est survenu dans les 4h30 précédentes.
· Traitement par des anticoagulants oraux à dose efficace (par exemple antagonistes de la vitamine K avec un INR > 1,3) (voir rubrique 4.4)
Contre-indications complémentaires dans l’indication d’accident vasculaire cérébral ischémique à la phase aiguë
· déficit neurologique mineur ou symptômes s’améliorant rapidement avant l’initiation du traitement,
· accident vasculaire cérébral jugé sévère d’après l’examen clinique (par exemple NIHSS > 25) et/ou l’examen d’imagerie réalisé à l’aide d’une technique adaptée ,
· antécédents connus ou suspicion d'hémorragie intracrânienne
· signes d’hémorragie intracrânienne (HIC) au scanner,
· symptômes suggérant une hémorragie sous-arachnoïdienne, même si la tomodensitométrie est normale ,
· traitement par des anticoagulants oraux à dose efficace (par exemple antagonistes de la vitamine K avec un INR > 1,7) (voir rubrique 4.4)
· administration d’héparine au cours des 48 heures précédentes avec TCA (temps de céphaline + activateur) dépassant la limite supérieure de la normale,
· antécédents d’AVC et de diabète concomitant ,
· antécédent d’accident vasculaire cérébral au cours des 3 derniers mois,
· numération plaquettaire inférieure à 100 000/mm3,
· pression artérielle systolique > 185 mm Hg ou pression artérielle diastolique > 110 mm Hg, lorsque la pression artérielle ne peut être réduite en dessous de ces limites par une prise en charge rigoureuse
· glycémie < 50 mg/dL (voir rubrique 4.4) ou > 400 mg/dL (< 2,8mmol/l ou > 22,2 mmol/l).
Utilisation chez l’enfant, l’adolescent
ACTILYSE n’est pas indiqué pour le traitement de l’accident vasculaire cérébral ischémique à la phase aiguë chez les enfants âgés de moins de 16 ans (pour les adolescents âgés de 16 ans ou plus, voir rubrique 4.4).
4.4. Mises en garde spéciales et précautions d'emploi
Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
La présentation appropriée d’ACTILYSE doit être choisie soigneusement et en accord avec l’utilisation prévue. Le flacon de 2 mg d’altéplase n’est pas adapté pour une utilisation à la phase aiguë de l’infarctus du myocarde, de l’embolie pulmonaire massive à la phase aiguë ou de l’accident vasculaire cérébral ischémique (en raison d’un risque de sous-dosage important). Seuls les flacons de 10 mg, 20 mg et 50 mg sont indiqués pour ces utilisations.
Il est recommandé d’administrer ACTILYSE au sein de structures disposant en permanence d’équipements et de traitements de réanimation.
Hypersensibilité
Les réactions d’hypersensibilité à médiation immunitaire associées à l’administration d’ACTILYSE peuvent être induites par la substance active altéplase ou à l’un des excipients.
Aucune formation durable d’anticorps dirigés contre la molécule recombinante d’activateur tissulaire du plasminogène humain n’a été observée après le traitement. Il n’y a pas de données relatives à une réadministration d’ACTILYSE.
Il existe également un risque de réactions d’hypersensibilité, médiées par un mécanisme non immunologique.
La réaction d’hypersensibilité la plus fréquemment rapportée avec ACTILYSE est l’angio-œdème. Le risque peut être accru dans le cas d’un accident vasculaire cérébral ischémique à la phase aiguë et/ou par la prise d’un traitement concomitant à base d’inhibiteurs de l’enzyme de conversion de l’angiotensine (voir rubrique 4.5). Le risque d’angio-œdème doit être surveillé chez les patients traités avec ACTILYSE , pendant la perfusion et dans les 24 heures suivant la perfusion.
En cas d’apparition d’une réaction d’hypersensibilité sévère (par ex. angio-œdème), la perfusion doit être interrompue et un traitement approprié instauré immédiatement. Cela peut inclure l’intubation.
Saignement
La complication la plus fréquemment rencontrée pendant le traitement par ACTILYSE est le saignement. L’utilisation concomitante d’autres substances actives agissant sur la coagulation ou la fonction plaquettaire peut contribuer aux saignements (voir rubrique 4.3) (voir rubrique 4.2 pour l’accident vasculaire cérébral (AVC) ischémique à la phase aiguë). Durant le traitement par ACTILYSE, la fibrine est lysée, pouvant causer des saignements aux sites de ponction récente. Ainsi, tout traitement thrombolytique nécessite une attention particulière à tous les sites potentiels de saignements (incluant ceux faisant suite à l’insertion d’un cathéter, points de ponction artérielle ou veineuse, sièges d’incision et points de piqûre). L’utilisation de cathéters rigides, les injections intramusculaires et interventions non essentielles doivent être évitées durant le traitement par ACTILYSE.
Le traitement fibrinolytique et le traitement concomitant par de l’héparine doit être interrompu immédiatement en cas de survenue d'un saignement potentiellement dangereux, en particulier d’une hémorragie cérébrale. En général, il n'est cependant pas nécessaire d'administrer des facteurs de coagulation en raison de la courte demi-vie de l'altéplase et de ses faibles effets sur ces facteurs de coagulation systémiques. Dans la plupart des cas, les saignements peuvent être contrôlés par une interruption des traitements thrombolytique et anticoagulant, par l’administration d’une solution de remplissage vasculaire ou par une pression manuelle sur le vaisseau lésé. On peut envisager de recourir à la protamine en cas d'administration d'héparine dans les 4 heures précédant la survenue de l’hémorragie. Chez les rares patients ne répondant pas à ces mesures conservatrices, l’utilisation appropriée de produits de transfusion peut être envisagée. Une transfusion de cryoprécipité, de plasma frais congelé ou de plaquettes peut être envisagée en surveillant les paramètres cliniques et biologiques après chaque administration. Le taux de fibrinogène à atteindre en cas de perfusion de cryoprécipité est de 1 g/l. Les antifibrinolytiques constituent la dernière alternative thérapeutique.
Le risque d'hémorragie intracrânienne est augmenté chez le sujet âgé, par conséquent, il y a lieu d'évaluer avec soin le rapport bénéfice/risque chez ce type de patient.
L'utilisation de l'altéplase doit prendre soigneusement en compte les risques éventuels et le bénéfice thérapeutique attendu, en particulier dans les cas suivants :
· traumatismes mineurs récents, tels que biopsies, ponction de gros vaisseaux, injections intramusculaires
· réanimation cardio-pulmonaire ou massage cardiaque prolongé (> 2 minutes) ou traumatique.
· patients recevant un anticoagulant par voie orale : l’utilisation d’ACTILYSE peut être envisagée lorsque la dose ou le délai depuis la dernière prise du traitement anticoagulant rend peu probable un effet résiduel et si le(s) test(s) approprié(s) de l’activité anticoagulante du/des produit(s) concerné(s) ne montrent pas d’activité cliniquement significative sur le système de la coagulation (par exemple, un INR ≤ 1,3 (IDM aigu et EP) ou INR ≤ 1,7 (AVC ischémique) pour les antagonistes de la vitamine K ou, pour les autres anticoagulants oraux, un résultat au[x] test[s] approprié[s] ne dépassant pas la limite supérieure à la normale).
Thromboembolie
L’utilisation d’ACTILYSE peut augmenter le risque d’événements thromboemboliques chez les patients présentant un thrombus, par exemple un thrombus ventriculaire gauche (sténose mitrale ou fibrillation auriculaire, etc.).
Population pédiatrique
Il y a peu d'expérience de l'utilisation d’ACTILYSE chez les enfants et les adolescents.
Lorsque ACTILYSE est envisagé comme traitement de l’accident vasculaire cérébral ischémique à la phase aiguë chez des adolescents âgés de 16 ans ou plus, sélectionnés avec attention, le bénéfice du traitement doit être attentivement évalué de manière individuelle, par rapport aux risques encourus. Ce choix doit être discuté avec le patient et les parents ou le tuteur le cas échéant. Les adolescents âgés de 16 ans ou plus doivent être traités selon les instructions du RCP données pour les patients adultes, après une technique d’imagerie appropriée permettant d’exclure les pathologies mimant l’accident vasculaire cérébral et confirmant l’occlusion artérielle, correspondant au déficit neurologique (voir rubrique 5.1).
Mises en garde spéciales et précautions particulières d’emploi complémentaires dans l’indication d’infarctus du myocarde à la phase aiguë et d’embolie pulmonaire massive à la phase aiguë
Ne pas administrer une dose d'altéplase supérieure à 100 mg en raison de la majoration du risque d'hémorragie intracrânienne.
Des précautions particulières doivent être prises pour s’assurer que la dose d’altéplase administrée est telle que décrite dans la rubrique 4.2.
L’utilisation de l’altéplase doit prendre soigneusement en compte les risques éventuels et le bénéfice thérapeutique attendu, en particulier chez les patients dont :
· la pression artérielle systolique est > 160 mm Hg (voir rubrique 4.3)
· le poids corporel est < 50 kg
· les personnes d’un âge avancé, par exemple chez les patients âgés de 75 ans ou plus, car cela peut augmenter le risque d’hémorragie intracérébrale.
Antagonistes des récepteurs GPIIb/IIIa : l’administration concomitante d’un antagoniste du récepteur GPIIb/IIIa accroît le risque hémorragique.
Mises en gardes spéciales et précautions particulières d’emploi en cas d’infarctus du myocarde à la phase aiguë
Arythmies :
Une thrombolyse coronarienne peut entraîner une arythmie de reperfusion.
Une arythmie de reperfusion peut causer un arrêt cardiaque, engager le pronostic vital et nécessiter l’utilisation de thérapies antiarythmiques conventionnelles.
Mises en garde spéciales et précautions particulières d’emploi complémentaires en cas d’accident vasculaire cérébral ischémique à la phase aiguë
Le traitement thrombolytique nécessite une surveillance adéquate. L’instauration et le suivi du traitement doivent être réalisés sous la responsabilité d’un médecin formé et expérimenté en pathologie neurovasculaire et sur l'utilisation des traitements thrombolytiques, disposant des installations nécessaires pour surveiller cette utilisation. Pour la vérification du choix de traitement, des mesures de diagnostic à distance (télémédecine) peuvent être considérées comme appropriées (voir rubriques 4.1 et 4 .2).
Mises en garde spéciales / Populations ayant une diminution du rapport bénéfice/risque
Saignements
L’hémorragie intracérébrale représente l’effet indésirable majeur du traitement de l’accident vasculaire cérébral ischémique à la phase aiguë (jusqu’à 15 % des patients sans augmentation de la mortalité globale et sans augmentation significative du critère combiné mortalité globale + handicap majeur, c’est-à-dire présentant un score sur l’échelle de Rankin modifiée (mRS) de 5 et 6).
Comparativement aux autres indications, les patients traités par ACTILYSE dans le cadre d’un accident vasculaire cérébral ischémique à la phase aiguë, présentent une augmentation marquée du risque d’hémorragie intracrânienne, les hémorragies survenant préférentiellement dans la zone de l’infarctus.
Cette mise en garde s’applique notamment aux cas suivants :
· plus le délai de traitement suivant l’apparition des symptômes de l’accident vasculaire cérébral augmente plus le bénéfice clinique net diminue. Par conséquent l’administration d’ACTILYSE ne doit pas être retardée.
· les patients ayant reçu au préalable un traitement par l’acide acétylsalicylique (AAS) peuvent présenter un risque accru d’hémorragie intracérébrale et/ou de mortalité, en particulier si le traitement par ACTILYSE est mis en place tardivement.
· par rapport aux patients plus jeunes, les patients ayant un âge avancé (âgés de plus de 80 ans) peuvent présenter de moins bons résultats cliniques indépendamment du traitement. Ils sont également susceptibles de faire davantage d’accident vasculaire cérébral sévère associé à un risque absolu plus élevé d’hémorragie intracérébrale en cas de traitement par thrombolyse par rapport à l’accident vasculaire cérébral modéré traité par thrombolyse ou par rapport aux patients non traités par thrombolyse. Bien que les données disponibles montrent que le bénéfice net d’ACTILYSE chez les patients âgés de plus de 80 ans est plus faible que chez les patients plus jeunes. ACTILYSE peut être utilisé chez les patients âgés de plus de 80 ans sur la base du bénéfice et du risque individuel (voir rubrique 5.1). Les patients ayant un âge avancé doivent être sélectionnés avec attention en prenant en compte à la fois l’état de santé général et le statut neurologique.
Populations ayant une diminution du rapport bénéfice/risque :
Le rapport bénéfice/risque de l'administration d'ACTILYSE doit être soigneusement évalué chez les patients atteints d'AVCI présentant les conditions suivantes :
· le bénéfice thérapeutique est diminué chez les patients ayant des antécédents d’accident vasculaire cérébral (voir également la rubrique 4.3) ou présentant un diabète non contrôlé. Chez ces patients, le rapport bénéfice-risque est jugé moins favorable, mais reste positif.
· chez les patients présentant une forme très légère d’accident vasculaire cérébral, les risques liés au traitement l’emportent sur le bénéfice attendu (voir rubrique 4.3).
· les patients ayant fait un accident vasculaire cérébral très sévère, présentent un risque plus important d’hémorragie intracrânienne et de décès et ne doivent pas être traités par ACTILYSE.
· les patients ayant fait des infarctus étendus ont un risque accru d’évolution défavorable (dont hémorragies sévères et décès). Le rapport bénéfice/risque doit être soigneusement évalué chez ces patients.
· chez les patients présentant un accident vasculaire cérébral, la probabilité d’évolution favorable diminue avec l’augmentation du délai de traitement depuis l’apparition des symptômes, avec l’âge, avec le degré de sévérité de l’atteinte et avec les taux élevés de la glycémie à l’admission, tandis que le risque de handicap sévère, de décès ou d’hémorragie intracrânienne augmente, indépendamment du traitement.
· convulsions au moment de la survenue de l’AVC. (Chez ces patients, un traitement thrombolytique doit être envisagé uniquement en l’absence de suspicion de stroke mimic [déficit neurologique brutal d’origine non vasculaire] ou de traumatisme crânien significatif.)
· chez les patients présentant initialement une glycémie < 50 mg/dL, la thrombolyse peut être envisagée après normalisation de la glycémie, si le diagnostic d’AVC ischémique aigu persiste (voir rubrique 4.3).
Surveillance de la pression artérielle
Une surveillance de la pression artérielle est nécessaire lors de l’administration du traitement et dans les premières 24h suivant le traitement par alteplase Si la pression artérielle systolique est > 180 mm Hg ou si la pression artérielle diastolique est > 105 mm Hg, un traitement antihypertenseur par voie intraveineuse est recommandé.
Autres mises en gardes spéciales
La reperfusion de la zone de l’ischémie peut entraîner un œdème cérébral dans la zone de l’infarctus. En raison d’un risque hémorragique accru, aucun traitement antiagrégant plaquettaire ne doit être initié dans les premières 24 heures suivant le traitement thrombolytique par l’altéplase.
Actilyse contient du polysorbate 80
Ce médicament contient 1,0 mg, 2,0 mg ou 5,0 mg de polysorbate 80 dans chaque flacon de 10 mg, 20 mg ou de 50 mg respectivement. Les polysorbates peuvent provoquer des réactions allergiques.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
+ Médicaments agissant sur la coagulation/fonction plaquettaire
Les produits agissant sur la coagulation ou ceux modifiant la fonction plaquettaire sont susceptibles d’accroître le risque hémorragique (qu’ils soient administrés avant, pendant ou après un traitement par alteplase). L’utilisation de ces produits doit être évitée dans les 24 heures suivant le traitement de l’AVC ischémique par ACTILYSE. Voir les rubriques 4.2, 4.3 et 4.4 pour l’utilisation de ces substances en prétraitement.
+ Inhibiteurs de l’enzyme de conversion de l’angiotensine
L’administration concomitante d’IEC peut augmenter le risque de survenue d’une réaction d’hypersensibilité, voir rubrique 4.4.
L’administration concomitante d’un antagoniste des récepteurs GPIIb/IIIa augmente le risque hémorragique.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données sur l'administration d'altéplase à des femmes enceintes sont limitées. Des études non-cliniques réalisées avec l’altéplase à des doses plus élevées que celles utilisées chez l’homme ont révélé une immaturité fœtale et/ou une embryotoxicité secondaires à l’activité pharmacologique connue du produit. L’altéplase n’est pas considéré comme tératogène (voir rubrique 5.3). En cas de menace du pronostic vital, il faut prendre en considération les bénéfices attendus et les risques éventuels.
Allaitement
On ne sait pas si l’altéplase est excrété dans le lait humain et les données sur l’excrétion dans le lait animal sont limitées.
Il convient d'être prudent lors de l’utilisation d’Actilyse chez la femme qui allaite et une décision concernant l’interruption de l'allaitement pendant les 24 premières heures suivant l'utilisation d'Actilyse doit être prise.
Fertilité
Il n’y a pas de données cliniques sur la fertilité disponibles pour ACTILYSE. Les études non-cliniques réalisées avec l’altéplase n’ont pas montré d’effet indésirable sur la fertilité (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables les plus fréquemment associés à l’administration d’ACTILYSE sont les hémorragies, sous différentes formes, associées à une chute de l'hématocrite et/ou de l'hémoglobinémie.
Les effets indésirables cités ci-dessous sont présentés par fréquence et par classe de systèmes d’organes. Les groupes de fréquence sont définis selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
A l’exception des cas d’hémorragies intracrâniennes comme effet indésirable pour le traitement de l’accident vasculaire cérébral et des cas d’arythmies de reperfusion pour le traitement de l’infarctus du myocarde à la phase aiguë, aucune raison médicale ne laisse supposer que le profil qualitatif et quantitatif des effets indésirables d’ACTILYSE puisse être différent dans le cadre du traitement de l’embolie pulmonaire massive à la phase aiguë et de l’accident vasculaire cérébral ou dans le cadre du traitement de l’infarctus du myocarde à la phase aiguë.
Tableau 1 : Effets indésirables dans les indications d’infarctus du myocarde à la phase aiguë, d’embolie pulmonaire massive à la phase aiguë et d’accident vasculaire cérébral ischémique à la phase aiguë
|
Classe de systèmes d’organes |
Effet indésirable |
|
Hémorragies |
|
|
Très fréquent |
L’hémorragie intracérébrale représente le principal effet indésirable dans le traitement de l’accident vasculaire cérébral ischémique aigu. Toute hémorragie, dont celles listées dans ce tableau (ex : hémorragie intracrânienne et non-intracrânienne) |
|
Fréquent |
Hémorragie intracérébrale (telle que hémorragie cérébrale, hématome cérébral, accident vasculaire cérébral hémorragique, transformation hémorragique d’un accident vasculaire cérébral, hématome intracrânien, hémorragie sous-arachnoïdienne) en cas de traitement d’un infarctus du myocarde aigu ou d’une embolie pulmonaire massive à la phase aiguë Hémorragie pharyngée Hémorragie gastro-intestinale (telle qu’hémorragie gastrique, hémorragie ulcéreuse gastrique, hémorragie du rectum, hématémèse, méléna, hémorragie buccale, saignements des gencives) Ecchymoses Hémorragie urogénitale (telle qu’hématurie, hémorragie des voies urinaires) Hémorragie au site d’injection (hémorragie au site de ponction, hématome au site du cathéter, hémorragie au site du cathéter) |
|
Peu fréquent |
Hémorragie pulmonaire (telle que hémoptysie, hémothorax, hémorragie des voies respiratoires) Epistaxis Otorragie |
|
Rare |
Saignements oculaires Hémopéricarde Hémorragie retropéritonéale (telle que hématome retropéritonéal) |
|
Fréquence indéterminée*** |
Saignements des organes parenchymateux (tel que hémorragie hépatique) |
|
Affections du système immunitaire* |
|
|
Rare |
Réactions d’hypersensibilité (par exemple éruption cutanée, urticaire, bronchospasme, angio-œdème, hypotension, choc)* |
|
Très rare |
Anaphylaxie grave |
|
Affections du système nerveux |
|
|
Très rare |
Evènements d’origine centrale (par exemple crise d’épilepsie, convulsions, aphasie, troubles de la parole, délires, troubles neuropsychiatriques aigus, agitation, confusion, dépression, psychose), souvent associés à des évènements cérébrovasculaires d’origine ischémique ou hémorragique |
|
Affections cardiaques** |
|
|
Très fréquent |
Ischémie myocardique/angor récurrent(e), hypotension et insuffisance cardiaque/ œdème pulmonaire |
|
Fréquent |
Choc cardiogénique, arrêt cardiaque et récidive d’infarctus |
|
Peu fréquent |
Arythmies de reperfusion (tel qu’arythmie, extrasystole, bloc auriculo-ventriculaire du 1er degré jusqu’au bloc complet, fibrillation/flutter auriculaire, bradycardie, tachycardie, arythmie ventriculaire, tachycardie/fibrillation ventriculaire, dissociation électromécanique) Régurgitation mitrale, embolie pulmonaire, autre embolie systémique/embolie cérébrale, anomalies du septum ventriculaire |
|
Affections vasculaires |
|
|
Rare |
Embolie pouvant avoir des conséquences dans les organes affectés |
|
Affections gastro-intestinales |
|
|
Rare |
Nausées |
|
Fréquence indéterminée*** |
Vomissements |
|
Investigations |
|
|
Peu fréquent |
Diminution de la pression artérielle |
|
Fréquence indéterminée*** |
Augmentation de la température corporelle
|
|
Lésions, intoxications et complications liées aux procédures |
|
|
Fréquence indéterminée*** |
Embolie graisseuse (embolie par des cristaux de cholestérol) pouvant avoir des conséquences dans les organes affectés |
|
Actes médicaux et chirurgicaux |
|
|
Fréquence indéterminée*** |
Nécessité d’une transfusion sanguine |
*Voir les rubriques 4.4 et 4.5.
**Affections cardiaques
Comme avec les autres agents thrombolytiques, les évènements décrits ci-dessus dans la section correspondante ont été rapportés en tant que séquelles d’un infarctus du myocarde et/ou d’un traitement thrombolytique. Ces événements cardiaques peuvent menacer le pronostic vital et entraîner le décès.
***Calcul des fréquences
Cet effet indésirable a été observé après la commercialisation. Avec 95% de certitude, la catégorie de fréquence n’est pas supérieure à « rare », mais pourrait être plus faible. L’estimation précise de la fréquence n’est pas possible car l’effet indésirable n’a pas été décrit dans la base de données des 8299 patients des essais cliniques.
Des décès et des handicaps irréversibles ont été rapportés chez des patients ayant présenté un accident vasculaire cérébral (y compris des saignements intracrâniens) ou d’autres épisodes de saignements graves.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Symptômes
Si la dose maximale recommandée est dépassée, le risque d’hémorragie intracrânienne augmente.
Malgré la relative spécificité de l'altéplase pour la fibrine, un surdosage peut entraîner une diminution cliniquement significative des taux de fibrinogène et des autres facteurs de la coagulation.
Traitement
Dans la plupart des cas, il suffit d'attendre la régénération physiologique de ces éléments après la fin du traitement par ACTILYSE.
Toutefois, si une hémorragie sévère se produit, la transfusion de plasma frais congelé est recommandée, ainsi que, si nécessaire, l'administration d'antifibrinolytiques de synthèse.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Thrombolytiques, code ATC : B01AD02.
Mécanisme d’action
Le principe actif d’ACTILYSE est l'altéplase, un activateur tissulaire du plasminogène humain recombinant, c’est-à-dire une glycoprotéine qui active la biotransformation du plasminogène en plasmine.
Après administration intraveineuse, l'altéplase circulante reste relativement inactive. Elle n'est activée qu'après liaison à la fibrine et induit alors la conversion du plasminogène en plasmine, entraînant ainsi la dissolution du caillot de fibrine.
Effets pharmacodynamiques
En raison de sa relative spécificité pour la fibrine, l’altéplase, à la dose de 100 mg, diminue faiblement les taux de fibrinogène circulant jusqu’à environ 60% à 4 heures, mais avec un rétablissement à plus de 80% au bout de 24 heures. Les concentrations de plasminogène et d’alpha-2-antiplasmine diminuent environ jusqu’à des taux de 20% et 35% respectivement quatre heures après le traitement mais augmentent à nouveau par la suite jusqu’à plus de 80% au bout de 24 heures. Une diminution marquée et prolongée du fibrinogène circulant ne s’observe que chez un petit nombre de patients.
Efficacité et sécurité clinique
Lors d'une étude portant sur plus de 40 000 patients présentant un infarctus du myocarde à la phase aiguë (GUSTO), l'administration de 100 mg d'altéplase en 90 minutes, avec perfusion intraveineuse concomitante d'héparine, a été associée à un taux de mortalité à 30 jours plus faible (6,3 %) que celui enregistré sous streptokinase (1,5 millions d'unités sur 60 minutes) et héparine intraveineuse ou sous-cutanée (7,3 %). Le taux de reperméabilisation du vaisseau lésé a été supérieur chez les patients sous altéplase que chez ceux traités par la streptokinase, 60 et 90 minutes après la thrombolyse.
Il n'y a cependant pas eu de différence entre les taux de reperméabilisation mesurés au bout de 180 minutes et au-delà.
La mortalité à 30 jours est plus faible sous altéplase que chez les patients ne recevant aucun traitement thrombolytique.
La libération d'alpha-hydroxybutyrate-deshydrogénase (HBDH) est diminuée. Par rapport à l'absence de traitement thrombolytique, le traitement par l'altéplase a montré qu'il préservait la fonction ventriculaire globale et la mobilité pariétale locale.
Infarctus du myocarde à la phase aiguë
Une étude contrôlée contre placebo menée chez des patients traités 6 à 12 heures après l'apparition des symptômes par 100 mg d'ACTILYSE en 3 heures (LATE), a montré une diminution du taux de mortalité à 30 jours.
En présence de signes évidents d'infarctus du myocarde, un traitement instauré jusqu'à 24 heures après l'apparition de la symptomatologie peut s'avérer bénéfique.
Embolie pulmonaire massive à la phase aiguë
En cas d'embolie pulmonaire massive aiguë avec instabilité des paramètres hémodynamiques, le traitement thrombolytique par ACTILYSE réduit rapidement la taille du thrombus et diminue la pression artérielle pulmonaire. On ne dispose pas d'éléments sur le taux de mortalité dans cette indication.
Accident vasculaire cérébral ischémique à la phase aiguë
Dans deux études réalisées aux Etats-Unis (NINDS A/B), une proportion significativement plus élevée a présenté une évolution favorable avec l’altéplase par comparaison avec le placebo (absence de handicap ou handicap mineur), par rapport au groupe placebo. Ces résultats ont été confirmés dans l’essai ECASS III (voir paragraphe ci-dessous), alors que deux études européennes et une étude américaine complémentaire réalisées entre-temps (dans des conditions non-conformes au RCP actuel du produit) n’avaient pas permis d’obtenir des preuves adéquates.
L’étude ECASS III était un essai en double insu contrôlé versus placebo, conduit en Europe chez des patients présentant un AVC ischémique à la phase aiguë, dans une fenêtre de temps allant de 3h à 4h30 après l’apparition des symptômes. L’administration du traitement dans l’étude ECASS III était conforme au RCP européen d’ACTILYSE dans l’indication AVC ischémique à la phase aiguë, mis à part la borne supérieure de la fenêtre de traitement, à savoir 4h30.
Le critère principal d’évaluation était le degré de handicap à 90 jours évalué par l’échelle de Rankin modifiée (mRS), classifié entre favorable (score mRS de 0 à 1) et défavorable (score mRS de 2 à 6). Un total de 821 patients (418 altéplase/403 placebo) a été randomisé. Un plus grand nombre de patients a obtenu un résultat clinique favorable avec l’altéplase (52,4%) vs placebo (45,2% ; odds ratio [OR], 1,34 ; IC95% [1,02 ; 1,76] ; p = 0,038). L’incidence des hémorragies intracrâniennes, symptomatiques et non symptomatiques, était plus élevée avec l’altéplase qu’avec le placebo (toutes hémorragies intracrâniennes : 27,0% vs 17,6%, p = 0,0012 ; hémorragies intracrâniennes symptomatiques selon la définition d’ECASS III : 2,4% vs 0,2%, p=0,008).
La mortalité était faible et non significativement différente entre l’altéplase (7,7%) et le placebo (8,4% ; p = 0,681). Les résultats de sous-groupe d’ECASS III confirment qu’un délai plus long entre l’apparition des symptômes et le début du traitement est associé à un risque majoré de mortalité et d’hémorragies intracrâniennes symptomatiques. Les résultats d’ECASS III montrent un bénéfice clinique net favorable pour ACTILYSE dans la fenêtre 3h-4h30, alors que les données groupées montrent que celui-ci n’est plus favorable au-delà de 4h30.
La sécurité d’emploi et l’efficacité d’ACTILYSE dans le traitement de l’AVC ischémique à la phase aiguë jusqu’à 4h30 après l’apparition des symptômes ont été évaluées dans le cadre d’un registre en cours (SITS-ISTR : « The Safe Implementation of Thrombolysis in Stroke Registry »). Dans cette étude observationnelle, les données de tolérance de 21 566 patients traités dans la fenêtre 0-3h ont été comparées avec les données de 2376 patients traités dans la fenêtre 3h-4h30 après le début de l’AVC ischémique à la phase aiguë. L’incidence des hémorragies intracrâniennes symptomatiques (selon la définition de l’étude SITS-MOST) a été plus élevée dans la fenêtre 3h-4h30 (2,2%) par rapport à la fenêtre 0-3h (1,7%). Les taux de mortalité à 3 mois ont été similaires, en comparant la fenêtre 3h-4h30 (12,0%) avec la fenêtre 0-3h (12,3%), avec un OR non ajusté de 0,97 (IC95% [0,84 ; 1,13], p=0,70) et un OR ajusté de 1,26 (IC95% [1,07 ; 1,49], p=0,005). Les données observationnelles SITS appuient les résultats des essais cliniques qui montrent que l’intervalle de temps entre l’apparition des symptômes et le début du traitement est un important facteur de prédiction de l’évolution suite au traitement par l’altéplase de l’AVC ischémique à la phase aiguë.
Patients âgés (> 80 ans)
Des méta-analyses ajustées sur des données individuelles, provenant de 6 756 patients, dont les patients âgés de plus de 80 ans, inclus dans 9 essais cliniques randomisés comparant l’altéplase à un placebo ou contrôlés en ouvert, ont été utilisées pour évaluer le rapport bénéfice-risque de l’altéplase chez des patients âgés de plus de 80 ans. La probabilité d’évolution favorable après un accident vasculaire cérébral (score mRS de 0 à 1 au 90ème jour/180) était augmentée et était associée à un meilleur bénéfice lorsque les patients étaient traités plus tôt, pour tous les groupes d’âges (p= 0,0203 pour l’interaction) et sont indépendantes de l’âge.
L’effet du traitement par altéplase était similaire pour les patients âgés de 80 ans ou plus jeunes [délai moyen de traitement 4,1 heures : 990/2512 (39%) patients traités par altéplase vs 853/2515 (34%) patients du groupe contrôle qui ont eu une évolution favorable après un accident vasculaire cérébral au 90ème jour/180 ; OR 1,25, 95% CI 1,10-1,42] et les patients âgés de plus de 80 ans [délai de traitement moyen 3,7 heures: 155/879 (18%) patients traités par altéplase vs 112/850 (13%) patients du groupe contrôle qui ont eu une évolution favorable après un accident vasculaire cérébral au 90ème jour/180 ; OR 1,56, 95% CI 1,17-2,08].
Chez les patients âgés de plus de 80 ans traités par altéplase dans les 3 heures ou moins, une évolution favorable après un accident vasculaire cérébral était retrouvée chez 55/302 patients (18,2%) vs 30/264 patients (11,4%) dans le groupe contrôle (OR 1,86, 95% CI 1,11-3,13) ; chez ceux traités par altéplase dans la fenêtre 3h-4h30, 58/342 (17%) des patients ont eu une évolution favorable après un accident vasculaire cérébral vs 50/364 (13,7%) patients dans le groupe contrôle (OR 1,36, 95% CI 0,87-2,14).
Dans les 7 jours suivant l’administration de l’altéplase une hémorragie parenchymateuse de type 2 est survenue chez 231 (6,8%) sur 3 391 patients versus 44 (1,3%) sur 3 365 patients dans le groupe contrôle (OR 5,55, 95%, CI 4,01-7,70).
Dans les 7 jours suivant l’administration de l’altéplase une hémorragie parenchymateuse de type 2 d’évolution fatale est survenue chez 91 (2,7%) patients versus 13 (0,4%) patients dans le groupe contrôle (OR 7,14, 95% CI 3,98-12,79).
Chez les patients âgés de plus de 80 ans traités par altéplase, une hémorragie intracrânienne d’évolution fatale est survenue dans les 7 jours après traitement chez 32/879 (3,6%) patients vs chez 4/850 (0,5%) patients du groupe contrôle (OR 7,95, 95% CI 2,79-22,60).
Sur un total de 8 658 patients âgés de plus de 80 ans et traités dans les 4h30 après le début de l’AVC, collectés dans le registre SITS-ISTR, les données de 2 157 patients traités dans la fenêtre 3h-4h30 après le début de l’accident vasculaire cérébral ont été comparées à celles de 6 501 patients traités en moins de 3h.
L’indépendance fonctionnelle à 3 mois (score mRS0-2) était de 36 vs 37% (OR ajusté 0,79, 95% CI 0,68-0,92), la mortalité était de 29,0% vs 29,6% (OR ajusté 1,10, 95% CI 0,95-1,28) et l’HICs (selon la définition de SITS-MOST) était de 2,7% vs 1,6% (OR ajusté 1,62, 95% CI 1,12-2,34).
Population pédiatrique
Des données observationnelles non-randomisées et non comparatives, chez les patients âgés de 16-17 ans qui ont présenté un AVC avec la confirmation d’un traitement par l’altéplase, ont été obtenues à partir du registre SITS-ISTR (Safe Implementation of Treatments in Stroke – International Stroke Thrombolysis Register, un registre international indépendant). Entre 2003 et fin 2017, un total de 25 patients âgés de 16 à 17 ans, avec une utilisation confirmée d’altéplase ont été enregistrés dans le registre SITS. La dose médiane d’altéplase utilisée dans ce groupe de patient était de 0,9 mg/kg (entre 0,83 et 0,99 mg/kg). Parmi ces 25 patients, 23 ont reçu le traitement dans les 4,5 heures après le début des symptômes de l’AVC (19 dans les 3h, 4 dans les 3 à 4,5 h, 1 dans les 5 à 5,5 h, 1 cas non rapporté). Le poids des patients était compris entre 56 et 90 kg. La plupart des patients présentaient, au départ, un AVC modéré ou modéré à sévère avec un score NIHSS médian de 9.0 (entre 1 et 30).
Quatre-vingt-dix jours après le traitement, les scores mRS étaient disponibles pour 21 patients sur 25 et sur ces 21 patients, 14 avaient un score mRS compris entre 0 et 1 (aucun symptôme ou pas d’handicap significatif) et 5 autres avaient un score mRS = 2 (handicap léger). Cela signifie que 19 patients sur 21 (plus de 90%) des patients ont eu un résultat favorable au 90ème jour selon le score mRS. L’issue du traitement rapportée pour les 2 patients restants était soit un handicap modérément sévère (mRS =4 ; n=1), soit le décès (mRS = 6) dans les 7 jours (n=1).
Pour 4 patients, le score mRS n’a pas été rapporté au 90ème jour. La dernière information disponible a montré que 2 patients sur 4 avaient un score mRS égale à 2 au 7ème jour et 2 patients sur 4 ont rapporté une nette amélioration globale au 7ème jour.
Des données de sécurité sur les évènements indésirables étaient également disponibles dans le registre pour les hémorragies et les œdèmes. Sur les 25 patients dans cette tranche d’âge de 16-17 ans, aucun n’a eu d’hémorragie intracérébrale symptomatique (HICs, saignement d’HIC de type PH2). Cinq cas ont développé un œdème cérébral après le traitement par altéplase. Sur ces 5 patients, 4 ont soit rapportés un score mRS compris entre 0 et 2 au 90ème jour, soit ont montré une amélioration globale au 7ème jour après le traitement. Un patient avait un score mRS = 4 (handicap modérément sévère) rapporté au 90ème jour. Aucun de ces cas n’a été d’issue fatale.
En résumé, il y a eu 25 cas rapportés à partir du registre SITS pour des patients âgés de 16 à 17 ans présentant un accident vasculaire cérébral ischémique à la phase aiguë et qui ont été traités par altéplase selon les recommandations données pour les adultes. Malgré le faible effectif de l’échantillon qui ne permet pas d’effectuer l’analyse statistique, les résultats globaux montrent une tendance positive à l’utilisation de la dose adulte respective chez ces patients. Les données ne semblent pas montrer d’augmentation du risque d’hémorragie intracérébrale symptomatique ou d’œdème, comparé à l’adulte.
5.2. Propriétés pharmacocinétiques
Dans des conditions physiologiques, la majeure partie de l'altéplase dans la circulation sanguine est liée à l'inhibiteur. La clairance hépatique de l'altéplase n'est pas limitée par la présence d'autres protéines, y compris les inhibiteurs de l'altéplase. Les complexes d'altéplase et de son inhibiteur sont éliminés sous forme d’altéplase libre. La demi-vie plasmatique est de 4 à 5 minutes, ainsi après 20 minutes, moins de 10% de la valeur initiale sont encore présents dans le plasma.
Une demi-vie d'élimination de 40 minutes environ a été calculée pour la fraction résiduelle située dans le compartiment profond.
5.3. Données de sécurité préclinique
Les tests de mutagenèse n'ont pas mis en évidence de potentiel mutagène.
Aucun effet tératogène n’a été observé après perfusion intraveineuse de doses pharmacologiquement actives chez la femelle gestante. L’administration de plus de 3 mg/kg/jour a induit une embryotoxicité (mortalité embryonnaire, retard de croissance) chez la lapine. Aucun effet sur le développement péri et post-natal et sur les paramètres de la fertilité n’a été observé à des doses allant jusqu’à 10 mg/kg/jour chez le rat.
Arginine
Acide phosphorique (E 338) (pour l’ajustement du pH)
Polysorbate 80 (E 433)
Solvant :
Eau pour préparations injectables.
La solution reconstituée peut être diluée dans une solution stérile de chlorure de sodium à 0,9 % jusqu’à une concentration minimale de 0,2 mg d’altéplase par ml.
En cas de nouvelle dilution, l’utilisation d’eau pour préparations injectables ou en général l’utilisation de solutions glucosées pour perfusion, dextrose par exemple, n’est pas recommandée en raison d’une formation accrue de turbidité dans la solution reconstituée.
ACTILYSE ne doit pas être mélangé avec d'autres médicaments (y compris avec l'héparine), que ce soit dans le même flacon pour perfusion, ou dans le même cathéter.
2 ans pour Actilyse 10 mg, poudre et solvant pour solution injectable et perfusion.
3 ans pour Actilyse 20 mg et 50 mg, poudre et solvant pour solution injectable et perfusion.
Solution reconstituée
La stabilité de la solution reconstituée a été démontrée pendant 24 heures entre 2°C et 8°C et pendant 8 heures à 25°C.
Du point de vue microbiologique, le produit doit être utilisé immédiatement après reconstitution. En cas d’utilisation non immédiate, les durées et conditions de conservation après reconstitution et avant utilisation relèvent de la seule responsabilité de l’utilisateur, et ne devraient normalement pas dépasser 24h entre 2°C et 8°C.
6.4. Précautions particulières de conservation
A conserver dans l’emballage d’origine, à l'abri de la lumière.
A conserver à une température ne dépassant pas + 25°C.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre :
Flacons (verre stérile) de 10, 20 ou 50 ml, munis de bouchons stériles (butylé, siliconé, gris) avec un capuchon de type « flip-off » (aluminium/plastique).
Solvant :
Pour les présentations de 10 mg, 20 mg et 50 mg, l’eau pour préparations injectables est conditionnée dans des flacons de 10 ml, 20 ml ou 50 ml, en fonction de la taille des flacons de poudre.
Les flacons d’eau pour préparations injectables sont fermés par un bouchon (caoutchouc) avec un capuchon (aluminium/plastique) de type « flip-off ».
Canules de transfert (fournies avec les présentations de 20 et 50 mg uniquement).
Présentations :
10 mg
Un flacon de 467 mg de poudre pour solution pour perfusion
Un flacon de 10 ml d’eau pour préparations injectables
20 mg
Un flacon de 933 mg de poudre pour solution pour perfusion
Un flacon de 20 ml d’eau pour préparations injectables
Une canule de transfert
50 mg
Un flacon de 2333 mg de poudre pour solution pour perfusion
Un flacon de 50 ml d’eau pour préparations injectables
Une canule de transfert
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
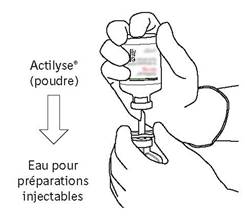
Afin d’obtenir une concentration finale de 1 mg d’altéplase par ml, le volume total de solvant fourni doit être introduit dans le flacon contenant la poudre d’ACTILYSE. Une canule de transfert est fournie à cet effet avec les présentations de 20 et 50 mg. Pour le flacon de 10 mg, une seringue doit être utilisée.
Afin d’obtenir une concentration finale de 2 mg d’altéplase par ml, seule la moitié du volume de solvant fourni doit être utilisée (voir tableau ci-dessous). Dans ce cas, une seringue doit toujours être utilisée pour introduire le volume requis de solvant dans le flacon contenant le lyophilisat d’ACTILYSE.
Dans des conditions rigoureuses d'asepsie, dissoudre l'altéplase (10, 20 ou 50 mg) dans un volume d'eau pour préparations injectables conformément au tableau suivant, afin d'obtenir une concentration finale soit de 1 mg d'altéplase/ml, soit de 2 mg d'altéplase/ml :
|
Quantité de poudre d’ACTILYSE |
10 mg |
20 mg |
50 mg |
|
(a) Volume d’eau stérile pour préparations injectables à ajouter à la poudre |
10 ml |
20 ml |
50 ml |
|
Concentration finale : |
1 mg d'altéplase/ml |
1 mg d'altéplase/ml |
1 mg d'altéplase/ml |
|
(b) Volume d’eau stérile pour préparations injectables à ajouter à la poudre |
5 ml |
10 ml |
25 ml |
|
Concentration finale : |
2 mg d'altéplase/ml |
2 mg d'altéplase/ml |
2 mg d'altéplase/ml |
La solution reconstituée doit alors être administrée par voie intraveineuse. La solution reconstituée de 1 mg/ml peut être diluée davantage avec une solution injectable stérile de chlorure de sodium à 9 mg/ml (0,9 %) jusqu'à une concentration minimale de 0,2 mg/ml car il ne peut être exclu l’apparition de turbidité dans la solution reconstituée. Il n’est pas recommandé de diluer davantage la solution reconstituée de 1 mg/ml au moyen d’eau pour préparations injectables ou d’un soluté sucré (dextrose par exemple, en raison d’une formation accrue de turbidité dans la solution reconstituée). ACTILYSE ne doit pas être mélangé à d’autres médicaments (dont l’héparine) dans le même flacon de perfusion.
Pour les incompatibilités, voir rubrique 6.2.
La solution reconstituée est destinée à un usage unique. Toute solution non utilisée ou déchet doit être éliminé conformément à la réglementation en vigueur.
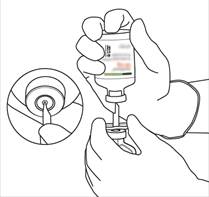
Instructions pour la reconstitution d’ACTILYSE
|
Reconstituez immédiatement avant administration. |
|
|
|
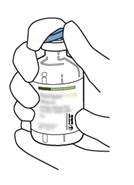
2 |
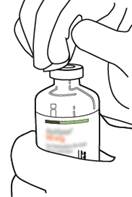
Retirez le capuchon protecteur des deux flacons contenant l’eau pour préparations injectables et la poudre d’ACTILYSE, en soulevant les capuchons avec le pouce. |
|
|
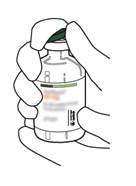
3 |
Essuyez le haut du caoutchouc de chaque flacon à l’aide d’un tampon imbibé d’alcool. |
|
|
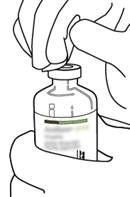
4 |
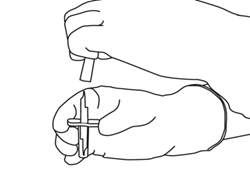
Sortez la canule de transfert* de son étui. Ne désinfectez pas ou ne stérilisez pas la canule de transfert, elle est stérile. Retirez l’un de ses capuchons. |
|
|
5 |
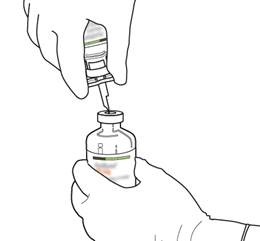
Tenez le flacon contenant l’eau pour préparations injectables en position verticale et sur une surface stable. Sur le dessus du flacon, percez le bouchon en caoutchouc avec la canule de transfert, de façon verticale et bien au centre du bouchon, en pressant doucement mais fermement, sans tourner. |
|
|
6 |
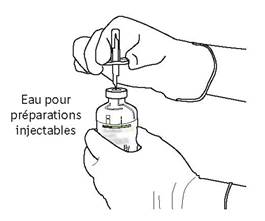
Tenez fermement le flacon contenant l’eau pour préparations injectables et la canule de transfert avec une seule main, à l’aide des deux rabats de chaque côté de la canule.
Retirez le capuchon restant sur le dessus de la canule de transfert. |
|
|
7 |
Tenez fermement le flacon contenant l’eau pour préparations injectables et la canule de transfert avec une seule main, à l’aide des deux rabats de chaque côté de la canule. Prenez le flacon d’ACTILYSE contenant la poudre et tenez-le verticalement au-dessus de la canule de transfert en positionnant l’extrémité pointue de la canule de transfert bien au centre du bouchon.
Poussez verticalement le flacon contenant la poudre sur la canule de transfert, doucement mais fermement et sans tourner, jusqu’à la perforation du caoutchouc du bouchon. |
|
|
8 |
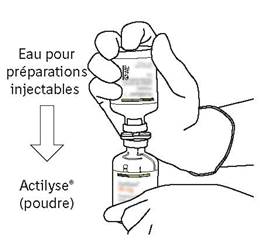
Retournez les deux flacons de manière à ce que l’eau pour préparations injectables puisse remplir complétement le flacon contenant la poudre. |
|
|
9 |
Retirez le flacon vide qui contenait l’eau pour préparations injectables en même temps que la canule de transfert. Ils peuvent être jetés. |
|
|
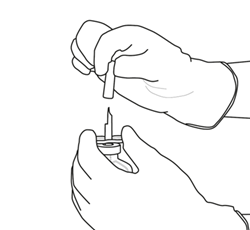
10 |
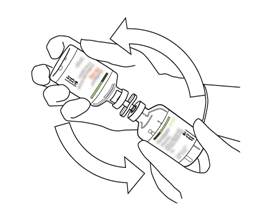
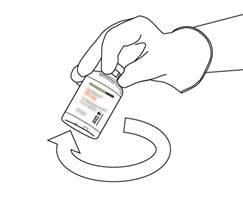
Prenez le flacon contenant la solution reconstituée d’ACTILYSE et agitez doucement jusqu’à dissoudre tout reste de poudre, mais ne remuez surtout pas, cela produira de la mousse.
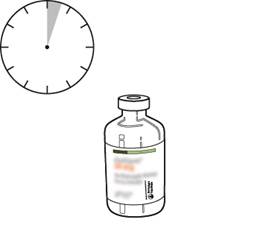
S’il reste des bulles, laissez la solution reposer quelques minutes et attendre la disparition des bulles. |
|
|
11 |
La solution reconstituée représente 1 mg/ml d’altéplase. Elle doit être claire et incolore voire jaune pâle et ne doit pas contenir de particules. |
|
|
12 |
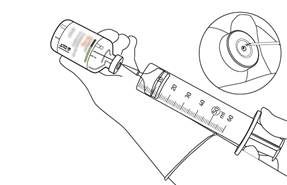
Prélevez la quantité nécessaire en utilisant uniquement une aiguille et une seringue. N’utilisez pas le même trou de perforation que celui de la canule de transfert pour éviter toute fuite. |
|
|
13 |
Utilisez immédiatement. Ne conservez pas la solution non utilisée. |
|
(*si une canule de transfert est incluse dans la boîte. La reconstitution peut aussi être faite avec une seringue et une aiguille.)
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
100-104 AVENUE DE FRANCE
75013 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 557 184 2 0 : poudre en flacon (verre) + 1 flacon (verre) de 10 ml de solvant.
· 34009 558 529 3 3 : poudre en flacon (verre) + 1 flacon (verre) de 20 ml de solvant + 1 canule de transfert.
· 34009 558 530 1 5 : poudre en flacon (verre) + 1 flacon (verre) de 50 ml de solvant + 1 canule de transfert.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament réservé à l’usage hospitalier et à l’usage en situation d’urgence selon l’article R.5121-96 du code de la santé publique.
ANSM - Mis à jour le : 16/04/2026
ACTILYSE, poudre et solvant pour solution injectable et perfusion
altéplase
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre infirmier/ère.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ACTILYSE, poudre et solvant pour solution injectable et perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser ACTILYSE, poudre et solvant pour solution injectable et perfusion?
3. Comment utiliser ACTILYSE, poudre et solvant pour solution injectable et perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ACTILYSE, poudre et solvant pour solution injectable et perfusion?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ACTILYSE, poudre et solvant pour solution injectable et perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Thrombolytiques - code ATC : B01AD02
La substance active présente dans ACTILYSE est l’altéplase.
Elle fait partie du groupe de médicaments nommés agents thrombolytiques. Ces médicaments agissent en dissolvant les caillots de sang qui se sont formés dans les vaisseaux sanguins.
ACTILYSE 10, 20 ou 50 mg sont utilisés dans le traitement d’un certain nombre d’affections provoquées par les caillots de sang qui se forment dans les vaisseaux sanguins, notamment :
· la crise cardiaque provoquée par des caillots sanguins présents dans les artères du cœur (infarctus du myocarde à la phase aiguë),
· les caillots sanguins présents dans les artères des poumons (embolie pulmonaire massive à la phase aiguë),
· l’accident vasculaire cérébral provoqué par un caillot sanguin présent dans une artère du cerveau (accident vasculaire cérébral ischémique aigu) s’il s’est écoulé moins de 4 heures 30 depuis la dernière fois où vous avez été vu sans symptômes de l’AVC en cours.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ACTILYSE, poudre et solvant pour solution injectable et perfusion ?
· si vous êtes allergique (hypersensible) à l’altéplase ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
· si vous présentez, ou avez récemment présenté, une maladie qui augmente vos risques de saignements (hémorragies), notamment :
o un trouble hémorragique ou une tendance au saignement (hémorragie),
o un saignement sévère ou dangereux dans une partie du corps,
o
o une tension artérielle très élevée, non contrôlée,
o une infection bactérienne ou une inflammation du cœur (endocardite), ou une inflammation des membranes qui entourent le cœur (péricardite),
o une inflammation du pancréas (pancréatite aiguë),
o un ulcère gastrique ou des ulcères de l’intestin,
o des veines variqueuses dans l’œsophage (varices œsophagiennes),
o une anomalie des vaisseaux sanguins, telles qu’un gonflement localisé d’une artère (anévrisme),
o certaines tumeurs,
o une maladie sévère du foie,
· si vous prenez un médicament servant à fluidifier le sang (anticoagulants) à moins que des tests appropriés aient confirmé l’absence d’activité cliniquement significative de ce médicament,
· si vous avez déjà subi une intervention chirurgicale au cerveau ou à la colonne vertébrale,
· si vous avez subi une chirurgie lourde ou une blessure significative au cours des 3 derniers mois,
· si vous avez eu récemment une ponction d'un vaisseau sanguin important,
· si vous avez eu un enfant au cours des 10 derniers jours.
Votre médecin n’utilisera pas ACTILYSE dans le traitement de crises cardiaques ou de caillots sanguins dans les artères des poumons
· si vous présentez ou avez déjà présenté un accident vasculaire cérébral provoqué par un saignement dans le cerveau (accident vasculaire cérébral hémorragique)
· ou un accident vasculaire cérébral de cause inconnue
· si vous avez présenté un accident vasculaire cérébral causé par un caillot sanguin dans une artère du cerveau (accident vasculaire cérébral ischémique) au cours des 6 derniers mois.
De plus, votre médecin n’utilisera pas ACTILYSE dans le traitement d’un accident vasculaire cérébral causé par un caillot sanguin présent dans une artère du cerveau (accident vasculaire cérébral ischémique aigu)
· si votre accident vasculaire cérébral ne provoque que de très légers symptômes,
· si un saignement est présent dans le cerveau ou dans le crâne,
· si vous avez eu un accident vasculaire cérébral dans les trois derniers mois,
· si les symptômes s’améliorent rapidement avant de recevoir ACTILYSE,
· si vous présentez un accident vasculaire cérébral très sévère,
· ,
· si votre temps de céphaline activée (TCA : examen du sang destiné à vérifier si votre sang coagule bien) est anormal. Cet examen est susceptible d’être anormal si vous avez reçu de l’héparine (un médicament utilisé pour fluidifier le sang) dans les 48 heures précédentes.
· si vous êtes diabétique et avez déjà eu un accident vasculaire cérébral dans le passé,
· si le nombre de plaquettes (thrombocytes) dans votre sang est très faible,
· si vous présentez une tension artérielle très élevée (supérieure à 185/110) qui ne peut être diminuée qu’avec une prise en charge rigoureuse,
· si la quantité de sucre (glucose) dans votre sang est très faible (inférieure à 50 mg/dL), ou
· très élevée (supérieure à 400 mg/dL),
· si vous êtes âgé(e) de moins de 16 ans. (Pour les adolescents âgés de 16 ans ou plus, voir la rubrique « Avertissements et précautions ».)
Avertissements et précautions
· si vous avez déjà eu une réaction allergique autre qu’une réaction allergique soudaine menaçant le pronostic vital (hypersensibilité sévère) à la substance active altéplase, ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6),
· si vous présentez ou avez récemment présenté une autre affection qui augmente votre risque de saignement, telle que :
o une petite lésion,
o une biopsie (intervention effectuée pour obtenir un échantillon de tissu),
o une ponction de vaisseaux importants,
o une injection intramusculaire,
o un massage cardiaque externe,
· si vous présentez une anomalie des valves cardiaques (sténose mitrale par exemple) avec un rythme cardiaque anormal (fibrillation atriale par exemple),
· si vous avez déjà reçu ACTILYSE avant,
· si vous êtes âgé(e) de plus de 75 ans,
Votre médecin prendra des précautions particulières avec ACTILYSE pour le traitement des crises cardiaques ou des caillots sanguins dans les artères pulmonaires.
· Si vous êtes hypertendu(e) ;
· Si vous pesez moins de 50 kg.
De plus, votre médecin prendra des précautions particulières avec ACTILYSE pour le traitement d'un accident vasculaire cérébral causé par un caillot sanguin dans une artère du cerveau (accident vasculaire cérébral ischémique aigu)
si les signes d'un accident vasculaire cérébral ischémique aigu persistent après la normalisation du taux de sucre dans votre sang, votre médecin peut encore envisager un traitement thrombolytique
si vous avez eu des crampes (convulsions) au début de votre accident vasculaire cérébral
si vous avez plus de 80 ans, vous pourrez présenter une évolution clinique mois favorable indépendamment du traitement avec ACTILYSE Cependant, d’une manière générale le rapport bénéfice-risque d’ACTILYSE chez les patients de plus de 80 ans demeure positif et l’âge seul n’est pas une barrière au traitement par ACTILYSE.
si vous souffrez d'hypertension artérielle
si vous êtes un adolescent âgé de 16 ans ou plus, le bénéfice du traitement de l’accident vasculaire cérébral ischémique à la phase aiguë sera évalué attentivement par rapport au risque et de manière individuelle.
Autres médicaments et ACTILYSE, poudre et solvant pour solution injectable et perfusion :
Informez votre médecin si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament y compris un médicament obtenu sans ordonnance.
Il est particulièrement important que vous informiez votre médecin si vous prenez ou avez pris récemment :
· des médicaments utilisés pour fluidifier le sang, notamment :
o de l’acide acétylsalicylique (aspirine),
o de la warfarine,
o de la coumarine,
o de l’héparine,
· certains médicaments utilisés pour traiter une tension artérielle élevée (inhibiteurs de l’enzyme de conversion).
ACTILYSE, poudre et solvant pour solution injectable et perfusion avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou allaitante, que vous pensez être enceinte ou souhaitez avoir un enfant, demandez conseil à votre médecin.
Votre médecin ne vous donnera ACTILYSE que si les bénéfices possibles l’emportent sur les risques possibles pour votre bébé.
Conduite de véhicules et utilisation de machines
Sans objet.
ACTILYSE contient du polysorbate 80
Ce médicament contient 1,0 mg, 2,0 mg ou 5,0 mg de polysorbate 80 dans chaque flacon de 10 mg, 20 mg ou de 50 mg respectivement. Les polysorbates peuvent provoquer des réactions allergiques. Informez votre médecin si vous avez déjà présenté une allergie.
3. COMMENT UTILISER ACTILYSE, poudre et solvant pour solution injectable et perfusion ?
Le traitement par ACTILYSE doit être instauré dès que possible après le début de vos symptômes.
Il existe trois maladies différentes pour lesquelles ce médicament peut être donné :
Crise cardiaque (infarctus du myocarde à la phase aiguë)
La dose qui vous est donnée est fonction de votre poids corporel. La dose maximale d’ACTILYSE est de 100 mg, mais celle-ci sera inférieure si vous pesez moins de 65 kg.
Elle peut être administrée de deux manières différentes :
a) La forme d’administration dite « des 90 minutes », pour les patients traités dans les 6 heures suivant le début de leurs symptômes. Celle-ci consiste en :
· une injection initiale d’une partie de la dose d’ACTILYSE dans une veine,
· des perfusions du reste de la dose pendant les 90 minutes qui suivent.
b) La forme d’administration dite « des 3 heures », pour les patients traités entre 6 et 12 heures après le début de leurs symptômes. Celle-ci consiste en :
· une injection initiale d’une partie de la dose d’ACTILYSE dans une veine,
· des perfusions du reste de la dose pendant les 3 heures qui suivent.
En plus d’ACTILYSE, votre médecin vous donnera un autre médicament destiné à arrêter la coagulation du sang. Ce médicament vous sera donné dès que possible après le début de vos douleurs à la poitrine.
Caillots sanguins présents dans les artères des poumons (embolie pulmonaire massive à la phase aiguë)
La dose qui vous est donnée est fonction de votre poids corporel. La dose maximale d’ACTILYSE est de 100 mg, mais celle-ci sera inférieure si vous pesez moins de 65 kg.
Le médicament est habituellement donné sous forme :
· d’une injection initiale d’une partie de la dose dans une veine,
· d’une perfusion du reste de la dose pendant les 2 heures qui suivent.
Après le traitement par ACTILYSE, votre médecin débutera (ou recommencera) un traitement avec de l’héparine (médicament destiné à fluidifier le sang).
Accident vasculaire cérébral provoqué par un caillot sanguin présent dans une artère du cerveau (accident vasculaire cérébral ischémique aigu)
ACTILYSE doit être donné s’il s’est écoulé moins de 4 heures 30 depuis la dernière fois où vous avez été vu sans symptômes de l’AVC en cours . Plus tôt vous recevez ACTILYSE, plus le traitement peut vous être bénéfique et plus la probabilité de survenue d’effets indésirables nocifs est faible. La dose qui vous est donnée est fonction de votre poids corporel. La dose maximale de ce médicament est de 90 mg, mais celle-ci sera inférieure si vous pesez moins de 100 kg. ACTILYSE est donné sous forme :
· d’une injection initiale d’une partie de la dose dans une veine,
· d’une perfusion du reste de la dose pendant les 60 minutes qui suivent.
Vous ne devez pas prendre d’acide acétylsalicylique pendant les 24 premières heures qui suivent votre traitement par ACTILYSE pour un accident vasculaire cérébral. Votre médecin pourra vous administrer une injection d’héparine si celle-ci se révèle nécessaire.
Si vous avez utilisé plus d’ACTILYSE, poudre et solvant pour solution injectable et perfusion que vous n’auriez dû :
Sans objet.
Si vous oubliez d’utiliser ACTILYSE, poudre et solvant pour solution injectable et perfusion :
Si vous arrêtez d’utiliser ACTILYSE, poudre et solvant pour solution injectable et perfusion :
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables décrits ci-dessous sont survenus chez des personnes ayant reçu ACTILYSE :
Très fréquent (se produit chez plus de 1 patient sur 10 recevant le médicament)
· insuffisance cardiaque - l’arrêt du traitement peut se révéler nécessaire,
· saignement dans le cerveau (hémorragie cérébrale) après le traitement d’un accident vasculaire cérébral provoqué par un caillot sanguin présent dans une artère du cerveau (accident vasculaire cérébral ischémique aigu) - l’arrêt du traitement peut se révéler nécessaire,
· présence de liquide dans les poumons (œdème pulmonaire),
· saignement du vaisseau sanguin endommagé (tel qu’un hématome),
· pression artérielle faible (hypotension),
· douleurs au thorax (angine de poitrine).
Fréquent (se produit chez moins de 1 patient sur 10 recevant le médicament)
· une nouvelle crise cardiaque,
· saignements dans le cerveau (hémorragie cérébrale) après le traitement de crises cardiaques (infarctus du myocarde) - l’arrêt du traitement peut se révéler nécessaire,
· arrêt du battement du cœur (arrêt cardiaque) - l’arrêt du traitement peut se révéler nécessaire,
· état de choc (pression artérielle extrêmement faible) dû à une insuffisance cardiaque - l’arrêt du traitement peut se révéler nécessaire,
· saignements dans la gorge,
· saignements dans l’estomac ou l’intestin, notamment vomissement de sang (hématémèse) ou présence de sang dans les selles (méléna ou hémorragie rectale), saignements des gencives,
· saignements dans les tissus corporels à l’origine de bleus violacés (ecchymose),
· saignements de l’appareil urinaire ou des organes de reproduction, qui peuvent conduire à une présence de sang dans les urines (hématurie),
· saignement ou apparition de bleu (hématome) à l’endroit où l’injection est administrée.
Peu fréquent (se produit chez moins de 1 patient sur 100 recevant le médicament)
· saignements liés au poumon, tels que flegme teinté de sang (hémoptysie) ou saignement des voies respiratoires - l’arrêt du traitement peut se révéler nécessaire,
· saignements de nez (epistaxis),
· battement du cœur irrégulier après le rétablissement de l’irrigation sanguine vers le cœur,
· lésions des valvules du cœur (régurgitation mitrale) ou de la paroi des cavités du cœur (perforation du septum interventriculaire) – l’arrêt du traitement peut se révéler nécessaire,
· blocage soudain d’une artère des poumons (embolie pulmonaire), du cerveau (embolie cérébrale), ou de toute autre région du corps (embolie systémique),
· saignement de l’oreille,
· diminution de la pression artérielle.
Rare (se produit chez moins de 1 patient sur 1 000 recevant le médicament)
· saignements dans le sac membraneux entourant le cœur (hémopéricarde) - l’arrêt du traitement peut se révéler nécessaire,
· saignement interne dans la partie postérieure de l’abdomen (saignement rétropéritonéal) - l’arrêt du traitement peut se révéler nécessaire,
· formation de caillots de sang dans les vaisseaux sanguins susceptibles de se déplacer vers les autres organes du corps (embolie). Les symptômes seront variables selon l’organe touché,
· réactions allergiques, par exemple, urticaire et éruptions cutanées, difficulté respiratoire allant jusqu’à l’asthme (bronchospasme), présence de liquide sous la peau et les muqueuses (angio-œdème), pression artérielle faible ou état de choc - l’arrêt du traitement peut se révéler nécessaire,
· saignements dans les yeux (hémorragie oculaire),
· dérangement de l’estomac (nausées).
Très rare (se produit chez moins de 1 patient sur 10 000 recevant le médicament)
· réaction allergique grave (par exemple, anaphylaxie menaçant le pronostic vital) – l’arrêt du traitement peut se révéler nécessaire,
· événements touchant le système nerveux tels que :
o crampes (convulsions, crises),
o problèmes de langage,
o confusion ou délire (confusion très sévère),
o anxiété accompagnée d’une nervosité (agitation),
o dépression,
o altération de la réflexion (psychose).
Ces troubles se produisent souvent en association avec un accident vasculaire cérébral provoqué par un caillot sanguin ou un saignement dans le cerveau.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
· saignements dans les organes internes, par exemple saignement dans le foie (hémorragie hépatique) - l’arrêt du traitement peut se révéler nécessaire,
· formation de cristaux de cholestérol agglomérés susceptibles de se déplacer vers les autres organes du corps (embolie de cristaux de cholestérol). Les symptômes seront variables selon l’organe touché - l’arrêt du traitement peut se révéler nécessaire,
· saignements nécessitant une transfusion sanguine,
· vomissements,
· température du corps augmentée (fièvre).
Des décès ou invalidités permanentes peuvent survenir suite à un saignement dans le cerveau ou d'autres événements hémorragiques graves.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ACTILYSE, poudre et solvant pour solution injectable et perfusion ?
En principe, il ne devrait pas vous être demandé de conserver ACTILYSE, ce médicament vous étant donné par votre médecin.
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette du flacon et de l’emballage après {EXP}. La date de péremption fait référence au dernier jour de ce mois.
Solution reconstituée
La stabilité de la solution reconstituée a été démontrée pendant 24 heures entre 2°C et 8°C et pendant 8 heures à 25°C.
Du point de vue microbiologique, le produit doit être utilisé immédiatement après reconstitution. En cas d’utilisation non immédiate, les durées et conditions de conservation après reconstitution et avant utilisation sont de la seule responsabilité de l’utilisateur, et ne devraient normalement pas dépasser 24 heures entre 2°C et 8°C.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ACTILYSE, poudre et solvant pour solution injectable et perfusion
· La substance active est :
altéplase................................................... 10 mg (correspondant à 5 800 000 UI), pour un flacon,
ou
altéplase................................................. 20 mg (correspondant à 11 600 000 UI), pour un flacon,
ou
altéplase................................................. 50 mg (correspondant à 29 000 000 UI), pour un flacon.
L’altéplase est produite par la technique de l’ADN recombinant dans une lignée cellulaire d’ovaire de hamster chinois.
· Les autres composants sont : l’arginine, l’acide phosphorique (E 338) (pour ajuster le pH) et le polysorbate 80 (E 433).
· Le solvant est : l’eau pour préparations injectables.
ACTILYSE se présente sous forme de poudre et solvant pour solution injectable et perfusion. Chaque boîte complète contient un flacon de poudre et un flacon de solvant.
ACTILYSE est disponible dans les présentations suivantes :
· Un flacon de poudre avec 10 mg d’altéplase et un flacon avec 10 ml de solvant.
· Un flacon de poudre avec 20 mg d’altéplase, un flacon avec 20 ml de solvant et une canule de transfert.
· Un flacon de poudre avec 50 mg d’altéplase, un flacon avec 50 ml de solvant et une canule de transfert.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
100-104 AVENUE DE FRANCE
75013 PARIS
Exploitant de l’autorisation de mise sur le marché
BOEHRINGER INGELHEIM FRANCE
100-104 AVENUE DE FRANCE
75013 PARIS
BOEHRINGER INGELHEIM PHARMA GMBH & CO.KG
BIRKENDORFER STRASSE 65
88397 BIBERACH AN DER RISS
ALLEMAGNE
BOEHRINGER INGELHEIM FRANCE
100-104 AVENUE DE FRANCE
75013 PARIS
FRANCE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé:
Traçabilité
Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Le flacon de 2 mg n’est pas adapté pour une utilisation à la phase aiguë de l’infarctus du myocarde, à la phase aiguë de l’embolie pulmonaire massive ou de l’accident vasculaire cérébral ischémique (en raison d’un risque de sous-dosage important). Seules les présentations de 10 mg, 20 mg et 50 mg sont adaptées pour une utilisation dans ces indications.
Reconstitution
Afin d’obtenir une concentration finale de 1 mg d’altéplase par ml, le volume total de solvant fourni doit être introduit dans le flacon contenant la poudre d’ACTILYSE. Une canule de transfert est fournie à cet effet avec les flacons de 20 et 50 mg. Pour les flacons de 10 mg, une seringue doit être utilisée.
Afin d’obtenir une concentration finale de 2 mg d’altéplase par ml, seule la moitié du volume de solvant fourni doit être utilisée (voir tableau ci-dessous). Dans ce cas, une seringue doit toujours être utilisée pour introduire le volume requis de solvant dans le flacon contenant le lyophilisat d’ACTILYSE.
Dans des conditions rigoureuses d'asepsie, dissoudre l'altéplase (10, 20 ou 50 mg) dans un volume d'eau pour préparations injectables conformément au tableau suivant, afin d'obtenir une concentration finale soit de 1 mg d'altéplase/ml, soit de 2 mg d'altéplase/ml :
|
Quantité de poudre d’ACTILYSE |
10 mg |
20 mg |
50 mg |
|
(a) Volume d’eau stérile pour préparations injectables à ajouter à la poudre |
10 ml |
20 ml |
50 ml |
|
Concentration finale : |
1 mg d'altéplase/ml |
1 mg d'altéplase/ml |
1 mg d'altéplase/ml |
|
(b) Volume d’eau stérile pour préparations injectables à ajouter à la poudre |
5 ml |
10 ml |
25 ml |
|
Concentration finale : |
2 mg d'altéplase/ml |
2 mg d'altéplase/ml |
2 mg d'altéplase/ml |
La solution reconstituée doit alors être administrée par voie intraveineuse. La solution reconstituée de 1 mg/ml peut être diluée davantage avec une solution injectable stérile de chlorure de sodium à 9 mg/ml (0,9 %) jusqu'à une concentration minimale de 0,2 mg/ml car il ne peut être exclu l’apparition d’un trouble dans la solution reconstituée. Il n’est pas recommandé de diluer davantage la solution reconstituée de 1 mg/ml au moyen d’eau pour préparations injectables ou d’un soluté sucré (dextrose par exemple, en raison d’une formation accrue d’un trouble dans la solution reconstituée).
ACTILYSE ne doit pas être mélangé à d’autres médicaments (dont l’héparine) dans le même flacon de perfusion.
Pour les conditions de conservation, voir la rubrique 5 de cette notice.
La solution reconstituée est destinée à un usage unique. Toute solution non utilisée doit être jetée.
Instructions pour la reconstitution d’ACTILYSE
|
1 |
Reconstituez immédiatement avant administration. |
|
|
2 |
Retirez le capuchon protecteur des deux flacons contenant l’eau pour préparations injectables et la poudre d’ACTILYSE, en soulevant les capuchons avec le pouce. |
|
|
3 |
Essuyez le haut du caoutchouc de chaque flacon à l’aide d’un tampon imbibé d’alcool. |
|
|
4 |
Sortez la canule de transfert * de son étui. Ne désinfectez pas ou ne stérilisez pas la canule de transfert, elle est stérile. Retirez l’un de ses capuchons. |
|
|
5 |
Tenez le flacon contenant l’eau pour préparations injectables en position verticale et sur une surface stable. Sur le dessus du flacon, percez le bouchon en caoutchouc avec la canule de transfert, de façon verticale et bien au centre du bouchon, en pressant doucement mais fermement, sans tourner. |
|
|
6 |
Tenez fermement le flacon contenant l’eau pour préparations injectables et la canule de transfert avec une seule main, à l’aide des deux rabats de chaque côté de la canule.
Retirez le capuchon restant sur le dessus de la canule de transfert. |
|
|
7 |
Tenez fermement le flacon contenant l’eau pour préparations injectables et la canule de transfert avec une seule main, à l’aide des deux rabats de chaque côté de la canule. Prenez le flacon d’ACTILYSE contenant la poudre et tenez-le verticalement au-dessus de la canule de transfert en positionnant l’extrémité pointue de la canule de transfert bien au centre du bouchon.
Poussez verticalement le flacon contenant la poudre sur la canule de transfert, doucement mais fermement et sans tourner, jusqu’à la perforation du caoutchouc du bouchon. |
|
|
8 |
Retournez les deux flacons de manière à ce que l’eau pour préparations injectables puisse remplir complétement le flacon contenant la poudre. |
|
|
9 |
Retirez le flacon vide qui contenait l’eau pour préparations injectables en même temps que la canule de transfert. Ils peuvent être jetés. |
|
|
10 |
Prenez le flacon contenant la solution reconstituée d’ACTILYSE et agitez doucement jusqu’à dissoudre tout reste de poudre, mais ne remuez surtout pas, cela produira de la mousse.
S’il reste des bulles, laissez la solution reposer quelques minutes et attendre la disparition des bulles. |
|
|
11 |
La solution reconstituée représente 1 mg/ml d’altéplase. Elle doit être claire et incolore voire jaune pâle et ne doit pas contenir de particules. |
|
|
12 |
Prélevez la quantité nécessaire en utilisant uniquement une aiguille et une seringue. N’utilisez pas le même trou de perforation que celui de la canule de transfert pour éviter toute fuite. |
|
|
13 |
Utilisez immédiatement. Ne conservez pas la solution non utilisée. |
|
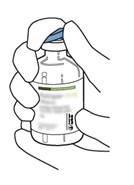
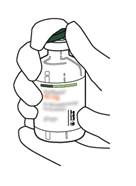
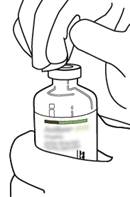
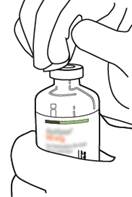
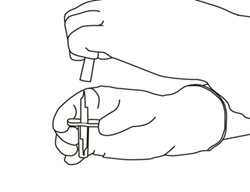
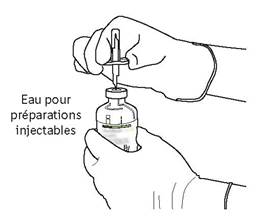
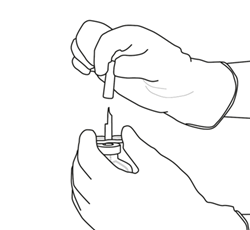
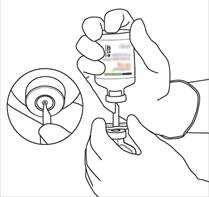
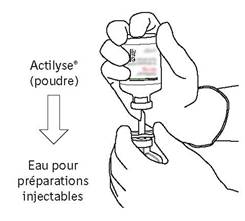
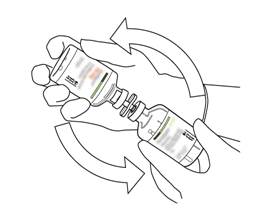
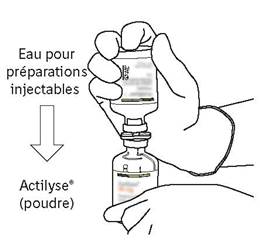
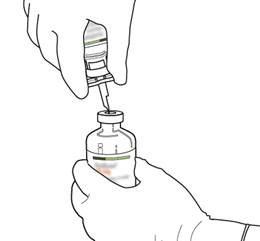
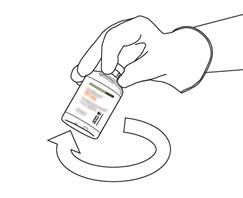
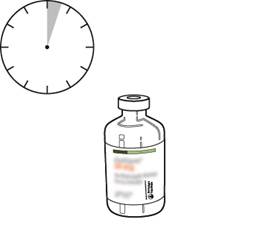
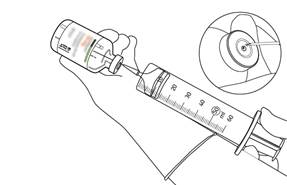
(*si une canule de transfert est incluse dans la boîte. La reconstitution peut aussi être faite avec une seringue et une aiguille.)
Posologie et mode d’administration
Infarctus du myocarde à la phase aiguë
Posologie
a) Schéma posologique dit "accéléré" (90 minutes) adapté aux patients à la phase aiguë de l’infarctus du myocarde pouvant être traités dans les 6 heures suivant l'apparition des symptômes.
Chez les patients de poids corporel ≥ 65 kg :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 15 mg, suivi immédiatement par |
15 ml |
7,5 ml |
|
Perfusion intraveineuse à débit constant de 50 mg sur les 30 premières minutes, immédiatement suivi par |
50 ml |
25 ml |
|
Perfusion intraveineuse à débit constant de 35 mg sur 60 minutes, sans dépasser la dose maximale totale de 100 mg |
35 ml |
17,5 ml |
Chez les patients de poids corporel < 65 kg, la dose totale doit être adaptée en fonction du poids selon le schéma d’administration suivant :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 15 mg, suivi immédiatement par |
15 ml |
7,5 ml |
|
Perfusion intraveineuse à débit constant de 0,75 mg/kg de poids corporel (pc) sur les 30 premières minutes, suivi immédiatement par |
0,75 ml/kg |
0,375 ml/kg |
|
Perfusion intraveineuse à débit constant de 0,5 mg/kg de poids corporel (pc) sur 60 minutes |
0,5 ml/kg |
0,25 ml/kg |
b) Schéma posologique dit "des 3 heures" adapté aux patients souffrant d’un infarctus du myocarde à la phase aigüe chez qui le traitement est mis en œuvre entre la 6e et la 12e heure suivant l'apparition des symptômes :
Chez les patients de poids corporel ≥ 65 kg :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 10 mg, suivi immédiatement par |
10 ml |
5 ml |
|
Perfusion intraveineuse à débit constant de 50 mg sur les 60 premières minutes, suivi immédiatement par |
50 ml |
25 ml |
|
Perfusions intraveineuses à débit constant de 40 mg sur 2 heures jusqu’à une dose maximale totale de 100 mg |
40 ml |
20 ml |
Chez les patients de poids corporel < 65 kg :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 10 mg, suivi immédiatement par |
10 ml |
5 ml |
|
Perfusion intraveineuse à débit constant sur 3 heures, jusqu’à la dose maximale totale de 1,5 mg/kg pc |
1,5 ml/kg |
0,75 ml/kg |
Traitements associés
Un traitement adjuvant anti-thrombotique est recommandé conformément aux recommandations internationales actuelles concernant la prise en charge des patients présentant un infarctus du myocarde avec sus-décalage du segment ST.
Mode d’administration
La solution reconstituée doit être administrée par voie intraveineuse et doit être utilisée immédiatement.
Le flacon de 2 mg d’altéplase n’est pas adapté pour une utilisation dans cette indication.
Embolie pulmonaire massive à la phase aiguë
Posologie
Chez les patients de poids corporel ≥ 65 kg :
Une dose totale de 100 mg d’altéplase doit être administrée en 2 heures. L'expérience acquise porte essentiellement sur le schéma posologique suivant :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 10 mg sur 1 à 2 minutes, suivi immédiatement par |
10 ml |
5 ml |
|
Perfusion intraveineuse à débit constant de 90 mg sur 2 heures jusqu’à une dose maximale totale de 100 mg |
90 ml |
45 ml |
Chez les patients de poids corporel <65 kg :
|
|
Volume à administrer en fonction de la concentration d'altéplase |
|
|
1 mg/ml |
2 mg/ml |
|
|
Bolus intraveineux de 10 mg sur 1 à 2 minutes, suivi immédiatement par |
10 ml |
5 ml |
|
Perfusion intraveineuse à débit constant sur 2 heures jusqu’à la dose maximale totale de 1,5 mg/kg pc |
1,5 ml/kg |
0,75 ml/kg |
Traitement associé
Après le traitement par ACTILYSE, une héparinothérapie doit être instaurée (ou reprise) si la valeur du TCA est inférieure à deux fois la limite supérieure de la normale. La perfusion doit être ajustée afin d'obtenir un TCA de 50 à 70 secondes (1,5 à 2,5 fois la valeur de référence).
Mode d’administration
La solution reconstituée doit être administrée par voie intraveineuse et doit être utilisée immédiatement.
Le flacon de 2 mg d’altéplase n’est pas adapté pour une utilisation dans cette indication.
Accident vasculaire cérébral ischémique à la phase aiguë
L’instauration et le suivi du traitement doivent être réalisés sous la responsabilité d’un médecin formé et expérimenté en pathologie neurovasculaire, voir RCP rubriques 4.3 Contre-indications et 4.4 Mises en garde spéciales et précautions d’emploi.
Le traitement par ACTILYSE doit être initié chez les adultes et les adolescents âgés de 16 ans ou plus (voir rubriques 4.3 et 4.4), le plus tôt possible dans les 4 heures 30 suivant le dernier moment où le patient a été vu en bonne santé, et après exclusion d’une hémorragie intracrânienne par des techniques d’imagerie adaptées. L’effet du traitement étant temps-dépendant, l’instauration précoce du traitement augmente les chances d’évolution favorable . Au-delà de 4h30 après l’apparition des symptômes, l’administration d’ACTILYSE est associée à un rapport bénéfice/risque défavorable, ACTILYSE ne doit donc pas être administré (voir RCP rubrique 5.1).
Posologie
La posologie totale recommandée est de 0,9 mg d’altéplase/kg de poids corporel (dose maximale de 90 mg) 10% de la dose totale devant être administrée initialement par bolus intraveineux, suivi immédiatement par une perfusion intraveineuse sur 60 minutes de la dose restante.
|
TABLE DES DOSES POUR L’AVC ISCHEMIQUE A LA PHASE AIGUË |
|||
|
En utilisant la concentration standard recommandée de 1 mg/ml, le volume (ml) à administrer est égal à la valeur de la dose recommandée (mg) |
|||
|
Poids
(kg) |
Dose totale
(mg) |
Dose en bolus
(mg) |
Dose en perfusion*
(mg) |
|
40 |
36,0 |
3,6 |
32,4 |
|
42 |
37,8 |
3,8 |
34,0 |
|
44 |
39,6 |
4,0 |
35,6 |
|
46 |
41,4 |
4,1 |
37,3 |
|
48 |
43,2 |
4,3 |
38,9 |
|
50 |
45,0 |
4,5 |
40,5 |
|
52 |
46,8 |
4,7 |
42,1 |
|
54 |
48,6 |
4,9 |
43,7 |
|
56 |
50,4 |
5,0 |
45,4 |
|
58 |
52,2 |
5,2 |
47,0 |
|
60 |
54,0 |
5,4 |
48,6 |
|
62 |
55,8 |
5,6 |
50,2 |
|
64 |
57,6 |
5,8 |
51,8 |
|
66 |
59,4 |
5,9 |
53,5 |
|
68 |
61,2 |
6,1 |
55,1 |
|
70 |
63,0 |
6,3 |
56,7 |
|
72 |
64,8 |
6,5 |
58,3 |
|
74 |
66,6 |
6,7 |
59,9 |
|
76 |
68,4 |
6,8 |
61,6 |
|
78 |
70,2 |
7,0 |
63,2 |
|
80 |
72,0 |
7,2 |
64,8 |
|
82 |
73,8 |
7,4 |
66,4 |
|
84 |
75,6 |
7,6 |
68,0 |
|
86 |
77,4 |
7,7 |
69,7 |
|
88 |
79,2 |
7,9 |
71,3 |
|
90 |
81,0 |
8,1 |
72,9 |
|
92 |
82,8 |
8,3 |
74,5 |
|
94 |
84,6 |
8,5 |
76,1 |
|
96 |
86,4 |
8,6 |
77,8 |
|
98 |
88,2 |
8,8 |
79,4 |
|
100+ |
90,0 |
9,0 |
81,0 |
*administré à une concentration de 1 mg/ml sur 60 minutes, en perfusion à débit constant.
Traitement associé
Médicaments affectant la coagulation / fonction plaquettaire
La tolérance et l’efficacité de ce protocole d’administration en association avec l’héparine ou un antiagrégant plaquettaire comme l’acide acétylsalicylique au cours des 24 premières heures suivant l’administration d’ACTILYSE n’ont pas été suffisamment étudiées. Par conséquent, l’administration d’héparine par voie intraveineuse ou d’un antiagrégant plaquettaire comme l’acide acétylsalicylique doit être évitée au cours des premières 24 heures suivant l’administration d’ACTILYSE en raison du risque d’hémorragie augmenté. Si l’administration d’héparine est rendue nécessaire pour d’autres indications (par exemple en prévention de thrombose veineuse profonde), la posologie ne doit pas dépasser 10 000 UI par jour, par voie sous-cutanée.
Mode d’administration
La solution reconstituée doit être administrée par voie intraveineuse et doit être utilisée immédiatement.
Le flacon de 2 mg d’altéplase n’est pas adapté pour une utilisation dans cette indication.
Population pédiatrique
L’expérience de l’utilisation d’ACTILYSE chez les enfants et les adolescents est limitée. ACTILYSE est contre-indiqué pour le traitement de l’accident vasculaire cérébral ischémique à la phase aiguë chez les enfants et les adolescents âgés de moins de 16 ans (voir RCP rubrique 4.3). La dose pour les adolescents âgés de 16 ans ou plus est la même que celle pour les adultes (voir rubrique 4.4 pour les recommandations sur les techniques d’imageries à utiliser en amont).
Les adolescents âgés de 16 ans ou plus devraient être traités selon les instructions données pour les patients adultes, après une technique d’imagerie appropriée permettant d’exclure les pathologies mimant l’accident vasculaire cérébral et confirmant l’occlusion artérielle, correspondant au déficit neurologique.