Dernière mise à jour le 01/06/2026
IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : L01EA01
IMATINIB SANDOZ est un médicament qui contient une substance active appelée imatinib. Ce médicament agit par inhibition de la croissance des cellules anormales des maladies décrites ci-dessous parmi lesquelles certains types de cancer.
IMATINIB SANDOZ est un traitement chez les adultes et les enfants de :
· Leucémie myéloïde chronique (LMC)
La leucémie est un cancer des globules blancs du sang. Ces globules blancs aident habituellement l’organisme à se défendre contre les infections. La leucémie myéloïde chronique est une forme de leucémie dans laquelle certains globules blancs anormaux, appelés cellules myéloïdes, commencent à se multiplier de manière incontrôlée.
· Leucémie aiguë lymphoïde chromosome Philadelphie positive (LAL Ph-positive)
La leucémie est un cancer des globules blancs du sang. Ces globules blancs aident habituellement l’organisme à se défendre contre les infections. La leucémie aiguë lymphoblastique est une forme de leucémie dans laquelle certains globules blancs anormaux (appelés lymphoblastes) commencent à se multiplier de manière incontrôlée. IMATINIB SANDOZ inhibe la croissance de ces cellules.
IMATINIB SANDOZ est également un traitement chez les adultes de :
· Syndromes myéloprolifératifs/myélodysplasiques (SMP/SMD)
Il s’agit d’un groupe de maladies du sang pour lesquelles des cellules du sang commencent à se multiplier de manière incontrôlée. IMATINIB SANDOZ inhibe la croissance de ces cellules dans un certain sous-groupe de ces maladies.
· Syndrome hyperéosinophilique (SHE) et/ou de la leucémie chronique à éosinophiles (LCE)
Ce sont des maladies pour lesquelles des cellules du sang (appelées éosinophiles) commencent à se multiplier de manière incontrôlée. IMATINIB SANDOZ inhibe la croissance de ces cellules dans un certain sous-groupe de ces maladies.
· Tumeurs stromales gastro-intestinales malignes (GIST)
GIST est un cancer de l'estomac et des intestins. Il résulte de la croissance cellulaire incontrôlée des tissus de support de ces organes. IMATINIB SANDOZ inhibe la croissance de ces cellules.
· Dermatofibrosarcome protuberans (DFSP)
Le dermatofibrosarcome est un cancer du tissu sous la peau dans lequel certaines cellules commencent à se multiplier de manière incontrôlée. IMATINIB SANDOZ inhibe la croissance de ces cellules.
Par la suite dans cette notice, les abréviations ci-dessus sont utilisées pour désigner ces maladies.
Si vous avez des questions sur la façon dont IMATINIB SANDOZ agit ou pourquoi ce médicament vous a été prescrit, adressez-vous à votre médecin.
Présentations
> plaquette(s) PVC polyéthylène PVDC aluminium de 60 comprimé(s)
Code CIP : 34009 300 483 0 6
Déclaration de commercialisation : 22/12/2016
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 307,79 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 308,81 €
- Taux de remboursement :100%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : SANDOZ
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière semestrielle
- prescription initiale réservée à certains spécialistes
- renouvellement de la prescription réservé aux médecins compétents en CANCEROLOGIE
- renouvellement de la prescription réservé aux spécialistes en HEMATOLOGIE
- renouvellement de la prescription réservé aux spécialistes en MEDECINE INTERNE
- renouvellement de la prescription réservé aux spécialistes en ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 342 803 9
ANSM - Mis à jour le : 25/07/2024
IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé pelliculé sécable contient 100 mg d’imatinib (sous forme de mésilate).
Pour la liste complète des excipients, voir rubrique 6.1.
Jaune très foncé à brun orangé, rond, biconvexe, à bords biseautés, portant l’inscription « NVR » gravée sur une face et « SA » sur l’autre face avec une barre de sécabilité séparant 2 lettres.
Diamètre : environ 9,2 mm.
Le comprimé pelliculé sécable peut être divisé en doses égales.
4.1. Indications thérapeutiques
IMATINIB SANDOZ est indiqué dans le traitement :
· des patients adultes et enfants atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie (bcr-abl) positive (Ph+) nouvellement diagnostiquée lorsque la greffe de moelle osseuse ne peut être envisagée comme un traitement de première intention,
· des patients adultes et enfants atteints de LMC Ph+ en phase chronique après échec du traitement par l’interféron alpha, ou en phase accélérée ou en crise blastique,
· des patients adultes et enfants atteints de leucémie aiguë lymphoïde chromosome Philadelphie positive (LAL Ph+) nouvellement diagnostiquée en association avec la chimiothérapie,
· des patients adultes atteints de LAL Ph+ réfractaire ou en rechute en monothérapie,
· des patients adultes atteints de syndromes myélodysplasiques/myéloprolifératifs (SMD/SMP)associés à des réarrangements du gène du PDGFR (platelet-derived growth factor receptor),
· des patients adultes atteints d’un syndrome hyperéosinophilique (SHE) à un stade avancé et/ou d’une leucémie chronique à éosinophiles (LCE) associés à un réarrangement du FIP1L1- PDGFRa.
L’effet de l’imatinib sur l’issue d’une greffe de moelle osseuse n’a pas été évalué.
IMATINIB SANDOZ est indiqué dans :
· le traitement des patients adultes atteints de tumeurs stromales gastro-intestinales (GIST - gastrointestinal stromal tumours) malignes Kit (CD 117) positives non résécables et/ou métastatiques,
· le traitement adjuvant des patients adultes présentant un risque significatif de rechute après résection d’une tumeur stromale gastro-intestinale GIST Kit (CD117) positive. Les patients qui présentent un faible ou très faible risque ne doivent pas être traités,
· le traitement des patients adultes atteints de dermatofibrosarcome protuberans (DFSP ou maladie de Darier-Ferrand) non résécable et patients adultes atteints de DFSP en rechute et/ou métastatique ne relevant pas d’un traitement chirurgical.
Chez l’adulte et les patients pédiatriques, l’efficacité de l’imatinib est basée sur les taux de réponses hématologiques et cytogénétiques globales et la survie sans progression dans la LMC, sur les taux de réponses hématologique et cytogénétique des LAL Ph+, des SMD/SMP, sur les taux de réponses hématologiques des SHE/LCE et sur les taux de réponses objectives des patients adultes dans les GIST et les DFSP non résécables et/ou métastatiques et la survie sans rechute dans le traitement adjuvant des GIST. L’expérience avec l’imatinib chez les patients atteints de SMD/SMP associés à des réarrangements du gène du PDGFR est très limitée (voir rubrique 5.1).
A l’exception de la LMC en phase chronique nouvellement diagnostiquée, il n’existe pas d’étude clinique contrôlée démontrant un bénéfice clinique ou une prolongation de la durée de vie, pour ces maladies.
4.2. Posologie et mode d'administration
Pour les doses de 400 mg et plus (voir les recommandations de doses ci-dessous), un comprimé pelliculé sécable à 400 mg est disponible.
La dose prescrite doit être administrée par voie orale avec un grand verre d'eau, au cours d'un repas pour réduire le risque d’irritations gastro-intestinales. Les doses de 400 mg ou 600 mg devront être administrées en une prise par jour, tandis que la dose journalière de 800 mg devra être répartie en deux prises de 400 mg par jour, matin et soir.
Pour les patients incapables d'avaler les comprimés pelliculés, il est possible de disperser ces comprimés dans un verre d'eau plate ou de jus de pomme. Le nombre de comprimés requis devra être placé dans un volume de boisson approprié (approximativement 50 mL pour un comprimé à 100 mg et 200 mL pour un comprimé à 400 mg) et être remué avec une cuillère. La suspension devra être administrée immédiatement après désagrégation complète du (des) comprimé(s).
Posologie dans la LMC chez l’adulte
Patients adultes en phase chronique : la posologie recommandée est de 400 mg/j. La phase chronique est définie par l’ensemble des critères suivants : blastes < 15 % dans le sang et la moelle osseuse, basophiles dans le sang < 20 %, plaquettes > 100 x 109/L.
Patients adultes en phase accélérée : la posologie recommandée est de 600 mg/j. La phase accélérée est définie par la présence d’un des critères suivants : blastes ³ 15 % mais < 30 % dans le sang ou la mœlle osseuse, blastes plus promyélocytes ³ 30 % dans le sang ou la moelle osseuse (à condition que blastes < 30 %), basophiles dans le sang ³ 20 %, plaquettes < 100 x 109/L indépendamment du traitement.
Patients adultes en crise blastique : la posologie recommandée est de 600 mg/j. La crise blastique est définie par la présence de blastes ≥ 30 % dans le sang ou la moelle osseuse ou un envahissement extra-médullaire autre qu’une hépatosplénomégalie.
Durée du traitement : dans les études cliniques, le traitement est poursuivi jusqu’à progression de la maladie. L’effet de l’arrêt du traitement après l’obtention d’une réponse cytogénétique complète n’a pas été étudié.
En l’absence d'effets indésirables sévères et de neutropénie ou de thrombopénie sévères non imputables à la leucémie, une augmentation de la dose peut être envisagée de 400 mg à 600 mg ou 800 mg, chez les patients en phase chronique, ou de 600 mg à un maximum de 800 mg (en deux prises de 400 mg par jour) chez les patients en phase accélérée ou en crise blastique, dans les circonstances suivantes : évolution de la maladie (à tout moment), absence de réponse hématologique satisfaisante après un minimum de 3 mois de traitement, absence de réponse cytogénétique après 12 mois de traitement, ou perte de la réponse hématologique et/ou cytogénétique obtenue auparavant. Les patients devront être surveillés étroitement après augmentation de la dose étant donnée la possibilité d’une incidence accrue des effets indésirables à plus fortes doses.
Posologie dans la LMC chez l’enfant
Chez l'enfant, la posologie devra être établie en fonction de la surface corporelle (mg/m2). La dose journalière recommandée chez l'enfant est de 340 mg/m2 dans la LMC en phase chronique et dans la LMC en phase avancée (ne doit pas dépasser une dose totale de 800 mg). Le traitement peut être administré en une prise quotidienne ou bien être divisé en deux prises (une le matin et une le soir). Ces recommandations posologiques reposent actuellement sur un faible nombre d’enfants (voir rubriques 5.1 et 5.2). On ne dispose d'aucune donnée chez l'enfant de moins de 2 ans.
L’augmentation de doses de 340 mg/m2 jusqu’à 570 mg/m2 par jour (sans dépasser la dose totale de 800 mg) peut être envisagée chez l’enfant en l’absence d’effets indésirables graves et de neutropénie ou thrombopénie sévères non liées à la leucémie dans les circonstances suivantes : progression de la maladie (à n’importe quel moment) ; absence de réponse hématologique satisfaisante après au moins 3 mois de traitement ; absence de réponse cytogénétique après 12 mois de traitement ; ou perte d’une réponse hématologique et/ou cytogénétique antérieure.
Les patients devront être surveillés attentivement au cours des escalades de doses compte tenu du risque accru d’effets indésirables à des doses plus élevées.
Posologie dans les LAL Ph+ chez l’adulte
La posologie recommandée d’imatinib est de 600 mg/jour chez les patients adultes atteints de LAL Ph+. Le traitement devrait être supervisé par des hématologues experts dans la prise en charge de cette maladie pour toutes les phases de traitement.
Schéma thérapeutique : sur la base des données existantes, l’imatinib s’est montré efficace et sûr lorsqu’il est administré à 600 mg/j en association à une chimiothérapie d’induction, de consolidation et d’entretien utilisée des LAL Ph+ nouvellement diagnostiquées de l’adulte (voir rubrique 5.1).
La durée de traitement par imatinib peut varier en fonction du traitement appliqué, mais généralement les traitements prolongés d’imatinib ont fourni de meilleurs résultats.
Chez les patients adultes atteints de LAL Ph+ en rechute ou réfractaire, une monothérapie par imatinib à la dose de 600 mg/j est sure, efficace et peut être poursuivie jusqu’à la progression de la maladie.
Posologie dans les LAL Ph+ chez l’enfant
Chez l’enfant, la posologie devra être établie en fonction de la surface corporelle (mg/m2). Dans les LAL Ph+, la dose journalière recommandée chez l’enfant est de 340 mg/m2 (sans dépasser une dose totale de 600 mg).
Posologie dans les SMD/SMP
La posologie recommandée d’imatinib est de 400 mg/jour chez les patients adultes atteints de SMD/SMP.
La durée de traitement : dans l’unique étude clinique menée à ce jour, le traitement par imatinib a été poursuivi jusqu’à la progression de la maladie (voir rubrique 5.1). A la date de l’analyse, la durée médiane de traitement était de 47 mois (24 jours à 60 mois).
Posologie dans les SHE/LCE
La dose recommandée d’imatinib est de 100 mg/jour chez les patients adultes atteints de SHE/LCE.
Une augmentation de dose de 100 mg à 400 mg chez ces patients peut être envisagée si la réponse au traitement est insuffisante et en l’absence d’effets indésirables.
Le traitement doit être poursuivi aussi longtemps qu’il est bénéfique pour le patient.
Posologie dans les GIST
Patients adultes atteints de GIST malignes non résécables et/ou métastatiques : la posologie recommandée est de 400 mg/j.
Les données concernant l’effet de l’augmentation des doses de 400 mg à 600 mg ou 800 mg chez des patients en progression lorsqu’ils sont traités à la plus faible dose sont limitées (voir rubrique 5.1).
Durée du traitement : dans les études cliniques menées chez des patients atteints de GIST, le traitement par imatinib a été poursuivi jusqu’à la progression de la maladie. A la date de l’analyse, la durée médiane de traitement était de 7 mois (7 jours à 13 mois). L’effet de l’arrêt du traitement après l’obtention d’une réponse n’a pas été étudié.
La dose recommandée d’imatinib est de 400 mg par jour dans le traitement adjuvant des patients adultes après résection d’une tumeur stromale gastro-intestinale (GIST). La durée optimale de traitement n’a pas encore été établie. La durée de traitement dans les essais cliniques dans cette indication était de 36 mois (voir rubrique 5.1).
Posologie dans le DFSP
La posologie recommandée d’imatinib est de 800 mg/jour chez les patients adultes atteints de DFSP.
Ajustement de la posologie en cas d'effets indésirables
Effets indésirables extra-hématologiques
En cas de survenue d’un effet indésirable extra-hématologique sévère lors d’un traitement par l’imatinib, ce dernier doit être interrompu jusqu'à résolution de l'événement. Le traitement peut ensuite être repris de manière appropriée en fonction de la sévérité initiale de l'événement.
En cas d’élévation de la bilirubine > 3 x la limite supérieure de la normale (LSN) fournie par le laboratoire d’analyses ou des transaminases > 5 x la LSN, l’imatinib doit être interrompu jusqu’à un retour de la bilirubine à un taux < 1,5 x la LSN et des transaminases à un taux < 2,5 x la LSN. Le traitement par l’imatinib peut alors être repris à dose quotidienne réduite. Chez l’adulte, la dose sera diminuée de 400 à 300 mg ou de 600 à 400 mg ou de 800 mg à 600 mg, et chez l’enfant et l’adolescent la dose sera diminuée de 340 à 260 mg/m2/jour.
Effets indésirables hématologiques
En cas de neutropénie ou thrombopénie sévères, il est recommandé de diminuer la dose ou d'interrompre le traitement conformément au tableau ci-dessous.
Ajustements de posologie en cas de neutropénie et de thrombocytopénie :
|
SHE/LCE (dose initiale de 100 mg) |
PN < 1,0 x 109/L et/ou plaquettes < 50 x 109/L |
1. Arrêter l’imatinib jusqu'à ce que PN ≥ 1,5 x 109/L et plaquettes ≥ 75 x 109/L. 2. Reprendre le traitement par l’imatinib à la dose antérieure (c’est à dire avant l’effet indésirable sévère). |
|
LMC en phase chronique, SMD/SMP et GIST (dose initiale 400 mg) SHE/LCE (à la dose de 400 mg) |
PN < 1,0 x 109/L et/ou plaquettes < 50 x 109/L |
1. Arrêter l’imatinib jusqu'à ce que PN ≥ 1,5 x 109/L et plaquettes ≥ 75 x 109/L. 2. Reprendre le traitement par l’imatinib à la dose antérieure (c’est à dire avant l’effet indésirable sévère). 3. En cas de récidive de PN < 1,0 x 109/L et/ou plaquettes < 50 x 109/L, répéter l'étape 1 puis reprendre l’imatinib à la dose de 300 mg. |
|
LMC en phase chronique en pédiatrie (à la dose de340 mg/m2) |
PN < 1,0 x 109/L et/ou plaquettes < 50 x 109/L |
1. Arrêter l’imatinib jusqu'à ce que PN≥ 1,5 x 109/L et plaquettes ≥ 75 x 109/L. 2. Reprendre le traitement par l’imatinib à la dose antérieure (c’est à dire avant l’effet indésirable sévère). 3. En cas de récidive de PN < 1,0 x 109/Let/ou plaquettes < 50 x 109/L, répéter l'étape 1 puis reprendre l’imatinib à la dose de 260 mg/m2. |
|
LMC en phase accélérée ou crise blastique et LAL Ph+ (dose initiale 600 mg) |
aPN < 0,5 x 109/L et/ou plaquettes < 10 x 109/L |
1. Vérifier si la cytopénie est imputable à la leucémie (ponction ou biopsie médullaire). 2. Si la cytopénie n'est pas imputable à la leucémie, diminuer la dose d’imatinib à 400 mg. 3. Si la cytopénie persiste pendant 2 semaines, diminuer encore la dose à 300 mg. 4. Si la cytopénie persiste pendant 4 semaines et n'est toujours pas imputable à la leucémie, arrêter l’imatinib jusqu'à ce que PN ≥1 x 109/L et plaquettes ≥ 20 x 109/L, puis reprendre le traitement à 300 mg. |
|
LMC en phase accélérée ou en crise blastique en pédiatrie (à la dose de 340 mg/m2) |
aPN < 0.5 x 109/L et/ou plaquettes < 10 x 109/L
|
1. Vérifier si la cytopénie est imputable à la leucémie (ponction ou biopsie médullaire). 2. Si la cytopénie n'est pas imputable à la leucémie, diminuer la dose d’imatinib à 260 mg/m2. 3. Si la cytopénie persiste pendant 2 semaines, diminuer encore la dose à200 mg/m2. 4. Si la cytopénie persiste pendant 4 semaines et n'est toujours pas imputable à la leucémie, arrêter l’imatinib jusqu'à ce que PN ≥ 1 x 109/L et plaquettes ≥ 20 x 109/L, puisreprendre le traitement à 200 mg/m2. |
|
DFSP (à la dose de 800 mg) |
PN < 1,0 x 109/L et/ou plaquettes < 50 x 109/L |
1. Arrêter l’imatinib jusqu'à ce que PN ≥ 1,5 x109/L et plaquettes ≥ 75 x 109/L. 2. Reprendre le traitement par l’imatinib à la dose de 600 mg. 3. En cas de récidive de PN < 1,0 x 109/Let/ou plaquettes < 50 x 109/L, répéter l'étape 1 puis reprendre l’imatinib à la dose réduite de 400 mg. |
|
PN = Nombre de Polynucléaires Neutrophiles en valeur absolue a survenant après au moins 1 mois de traitement |
||
Populations spéciales
Enfant : il n’y a pas d’expérience chez l’enfant de moins de 2 ans atteint de LMC et chez l’enfant de moins d’un an atteint de LAL Ph+ (voir rubrique 5.1). L’expérience est très limitée chez l’enfant atteint de SMD/SMP, de DFSP, de GIST et de SHE/LCE.
La sécurité et l’efficacité de l’imatinib chez les enfants âgés de moins de 18 ans atteints de SMD/SMP, DFSP, GIST et SHE/LCE n’ont pas été établies par des études cliniques. Les données publiées actuellement disponibles sont résumées dans la rubrique 5.1 mais aucune recommandation sur la posologie ne peut être donnée.
Insuffisance hépatique : L’imatinib est principalement métabolisé par le foie. Les patients présentant une altération de la fonction hépatique, légère, modérée ou importante devraient être traités à la dose minimale recommandée 400 mg. La dose peut être réduite si elle est mal tolérée (voir rubrique 4.4, 4.8 et 5.2).
Classification des altérations hépatiques :
|
Altération de la fonction hépatique |
Tests de la fonction hépatique |
|
Légère |
Augmentation de la bilirubine totale de 1,5 fois la LSN ASAT : > LSN (peut être normale ou < LSN si la bilirubine totale est > LSN) |
|
Modérée |
Augmentation de la bilirubine totale > à 1,5 fois la LSN et < 3,0 fois la LSN ASAT quelle que soit la valeur |
|
Importante |
Augmentation de la bilirubine totale > 3,0 fois la LSN et < 10 fois la LSN ASAT quelle que soit la valeur |
LSN : Limite supérieure de la normale du laboratoire
ASAT : aspartate aminotransférase
Insuffisance rénale : chez les patients présentant une altération de la fonction rénale ou sous dialyse, la dose initiale de traitement de 400 mg par jour est recommandée.
Toutefois, la prudence est recommandée chez ces patients. La dose peut être réduite si elle est mal tolérée. Si elle est tolérée, la dose peut être augmentée en l’absence d’efficacité (voir rubrique 4.4 et 5.2).
Personnes âgées : la pharmacocinétique de l’imatinib n’a pas été spécifiquement étudiée chez les personnes âgées. Aucune différence significative de pharmacocinétique n’a été observée en fonction de l’âge chez les patients adultes inclus dans les études cliniques dont plus de 20 % étaient âgés de 65 ans et plus. Par conséquent, aucune recommandation particulière sur la posologie n’est requise chez les personnes âgées.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
L’utilisation concomitante d’imatinib et de médicaments inducteurs du CYP3A4 (par exemple : dexaméthasone, phénytoïne, carbamazépine, rifampicine, phénobarbital, ou Hypericum perforatum (millepertuis), peut réduire significativement l’exposition systémique à l’imatinib et augmenter potentiellement le risque d’échec thérapeutique. Par conséquent, l’utilisation concomitante d’imatinib avec des inducteurs puissants du CYP3A4 devra être évitée (voir rubrique 4.5).
Hypothyroïdie
Des cas cliniques d’hypothyroïdie ont été rapportés chez des patients traités par l’imatinib, ayant subi une thyroïdectomie et recevant un traitement par lévothyroxine (voir rubrique 4.5). Les taux de l’hormone thyréotrope (TSH) devront être étroitement surveillés chez ces patients.
Hépatotoxicité
Le métabolisme de l’imatinib est principalement hépatique, et seulement 13 % de l’excrétion est rénale. Chez les patients présentant une altération de la fonction hépatique (légère, modérée ou importante) la numération formule sanguine et les enzymes hépatiques devront être étroitement surveillées (cf. rubrique 4.2, 4.8 et 5.2). Il est à noter que les patients atteints de GIST peuvent présenter des métastases hépatiques qui pourraient entraîner une insuffisance hépatique.
Des cas d’altérations de la fonction hépatique, y compris des cas d’insuffisance hépatique et de nécrose hépatique ont été observés avec l’imatinib. Lorsque l’imatinib est associé à des chimiothérapies à fortes doses, une augmentation des réactions hépatiques graves a été mise en évidence. Une surveillance étroite de la fonction hépatique est recommandée lorsque l’imatinib est associé à une chimiothérapie connue comme pouvant être associée à une altération de la fonction hépatique (voir rubriques 4.5 et 4.8).
Rétention hydrique
Des cas de rétention hydrique sévère (épanchement pleural, œdème, œdème pulmonaire, ascite, œdème superficiel) ont été décrits chez environ 2,5 % des patients atteints de LMC nouvellement diagnostiqués traités par l’imatinib. Il est donc fortement recommandé de peser régulièrement les patients.
Une prise de poids rapide et inattendue devra faire l'objet d'un examen plus approfondi et, si nécessaire, l’instauration d'un traitement symptomatique et des mesures thérapeutiques devront être entreprises. Dans les études cliniques, l’incidence de ces effets indésirables était augmentée chez les personnes âgées ainsi que chez ceux ayant des antécédents cardiaques. La prudence est donc recommandée chez des patients présentant un dysfonctionnement cardiaque.
Patients présentant des pathologies cardiaques
Les patients présentant des pathologies cardiaques, des facteurs de risque de survenue d’insuffisance cardiaque ou des antécédents d’insuffisance rénale devront être étroitement surveillés, et tout patient présentant des signes ou des symptômes évocateurs d’une insuffisance cardiaque ou rénale doit faire l’objet d’une évaluation et être traités.
Chez des patients présentant un syndrome hyperéosinophilique (HES) avec infiltration de cellules HES dans le myocarde, des cas isolés de choc cardiogénique et d’altération de la fonction ventriculaire gauche ont été associés à la dégranulation de cellules HES lors de l’instauration d’un traitement par imatinib. Cette situation s’est montrée réversible après l’administration d’une corticothérapie systémique, des mesures d’assistance circulatoire et l’interruption temporaire de l’imatinib. Comme des effets indésirables cardiaques ont été observés peu fréquemment avec l’imatinib, une évaluation du rapport bénéfices/risques du traitement par l’imatinib devra être envisagée chez les patients atteints de SHE/LCE avant l’instauration du traitement.
Les syndromes myélodysplasiques/myéloprolifératifs associés à des réarrangements du gène du récepteur PDGFR pourront être associés à des taux élevés d’éosinophiles. La prise en charge par un cardiologue, la réalisation d’un échocardiogramme et le dosage sérique de la troponine devront donc être envisagés chez les patients atteints de SHE/LCE et chez les patients atteints de SMD/SMP associés à des taux élevés d’éosinophiles, avant l’administration de l’imatinib. Si l’un de ces examens est anormal, le suivi par un cardiologue et l’administration prophylactique d’une corticothérapie systémique (1-2 mg/kg) pendant une à deux semaines en association avec l’imatinib devra être envisagée lors de l’instauration du traitement.
Hémorragies gastro-intestinales
Dans l’étude clinique menée chez des patients atteints de GIST non résécables et/ou métastatiques, des hémorragies gastro-intestinales et intratumorales ont été rapportées (voir rubrique 4.8). Sur la base des données disponibles, aucun facteur (ex. taille de la tumeur, localisation de la tumeur, troubles de la coagulation) n’a été identifié, prédisposant les patients atteints de GIST à un risque plus élevé de développer l’un ou l’autre des deux types d’hémorragies. Puisqu’une augmentation de la vascularisation et une propension aux saignements font partie de la nature et l’évolution clinique de la maladie, des modalités standardisées de suivi et de prise en charge des hémorragies devront être adoptées pour tous les patients.
De plus, des ectasies vasculaires de l’antre gastrique (EVAG), une cause rare d’hémorragies gastro- intestinales, ont été rapportées depuis la mise sur le marché chez des patients atteints de LMC, de LAL et d’autres pathologies (voir rubrique 4.8). Lorsque cela est nécessaire, l’arrêt du traitement par IMATINIB SANDOZ doit être envisagé.
Syndrome de lyse tumorale
En raison de la survenue possible de syndrome de lyse tumorale (SLT), il est recommandé de corriger toute déshydratation cliniquement significative et de traiter l’hyperuricémie avant l’initiation du traitement par l’imatinib (voir rubrique 4.8).
Réactivation de l'hépatite B
Des cas de réactivation du virus l’hépatite B ont été rapportés chez des patients porteurs chroniques du virus et traités par des inhibiteurs de la tyrosine kinase BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante requérant une transplantation hépatique ou dont l’issue a été fatale.
Tous les patients doivent faire l’objet d’un dépistage d’une infection par le VHB avant l’initiation d’un traitement par IMATINIB SANDOZ. Un médecin spécialisé en hépatologie doit être consulté avant instauration du traitement chez les patients porteurs de marqueurs sérologiques positifs (y compris ceux ayant une hépatite B active) et chez les patients dont la sérologie devient positive en cours du traitement. Les patients porteurs du VHB doivent être étroitement suivis tout au long du traitement par IMATINIB SANDOZ et plusieurs mois après la fin du traitement (voir rubrique 4.8).
Phototoxicité
L'exposition directe au soleil doit être évitée ou réduite en raison du risque de phototoxicité associé au traitement par imatinib. Il doit être conseillé aux patients d’utiliser des mesures telles que le port de vêtements de protection et l’utilisation d’un écran solaire avec un facteur de protection solaire (FPS) élevé.
Microangiopathie thrombotique
Les inhibiteurs de la tyrosine kinase (ITK) BCR-ABL ont été associés à des microangiopathies thrombotiques (MAT), et des cas individuels ont été rapportés avec l’imatinib(voir rubrique 4.8). Si des résultats biologiques ou cliniques associés à une MAT surviennent chez un patient traité par IMATINIB SANDOZ, le traitement doit être interrompu et une évaluation approfondie de la MAT doit être réalisée, incluant la détermination de l’activité ADAMTS13 et des anticorps anti-ADAMTS13.
Si le taux d’anticorps anti-ADAMTS13 est élevé conjointement à une activité ADAMTS13 faible, le traitement par IMATINIB SANDOZ ne doit pas être repris.
Analyses biologiques
Des numérations formules sanguines doivent être effectuées régulièrement pendant le traitement par l’imatinib. Le traitement par l’imatinib de patients atteints de LMC a été associé à une neutropénie ou une thrombopénie. Cependant, la survenue de ces cytopénies est vraisemblablement liée au stade de la maladie traitée : elles ont été plus fréquemment retrouvées chez les patients en phase accélérée ou en crise blastique que chez ceux en phase chronique de la LMC. Le traitement par l’imatinib peut alors être interrompu ou la dose réduite, selon les recommandations de la rubrique 4.2.
La fonction hépatique (transaminases, bilirubine, phosphatases alcalines) doit faire l’objet d’une surveillance régulière chez les patients traités par l’imatinib.
Chez les patients présentant une altération de la fonction rénale, l’exposition plasmatique à l’imatinib semble être supérieure à celle des patients présentant une fonction rénale normale, probablement en raison d’un taux plasmatique élevé de l’alpha-glycoprotéine acide, une protéine plasmatique liée à l’imatinib, chez ces patients. Chez les patients présentant une altération de la fonction rénale, la dose la plus faible devra être utilisée à l’initiation du traitement. Les patients présentant une altération sévère de la fonction rénale devront être traités avec attention. La dose peut être réduite si elle est mal tolérée (voir rubrique 4.2 et 5.2).
Un traitement au long terme par imatinib peut être associé à une détérioration cliniquement significative de la fonction rénale. La fonction rénale doit donc être évaluée avant le début du traitement par imatinib et étroitement surveillée pendant le traitement, en portant une attention particulière aux patients présentant des facteurs de risque d’altération de la fonction rénale. Si une altération de la fonction rénale est observée, une prise en charge et un traitement adaptés doivent être mis en place en accord avec les recommandations standards de traitement.
Population pédiatrique
Des cas de retard de croissance chez les enfants et pré-adolescents recevant de l’imatinib ont été rapportés. Dans une étude observationnelle chez les patients pédiatriques atteints de LMC, une diminution statistiquement significative (mais d’une pertinence clinique incertaine) des valeurs d’écart-type de la taille médiane après 12 et 24 mois de traitement a été reportée dans deux sous-groupes de taille limitée, indépendamment du statut pubertaire ou du sexe. Une surveillance étroite de la croissance chez les enfants traités par imatinib est recommandée (voir rubrique 4.8).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Substances actives pouvant augmenter les concentrations plasmatiques d’imatinib
Les substances inhibant l’activité de l’isoenzyme CYP3A4 du cytochrome P450 (par exemple : inhibiteurs de protéase tels qu’indinavir, lopinavir/ritonavir, ritonavir, saquinavir, télaprévir, nelfinavir, bocéprévir ; antifongiques azolés tels que kétoconazole, itraconazole, posaconazole, voriconazole ; certains macrolides tels qu’érythromycine, clarithromycine et télithromycine) pourraient diminuer le métabolisme de l’imatinib et donc augmenter les concentrations plasmatiques de l’imatinib.
Une augmentation significative de l’exposition systémique à l’imatinib (la valeur moyenne de la Cmax et de l’ASC (Aire sous la courbe) ont respectivement été augmentées de 26 % et 40 %) a été observée chez des volontaires sains lors de l’administration d’une dose unique de kétoconazole (un inhibiteur du CYP3A4). La prudence est requise lorsque l’imatinib est administré avec des inhibiteurs du CYP3A4.
Substances actives pouvant diminuer les concentrations plasmatiques d’imatinib
Les substances agissant comme inducteurs de l’activité du CYP3A4 (par exemple : dexaméthasone, phénytoïne, carbamazépine, rifampicine, phénobarbital, fosphénytoïne, primidone, Hypericum perforatum (millepertuis)) pourraient réduire significativement l’exposition systémique à l’imatinib, et potentiellement augmenter le risque d’échec thérapeutique. Un traitement préalable par 600 mg de rifampicine à doses multiples suivies d'une dose unique de 400 mg d’imatinib, a entraîné une diminution de Cmax et de l’ASC(0-24) d’au moins 54 % et 74 %, par rapport à leurs valeurs respectives sans traitement par rifampicine. Des résultats similaires ont été observés chez des patients atteints de gliomes malins traités par l’imatinib et avec des antiépileptiques inducteurs enzymatiques tels que la carbamazépine, l’oxcarbazépine et la phénytoïne. L’ASC plasmatique de l’imatinib a diminué de 73 % par rapport à celle des patients non traités par antiépileptiques inducteurs enzymatiques. L’utilisation concomitante d’imatinib avec des inducteurs puissants du CYP3A4 devra être évitée.
Substances actives dont la concentration plasmatique peut être modifiée par l’imatinib
L’imatinib augmente la valeur moyenne de la Cmax et l’ASC de la simvastatine (substrat du CYP3A4), respectivement 2 fois et 3,5 fois, indiquant ainsi une inhibition du CYP3A4 par l’imatinib. L’imatinib doit donc être associé avec prudence à des substrats du CYP3A4 dont l’index thérapeutique est étroit (par exemple : ciclosporine, pimozide, tacrolimus, sirolimus, ergotamine, diergotamine, fentanyl, alfentanil, terfénadine, bortézomib, docétaxel et quinidine). Par ailleurs, l’imatinib peut augmenter la concentration plasmatique d’autres médicaments métabolisés par le CYP3A4 (par exemple triazolo-benzodiazépines, inhibiteurs calciques de type dihydropyridine, certains inhibiteurs de l’HMG-CoA réductase, c’est à dire les statines, etc.).
En raison des risques connus d’augmentation des saignements associés à l’utilisation de l’imatinib (par exemple hémorragie), les patients nécessitant un traitement anticoagulant devront recevoir de l’héparine standard ou de bas poids moléculaire au lieu de dérivés de la coumarine tels que la warfarine.
In vitro, l’imatinib inhibe l’activité de l’isoenzyme CYP2D6 du cytochrome P450 à des concentrations similaires à celles affectant l’activité du CYP3A4. L’imatinib à 400 mg deux fois par jour avait un effet inhibiteur sur le métabolisme de métoprolol via le CYP2D6, avec une augmentation approximativement de 23 % du Cmax et de l’ASC du métoprolol (IC 90 % [1,16-1,30]. Il ne semble pas nécessaire d’adapter les doses lorsque l’imatinib est administré avec des substrats du CYP2D6, toutefois la prudence est recommandée avec les substrats du CYP2D6 présentant une fenêtre thérapeutique étroite tel que le métoprolol. Chez les patients traités par métoprolol, la surveillance clinique devra être envisagée.
In vitro, l’imatinib inhibe l’O-glucuronidation du paracétamol avec un Ki de 58,5 µmol/L. Cette inhibition n’a pas été observée in vivo après l’administration de 400 mg d’imatinib et 1000 mg de paracétamol. Des doses plus élevées d’imatinib et de paracétamol n’ont pas été étudiées.
La prudence est donc requise lors de l’utilisation concomitante de fortes doses d’imatinib et de paracétamol.
Des cas cliniques d’hypothyroïdie ont été rapportés chez des patients traités par l’imatinib, ayant subi une thyroïdectomie et recevant un traitement par lévothyroxine (voir rubrique 4.4). La prudence est donc recommandée. Toutefois, le mécanisme de cette interaction observée est à ce jour inconnu.
Chez les patients atteints de LAL Ph+, on dispose d’une expérience clinique de l’administration concomitante d’imatinib avec une chimiothérapie (voir rubrique 5.1), cependant les interactions médicamenteuses entre l’imatinib et les schémas thérapeutiques de chimiothérapie n’ont pas été clairement identifiés.
Les effets indésirables de l’imatinib tels qu’une hépatotoxicité, une myélosuppression ou d’autres effets, peuvent augmenter et il a été rapporté qu’une utilisation concomitante de la L-asparaginase pourrait être associée à une hépatotoxicité augmentée (voir rubrique 4.8). Ainsi, l’administration d’imatinib en association nécessite des précautions particulières.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Les femmes en âge de procréer doivent être informées qu’elles doivent utiliser une contraception efficace pendant le traitement et pendant au moins 15 jours après l’arrêt du traitement par imatinib
Grossesse
Les données concernant l'utilisation de l'imatinib chez la femme enceinte sont limitées. Depuis la mise sur le marché, des cas d’avortements spontanés et d’anomalies congénitales chez le nourrisson ont été rapportés chez des femmes ayant été traitées par imatinib. Les études effectuées chez l'animal ont toutefois mis en évidence une toxicité sur la reproduction (voir rubrique 5.3) et le risque potentiel sur le fœtus en clinique n’est pas connu. L’imatinib ne doit pas être utilisé pendant la grossesse à moins d’une nécessité absolue. S’il est utilisé au cours de la grossesse, la patiente doit être prévenue du risque potentiel pour le fœtus.
Les informations concernant la distribution de l’imatinib dans le lait maternel sont limitées. Les études chez deux patientes qui allaitaient ont montré que l’imatinib et son métabolite actif peuvent être distribués dans le lait maternel. Le rapport lait/plasma des concentrations d’imatinib mesuré chez une patiente était de 0,5 et celui du métabolite était de 0,9, suggérant une distribution plus élevée du métabolite dans le lait. En considérant la concentration de l’imatinib associée à celle de son métabolite et la quantité de lait journalière ingérée par les nourrissons, l’exposition totale attendue devrait être faible (environ 10 % de la dose thérapeutique).
Cependant, les effets d’une exposition de faible dose chez le nourrisson n’étant pas connus, les femmes ne doivent pas allaiter pendant le traitement et pendant au moins 15 jours après l’arrêt du traitement par imatinib.
Fertilité
La fertilité des rats mâles et femelles n’a pas été affectée dans les études précliniques, bien que des effets sur les paramètres de la reproduction aient été observés (voir rubrique 5.3). Aucune étude n'a été effectuée sur des patients recevant l’imatinib et sur son effet sur la fécondité et la gamétogenèse. Les patients préoccupés par leur fécondité sous traitement par l’imatinib doivent consulter leur médecin.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les patients atteints de pathologies malignes à un stade avancé peuvent présenter des affections intercurrentes. Ces affections peuvent rendre difficile l’évaluation du lien entre l’administration de l’imatinib et la survenue d’événements indésirables en raison de la variété des symptômes liés à la maladie sous-jacente, à sa progression ou à la co-administration de nombreux médicaments.
Au cours des études cliniques menées dans la LMC, un arrêt du traitement motivé par des effets indésirables imputables au médicament a été observé chez 2,4 % des patients nouvellement diagnostiqués, 4 % des patients en phase chronique tardive après échec du traitement par l’interféron, 4 % des patients en phase accélérée après échec du traitement par l’interféron et 5 % des patients en crise blastique après échec du traitement par l’interféron. Dans les GIST, le produit étudié a été arrêté en raison d'effets indésirables imputables au traitement chez 4 % des patients.
Les effets indésirables ont été comparables dans toutes les indications, à deux exceptions près. Il y a eu plus de myélosuppressions observées chez les patients atteints de LMC que chez ceux atteints de GIST, ce qui est probablement dû à la maladie sous-jacente. Dans l’étude clinique menée chez des patients atteints de GIST non résécables et/ou métastatiques, 7 (5 %) patients ont présenté des saignements de grade 3 / 4 selon la classification CTC (Common Toxicity Criteria) : saignements gastro-intestinaux (3 patients), saignements intra-tumoraux (3 patients), les deux types (1 patient).
La localisation de la tumeur gastro-intestinale peut avoir été à l’origine des saignements gastro-intestinaux (voir rubrique 4.4). Les saignements gastro-intestinaux et intra-tumoraux peuvent être sérieux et dans certains cas fatals. Les effets indésirables les plus fréquemment rapportés (≥ 10 %) pouvant être imputables au traitement par imatinib dans les deux indications ont été des nausées modérées, vomissements, diarrhée, douleur abdominale, fatigue, myalgies, crampes musculaires et rash. Des œdèmes superficiels ont été très fréquemment observés dans toutes les études cliniques et décrits principalement comme des œdèmes péri-orbitaux ou des membres inférieurs.
Toutefois, ces œdèmes ont été rarement sévères et ont pu être contrôlés par des diurétiques, d’autres mesures symptomatiques ou en réduisant la dose d’imatinib.
Lorsque l’imatinib était associé à des doses élevées de chimiothérapie chez des patients atteints de LAL Ph+, une toxicité hépatique transitoire se traduisant par une élévation des transaminases et une hyperbilirubinémie a été observée. Au vu des données limitées de tolérance, les effets indésirables rapportés ci-après chez l’enfant sont cohérents avec le profil de tolérance observé chez l’adulte atteint de LAL Ph+. Les données de tolérance chez l’enfant atteint de LAL Ph+ sont très limitées bien qu’aucun nouveau problème de sécurité n’ait été identifié.
Divers effets indésirables tels qu’épanchement pleural, ascite, œdème pulmonaire, prise de poids rapide avec ou sans œdème superficiel ont été décrits dans le cadre de rétention hydrique. Ces effets peuvent habituellement être contrôlés par l’interruption temporaire d’imatinib et par l’utilisation de diurétiques et d’autres traitements symptomatiques appropriés. Cependant, certains de ces effets peuvent être graves, voire mettre en jeu le pronostic vital : plusieurs patients en crise blastique sont décédés, avec un tableau clinique complexe associant un épanchement pleural, une insuffisance cardiaque congestive et une insuffisance rénale. Les études cliniques menées chez l’enfant n’ont pas révélé de données de tolérance particulière à cette population.
Effets indésirables
Les effets indésirables, en dehors des cas isolés, sont repris ci-dessous par organe et par ordre de fréquence. Les catégories de fréquence sont définies selon la convention suivante : très fréquent (≥1/10), fréquent (≥1/100, <1/10), peu fréquent (≥1/1 000, <1/100), rare (≥1/10 000, <1/1 000), très rare (<1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables doivent être présentés suivant un ordre de fréquence, le plus fréquent en premier.
Les effets indésirables et leurs fréquences sont présentés dans le Tableau 1.
Tableau 1 : Tableau de synthèse des effets indésirables
|
Infections et infestations |
|
|
Peu fréquent : |
Zona, herpès simplex, inflammation nasopharyngée, pneumonie1, sinusite, cellulite, infection des voies respiratoires hautes, grippe, infection des voies urinaires, gastroentérite, septicémie |
|
Rare : |
Infection fongique |
|
Fréquence indéterminée : |
Réactivation de l’hépatite B11 |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
|
|
Rare : |
Syndrome de lyse tumorale |
|
Fréquence indéterminée : |
Hémorragie tumorale/nécrose tumorale* |
|
Affections du système immunitaire |
|
|
Fréquence indéterminée : |
Choc anaphylactique* |
|
Affections hématologiques et du système lymphatique |
|
|
Très fréquent : |
Neutropénie, thrombopénie, anémie |
|
Fréquent : |
Pancytopénie, neutropénie fébrile |
|
Peu fréquent : |
Thrombocythémie, lymphopénie, aplasie médullaire, éosinophilie, lymphadénopathie |
|
Rare : |
Anémie hémolytique, microangiopathie thrombotique |
|
Troubles du métabolisme et de la nutrition |
|
|
Fréquent : |
Anorexie |
|
Peu fréquent : |
Hypokaliémie, augmentation de l’appétit, hypophosphatémie, diminution de l’appétit, déshydratation, goutte, hyperuricémie, hypercalcémie, hyperglycémie, hyponatrémie |
|
Rare : |
Hyperkaliémie, hypomagnésémie |
|
Affections psychiatriques |
|
|
Fréquent : |
Insomnie |
|
Peu fréquent : |
Dépression, diminution de libido, anxiété |
|
Rare : |
Confusion |
|
Affections du système nerveux |
|
|
Très fréquent : |
Céphalées2 |
|
Fréquent : |
Sensations vertigineuses, paresthésies, troubles du goût, hypoesthésie |
|
Peu fréquent : |
Migraine, somnolence, syncope, neuropathie périphérique, troubles de la mémoire, sciatique, syndrome des jambes sans repos, tremblement, hémorragie cérébrale |
|
Rare : |
Hypertension intracrânienne, convulsions, névrite optique |
|
Fréquence indéterminée : |
Œdème cérébral* |
|
Affections oculaires |
|
|
Fréquent : |
Œdème des paupières, sécrétions lacrymales augmentées, hémorragie conjonctivale, conjonctivite, yeux secs, vision trouble |
|
Peu fréquent : |
Irritation oculaire, douleur oculaire, œdème orbitaire, hémorragie sclérale, hémorragie rétinienne, blépharite, œdème maculaire |
|
Rare : |
Cataracte, glaucome, œdème papillaire |
|
Fréquence indéterminée : |
Hémorragie du corps vitré* |
|
Affections de l’oreille et du labyrinthe |
|
|
Peu fréquent : |
Vertiges, acouphènes, perte auditive |
|
Affections cardiaques |
|
|
Peu fréquent : |
Palpitations, tachycardie, insuffisance cardiaque congestive3, œdème pulmonaire |
|
Rare : |
Arythmie, fibrillation auriculaire, arrêt cardiaque, infarctus du myocarde, angine de poitrine, épanchement péricardique |
|
Fréquence indéterminée : |
Péricardite*, tamponnade* |
|
Affections vasculaires4 |
|
|
Fréquent : |
Bouffées vasomotrices, hémorragie |
|
Peu fréquent : |
Hypertension, hématome, hématome sous-dural, extrémités froides, hypotension, syndrome de Raynaud |
|
Fréquence indéterminée : |
Thrombose/embolie* |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Fréquent : |
Dyspnée, épistaxis, toux |
|
Peu fréquent : |
Epanchement pleural5, douleur pharyngolaryngée, pharyngite |
|
Rare : |
Douleur pleurale, fibrose pulmonaire, hypertension pulmonaire, hémorragie pulmonaire |
|
Fréquence indéterminée : |
Insuffisance respiratoire aiguë10*, pneumopathie interstitielle* |
|
Affections gastro-intestinales |
|
|
Très fréquent : |
Nausées, diarrhée, vomissements, dyspepsie, douleur abdominale6 |
|
Fréquent : |
Flatulence, ballonnements, reflux gastro-œsophagien, constipation, sécheresse de la bouche, gastrite |
|
Peu fréquent : |
Stomatite, mucite, hémorragie gastro-intestinale7, éructation, méléna, œsophagite, ascite, ulcère gastrique, hématémèse, chéilite, dysphagie, pancréatite |
|
Rare : |
Colite, iléus, affection abdominale inflammatoire |
|
Fréquence indéterminée : |
Iléus/occlusion intestinale*, perforation gastro-intestinale*, diverticulite*, ectasie vasculaire de l’antre gastrique (EVAG)* |
|
Affections hépatobiliaires |
|
|
Fréquent : |
Elévation des enzymes hépatiques |
|
Peu fréquent : |
Hyperbilirubinémie, hépatite, ictère |
|
Rare : |
Insuffisance hépatique8, nécrose hépatique |
|
Affections de la peau et du tissu sous-cutané |
|
|
Très fréquent : |
Œdème péri-orbitaire, dermatite/eczéma/rash |
|
Fréquent : |
Prurit, œdème de la face, peau sèche, érythème, alopécie, sueurs nocturnes, réaction de photosensibilité |
|
Peu fréquent : |
Rash pustuleux, contusion, hypersudation, urticaire, ecchymose, tendance augmentée aux ecchymoses, hypotrichose, hypopigmentation cutanée, dermatite exfoliative, ongles cassants, folliculite, pétéchies, psoriasis, purpura, hyperpigmentation cutanée, éruption bulleuse, panniculite (y compris l’érythème noueux) pannic
pannic
|
|
Rare : |
Dermatose aiguë fébrile neutrophilique (syndrome de Sweet), décoloration des ongles, œdème de Quincke, rash vésiculaire, érythème polymorphe, vascularite leucocyclasique, syndrome de Stevens-Johnson, pustulose exanthémateuse aiguë généralisée, pemphigus* |
|
Fréquence indéterminée : |
Erythrodysesthésie palmo-plantaire*, kératose lichenoïde*, lichen plan*, nécrolyse épidermique toxique*, syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (syndrome DRESS)*, pseudoporphyrie* |
|
Affections musculo-squelettiques et systémiques |
|
|
Très fréquent : |
Crampes et spasmes musculaires, douleurs musculo-squelettiques incluant les myalgies12, arthralgies, douleurs osseuses9 |
|
Fréquent : |
Gonflement des articulations |
|
Peu fréquent : |
Raideur articulaire et musculaire, ostéonécrose* |
|
Rare : |
Faiblesse musculaire, arthrite, rhabdomyolyse/myopathie |
|
Fréquence indéterminée : |
Retard de croissance chez l’enfant* |
|
Affections du rein et des voies urinaires |
|
|
Peu fréquent : |
Douleur rénale, hématurie, insuffisance rénale aiguë, pollakiurie |
|
Fréquence indéterminée : |
Insuffisance rénale chronique |
|
Affections des organes de reproduction et du sein |
|
|
Peu fréquent : |
Gynécomastie, dysfonctionnement érectile, ménorragie, menstruation irrégulière, troubles sexuels, douleur des mamelons, gonflement des seins, œdème du scrotum |
|
Rare : |
Corps jaune hémorragique, kyste ovarien hémorragique |
|
Troubles généraux et anomalies au site d’administration |
|
|
Très fréquent : |
Rétention hydrique et œdème, fatigue |
|
Fréquent : |
Faiblesse, pyrexie, anasarque, frissons, rigidité |
|
Peu fréquent : |
Douleur thoracique, malaise |
|
Investigations |
|
|
Très fréquent : |
Prise de poids |
|
Fréquent : |
Perte de poids |
|
Peu fréquent : |
Augmentation de la créatininémie, augmentation de la créatine phosphokinase, augmentation de la lacticodéshydrogénase, augmentation des phosphatases alcalines |
|
Rare : |
Augmentation de l’amylasémie |
*Ces effets indésirables ont été principalement rapportés après la mise sur le marché de l’imatinib
Ceci inclut les cas issus de la notification spontanée ainsi que les événements indésirables graves des études cliniques en cours, des programmes d’accès élargi, des études de pharmacologie clinique et des études exploratoires menées dans le cadre d’indications thérapeutiques non approuvées. Etant donné que ces évènements sont issus d’une population dont la taille n’est pas déterminée, il n’est pas toujours possible d’estimer de manière fiable leur fréquence ou d’établir la relation de causalité avec l’exposition à l’imatinib.
1 La pneumonie a été le plus fréquemment observée chez les patients atteints de LMC en transformation et les patients atteints de GIST.
2 Les céphalées ont été le plus fréquemment observées chez les patients atteints de GIST.
3 Selon l’unité de mesure « patient-année », les effets cardiaques incluant l’insuffisance cardiaque congestive ont été plus fréquemment observés chez les patients ayant une LMC en transformation que chez ceux ayant une LMC en phase chronique.
4 Les bouffées vasomotrices ont été le plus fréquemment observées chez les patients atteints de GIST et les saignements (hématomes et hémorragies) ont été le plus fréquemment observés chez les patients atteints de GIST et les patients atteints de LMC en transformation (LMC en phase accélérée et LMC en crise blastique).
5 L’épanchement pleural a été rapporté plus fréquemment chez les patients atteints de GIST et les patients ayant une LMC en transformation (LMC en phase accélérée et LMC en crise blastique) que chez les patients en phase chronique.
6+7 Les douleurs abdominales et les hémorragies gastro-intestinales ont été le plus fréquemment observées chez les patients atteints de GIST.
8 Des cas mortels d’insuffisance hépatique et de nécrose hépatique ont été rapportés.
9 Les douleurs musculosquelettiques et les effets reliés à ces douleurs ont été plus fréquemment observés chez les patients atteints de LMC que chez les patients atteints de GIST.
10 Des cas d’évolution fatale ont été rapportés chez des patients à un stade avancé de la maladie, présentant des infections sévères, des neutropénies sévères et d’autres troubles cliniques concomitants sévères.
11 Des cas de réactivation du virus de l’hépatite B ont été rapportés chez des patients traités par des inhibiteurs de la tyrosine kinase BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante requérant une transplantation hépatique ou dont l’issue a été fatale (voir rubrique 4.4).
12 Depuis la commercialisation, des douleurs musculo-squelettiques ont été rapportées pendant le traitement ou après l’arrêt du traitement par imatinib.
Anomalies biologiques :
Paramètres hématologiques
Dans les LMC, des cytopénies, en particulier des neutropénies et des thrombopénies, ont été régulièrement rapportées dans toutes les études cliniques avec une fréquence plus élevée aux fortes doses ≥ 750 mg (étude de phase I). Cependant, la survenue des cytopénies dépendait aussi nettement du stade de la maladie : la fréquence des neutropénies et des thrombopénies de grade 3 ou 4 (PN < 1,0 x 109/L ; taux de plaquettes < 50 x 109/L) étant 4 à 6 fois plus élevée dans les LMC en crise blastique ou en phase accélérée (respectivement 59– 64 % pour les neutropénies et 44– 63 % pour les thrombopénies) que dans les LMC en phase chronique nouvellement diagnostiquées (16,7 % de neutropénie et 8,9 % de thrombopénie). Dans les LMC en phase chronique nouvellement diagnostiquées, les neutropénies et les thrombopénies de grade 4 (PN< 0,5 x 109/L ; plaquettes < 10 x 109/L) ont été observées chez 3,6 % et < 1 % des patients respectivement.
La durée médiane des épisodes de neutropénie est habituellement de l’ordre de 2 à 3 semaines, et de 3 à 4 semaines pour les épisodes de thrombopénie. Ces événements peuvent être habituellement contrôlés soit par une réduction de la dose soit par une interruption du traitement par l’imatinib, mais peuvent, dans de rares cas, conduire à une interruption définitive du traitement. En pédiatrie, chez les enfants atteints de LMC, les toxicités les plus fréquemment observées étaient des cytopénies de grade 3 ou 4 incluant des neutropénies, des thrombocytopénies et des anémies. Elles surviennent principalement dans les premiers mois de traitement.
Dans l’étude menée chez des patients atteints de GIST non résécables et/ou métastatiques, une anémie de grade 3 ou 4 a été rapportée respectivement chez 5,4 % et 0,7 % des patients. Ces cas d'anémies pouvaient être liés à un saignement gastro-intestinal ou intra-tumoral, au moins chez certains de ces patients. Des neutropénies de grade 3 ou 4 ont été rapportées respectivement chez 7,5 % et 2,7 % des patients et une thrombopénie de grade 3 chez 0,7 % des patients. Aucun patient n'a développé de thrombopénie de grade 4. Des diminutions du nombre de leucocytes et de neutrophiles ont principalement été observées au cours des six premières semaines du traitement, les valeurs demeurant relativement stables par la suite.
Paramètres biochimiques
Des augmentations importantes des transaminases (< 5 %) ou de la bilirubine (< 1 %) ont été observées chez des patients atteints de LMC et ont été habituellement contrôlées par une réduction de la dose ou une interruption du traitement (la durée médiane de ces épisodes était d’environ une semaine). Le traitement a été interrompu définitivement en raison d’anomalies biologiques hépatiques chez moins de 1 % des patients atteints de LMC.
Chez les patients atteints de GIST (étude B2222), on a observé 6,8 % d’augmentations de grade 3 à 4 des ALAT (alanine aminotransférase) et 4,8 % d’augmentations de grade 3 à 4 des ASAT (aspartate aminotransférase). L’augmentation de la bilirubine était inférieure à 3 %.
Il y a eu des cas d’hépatite cytolytique et cholestatique et de défaillance hépatique. Dans certains cas l’issue fut fatale, notamment pour un patient sous dose élevée de paracétamol.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
L’expérience avec des doses supérieures aux doses thérapeutiques recommandées est limitée. Des cas spontanés de surdosage avec imatinib ont été rapportés et publiés dans la littérature. En cas de surdosage, le patient doit être surveillé et un traitement symptomatique approprié doit lui être administré. L’évolution rapportée de ces cas était une « amélioration » ou un « rétablissement ». Les événements qui ont été rapportés à des doses différentes sont les suivants :
Population adulte
1200 à 1600 mg (sur une durée allant de 1 à 10 jours) : nausée, vomissements, diarrhées, rash, érythème, œdème, gonflement, fatigue, crampes musculaires, thrombopénie, pancytopénie, douleurs abdominales, céphalées, diminution de l’appétit.
1800 à 3200 mg (jusqu’à 3200 mg par jour pendant 6 jours) : faiblesse, myalgie, taux de créatine- phosphokinase augmenté, taux de bilirubine augmenté, douleur gastro-intestinale.
6400 mg (dose unique) : un cas rapporté dans la littérature d’un patient qui a présenté nausées, vomissements, douleurs abdominales, fièvre, œdème du visage, diminution du taux de neutrophiles, augmentation des transaminases.
8 à 10 g (dose unique) : vomissements et douleurs gastro-intestinales ont été rapportés.
Population pédiatrique
Un garçon âgé de 3 ans ayant pris une dose unique de 400 mg a présenté des vomissements, une diarrhée et une anorexie et un autre garçon âgé de 3 ans ayant pris une dose unique de 980 mg a présenté une diminution du taux de globules blancs et une diarrhée.
En cas de surdosage, le patient devra être placé sous surveillance et recevoir un traitement symptomatique approprié.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : inhibiteur de protéine-tyrosine kinase, Code ATC : L01EA01
Mécanisme d'action
L'imatinib est une petite molécule chimique inhibitrice de protéine tyrosine kinase qui inhibe puissamment l'activité de la tyrosine kinase (TK) Bcr-Abl ainsi que plusieurs récepteurs TK : Kit, le récepteur du SCF (stem cell factor) codé par le proto-oncogène c-Kit, les récepteurs du domaine discoidine (DDR1 et DDR2), le CSF-1R (récepteur du colony stimulating factor) et les récepteurs alpha et bêta du PDGF (platelet-derived growth factor) (PDGFR-alpha et PDGFR-bêta). L'imatinib peut également inhiber les processus cellulaires médiés par l'activation des kinases de ces récepteurs.
Effets pharmacodynamiques
L’imatinib est un inhibiteur de protéine tyrosine kinase qui inhibe puissamment la tyrosine kinase Bcr- Abl au niveau cellulaire in vitro et in vivo. Le produit inhibe sélectivement la prolifération et induit une apoptose dans les lignées cellulaires Bcr-Abl positives ainsi que dans les cellules leucémiques fraîches provenant de patients atteints de LMC ou de leucémie aiguë lymphoblastique (LAL) chromosome Philadelphie positives.
In vivo, le produit présente une activité anti-tumorale lorsqu'il est administré en monothérapie chez l'animal porteur de cellules tumorales Bcr-Abl positives.
L’imatinib est également un inhibiteur des tyrosines kinases du récepteur du PDGF (platelet-derived growth factor), PDGF-R et du SCF (stem cell factor) c-Kit et il inhibe les processus cellulaires médiés par le PDGF et le SCF. In vitro, l'imatinib inhibe la prolifération et induit une apoptose des cellules de tumeur stromale gastro-intestinale (GIST), qui expriment une mutation activatrice du kit.
L’activation constitutive du récepteur du PDGF ou des tyrosines kinases Abl, résultant de la fusion à différentes protéines partenaires ou de la production constitutive de PDGF, sont impliquées dans la pathogénèse des SMD/SMP, des SHE/LCE et du DFSP. L’imatinib inhibe la signalisation et la prolifération des cellules sensibles à l’activité dérégulée des kinases Abl ou PDGFR.
Etudes cliniques dans la leucémie myéloïde chronique
L’efficacité de l’imatinib est basée sur les taux de réponses cytogénétiques et hématologiques globales et la survie sans progression. A l’exception de la LMC en phase chronique nouvellement diagnostiquée, il n’existe pas actuellement d’étude contrôlée démontrant un bénéfice clinique tel qu’une amélioration des symptômes liés à la maladie ou une prolongation de la durée de vie.
Trois grandes études ouvertes internationales de phase II, non contrôlées, ont été menées chez des patients atteints de LMC chromosome Philadelphie positive (Ph+) en phase avancée, crise blastique ou phase accélérée, ainsi que chez des patients atteints d'autres leucémies Ph+ ou de LMC en phase chronique en échec d’un traitement antérieur par interféron alpha (IFN). Une vaste étude randomisée ouverte, multicentrique, internationale de phase III a été menée chez des patients présentant une LMC Ph+ nouvellement diagnostiquée. De plus, des enfants ont été traités dans deux études de phase I et une étude de phase II.
Dans toutes les études cliniques, 38– 40 % des patients avaient ≥ 60 ans et 10– 12 % des patients avaient ≥ 70 ans.
Phase chronique nouvellement diagnostiquée : Cette étude de phase III, chez des patients adultes, a comparé l'administration de l’imatinib en monothérapie à une association d'interféron-alpha (IFN) et de cytarabine (Ara-C). Les patients présentant une absence de réponse [absence de réponse hématologique complète (RHC) à 6 mois, augmentation des leucocytes, absence de réponse cytogénétique majeure (RCyM) à 24 mois], une perte de la réponse (perte de la RHC ou de la RCyM) ou une intolérance sévère au traitement ont été autorisés à permuter dans l'autre groupe de traitement. Dans le groupe recevant imatinib, les patients ont été traités par 400 mg/jour. Dans le groupe recevant IFN, les patients ont reçu une dose cible d'IFN de 5 MUI/m2/jour par voie sous-cutanée, en association à une dose sous-cutanée d'Ara-C de 20 mg/m2/jour pendant 10 jours/mois.
Au total, 1 106 patients ont été randomisés, soit 553 patients par groupe de traitement. Les caractéristiques initiales étaient bien équilibrées dans les deux groupes de traitement. L'âge médian était de 51 ans (extrêmes : 18– 70 ans) dont 21,9 % des patients âgés de 60 ans ou plus. Les groupes étaient composés de 59 % d'hommes et 41 % de femmes, de 89,9 % de blancs et 4,7 % de noirs. Sept ans après l’inclusion du dernier patient, la durée médiane de traitement en première ligne était respectivement de 82 et de 8 mois dans les bras traités par imatinib et par l’IFN.
La durée médiane de traitement en seconde ligne par imatinib était de 64 mois. Au total, chez les patients recevant de l’imatinib en première ligne, la dose moyenne quotidienne dispensée était de 406 ± 76 mg. Dans cette étude, le paramètre d'évaluation principal de l'efficacité est la survie sans progression de la maladie. La progression était définie par l’un des événements suivants : évolution vers une phase accélérée ou une crise blastique, décès, perte de la RHC ou de la RCyM, ou, chez les patients n'obtenant pas une RHC, augmentation des leucocytes malgré une prise en charge thérapeutique adaptée. La réponse cytogénétique majeure, la réponse hématologique, la réponse moléculaire (évaluation de la maladie résiduelle), le délai avant apparition d'une phase accélérée ou d'une crise blastique et la survie sont les principaux critères d'évaluation secondaires. Le Tableau 2 présente les données relatives aux types de réponses.
Tableau 2 : Réponses observées dans l'étude portant sur les LMC de diagnostic récent (données à 84 mois)
|
(Meilleurs taux de réponse) |
Imatinib n=553 |
IFN+Ara-C n=553 |
|
Réponse hématologique |
||
|
Taux de RHC n (%) |
534 (96,6 %)* |
313 (56,6 %)* |
|
[95 % IC] |
[94,7 %, 97,9 %] |
[52,4 %, 60,8 %] |
|
Réponse cytogénétique |
||
|
Réponse majeure n (%) |
490 (88,6 %)* |
129 (23,3 %)* |
|
[95 % IC] |
[85,7 %, 91,1 %] |
[19,9 %, 27,1 %] |
|
RCy complète n (%) |
456 (82,5 %)* |
64 (11,6 %)* |
|
RCy partielle n (%) |
34 (6,1 %) |
65 (11,8 %) |
|
Réponse moléculaire** |
||
|
Réponse moléculaire** |
153/305=50,2 % |
8/83=9,6 % |
|
Réponse majeure à 24 mois (%) |
73/104=70,2 % |
3/12=25 % |
|
Réponse majeure à 84 mois (%) |
102/116=87,9 % |
3/4=75 % |
|
* p<0,001, Fischer’s exact test ** les pourcentages de réponses moléculaires sont basés sur les prélèvements disponibles Critères de réponse hématologique (toutes les réponses devaient être confirmées après ≥ 4 semaines) : Leucocytes< 10 x 109/L, plaquettes < 450 x 109/L, myélocytes+métamyélocytes < 5 % dans le sang, aucun blaste ni promyélocyte dans le sang, basophiles < 20 %, aucun envahissement extramédullaire. Critères de réponse cytogénétique : complète (0 % de métaphases Ph+), partielle (1-35 %), mineure (36-65 %) ou minime (66-95 %). Une réponse majeure (0-35 %) associe les réponses complètes ou partielles. Critères de réponse moléculaire majeure : dans le sang périphérique, réduction d’au moins 3 log du taux de transcrit BCR-ABL (mesuré par un test RT-PCR quantitatif en temps réel) par rapport à une valeur de base standardisée. |
||
Les taux de réponse hématologique complète, de réponse cytogénétique majeure et de réponse cytogénétique complète au traitement en première ligne ont été estimés selon la méthode de Kaplan- Meier, avec laquelle les non-réponses sont censurées à la date du dernier examen. En utilisant cette approche, les taux estimés de réponse cumulée au traitement par imatinib en première ligne se sont améliorés de 12 mois à 84 mois respectivement comme suit : RHC de 96,4 % à 98,4 % et RCyC de 69,5 % à 87,2 %.
Dans les 7 ans de suivi, 93 (16,8 %) progressions ont été observées dans le bras traité par imatinib : 37 (6,7 %) étaient des progressions vers une phase accélérée ou une crise blastique, 31 (5,6 %) étaient une perte de la réponse cytogénétique majeure, 15 (2,7 %) étaient une perte de la réponse hématologique complète ou une augmentation du taux de leucocytes et 10 (1,8 %) étaient des décès non liés à la LMC. Par opposition, dans le bras traité par l’IFN+Ara-C 165 (29,8 %) événements ont été observés dont 130 sont apparus au cours du traitement en première ligne par IFN+Ara-C.
Le taux estimé de patients sans progression vers une phase accélérée ou une crise blastique à 84 mois est significativement plus élevé dans le bras avec imatinib par rapport au bras IFN (92,5 % versus 85,1 %, p<0,001). Le taux annuel de progression vers une phase accélérée ou une crise blastique a diminué avec la durée de traitement, il était de moins de 1 % par an dans la quatrième et cinquième année de traitement. Le taux estimé de survie sans progression à 84 mois était de 81,2 % avec imatinib contre 60,6 % dans le groupe témoin (p<0,001). Les taux annuels de progression, quel que soit le type de progression, par année de traitement par imatinib ont aussi diminué avec le temps.
Il y a eu au total 71 (12,8 %) et 85 (15,4 %) décès rapportés respectivement dans les groupes imatinib et IFN+Ara-C. A 84 mois, le taux de survie globale estimé est, de 86,4 % (83, 90) et 83,3 % (80, 87) dans les groupes randomisés traités par imatinib ou IFN+Ara-C (p=0,073, test log rank). Ce critère « délai jusqu’à l’événement » est fortement influencé par le taux élevé de cross-over du bras IFN+Ara C vers le bras imatinib. L’effet du traitement par imatinib sur la survie des LMC en phase chronique nouvellement diagnostiquées a fait l’objet d’une évaluation supplémentaire avec une analyse rétrospective des données de l’imatinib rapportées ci-dessus avec celles issues d’une autre étude de phase III étudiant IFN+Ara-C (n=325) selon un schéma thérapeutique identique. Dans cette analyse rétrospective, la supériorité de l’imatinib par rapport à l’IFN+Ara-C sur la survie globale a été démontrée (p<0,001) ; à l’issue des 42 mois de suivi, 47 (8,5 %) patients traités par imatinib et 63 (19,4 %) patients traités par l’IFN+Ara-C sont décédés.
Le degré de réponse cytogénétique et de réponse moléculaire a montré un effet net sur le devenir à long terme des patients traités par imatinib. Alors que 96 % (93 %) des patients présentant une RCy complète (RCy partielle) à 12 mois n’avaient pas progressé vers une phase accélérée/crise blastique, uniquement 81 % de patients ne présentant pas de RCy majeure à 12 mois, n’ont pas progressé vers le stade avancé de la LMC à 84 mois (p<0,001 globale, p=0,25 entre RCy complète et RCy partielle). Chez les patients présentant une réduction d’au moins 3 log du taux de transcrit Bcr-Abl à 12 mois la probabilité de maintenir l’absence de progression vers la phase accélérée/crise blastique était de 99 % à 84 mois. Des résultats similaires ont été mis en évidence avec une analyse landmark à 18 mois.
Dans cette étude, des augmentations de dose de 400 mg à 600 mg par jour, puis de 600 mg à 800 mg par jour ont été autorisées. Après 42 mois de suivi, une perte confirmée (dans les 4 semaines) de la réponse cytogénétique a été observée chez 11 patients. Parmi ces 11 patients, 4 patients ont eu une augmentation de dose jusqu’à 800 mg par jour et 2 d’entre eux ont obtenu de nouveau une réponse cytogénétique (1 partielle et 1 complète, cette dernière associée à une réponse moléculaire), tandis que parmi les 7 patients qui n’ont pas eu d’augmentation de doses, un seul patient a présenté de nouveau une réponse cytogénétique complète. Le pourcentage de certains effets indésirables observés était plus élevé chez les 40 patients dont la dose a été augmentée jusqu’à 800 mg par rapport aux patients observés avant toute augmentation de la dose (n=551). Les effets indésirables les plus fréquents ont été des hémorragies gastro-intestinales, des conjonctivites et une augmentation des transaminases ou de la bilirubine. D’autres effets indésirables ont été rapportés à des fréquences plus faibles ou équivalentes.
Phase chronique, échec de l'interféron : 532 patients adultes ont été traités avec une dose initiale de 400 mg. Les patients ont été répartis en trois catégories principales : échec hématologique (29 %), échec cytogénétique (35 %) ou intolérance à l'interféron (36 %). Les patients avaient précédemment reçu un traitement par l’IFN à des doses ³ 25 x 106 UI/semaine pendant une durée médiane de 14 mois et étaient tous en phase chronique tardive, avec un diagnostic établi depuis une durée médiane de 32 mois. Le critère principal d'efficacité de l'étude était le taux de réponse cytogénétique majeure (réponses complètes plus réponses partielles : 0 à 35 % de métaphases Ph+ dans la moelle osseuse).
Dans cette étude, 65 % des patients ont obtenu une réponse cytogénétique majeure, la réponse étant complète chez 53 % (confirmée 43 %) des patients (Tableau 3). Une réponse complète hématologique a été observée chez 95 % des patients.
Phase accélérée : L'étude a inclus 235 patients adultes en phase accélérée. Les 77 premiers patients ont commencé le traitement à 400 mg. Par la suite, le protocole a été amendé pour autoriser une posologie plus élevée et les 158 patients suivants ont commencé le traitement à 600 mg.
Le critère principal d'efficacité était le taux de réponse hématologique, défini comme une réponse complète hématologique, ou bien une disparition des signes de leucémie (c’est à dire : disparition des blastes de la moelle osseuse et du sang, mais sans la récupération hématologique périphérique totale observée dans le cas de réponse complète), ou encore un retour en phase chronique de la LMC. Une réponse hématologique confirmée a été obtenue chez 71,5 % des patients (Tableau 3).
Fait important, 27,7 % des patients ont également présenté une réponse cytogénétique majeure, qui a été complète chez 20,4 % des patients (confirmée 16 %). Pour les patients ayant reçu une dose de 600 mg, les estimations actuelles de la médiane de survie sans progression et de la survie globale ont été de 22,9 mois et 42,5 mois respectivement.
Crise blastique myéloïde : 260 patients en crise blastique myéloïde ont été inclus. 95 patients (37 %) avaient reçu une chimiothérapie (« patients prétraités ») comme traitement antérieur d’une phase accélérée ou d’une crise blastique alors que 165 (63 %) n'en avaient pas reçu (« patients non prétraités »). Les 37 premiers patients ont débuté le traitement à 400 mg. Le protocole a ensuite été amendé pour permettre une posologie plus élevée, et les 223 patients suivants ont reçu une dose initiale de 600 mg.
Le critère principal d'efficacité était le taux de réponse hématologique, défini comme soit une réponse complète hématologique, soit une disparition des signes de leucémie ou encore un retour en phase chronique de la LMC selon les mêmes critères que ceux de l’étude menée chez des patients en phase accélérée. Dans cette étude, 31 % des patients ont obtenu une réponse hématologique (36 % chez les patients non prétraités et 22 % chez les patients prétraités). Le taux de réponse a également été supérieur chez les patients traités par 600 mg (33 %) par rapport aux patients traités par 400 mg (16 %, p=0,0220).
L'estimation actuelle de la médiane de survie des patients non prétraités est de 7,7 mois, contre 4,7 mois chez les patients prétraités.
Crise blastique lymphoïde : un nombre limité de patients a été inclus dans les études de phase I (n=10). Le taux de réponse hématologique était de 70 % sur une durée de 2 à 3 mois.
Tableau 3 : Réponse dans les études LCM chez l’adulte
|
Etude 0110 Données à 37 mois Phase chronique, échec IFN (n=532) |
Etude 0109 Données à 40,5 mois Phase accélérée (n=235) |
Etude 0102 Données à 38 mois Crise blastique myéloïde (n=260) |
|
|
% de patients (CI95 %) |
|||
|
Réponse hématologique1 Réponse complète hématologique (RCH) Absence de signe de leucémie (ASL) Retour à la phase chronique (RPC) |
95 % (92,3-96,3) 95 %
Sans objet
Sans objet |
71 % (65,3-77,2) 42 %
12 %
17 % |
31 % (25,2-36,8) 8 %
5 %
18 % |
|
Réponse cytogénétique majeure2 Complète (Confirmée3) [IC 95 %] Partielle |
65 % (61,2-69,5)
53 % (43 %) [38,6-47,2] 12 % |
28 %(22,0-33,9) 20 % (16 %) [11,3-21,0] 7 % |
15 % (11,2-20,4) 7 % (2 %) [0,6-4,4] 8 % |
|
1 Critères de réponse hématologique (toutes les réponses étaient à confirmer après ≥ 4 semaines) : RCH : Etude 0110 [GB < 10 x 109/L, plaquettes < 450 x 109/L, myélocytes+métamyélocytes < 5 % dans le sang, absence de cellules blastiques et de promyélocytes dans le sang, basophiles < 20 %, absence d'atteinte extramédullaire] ; dans les études 0102 et 0109 [PN ≥ 1,5 x 109/L, plaquettes ≥ 100 x 109/L, absence de cellules blastiques dans le sang, présence de blastes dans la MO < 5 %, absence d'atteinte extramédullaire] ASL : Mêmes critères que pour RCH mais PN ≥ 1 x 109/L et plaquettes ≥ 20 x 109/L (uniquement pour les études 0102 et 0109) RPC < 15 % de blastes dans la MO et le SP, < 30 % blastes+promyélocytes dans la MO et le SP, <20 % basophiles dans le SP, absence d'atteinte extramédullaire en dehors de la rate et du foie (uniquement pour les études 0102 et 0109). MO = moelle osseuse, SP = sang périphérique 2 Critères de réponse cytogénétique : une réponse majeure englobe les réponses complètes et partielles : complète (0 % métaphases Ph+), partielle (1 à 35 %) 3 Réponse complète cytogénétique confirmée par une seconde évaluation cytogénétique de la moelle osseuse réalisée au moins un mois après l’étude initiale de moelle osseuse. |
|||
Patients pédiatriques : Un total de 26 patients pédiatriques âgés de moins de 18 ans, présentant une LMC en phase chronique (n=11) ou une LMC en crise blastique ou une leucémie aiguë Ph+ (n=15), ont été inclus dans une étude de phase I avec escalade de doses. Il s'agissait d'une population de patients lourdement prétraités, dans la mesure où 46 % avaient déjà bénéficié d'une transplantation médullaire et 73 % d'une polychimiothérapie.
Les doses d'imatinib administrées étaient de 260 mg/m2/jour (n=5), 340 mg/m2/jour (n=9), 440 mg/m2/jour (n=7) et 570 mg/m2/jour (n=5). Parmi les 9 patients atteints de LMC en phase chronique dont les données cytogénétiques sont disponibles, 4 (44 %) ont obtenu une réponse cytogénétique complète et 3 (33 %) une réponse cytogénétique partielle, pour un taux de RCyM de 77 %.
Un total de 51 patients pédiatriques atteints d’une LMC en phase chronique nouvellement diagnostiquée non traitée ont été inclus dans une étude de phase II avec un seul bras, multicentrique en ouvert. Ces enfants étaient traités par l’imatinib à la dose de 340 mg/m2/jour, sans interruption de traitement en l’absence de toxicité dose limitante.
Le traitement par l’imatinib induit une réponse rapide chez les patients pédiatriques atteints de LMC nouvellement diagnostiqués avec une RCH de 78 % après 8 semaines de traitement. Le taux élevé de RCH s’accompagne d’une réponse complète cytogénétique de 65 % qui est comparable aux résultats observés chez les adultes. De plus, une réponse cytogénétique partielle (RCyP) était observée à un taux de 16 % pour un taux de 81 % de réponses cytogénétiques majeures (RCyM). La majorité des patients qui ont atteint une réponse cytogénétique complète ont développé cette réponse entre 3 et 10 mois avec une estimation selon Kaplan-Meier de la durée médiane de réponse à 5,6 mois.
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec imatinib dans tous les sous-groupes de la population pédiatrique atteinte de leucémie myéloïde chronique chromosome Philadelphie positive (translocation bcr-abl) (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Etudes cliniques dans la LAL Ph+
LAL Ph+ nouvellement diagnostiquée : dans une étude contrôlée (ADE10) comparant l’imatinib versus chimiothérapie d’induction, chez 55 patients nouvellement diagnostiqués âgés de 55 ans et plus, l’imatinib utilisé seul a induit un taux significativement plus élevé de réponse hématologique complète par rapport à la chimiothérapie (96,3 % versus 50 % ; p = 0,0001). Lorsque le traitement de rattrapage par l’imatinib a été administré aux patients qui n’avaient pas répondu ou avaient mal répondu à la chimiothérapie, 9 patients (81,8 %) sur les 11 ont atteint une réponse hématologique complète. Cet effet clinique était associé à une plus forte réduction des taux de transcrits bcr-abl après deux semaines de traitement (p = 0,02) chez les patients traités par l’imatinib par rapport aux patients traités par chimiothérapie. Tous les patients ont reçu l’imatinib et une chimiothérapie de consolidation (voir Tableau 4) après le traitement d’induction, et les taux de transcrits bcr-abl étaient identiques entre les deux bras après huit semaines de traitement.
Comme on pouvait s’y attendre compte tenu du schéma de l’étude, aucune différence n’a été observée en termes de durée de rémission, de survie sans maladie ou de survie globale, même s’il faut noter que les patients en réponse moléculaire complète chez qui persistait une maladie résiduelle minime avait un pronostic plus favorable tant en termes de durée de rémission (p = 0,01) que de survie sans maladie (p = 0,02).
Les résultats observés dans quatre études cliniques non contrôlées (AAU02, ADE04, AJP01 et AUS01), dans une population de 211 patients atteints de LAL Ph+ nouvellement diagnostiquée, sont consistants avec les résultats décrits ci-dessus. L’imatinib en association avec la chimiothérapie d’induction (voir Tableau 4) a permis d’obtenir un taux de réponse complète hématologique de 93 % (147 sur 158 patients évaluables) et un taux de réponse cytogénique majeure de 90 % (19 sur 21 patients évaluables). Le taux de réponse moléculaire complète était de 48 % (49 sur 102 patients évaluables). La survie sans maladie et la survie globale ont constamment dépassé un an et elles étaient supérieures aux données historiques des groupes contrôles (survie sans maladie p<0,001 ; survie globale p<0,0001) dans les deux études (AJP01 et AUS01).
Tableau 4 : Schéma des chimiothérapies utilisées en association avec l’imatinib
|
Etude ADE10 |
|
|
Pré-phase |
DEX 10 mg/m2 oral, jours 1 à 5 ; CP 200 mg/m2 i.v., jours 3, 4, 5 ; MTX 12 mg intrathécal, jour 1 |
|
Traitement d’induction (rémission) |
DEX 10 mg/m2 oral, jours 6 et 7, 13 à 16 ; VCR 1 mg i.v., jours 7, 14 ; IDA 8 mg/m2 i.v. (0,5 h), jours 7, 8, 14, 15 ; CP 500 mg/m2 i.v. (1 h) jour 1 ; Ara-C 60 mg/m2 i.v., jours 22 à 25, 29 à 32 |
|
Traitement de consolidation I, III, V |
MTX 500 mg/m2 i.v. (24 h), jours 1, 15 ; 6-MP 25 mg/m2 oral, jours 1 à 20 |
|
Traitement de consolidation II, IV |
Ara-C 75 mg/m2 i.v. (1 h), jours 1 à 5 ; VM26 60 mg/m2 i.v. (1 h), jours 1 à 5 |
|
Etude AAU02 |
|
|
Traitement d’induction (LAL Ph+ de novo) |
Daunorubicine 30 mg/m2 i.v., jours 1 à 3, 15 et 16 ; VCR dose totale 2 mg i.v., jours 1, 8, 15, 22 ; CP 750 mg/m2 i.v., jours 1, 8 ; Prednisone 60 mg/m2 oral, jours 1 à 7, 15 à 21 ; IDA 9 mg/m2 oral, jours 1 à 28 ; MTX 15 mg intrathécal, jours 1, 8, 15, 22 ; Ara-C 40 mg intrathécal, jours 1, 8,15, 22 ; Méthylprednisolone 40 mg intrathécal, jours 1, 8, 15, 22 |
|
Consolidation (LAL Ph+ de novo) |
Ara-C 1 g/m2 toutes les 12 h i.v. (en 3 h), jours 1 à 4 ; Mitoxantrone 10 mg/m2 i.v. jours 3 à 5 ; MTX 15 mg intrathécal, jour 1 ; Méthylprednisolone 40 mg intrathécal, jour 1 |
|
Etude ADE04 |
|
|
Pré-phase |
DEX 10 mg/m2 oral, jours 1 à 5 ; CP 200 mg/m2 i.v., jours 3 à 5 ; MTX 15 mg intrathécal, jour 1 |
|
Traitement d’induction I |
DEX 10 mg/m2 oral, jours 1 à 5 ; VCR 2 mg i.v., jours 6, 13, 20 ; Daunorubicine 45 mg/m2 i.v., jours 6 à 7, 13, 14 |
|
Traitement d’induction II |
CP 1 g/m2 i.v. (1 h), jours 26, 46 ; Ara-C 75 mg/m2 i.v. (1 h), jours 28 à 31, 35 à 38, 42 à 45 ; 6-MP 60 mg/m2 oral, jours 26 à 46 |
|
Traitement de consolidation |
DEX 10 mg/m2 oral, jours 1 à 5 ; Vindésine 3 mg/m2 i.v., jour 1 ; MTX 1,5 g/m2 i.v. (24 h), jour 1 ; Etoposide 250 mg/m2 i.v. (1 h) jours 4 à 5 ; Ara-C 2x 2 g/m2 i.v. (en 3 h, toutes les 12 h), jour 5 |
|
Etude AJP01 |
|
|
Traitement d’induction |
CP 1,2 g/m2 i.v. (3 h), jour 1 ; Daunorubicine 60 mg/m2 i.v. (1 h), jours 1 à 3 ; Vincristine 1,3 mg/m2 i.v., jours 1, 8, 15, 21; Prednisolone 60 mg/m2/jour oral |
|
Traitement de consolidation |
Régime de chimiothérapie en alternance : chimiothérapie à hautes doses de MTX 1 g/m2 i.v. (24 h), jour 1, et Ara-C 2 g/m2 i.v. (toutes les 12 h), jours 2 et 3, pour 4 cycles |
|
Traitement d’entretien |
VCR 1,3 g/m2 i.v., jour 1 ; Prednisolone 60 mg/m2 oral, jours 1 à 5 |
|
Etude AUS01 |
|
|
Traitement d’induction et de consolidation |
Protocole Hyper-CVAD : CP 300 mg/m2 i.v. (en 3 h, toutes les 12 h), jours 1 à 3 ; Vincristine 2 mg i.v., jours 4 et 11 ; Doxorubicine 50 mg/m2 i.v. (24 h), jour 4 ; DEX 40 mg/jour jours 1 à 4 et 11 à 14, en alternance avec MTX 1 g/m2 i.v. (24 h), jour 1, Ara-C 1 g/m2 i.v. (en 2 h, toutes les 12 h), jours 2 et 3 (pour un total de 8 cycles) |
|
Traitement d’entretien |
VCR 2 mg i.v. une fois par mois pendant 13 mois ; Prednisolone 200 mg oral, 5 jours par mois pendant 13 mois |
Tous les schémas thérapeutiques comprennent l’administration de corticoïdes en prophylaxie neuroméningée.
Ara-C : cytarabine ; CP : cyclophosphamide ; DEX : dexaméthasone ; MTX : méthotrexate ;
6-MP : 6-mercaptopurine ; VM26 : téniposide ; VCR : vincristine ; IDA : idarubicine ; i.v.: Intraveineux.
Population pédiatrique
Dans l’étude I2301, de phase III, non randomisée, en ouvert, multicentrique, en cohortes séquentielles, un total de 93 enfants, adolescents et jeunes adultes (âgés de 1 à 22 ans) atteints d’une LAL Ph+ ont été inclus et traités par l’imatinib (340 mg/m²/jour) en association avec une chimiothérapie d’intensification après une thérapie d’induction. L’imatinib a été administré par intermittence dans les cohortes 1 à 5, avec une augmentation de la durée et une administration plus précoce de cohorte en cohorte : la cohorte 1 a reçu la plus faible intensité et la cohorte 5 a reçu la plus forte intensité de l’imatinib (la plus longue durée en jours avec une administration continue de la dose de l’imatinib durant les premiers cycles de chimiothérapie). Comparativement aux groupes contrôles historiques (n=120) qui avaient reçu une chimiothérapie standard sans l’imatinib, l’exposition journalière continue et précoce à l’imatinib dans le cycle de traitement en association à la chimiothérapie chez les patients de la cohorte 5 (n=50) a augmenté la survie sans événement à 4 ans (69,6 % vs. 31,6 %, respectivement). La survie globale estimée à 4 ans était de 83,6 % chez les patients de la cohorte 5 versus 44,8 % chez ceux des groupes contrôles historiques. Vingt patients sur 50 (40 %) de la cohorte 5 ont reçu une transplantation de cellules souches hématopoïétiques.
Tableau 5 : Schéma des chimiothérapies utilisées en association avec l’imatinib dans l’étude I2301
|
Bloc de consolidation 1 (3 semaines) |
VP-16 (100 mg/m2/jour, IV) : jours 1-5 Ifosfamide (1,8 g/m2/jour, IV) : jours 1-5 MESNA (360 mg/m2/dose en 3 heures, x 8 doses/jour, IV) : jours 1-5 G-CSF (5 μg/kg, SC) : jours 6-15 ou jusqu’à un taux de neutrophile absolu > 1500 post nadir Méthotrexate IT (ajusté à l’âge) : jour 1 SEULEMENT Triple thérapie intrathécale (ajustée à l’âge) : jours 8 et 15 |
|
Bloc de consolidation 2 (3 semaines) |
Méthotrexate (5 g/m2 sur 24 heures, IV) : jour 1 Leucovorine (75 mg/m2 à heure 36, IV ; 15 mg/m2 IV ou PO toutes les 6 heures x 6 doses) iii: jour 2 et 3 Triple thérapie intrathécale (ajustée à l’âge) : jour 1 ARA-C (3 g/m2/dose toutes les 12 heures x 4, IV) : jours 2 et 3 G-CSF (5 μg/kg, SC) : jours 4-13 ou jusqu’à un taux de neutrophile absolu > 1500 post nadir |
|
Bloc de réinduction 1 (3 semaines) |
VCR (1,5 mg/ m2/jour, IV) : jours 1, 8, et 15 DAUN (45 mg/ m2jour bolus, IV) : jours 1 et 2 CPM (250 mg/ m2/dose toutes les 12 heures x 4 doses, IV) : jours 3 et 4 PEG-ASP (2500 UI/ m2, IM) : jour 4 G-CSF (5 μg/kg, SC) : jours 5-14 ou jusqu’à un taux de neutrophile absolu > 1500 post nadir Triple thérapie intrathécale (ajustée à l’âge) : jours 1 et 15 DEX (6 mg/ m2/jour, PO) : jours 1-7 et 15-21 |
|
Bloc d’intensification 1 (9 semaines) |
Méthotrexate (5 g/ m2 sur 24 heures, IV) : jours 1 et 15 Leucovorine (75 mg/ m2 à heure 36, IV ; 15 mg/m2 IV ou PO toutes les 6 heures x 6 doses) iii : jours 2, 3, 16, et 17 Triple thérapie intrathécale (ajustée à l’âge) : jours 1 et 22 VP-16 (100 mg/ m2/jour, IV) : jours 22-26 CPM (300 mg/ m2/jour, IV) : jours 22-26 MESNA (150 mg/ m2/jour, IV) : jours 22-26 G-CSF (5 μg/kg, SC) : jours 27-36 ou jusqu’à un taux de neutrophile absolu > 1500 post nadir ARA-C (3 g/ m2, toutes les 12 heures, IV) : jours 43 et 44 L-ASP (6000 UI/ m2, IM) : jour 44 |
|
Bloc de réinduction 2 (3 semaines) |
VCR (1,5 mg/m2/jour, IV) : jours 1, 8 et 15 DAUN (45 mg/m2/jour bolus, IV) : jours 1 et 2 CPM (250 mg/ m2/dose toutes les 12 heures x 4 doses, iv) : jours 3 et 4 PEG-ASP (2500 UI/m2, IM) : jour 4 G-CSF (5 μg/kg, SC) : jours 5-14 ou jusqu’à un taux de neutrophile absolu > 1500 post nadir Triple thérapie intrathécale (ajustée à l’âge) : jours 1 et 15 DEX (6 mg/m2/jour, PO) : jours 1-7 et 15-21 |
|
Bloc d’intensification 2 (9 semaines) |
Méthotrexate (5 g/m2 sur 24 heures, IV) : jours 1 et 15 Leucovorine (75 mg/m2 à heure 36, IV ; 15 mg/m2 IV ou PO toutes les 6 heures x 6 doses) iii : jours 2, 3, 16, et 17 Triple thérapie intrathécale (ajustée à l’âge) : jours 1 et 22 VP-16 (100 mg/m2/jour, IV) : jours 22-26 CPM (300 mg/m2/jour, IV) : jours 22-26 MESNA (150 mg/m2/jour, IV) : jours 22-26 G-CSF (5 μg/kg, SC) : jours 27-36 ou jusqu’à un taux de neutrophile absolu > 1500 post nadir ARA-C (3 g/m2, toutes les 12 heures, IV) : jours 43 et 44 L-ASP (6000 UI/m2, IM) : jour 44 |
|
Maintenance (cycles de 8 semaines) Cycles 1-4 |
Méthotrexate (5 g/m2 sur 24 heures, IV) : jour 1 Leucovorine (75 mg/m2 à heure 36, IV ; 15 mg/ m2 IV ou PO toutes les 6 heures x 6 doses) iii : jours 2 et 3 Triple thérapie intrathécale (ajustée à l’âge) : jours 1 et 29 VCR (1,5 mg/m2, IV) : jours 1 et 29 DEX (6 mg/m2/jour PO) : jours 1-5 ; 29-33 6-MP (75 mg/m2/jour, PO) : jours 8-28 Méthotrexate (20 mg/m2/semaine, PO) : jours 8, 15, 22 VP-16 (100 mg/m2, IV) : jours 29-33 CPM (300 mg/m2, IV) : jours 29-33 MESNA (IV) : jours 29-33 G-CSF (5 μg/kg, SC) : jours 34-43 |
|
Maintenance (cycles de 8 semaines) Cycle 5 |
Irradiation crânienne (cycle 5 uniquement) 12 Gy en 8 fractions pour tous les patients étant SNC1 et SNC2 au diagnostic 18 Gy en 10 fractions pour les patients étant SNC3 au diagnostic VCR (1,5 mg/m2/jour, IV) : jours 1, 29 DEX (6 mg/m2/jour, PO) : jours 1-5 ; 29-33 6-MP (75 mg/m2/jour, PO) : jours 11-56 (suspendre le 6-MP durant les jours 6-10 de l’irradiation crânienne commençant le jour 1 du cycle 5. Commencer le 6-MP le 1er jour après la fin de l’irradiation crânienne.) Méthotrexate (20 mg/m2/semaine, PO) : jours 8, 15, 22, 29, 36, 43, 50 |
|
Maintenance (cycles de 8 semaines) Cycles 6-12 |
VCR (1,5 mg/m2/jour, IV) : jours 1 et 29 DEX (6 mg/m2/jour, PO) : jours 1-5 ; 29-33 6-MP (75 mg/m2/jour, PO) : jours 1-56 Méthotrexate (20 mg/m2/semaine, PO) : jours 1, 8, 15, 22, 29, 36, 43, 50 |
G-CSF= granulocyte colony stimulating factor, VP-16 = étoposide, MTX = méthotrexate, IV = intraveineux, SC = sous-cutané, IT = intrathécal, PO = per os, IM = intramusculaire, Ara-C = cytarabine, CPM = cyclophosphamide, VCR = vincristine, DEX = dexaméthasone, DAUN = daunorubicine, 6-MP = 6-mercaptopurine, E.coli L-ASP = L-asparaginase, PEG-ASP = asparaginase pégylée, MESNA = 3-mercaptoéthane sulfonate sodium, iii = ou jusqu’à ce que le taux de méthotrexate soit < 0,1 µM, Gy = Gray.
L’étude AIT07 était une étude de phase II/III multicentrique, en ouvert, randomisée qui a inclus 128 patients (de 1 à moins de 18 ans) traités avec l’imatinib en association à la chimiothérapie. Les données de tolérance de cette étude semblent correspondre au profil de tolérance de l’imatinib chez les patients atteints de LAL Ph+.
LAL Ph+ en rechute ou réfractaire : Lorsque l’imatinib a été utilisé en monothérapie chez des patients atteints de LAL Ph+ en rechute ou réfractaire, il a été observé un taux de réponse hématologique de 30 % (9 % réponse complète) et un taux de réponse cytogénétique majeure de 23 % parmi les 53 patients évaluables pour la réponse sur un effectif total de 411 patients. (A noter que sur ces 411 patients, 353 avaient été traités dans le cadre d’un programme d’accès élargi au cours duquel la réponse primaire n’était pas collectée). La durée médiane jusqu’à la progression de la maladie dans la population globale de 411 patients atteints de LAL Ph+ en rechute ou réfractaire était de 2,6 à 3,1 mois, avec une médiane de survie globale allant de 4,9 à 9 mois chez 401 patients évaluables. Les données étaient identiques lorsque l’analyse a été de nouveau réalisée en prenant en compte uniquement les patients âgés de 55 ans et plus.
Etudes cliniques dans les SMD/SMP
L’expérience avec l’imatinib dans cette indication est très limitée, elle est basée sur les taux de réponse hématologique et cytogénétique. Il n’y a pas d’étude contrôlée démontrant un bénéfice clinique telles que l’amélioration des symptômes liés à la maladie ou l’augmentation de la survie. Une étude ouverte multicentrique de phase II (étude B2225) a été menée avec l’imatinib chez des patients atteints de diverses maladies impliquant les tyrosines kinases Abl, Kit ou PDGFR et menaçant le pronostic vital.
Cette étude a inclus 7 patients atteints de SMD/SMP traités par l’imatinib à 400 mg/jour. Trois patients ont présenté une réponse complète hématologique (RCH) et un patient a présenté une réponse partielle hématologique (RPH). A la date de l’analyse, trois des quatre patients qui avaient des réarrangements du gène du PDGFR ont présenté une réponse hématologique (2 réponses hématologiques complètes et 1 réponse hématologique partielle). L’âge des patients allait de 20 à 72 ans.
Un registre observationnel (étude L2401) a été mis en place pour collecter des données de sécurité et d’efficacité à long terme chez des patients souffrant de néoplasmes myéloprolifératifs avec réarrangement de PDGFR- β et ayant été traités par imatinib. Les 23 patients inclus dans ce registre ont reçu une dose journalière médiane d’imatinib de 264 mg (comprise entre 100 et 400 mg) pendant une durée médiane de 7,2 ans (comprise entre 0,1 et 12,7 ans). En raison du caractère observationnel de ce registre, les données d’évaluation hématologique, cytogénétique et moléculaire ne sont disponibles respectivement que pour 22, 9 et 17 des 23 patients inclus. En présumant que les patients dont les données sont manquantes étaient non-répondeurs, une réponse hématologique complète a été observée chez 20/23 patients (87 %), une réponse cytogénétique complète chez 9/23 patients (39,1%) et une réponse moléculaire complète chez 11/23 patients (47,8 %). Lorsque le taux de réponse est calculé chez les patients avec au moins une évaluation validée, le taux de réponse complète hématologique, cytogénétique et moléculaire était respectivement de 20/22 (90,9 %), 9/9 (100 %) et 11/17 (64,7 %).
De plus, 24 patients supplémentaires atteints de SMD/SMP ont été rapportés dans 13 publications. 21 patients ont été traités par l’imatinib à 400 mg/j, alors que les 3 autres patients ont reçu des doses plus faibles. Chez les 11 patients pour lesquels un réarrangement du gène du récepteur PDGFR a été mis en évidence, 9 d’entre eux ont présenté une réponse hématologique complète et 1 patient une réponse hématologique partielle. L’âge allait de 2 à 79 ans. Dans une publication récente, la mise à jour du suivi sur 6 de ces 11 patients, a montré que tous restaient en rémission cytogénétique (suivi de 32-38 mois).
La même publication rapportait des données du suivi à long terme de 12 patients atteints de SMD/SMP associés à des réarrangements du gène du récepteur PDGFR (dont 5 patients de l’étude clinique B2225). Ces patients ont reçu de l’imatinib sur une durée médiane de 47 mois (24 jours à 60 mois). Chez 6 de ces patients, le suivi à ce jour est supérieur à 4 ans. 11 patients ont atteint une réponse hématologique complète rapide ; 10 ont présenté une résolution complète des anomalies cytogénétiques et une diminution ou une disparition du transcript de fusion (mesuré par un test RT-PCR). Les réponses hématologiques et cytogénétiques ont été respectivement maintenues sur une durée médiane de 49 mois (19 à 60 mois) et 47 mois (16 à 59 mois). La survie globale est de 65 mois à partir du diagnostic (25 à 234 mois). L’administration d’imatinib chez des patients sans translocation génétique n’a pas généralement entrainé d’amélioration.
Il n’existe pas d’étude clinique contrôlée chez les patients pédiatriques atteints de SMD/SMP. Cinq (5) cas de patients atteints de SMD/SMP associés à des réarrangements du gène PDGFR ont été rapportés dans 4 publications. L’âge de ces patients allait de 3 mois à 4 ans et l’imatinib était administré à une posologie de 50 mg par jour ou comprise entre 92,5 et 340 mg/m² par jour. Tous les patients ont atteint une réponse hématologique complète, une réponse cytogénétique et/ou une réponse clinique.
Etudes cliniques dans les SHE/LCE
Une étude ouverte multicentrique de phase II (étude B2225) a été menée avec l’imatinib chez des patients atteints de diverses maladies impliquant les tyrosines kinases Abl, Kit ou PDGFR et menaçant le pronostic vital. Dans cette étude, 14 de ces patients atteints de SHE/LCE ont été traités par l’imatinib à la dose de 100 mg à 1 000 mg par jour. 162 patients supplémentaires atteints de SHE/LCE, rapportés dans 35 publications sous la forme d’observations individuelles, ont reçu l’imatinib à la dose allant de 75 mg à 800 mg par jour. Les anomalies cytogénétiques ont été évaluées chez 117 patients sur un total de 176 patients. La protéine de fusion FIP1L1-PDGFRα a été identifiée chez 61 des 117 patients. Quatre autres patients atteints de SHE rapportés dans 3 publications étaient FIP1L1-PDGFRα positifs.
Les 65 patients avec la protéine de fusion FIP1L1-PDGFRα ont atteint une RHC maintenue pendant des mois (de plus d’un mois à 44 mois censurés à la date du rapport). Comme cela a été rapporté dans une publication récente, 21 des 65 patients ont aussi présenté une rémission moléculaire avec une durée médiane de suivi de 28 mois (13 à 67 mois). L’âge de ces patients allait de 25 à 72 ans. De plus, les investigateurs ont rapporté dans ces observations individuelles des améliorations de la symptomatologie et des dysfonctionnements d’autres organes. Les améliorations ont été observées sur les groupe d’organe cardiaque, nerveux, cutané/sous-cutané, respiratoire/thoracique/médiastinal, musculo-squelettique/tissu conjonctif/vasculaire, gastro-intestinal.
Il n’existe pas d’étude clinique contrôlée chez les patients pédiatriques atteints de SHE/LCE. Trois (3) cas de patients atteints de SHE/LCE associés à des réarrangements du gène PDGFR ont été rapportés dans 3 publications. L’âge de ces patients allait de 2 à 16 ans et l’imatinib était administré à une posologie de 300 mg/m² par jour ou comprise entre 200 et 400 mg par jour. Tous les patients ont atteint une réponse hématologique complète, une réponse cytogénétique complète, et/ou une réponse moléculaire complète.
Etudes cliniques dans les GIST non resécables et/ou métastatiques
Une étude de Phase II, internationale, randomisée, en ouvert et non contrôlée a été conduite chez des patients atteints de tumeurs stromales gastro-intestinales (GIST) malignes non résécables ou métastatiques. Dans cette étude, 147 patients ont été inclus et randomisés pour recevoir soit 400 mg soit 600 mg d’imatinib par jour par voie orale pour une durée pouvant atteindre 36 mois. Ces patients étaient âgés de 18 à 83 ans et ils avaient un diagnostic pathologique de GIST malignes Kit-positives, non résécables et/ou métastatiques. Le dosage immunohistochimique a été réalisé en routine avec des anticorps antiKit (A-4502, antisérum polyclonal de lapin, 1:100 ; DAKO Corporation, Carpinteria, CA) par la méthode utilisant le complexe peroxydase-biotine-avidine après marquage de l’antigène.
Le critère principal d'efficacité était basé sur les taux de réponses objectives. Les tumeurs devaient être mesurables pour au moins un site de la maladie et la caractérisation de la réponse était basée sur les critères du Southwestern Oncology Group (SWOG). Les résultats sont présentés dans le Tableau 6.
Tableau 6 : Meilleure réponse tumorale dans l'essai STIB2222 (GIST)
|
Meilleure réponse |
Toutes les doses (n=147) 400 mg (n=73) 600 mg (n=74) n (%) |
|
Réponse complète Réponse partielle Stabilisation de la maladie Progression de la maladie Non évaluable Inconnu |
1 (0,7) 98 (66,7) 23 (15,6) 18 (12,2) 5 (3,4) 2 (1,4) |
Aucune différence des taux de réponses n'a été observée entre les deux groupes de dose. Un nombre important de patients qui présentaient une stabilisation de la maladie au moment de l'analyse intermédiaire a atteint une réponse partielle après une durée plus longue de traitement (médiane de suivi de 31 mois). La durée médiane pour obtenir une réponse était de 13 semaines (95 %IC 12– 23). La durée médiane jusqu’à échec du traitement chez les répondeurs était de 122 semaines (95 %IC 106– 147) alors que pour la population totale de l’étude, elle était de 84 semaines (95 %IC 71– 109). La durée médiane de survie globale n’a pas été atteinte. Après un suivi de 36 mois l’estimation selon Kaplan-Meier du taux de survie est de 68 %.
Dans les deux études cliniques (étude B2222 et étude intergroupe S0033), la dose quotidienne d’imatinib a été augmentée à 800 mg chez les patients qui progressaient aux plus faibles doses de 400 mg ou 600 mg par jour. Au total 103 patients ont eu une augmentation de doses à 800 mg : 6 patients ont atteint une réponse partielle et 21 une stabilisation de leur maladie après augmentation de la dose pour un bénéfice clinique global de 26 %. Sur la base des données de tolérance disponibles, l’augmentation de dose quotidienne à 800 mg chez des patients progressant aux plus faibles doses de 400 mg ou 600 mg par jour ne semble pas affecter le profil de sécurité d’emploi d’imatinib.
Etudes cliniques dans le traitement adjuvant des GIST
Dans le cadre du traitement adjuvant, imatinib a été étudié dans une étude clinique de phase III multicentrique contrôlée menée en double aveugle versus placebo (Z9001) impliquant 773 patients. Leur âge était de 18 à 91 ans. Les patients inclus avaient eu un diagnostic histologique confirmé de GIST avec immunohistochimie positive pour Kit et une taille tumorale ≥ 3 cm au maximum, avec une résection complète de la masse tumorale dans les 14 à 70 jours précédant l’inclusion. Après la résection du GIST primaire, les patients étaient randomisés dans l’un des deux bras : imatinib à 400 mg/j ou le placebo correspondant pendant un an.
Le critère primaire de l’étude était la survie sans rechute, définie comme le délai entre la date de randomisation jusqu’à la date d’une rechute ou d’un décès quelle que soit la cause.
Imatinib a prolongé significativement la survie sans rechute, 75 % des patients étaient sans rechute à 38 mois dans le groupe imatinib versus 20 mois dans le groupe placebo (95 % ICs [30- non estimable] ; [14- non estimable], respectivement) : (hazard ratio = 0,398 [0,259-0,610], p<0.0001). A un an, la survie sans rechute globale était significativement meilleure pour imatinib (97,7 %) versus placebo (82,3 %), (p<0,0001). Le risque de rechute était réduit de 89 % approximativement par comparaison au placebo (hazard ratio = 0,113 [0,049-0,264]).
Le risque de rechute après chirurgie chez des patients atteints de GIST a été évalué rétrospectivement en fonction des facteurs pronostiques suivants : taille de la tumeur, index mitotique, localisation de la tumeur. Les valeurs de l’index mitotique étaient disponibles chez 556 patients sur les 713 patients de la population en intention de traiter (ITT). Les résultats de l’analyse en sous-groupes selon les classifications de risque NIH « United States National Institutes of Health » et AFIP « Armed Forces Institute of Pathology » sont présentés dans le Tableau 7. Aucun bénéfice n’a été observé dans les groupes à faible et très faible risque. Il n’a pas été observé de bénéfice sur la survie globale.
Tableau 7 : Résumé des résultats de l’analyse de la survie sans rechute selon les classifications NIH et AFIP de l’étude Z9001
|
Critères de risque |
Risque |
% de patients |
Nombre d’événements / Nombre de patients |
Hazard ratio global (95%IC)* |
Taux de survie sans rechute (%) |
|
|
12 mois |
24 mois |
|||||
|
imatinib vs placebo |
imatinib vs placebo |
imatinib vs placebo |
||||
|
NIH |
Faible |
29,5 |
0/86 vs. 2/90 |
N.E. |
100 vs. 98,7 |
100 vs. 95,5 |
|
Intermédiaire |
25,7 |
4/75 vs. 6/78 |
0,59 (0,17 ; 2,10) |
100 vs. 94,8 |
97,8 vs. 89,5 |
|
|
Elevé |
44,8 |
21/140 vs. 51/127 |
0,29 (0,18 ; 0,49) |
94,8 vs. 64,0 |
80,7 vs. 46,6 |
|
|
AFIP |
Très faible |
20,7 |
0/52 vs. 2/63 |
N.E. |
100 vs. 98,1 |
100 vs. 93,0 |
|
Faible |
25,0 |
2/70 vs. 0/69 |
N.E. |
100 vs. 100 |
97,8 vs. 100 |
|
|
Modéré |
24,6 |
2/70 vs. 11/67 |
0,16 (0,03 ; 0,70) |
97,9 vs. 90,8 |
97,9 vs. 73,3 |
|
|
Elevé |
29,7 |
16/84 vs. 39/81 |
0.27 (0,15 ; 0,48) |
98,7 vs. 56,1 |
79,9 vs. 41,5 |
|
* Période complète de suivi ; NE – Non estimable
Une seconde étude de phase III multicentrique, en ouvert (SSG XVIII/AIO), a comparé un traitement par imatinib 400 mg/jour pendant 12 mois à un traitement pendant 36 mois chez des patients après résection chirurgicale d'une tumeur stromale gastro-intestinale (GIST) présentant l'une des caractéristiques suivantes : diamètre de la tumeur > 5 cm et index mitotique > 5/50 HPF (high power fields) ; ou diamètre de la tumeur > 10 cm quel que soit l’index mitotique ou tumeur de toute taille avec un index mitotique > 10/50 HPF ou tumeur rompue dans la cavité péritonéale. Au total, 397 patients ont donné leur consentement pour participer à l'étude et ont été randomisés (199 patients dans le groupe de 12 mois de traitement et 198 patients dans le groupe de 36 mois de traitement), leur âge médian était de 61 ans (extrêmes : 22 et 84 ans). La durée médiane du suivi a été de 54 mois (depuis la date de la randomisation jusqu'à la date limite de recueil des données), avec un total de 83 mois entre le premier patient randomisé et la date limite de recueil des données.
Le critère primaire de l’étude était la survie sans rechute, définie comme le délai entre la date de randomisation jusqu’à la date d’une rechute ou d’un décès quelle que soit la cause.
Un traitement par imatinib sur une durée de 36 mois a prolongé significativement la survie sans rechute comparativement à un traitement de 12 mois (avec un hazard ratio (HR) global = 0,46 [0,32, 0,65], p<0,0001) (Tableau 8, figure 1).
De plus, le traitement par imatinib pendant 36 mois a prolongé significativement la survie globale comparativement à un traitement de 12 mois (HR = 0,45 [0,22, 0,89], p = 0,0187) (Tableau 8, figure 2).
Un traitement de plus longue durée (> 36 mois) pourrait retarder la survenue de rechutes ultérieures ; cependant, l’impact de ces données sur la survie globale reste inconnu.
Le nombre total de décès a été de 25 dans le groupe de traitement de 12 mois et de 12 dans le groupe de traitement de 36 mois.
Un traitement par imatinib pendant 36 mois était supérieur à un traitement pendant 12 mois, selon l’analyse en ITT, c’est-à-dire en incluant la population entière de l’étude. Dans l’analyse planifiée par sous-groupes par type de mutation, chez les patients ayant des mutations de l’exon 11, le hazard ratio de la survie sans rechute était de 0,35 [95 % IC :0,22, 0,56] en faveur d’un traitement pendant 36 mois. On ne peut pas tirer de conclusion pour les autres sous-groupes ayant des mutations moins fréquentes en raison du faible nombre d’évènements observés.
Tableau 8 : Traitement de 12 mois et de 36 mois par imatinib (essai SSGXVIII/AIO)
|
Survie sans rechute |
Groupe de traitement de 12 mois %(IC) |
Groupe de traitement de 36 mois %(IC) |
|
12 mois |
93,7 (89,2-96,4) |
95,9 (91,9-97,9) |
|
24 mois |
75,4 (68,6-81,0) |
90,7 (85,6-94,0) |
|
36 mois |
60,1 (52,5-66,9) |
86,6 (80,8-90,8) |
|
48 mois |
52,3 (44,0-59,8) |
78,3 (70,8-84,1) |
|
60 mois |
47,9 (39,0-56,3) |
65,6 (56,1-73,4) |
|
Survie |
|
|
|
36 mois |
94,0 (89,5-96,7) |
96,3 (92,4-98,2) |
|
48 mois |
87,9 (81,1-92,3) |
95,6 (91,2-97,8) |
|
60 mois |
81,7 (73,0-87,8) |
92,0 (85,3-95,7) |
Figure 1 : Estimations de Kaplan-Meier pour le critère principal d'évaluation : la survie sans rechute (population ITT)
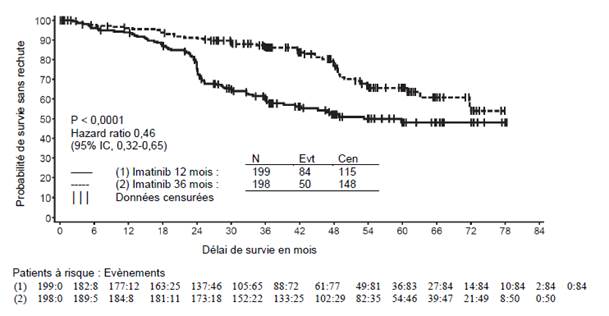
Figure 2 : Estimations de Kaplan-Meier pour la survie globale (population ITT)
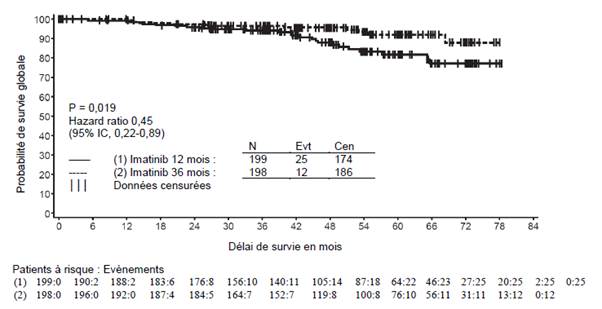
Il n’existe pas d’étude clinique contrôlée chez les patients pédiatriques atteints de GIST c-Kit positive. Dix-sept (17) cas de patients atteints de GIST (avec ou sans mutation de Kit ou PDGFR) ont été rapportés dans 7 publications. L’âge de ces patients allait de 8 à 18 ans et l’imatinib était administré en adjuvant et en situation métastatique à une posologie comprise entre 300 et 800 mg par jour. Les données confirmant les mutations c-Kit ou PDGFR manquaient chez la majorité des patients pédiatriques, ce qui a pu conduire à des résultats cliniques mitigés.
Etudes cliniques dans le DFSP
Une étude ouverte multicentrique de phase II (étude B2225) a été menée incluant 12 patients atteints de DFSP traité par l’imatinib à 800 mg/jour. L’âge des patients atteints de DFSP allait de 23 à 75 ans ; leur maladie était métastatique ou en rechute locale après une chirurgie d’exérèse initiale et n’était pas considérée comme relevant d’une chirurgie d’exérèse supplémentaire au moment de l’entrée dans l’étude. Le critère primaire d’efficacité reposait sur les taux de réponse objective. Parmi les 12 patients inclus, 9 ont répondu, 1 réponse complète et 8 réponses partielles. Trois des répondeurs partiels ont été rendu indemnes de maladie par chirurgie. La durée médiane de traitement dans l’étude B2225 était de 6,2 mois, avec une durée maximale de 24,3 mois. 6 autres patients atteints de DFSP et traités par imatinib ont été rapportés sous la forme de 5 observations individuelles, leur âge allait de 18 mois à 49 ans. Les patients adultes décrits dans la littérature ont été traités par imatinib soit à la posologie de 400 mg/jour (4 cas) soit de 800 mg/jour (1 cas). Cinq (5) patients ont répondu, 3 complètement et 2 partiellement. La durée médiane de traitement dans la littérature allait de 4 semaines à plus de 20 mois. La translocation t(17 :22) [(q22 : q13)], ou la protéine issue de ce gène hybride était présente chez pratiquement tous les répondeurs au traitement par l’imatinib.
Il n’existe pas d’étude clinique chez les patients pédiatriques atteints de DFSP. Cinq (5) cas de patients atteints de DFSP associés à des réarrangements du gène PDGFR ont été rapportés dans 3 publications. L’âge de ces patients allait du nouveau-né à 14 ans et l’imatinib était administré à une posologie de 50 mg par jour ou comprise entre 400 et 520 mg/m² par jour. Tous les patients ont atteint une réponse partielle et/ou complète.
5.2. Propriétés pharmacocinétiques
Paramètres pharmacocinétiques de l’imatinib
La pharmacocinétique de l’imatinib a été évaluée à des doses comprises entre 25 et 1 000 mg. Les profils pharmacocinétiques plasmatiques ont été analysés à J1, puis à J7 ou J28, au moment où les concentrations plasmatiques ont atteint un état d'équilibre.
Absorption
La biodisponibilité absolue moyenne de l’imatinib est de 98 %. Il existe une forte variabilité inter- patient de l’ASC de l’imatinib plasmatique après une prise orale. Lorsqu’il est pris au cours d’un repas riche en lipides, le taux d'absorption de l'imatinib est peu réduit (diminution de 11 % de la Cmax et prolongation de 1,5 h de tmax), avec une légère diminution de l’ASC (7,4 %) comparée à une prise à jeun. L’effet d’une chirurgie gastro-intestinale antérieure sur l’absorption du produit n’a pas été étudié.
Distribution
A des concentrations d’imatinib cliniquement significatives, la fraction liée aux protéines plasmatiques est approximativement de 95 %, sur la base des études in vitro ; il s’agit principalement d’une liaison à l’albumine et aux alpha-glycoprotéines acides, et dans une faible mesure aux lipoprotéines.
Biotransformation
Chez l’homme, le principal métabolite circulant est le dérivé pipérazine N-déméthylé qui présente in vitro une activité similaire à l’imatinib. L'ASC plasmatique de ce métabolite n’atteint que 16 % de l'ASC de l'imatinib. L’affinité pour les protéines plasmatiques du métabolite N-déméthylé est similaire à celle de la molécule mère.
L’imatinib et le métabolite N-déméthylé représentent au total environ 65 % du taux circulant de radioactivité (ASC(0-48h)). Le taux circulant de radioactivité restant correspond à un nombre de métabolites mineurs.
Les tests in vitro montrent que le CYP3A4 est la principale enzyme du cytochrome P450 humain catalysant la biotransformation de l’imatinib. Parmi un éventail de médicaments potentiellement co- administrés (paracétamol, aciclovir, allopurinol, amphotéricine, cytarabine, érythromycine, fluconazole, hydroxyurée, norfloxacine, pénicilline V) seuls l’érythromycine (IC50 50 µM) et le fluconazole (IC50 118 µM) ont montré une inhibition du métabolisme de l’imatinib pouvant être cliniquement significative.
In vitro, l’imatinib est un inhibiteur compétitif des substrats marqués du CYP2C9, des CYP2D6 et des CYP3A4/5 avec des valeurs de Ki de 27, 7,5 et 7,9 respectivement obtenues sur les microsomes hépatiques humains. Les concentrations plasmatiques maximales de l’imatinib sont de 2– 4 µmol/l. Par conséquent, une inhibition du métabolisme de produits co-administrés mettant en jeu les CYP2D6 et CYP3A4/5 est possible. L’imatinib n’interfère pas avec la biotransformation du 5-fluorouracile mais inhibe le métabolisme du paclitaxel par inhibition compétitive du CYP2C8 (Ki = 34,7). Cette valeur de Ki est de loin supérieure aux taux plasmatiques d’imatinib prévisibles chez les patients. Par conséquent, aucune interaction n’est attendue en cas de co-administration de l’imatinib avec le 5-fluorouracile ou le paclitaxel.
Élimination
Après administration d'une dose orale d'imatinib marqué au 14C, environ 81 % de la dose est éliminée au bout de 7 jours (68 % dans les fèces et 13 % dans les urines). La forme inchangée représente 25 % de la dose (5 % dans les urines, 20 % dans les fèces), le reste étant composé de métabolites.
Pharmacocinétique plasmatique
Après administration par voie orale chez le volontaire sain, la demi-vie, d’environ 18 h, est compatible avec une prise quotidienne unique. L'augmentation de l'ASC moyenne de l'imatinib est linéaire et proportionnelle à la dose administrée à des doses orales allant de 25 à 1 000 mg. Lors d’administrations répétées en prise quotidienne unique, la cinétique de l'imatinib n’est pas modifiée, mais son accumulation, à l’état d'équilibre, est augmentée d’un facteur de 1,5 à 2,5.
Pharmacocinétique chez des patients atteints de GIST
Chez des patients atteints de GIST, l’exposition à l’état d’équilibre était 1,5 fois supérieure à celle observée à la même dose (400 mg/jour) chez des patients atteints de LMC. Sur la base d’une analyse préliminaire de pharmacocinétique de population de patients atteints de GIST, on a identifié 3 variables (albumine, taux de globules blancs et bilirubine) qui présentaient une relation statistiquement significative avec la pharmacocinétique de l’imatinib. Des valeurs diminuées de l’albumine étaient associées à une diminution de la clairance (Cl/f) et les valeurs plus élevées du taux de globules blancs étaient associées à une réduction de la Cl/f. Toutefois, ces associations n’étaient pas suffisantes pour justifier un ajustement des doses. Dans cette population de patients, la présence de métastases hépatiques pourrait potentiellement conduire à une insuffisance hépatique et à une diminution du métabolisme.
Pharmacocinétiques de population
Une analyse de pharmacocinétique de population de patients atteints de LMC a montré une légère influence de l'âge sur le volume de distribution (augmentation de 12 % chez les patients > 65 ans), mais cette variation ne semble pas cliniquement significative. Bien que l’effet du poids corporel sur la clairance de l'imatinib laisse attendre une clairance moyenne de 8,5 L/h pour un patient pesant 50 kg, contre 11,8 l/h pour un patient pesant 100 kg, une adaptation de la posologie en fonction du poids n’est pas requise. Le sexe n'a aucune influence sur les paramètres cinétiques de l'imatinib.
Pharmacocinétique chez l'enfant
Comme chez l'adulte, l'imatinib a été rapidement absorbé après administration orale chez le patient pédiatrique dans des études de phase I et de phase II. Chez l'enfant, l'administration de doses de 260 et 340 mg/m2/jour a permis d'obtenir des concentrations plasmatiques équivalentes aux doses respectivement de 400 mg et 600 mg chez l'adulte. La comparaison de l'ASC(0-24) à J 8 et J 1 pour une dose de 340 mg/m2/jour a révélé une accumulation de 1,7 fois après des prises uniques quotidiennes itératives.
Des analyses poolées de données de pharmacocinétique de population chez les enfants atteints d’affections hématologiques (LMC, LAL Ph+, ou autres affections hématologiques traitées par l’imatinib) ont montré que la clairance de l’imatinib augmente parallèlement à celle de la surface corporelle (SC). Après correction de l’effet de la SC, d’autres caractéristiques démographiques telles que l’âge, le poids corporel, et l’indice de masse corporelle n’avaient pas d’effet cliniquement significatif sur l’exposition à l’imatinib. L’analyse a confirmé que l’exposition à l’imatinib chez les enfants recevant 260 mg/m² une fois par jour (sans dépasser 400 mg une fois par jour) ou 340 mg/m² une fois par jour (sans dépasser 600 mg une fois par jour) était comparable à celle des adultes qui ont reçu 400 mg ou 600 mg d’imatinib une fois par jour.
Altération des fonctions organiques
L’imatinib et ses métabolites ne sont pas excrétés de façon significative par le rein. Les patients ayant une altération de la fonction rénale légère à modérée présentent une exposition plasmatique supérieure à celle des patients présentant une fonction rénale normale. L’augmentation est approximativement 1,5 à 2 fois plus, correspondant à une augmentation de 1,5 fois le taux plasmatique d’AGP à laquelle l’imatinib est fortement lié.
La clairance de l’imatinib libre chez les patients ayant une altération de la fonction rénale est probablement similaire à celle des patients avec une fonction rénale normale, puisque l’excrétion rénale représente une voie d’élimination mineure de l’imatinib (voir rubrique 4.2 et 4.4).
Bien que l’analyse des résultats pharmacocinétiques ait montré une variabilité interindividuelle considérable, l’exposition moyenne à l’imatinib n’était pas augmentée chez des patients qui présentaient une altération de la fonction hépatique à des degrés variables par comparaison aux patients ayant une fonction hépatique normale (voir les rubriques 4.2, 4.4 et 4.8).
5.3. Données de sécurité préclinique
Le profil de tolérance préclinique de l’imatinib a été évalué chez le rat, le chien, le singe et le lapin.
Des études de toxicité à doses multiples ont mis en évidence des modifications hématologiques légères à modérées chez le rat, le chien et le singe, avec des modifications de la moelle osseuse chez le rat et le chien.
Le foie est un organe cible chez le rat et le chien. Des augmentations faibles à modérées des transaminases et de légères diminutions des taux de cholestérol, triglycérides, protéines totales et albumine ont été observées chez les deux espèces. Aucune modification histopathologique n’a été mise en évidence sur le foie de rat. Une toxicité hépatique sévère a été observée chez des chiens traités pendant deux semaines, avec une élévation des enzymes hépatiques, une nécrose hépato-cellulaire, une nécrose des canaux biliaires et une hyperplasie des canaux biliaires.
Une toxicité rénale a été observée chez des singes traités pendant deux semaines, avec une minéralisation et une dilatation focales des tubules rénaux et une néphrose tubulaire. Une augmentation de la créatinine et de l’azotémie a été observée chez plusieurs de ces animaux. Chez les rats une hyperplasie de l’épithélium de transition dans la papille rénale et dans la vessie a été observée à des doses > 6 mg/kg dans l’étude de 13 semaines, sans modification des paramètres urinaires et sanguins. Une augmentation du nombre d’infections opportunistes a été observée avec le traitement chronique par l’imatinib.
Dans une étude de 39 semaines chez le singe, la dose dépourvue d’effet indésirable observable n’a pu être définie avec la plus faible dose de 15 mg/kg, correspondant approximativement à un tiers de la dose maximale de 800 mg chez l’homme basée sur la surface corporelle. Le traitement a entraîné une aggravation des infections paludéennes normalement réprimées chez ces animaux.
L’imatinib n’a pas été considéré comme génotoxique dans un test sur cellules bactériennes in vitro (test d’AMES), dans un test sur cellules de mammifères in vitro (lymphome de souris) et dans un test sur micronoyaux de rat in vivo. Toutefois, des effets génotoxiques positifs ont été obtenus avec l’imatinib dans un test de clastogenèse (aberration chromosomique) sur cellules de mammifères in vitro (cellules ovariennes de hamster chinois) avec activation métabolique. Deux intermédiaires de synthèse, présents dans le produit final, sont positifs au test de mutagenèse d’AMES. L’un de ces intermédiaires était aussi positif dans le test sur le lymphome de souris.
Dans une étude de fertilité, chez le rat mâle traité pendant 70 j avant accouplement, le poids des testicules et de l’épididyme et le pourcentage de mobilité des spermatozoïdes ont diminué à la dose de 60 mg/kg, approximativement équivalente à la dose clinique maximale de 800 mg/j, basée sur la surface corporelle. Cela n’a pas été observé à des doses ≤ 20 mg/kg. Une réduction légère à modérée de la spermatogenèse a aussi été observée chez le chien à des doses orales ≥ 30 mg/kg. Chez des rats femelles traités pendant 14 jours avant accouplement et pendant 6 jours de gestation, aucun effet n’a été observé sur l’accouplement ou sur le nombre de femelles gestantes. Par contre, à la dose de 60 mg/kg, les rats femelles ont présenté une perte fœtale post-implantation significative et un nombre de fœtus vivants réduit significativement. Ceci n’a pas été observé à des doses ≤ 20 mg/kg.
Après administration orale au cours d'une étude sur le développement prénatal et post-natal chez le rat, un écoulement vaginal rouge a été observé dans le groupe sous 45 mg/kg/jour au 14-15ème jour de gestation. A la même dose, le nombre de ratons mort-nés ou décédant au cours des 4 premiers jours du post-partum était plus élevé. Dans la descendance F1, à la même dose, les poids moyens étaient réduits de la naissance jusqu’au sacrifice final et le nombre de portées atteignant le critère de séparation prépuciale était légèrement plus faible.
La fertilité de la descendance F1 n'était pas modifiée alors qu'un nombre accru de résorptions fœtales et une diminution du nombre de fœtus viables était observé à 45 mg/kg/jour. La dose sans effet observable (DSEO) pour les mères et la génération F1 était de 15 mg/kg/jour (soit un quart de la dose maximale humaine de 800 mg).
L’imatinib est tératogène chez les rats lorsqu’il est administré au cours de l’organogenèse, à des doses ≥ 100 mg/kg, approximativement équivalentes à la dose clinique maximale de 800 mg/j, basée sur la surface corporelle. Les effets tératogènes observés sont : une exencéphalie, une encéphalocèle, une réduction/absence de l’os frontal et une absence des os pariétaux. Ces effets n’ont pas été observés à des doses ≤ 30 mg/kg.
Au cours d’une étude de toxicité sur le développement du rat juvénile (jours 10 à 70 post-partum), aucun nouvel organe-cible n’a été identifié par rapport aux organes cibles connus chez le rat adulte. Dans l’étude de toxicité réalisée chez les rats juvéniles, des effets sur la croissance, un retard de l’ouverture vaginale et de la séparation prépuciale ont été observés à la plus haute dose recommandée de 340 mg/m2 correspondant à environ 0,3 à 2 fois l’exposition pédiatrique moyenne. De plus, des décès ont été observés chez les animaux juvéniles (autour de la phase de sevrage) à la dose la plus haute recommandée de 340 mg/m2 correspondant à environ 2 fois l’exposition pédiatrique moyenne.
Dans une étude de carcinogénicité d’une durée de deux ans menée chez le rat avec l’imatinib administré à la dose de 15, 30 et 60 mg/kg/j, une réduction statistiquement significative de la longévité a été observée chez les mâles à la dose de 60 mg/kg/j et chez les femelles à une dose ³ 30 mg/kg/j. L’examen histo-pathologique des animaux a mis en évidence comme cause principale de décès ou de sacrifice, des cardiomyopathies (pour les deux sexes), des néphropathies chroniques en progression (chez les femelles) et des papillomes des glandes prépuciales. Les organes cibles des modifications néoplasiques étaient les reins, la vessie, l’urètre, les glandes prépuciales et clitoridiennes, l’intestin grêle, les glandes parathyroïdes, les glandes surrénales, et l’estomac (hors tissu glandulaire).
Des papillomes/carcinomes des glandes prépuciales et clitoridiennes ont été observés à partir des doses de 30 mg/kg/j représentant approximativement 0,5 ou 0,3 fois l’exposition journalière (basée sur l’ASC) chez l’homme traité par 400 mg/j ou 800 mg/j respectivement et 0,4 fois l’exposition journalière (basée sur l’ASC) chez l’enfant traité par 340 mg/m2/jour. La dose sans effet observable (DSEO) était de 15 mg/kg/j. Les adénomes/carcinomes rénaux et les papillomes de la vessie et de l’urètre, les adénocarcinomes de l’intestin grêle, les adénomes des parathyroïdes, les tumeurs médullaires bénignes et malignes des glandes surrénales et les carcinomes/papillomes de l’estomac (hors tissu glandulaire) ont été observés à la dose de 60 mg/kg/j, représentant approximativement 1,7 ou 1 fois l’exposition journalière (basée sur l’ASC) chez l’homme traité par 400 mg/j ou 800 mg/j respectivement et 1,2 fois l’exposition journalière (basée sur l’ASC) chez l’enfant traité par 340 mg/m2/jour. La dose sans effet observable (DSEO) était de 30 mg/kg/j.
Le mécanisme et la pertinence chez l’homme des résultats de l’étude de carcinogénicité menée chez le rat ne sont pas encore clarifiés.
Des lésions non-néoplasiques qui n’avaient pas été identifiées au cours d’études précliniques antérieures, ont été observées sur le système cardiovasculaire, le pancréas, les glandes endocrines et les dents. Les modifications les plus importantes comprenaient l’hypertrophie et la dilatation cardiaque responsables de signes d’insuffisance cardiaque.
La substance active imatinib présente un risque environnemental pour les organismes vivant dans les sédiments.
Cellulose microcristalline, crospovidone (type A), hypromellose, stéarate de magnésium, silice colloïdale anhydre
Pelliculage du comprimé :
Oxyde de fer rouge (E172), oxyde de fer jaune (E172), macrogol 4000, talc, hypromellose.
3 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30°C.
A conserver dans l’emballage d'origine à l’abri de l’humidité.
6.5. Nature et contenu de l'emballage extérieur
Les comprimés pelliculés sécables sont présentés sous plaquettes (PVC/Aluminium) ou (PVC/PE/PVDC/Aluminium).
Boîte de 20, 30, 50, 60, 80, 90 et 120 comprimés pelliculés sécables.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
49 AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 969 3 2 : 20 comprimés pelliculés sécables sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 300 969 4 9 : 30 comprimés pelliculés sécables sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 300 969 5 6 : 50 comprimés pelliculés sécables sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 300 483 0 6 : 60 comprimés pelliculés sécables sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 300 969 6 3 : 80 comprimés pelliculés sécables sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 300 969 8 7 : 90 comprimés pelliculés sécables sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 300 483 1 3 : 120 comprimés pelliculés sécables sous plaquettes (PVC/PE/PVDC/Aluminium).
· 34009 300 969 9 4 : 20 comprimés pelliculés sécables sous plaquettes (PVC/Aluminium).
· 34009 300 970 0 7 : 30 comprimés pelliculés sécables sous plaquettes (PVC/Aluminium).
· 34009 300 970 1 4 : 50 comprimés pelliculés sécables sous plaquettes (PVC/Aluminium).
· 34009 300 483 2 0 : 60 comprimés pelliculés sécables sous plaquettes (PVC/Aluminium).
· 34009 300 970 2 1 : 80 comprimés pelliculés sécables sous plaquettes (PVC/Aluminium).
· 34009 300 970 3 8 : 90 comprimés pelliculés sécables sous plaquettes (PVC/Aluminium).
· 34009 300 483 3 7 : 120 comprimés pelliculés sécables sous plaquettes (PVC/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription initiale hospitalière de 6 mois.
Prescription initiale et renouvellement réservés aux spécialistes en hématologie, en oncologie, en médecine interne, ou aux médecins compétents en cancérologie.
ANSM - Mis à jour le : 25/07/2024
IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable
Imatinib
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable ?
3. Comment prendre IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : L01EA01
IMATINIB SANDOZ est un médicament qui contient une substance active appelée imatinib. Ce médicament agit par inhibition de la croissance des cellules anormales des maladies décrites ci-dessous parmi lesquelles certains types de cancer.
IMATINIB SANDOZ est un traitement chez les adultes et les enfants de :
· Leucémie myéloïde chronique (LMC)
La leucémie est un cancer des globules blancs du sang. Ces globules blancs aident habituellement l’organisme à se défendre contre les infections. La leucémie myéloïde chronique est une forme de leucémie dans laquelle certains globules blancs anormaux, appelés cellules myéloïdes, commencent à se multiplier de manière incontrôlée.
· Leucémie aiguë lymphoïde chromosome Philadelphie positive (LAL Ph-positive)
La leucémie est un cancer des globules blancs du sang. Ces globules blancs aident habituellement l’organisme à se défendre contre les infections. La leucémie aiguë lymphoblastique est une forme de leucémie dans laquelle certains globules blancs anormaux (appelés lymphoblastes) commencent à se multiplier de manière incontrôlée. IMATINIB SANDOZ inhibe la croissance de ces cellules.
IMATINIB SANDOZ est également un traitement chez les adultes de :
· Syndromes myéloprolifératifs/myélodysplasiques (SMP/SMD)
Il s’agit d’un groupe de maladies du sang pour lesquelles des cellules du sang commencent à se multiplier de manière incontrôlée. IMATINIB SANDOZ inhibe la croissance de ces cellules dans un certain sous-groupe de ces maladies.
· Syndrome hyperéosinophilique (SHE) et/ou de la leucémie chronique à éosinophiles (LCE)
Ce sont des maladies pour lesquelles des cellules du sang (appelées éosinophiles) commencent à se multiplier de manière incontrôlée. IMATINIB SANDOZ inhibe la croissance de ces cellules dans un certain sous-groupe de ces maladies.
· Tumeurs stromales gastro-intestinales malignes (GIST)
GIST est un cancer de l'estomac et des intestins. Il résulte de la croissance cellulaire incontrôlée des tissus de support de ces organes. IMATINIB SANDOZ inhibe la croissance de ces cellules.
· Dermatofibrosarcome protuberans (DFSP)
Le dermatofibrosarcome est un cancer du tissu sous la peau dans lequel certaines cellules commencent à se multiplier de manière incontrôlée. IMATINIB SANDOZ inhibe la croissance de ces cellules.
Par la suite dans cette notice, les abréviations ci-dessus sont utilisées pour désigner ces maladies.
Si vous avez des questions sur la façon dont IMATINIB SANDOZ agit ou pourquoi ce médicament vous a été prescrit, adressez-vous à votre médecin.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable ?
Suivez attentivement toutes les instructions qui vous sont données par votre médecin même si elles diffèrent des informations générales qui sont précisées dans cette notice.
Ne prenez jamais IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable :
· si vous êtes allergique à l’imatinib ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Si vous êtes concerné, parlez-en à votre médecin avant de prendre IMATINIB SANDOZ.
Si vous pensez que vous pouvez être allergique mais vous n’êtes pas sûr, demandez l’avis de votre médecin.
Avertissements et précautions
Adressez-vous à votre médecin avant de prendre IMATINIB SANDOZ :
· si vous avez ou avez eu un problème au foie, aux reins ou au cœur,
· si vous recevez un traitement par lévothyroxine car vous avez subi une chirurgie de la thyroïde,
· si vous avez déjà eu ou pourriez avoir actuellement une hépatite B. En effet, IMATINIB SANDOZ pourrait réactiver votre hépatite B, ce qui peut être fatal dans certains cas. Les patients seront étroitement surveillés par leur médecin afin de détecter tout signe d’infection avant l’instauration du traitement,
· si vous présentez des ecchymoses, des saignements, de la fièvre, de la fatigue et une confusion quand vous prenez IMATINIB SANDOZ, contactez votre médecin. Cela peut être un signe de lésion des vaisseaux sanguins connue sous le nom de microangiopathie thrombotique (MAT).
Si l’une de ces situations vous concerne, parlez-en à votre médecin avant de prendre IMATINIB SANDOZ.
Vous pourriez devenir plus sensible au soleil pendant votre traitement par IMATINIB SANDOZ. Il est important de couvrir les zones de la peau exposées au soleil et d’utiliser un écran solaire avec un facteur de protection solaire (FPS) élevé. Ces précautions doivent également être appliquées chez les enfants.
Au cours de votre traitement avec IMATINIB SANDOZ, informez immédiatement votre médecin si vous prenez du poids très rapidement. IMATINIB SANDOZ peut faire que votre corps va retenir plus d’eau (rétention d’eau sévère).
Pendant votre traitement par IMATINIB SANDOZ, votre médecin surveillera régulièrement si ce médicament agit. Des examens sanguins seront réalisés et vous serez régulièrement pesé.
Enfants et adolescents
IMATINIB SANDOZ est aussi un traitement de la LMC chez l’enfant et l’adolescent. Aucune expérience de ce médicament n’est disponible chez l’enfant de moins de 2 ans souffrant de LMC.
L’expérience est limitée chez les enfants et adolescents ayant une LAL Ph-positive et très limitée chez les enfants et adolescents ayant un SMP/SMD, DFSP, GIST et SHE/LCE.
Certains enfants ou adolescents traités par IMATINIB SANDOZ peuvent avoir un retard de croissance. Le médecin surveillera régulièrement la croissance de votre enfant lors des visites prévues.
Autres médicaments et IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance (tel que le paracétamol) et y compris les médicaments à base de plantes (tels que le millepertuis). Certains médicaments peuvent interagir sur l’effet d’IMATINIB SANDOZ lorsqu’ils sont pris en même temps. Ils peuvent augmenter ou diminuer l’effet d’IMATINIB SANDOZ en menant à une augmentation des effets indésirables ou à une moindre efficacité d’IMATINIB SANDOZ. IMATINIB SANDOZ peut agir de la même façon sur d’autres médicaments.
Prévenez votre médecin si vous utilisez d’autres médicaments qui empêchent la formation de caillots sanguins.
IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
· IMATINIB SANDOZ ne doit pas être utilisé au cours de la grossesse à moins d’une nécessité absolue car il pourrait nuire à votre bébé. Votre médecin discutera avec vous des risques possibles de prendre IMATINIB SANDOZ au cours de la grossesse.
· Chez les femmes pouvant être enceintes, une contraception efficace doit être conseillée pendant le traitement et pendant les 15 jours suivant l’arrêt du traitement.
· N’allaitez pas au cours du traitement par IMATINIB SANDOZ et pendant les 15 jours suivant l’arrêt du traitement, car cela pourrait être nocif pour votre bébé.
· Il est conseillé aux patients qui s'inquièteraient de leur fécondité lors de la prise d’IMATINIB SANDOZ de consulter leur médecin.
Conduite de véhicules et utilisation de machines
Vous pouvez avoir des vertiges, des étourdissements ou des troubles de la vision lorsque vous prenez ce traitement. Si cela se produit, ne conduisez pas ou n’utilisez pas de machines jusqu’à ce que vous vous sentiez de nouveau bien.
3. COMMENT PRENDRE IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable ?
Votre médecin vous a prescrit IMATINIB SANDOZ car vous souffrez d’une maladie grave. IMATINIB SANDOZ peut vous aider à lutter contre cette maladie.
Toutefois, veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Il est important de le faire aussi longtemps que votre médecin ou votre pharmacien vous le dit. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Vous ne devez pas arrêter de prendre IMATINIB SANDOZ à moins que votre médecin vous l’ait dit. Si vous ne pouvez pas prendre ce médicament alors que votre médecin vous l’a prescrit ou si vous pensez que vous n’en avez plus besoin, contactez immédiatement votre médecin.
Quelle dose d’IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable prendre ?
Utilisation chez les adultes
Votre médecin vous dira exactement combien de comprimés vous devrez prendre.
· Si vous êtes traité(e) pour une LMC :
En fonction de votre état, la dose usuelle pour débuter est soit 400 mg, soit 600 mg :
- pour 400 mg prendre 4 comprimés une fois par jour,
- pour 600 mg prendre 6 comprimés une fois par jour.
· Si vous êtes traité(e) pour un GIST :
La dose initiale est de 400 mg, soit prendre 4 comprimés une fois par jour.
Pour les LMC et les GIST, votre médecin peut prescrire une dose plus élevée ou plus faible en fonction de votre réponse au traitement. Si votre dose est de 800 mg par jour (8 comprimés), vous devrez prendre 4 comprimés le matin et 4 comprimés le soir.
· Si vous êtes traité(e) pour une LAL Ph-positive :
La dose initiale est de 600 mg, soit prendre 6 comprimés une fois par jour.
· Si vous êtes traité(e) pour un SMD/SMP :
La dose initiale est de 400 mg, soit prendre 4 comprimés une fois par jour.
· Si vous êtes traité(e) pour un SHE/LCE :
La dose initiale est de 100 mg, soit prendre un comprimé à 100 mg une fois par jour. Votre médecin peut décider d’augmenter la dose initiale à 400 mg, soit 4 comprimés à prendre en fonction de votre réponse au traitement.
· Si vous êtes traité(e) pour un DFSP :
La dose est de 800 mg par jour (8 comprimés), soit prendre 4 comprimés le matin et 4 comprimés le soir.
Utilisation chez les enfants et les adolescents
Votre médecin vous dira combien d’IMATINIB SANDOZ donner à votre enfant. La dose d’IMATINIB SANDOZ à avaler dépendra de l’état de votre enfant, de sa taille et de son poids. La dose quotidienne totale à avaler chez l’enfant ne doit pas dépasser 800 mg dans la LMC et 600 mg dans la LAL Ph-positive. Le traitement peut être donné à votre enfant soit en une seule prise par jour ou bien la dose quotidienne peut être répartie en deux prises (la moitié le matin et l’autre moitié le soir).
Le comprimé pelliculé sécable peut être divisé en doses égales.
Quand et comment prendre IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable ?
· prenez IMATINIB SANDOZ au cours d’un repas, cela vous aidera à protéger votre estomac des problèmes avec IMATINIB SANDOZ,
· avalez les comprimés entiers avec un grand verre d’eau.
Si vous ne pouvez pas avaler les comprimés, vous pouvez les dissoudre dans un verre d’eau plate ou de jus de pomme :
· prenez environ 50 mL pour chaque comprimé de 100 mg,
· remuez avec une cuillère jusqu’à dissolution complète des comprimés,
· lorsque les comprimés sont complètement dissous, buvez tout de suite la totalité du contenu du verre. Des traces des comprimés dissous peuvent rester déposées dans le verre.
Combien de temps prendre IMATINIB SANDOZ ?
Continuez à prendre IMATINIB SANDOZ tous les jours aussi longtemps que votre médecin vous le dira.
Si vous avez pris plus de IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable que vous n’auriez dû :
Si vous avez pris accidentellement trop de comprimés, informez-en immédiatement votre médecin.
Vous pourriez nécessiter une surveillance médicale. Emportez avec vous la boîte de votre traitement.
Si vous oubliez de prendre IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable :
· si vous oubliez de prendre une dose, prenez-la dès que vous vous en apercevez. Toutefois s’il est presque le moment de prendre la dose suivante, ne prenez pas la dose oubliée,
· continuez votre traitement selon la posologie habituelle,
· ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Certains effets peuvent être graves. Prévenez immédiatement votre médecin si vous présentez l’un des effets suivants :
Très fréquents (pouvant affecter plus d’1 personne sur 10) ou fréquents (pouvant affecter jusqu’à 1 personne sur 10) :
· prise rapide de poids. IMATINIB SANDOZ peut faire que votre corps va retenir plus d’eau (rétention hydrique sévère),
· signes d’infection tels que fièvre, frissons intenses, maux de gorge ou ulcérations de la bouche. IMATINIB SANDOZ peut réduire le nombre de globules blancs sanguins, aussi vous pouvez être plus sensible aux infections,
· saignements ou bleus sans raison évidente (lorsque vous ne vous êtes pas fait mal vous-même).
Peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) ou rares (pouvant affecter jusqu’à 1 personne sur 1000) :
· douleur thoracique ou rythme cardiaque irrégulier (signes de problèmes cardiaques),
· toux, difficultés ou douleurs respiratoires (signes de problèmes pulmonaires),
· sensations d’étourdissements, sensations vertigineuses ou un évanouissement (signes d’une baisse de la pression artérielle),
· mal au cœur (nausées) avec perte de l’appétit, coloration foncée des urines, jaunissement de la peau ou des yeux (signes de problèmes hépatiques),
· éruption, rougissement de la peau avec des cloques sur la peau, les lèvres, la bouche ou les yeux, la peau qui pèle, fièvre, survenue de taches rouges ou pourpres, démangeaisons, sensations de brûlures, éruption pustuleuse (signes de problèmes cutanés),
· nodules rouges et douloureux sur la peau, douleurs cutanées, rougeurs cutanées (inflammation du tissu adipeux sous la peau),
· douleur abdominale sévère, sang dans les vomissements, les selles ou les urines, ou selles noires (signes de problèmes gastro-intestinaux),
· réduction importante du volume urinaire, sensation de soif (signes de problèmes rénaux),
· mal au cœur (nausée) avec diarrhée et vomissements, douleurs abdominales ou fièvre (signes de problèmes intestinaux),
· maux de tête sévères, faiblesse ou paralysie des membres ou de la face, difficultés pour parler, perte de connaissance soudaine (signes de problèmes du système nerveux tels qu’un saignement ou un gonflement au niveau du crâne ou du cerveau),
· pâleur, sensation de fatigue, essoufflements, urines foncées (signes de faibles taux des globules rouges du sang),
· douleur au niveau des yeux ou détérioration de la vue, saignement dans les yeux,
· douleurs au niveau des os ou des articulations (signes d’ostéonécrose),
· cloques sur la peau ou les muqueuses (signes de pemphigus),
· orteils ou doigts froids ou engourdis (signes du syndrome de Raynaud),
· gonflement soudain et rougeur de la peau (signes d’une infection de la peau appelée cellulite),
· difficulté à entendre,
· faiblesse musculaire et spasmes musculaires avec un rythme cardiaque irrégulier (signes d’une variation de la quantité de potassium dans le sang),
· contusion (bleus),
· douleurs d’estomac, avec mal au cœur (nausée),
· crampes musculaires avec fièvre, urines rouge-brun, douleur ou faiblesse de vos muscles (signes de problèmes musculaires),
· douleurs pelviennes quelquefois accompagnées de nausées et de vomissements, avec des saignements vaginaux inattendus, des sensations de vertiges ou d’évanouissement en raison d’une pression artérielle basse (signes de problèmes des ovaires ou de l’utérus),
· nausées, essoufflement, pouls irrégulier, urine trouble, fatigue et/ou douleurs articulaires associées à des anomalies des résultats des analyses de sang (par exemple: hyperkaliémie, hyperurémie, hypercalcémie et hypophosphorémie),
· caillots de sang dans les petits vaisseaux sanguins (microangiopathie thrombotique).
Fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles) :
· association d’une éruption cutanée sévère et généralisée, de nausées, de fièvre, d’un taux élevé de certains globules blancs dans le sang (éosinophiles) ou d’une coloration jaune de la peau ou des yeux (signes d’une jaunisse) avec essoufflement, douleur ou gêne dans la poitrine, diminution importante du débit urinaire et sensation de soif, etc. (signes d’une réaction allergique liée au traitement : syndrome DRESS),
· insuffisance rénale chronique,
· réapparition (réactivation) de l’hépatite B si vous avez déjà eu une hépatite B dans le passé (infection hépatique).
Si vous présentez l’un des effets décrits ci-dessus, parlez-en immédiatement à votre médecin.
Les autres effets indésirables peuvent être :
Très fréquents (pouvant affecter plus d’1 personne sur 10) :
· maux de tête ou sensation de fatigue,
· mal au cœur (nausées), envie de vomir (vomissements), diarrhée ou indigestion,
· éruption,
· crampes musculaires ou articulaires, douleurs osseuses ou musculaires pendant le traitement par IMATINIB SANDOZ ou après l’arrêt du traitement,
· œdèmes tels que gonflement des chevilles ou gonflement des yeux,
· prise de poids.
Si l’un de ces effets vous affecte sévèrement, informez votre médecin.
Fréquents (pouvant affecter jusqu’à 1 personne sur 10) :
· anorexie, perte de poids ou trouble du goût,
· sensations vertigineuses ou faiblesse,
· difficulté d’endormissement (insomnie),
· écoulement de l’œil avec démangeaisons, rougeur ou gonflement (conjonctivite), yeux larmoyants ou vision trouble,
· saignement du nez,
· douleur ou gonflement de l’abdomen, flatulence, brûlures d’estomac ou constipation,
· démangeaisons,
· perte inhabituelle ou raréfaction des cheveux,
· engourdissement des mains ou des pieds,
· aphtes,
· gonflement des articulations et douleurs articulaires,
· bouche sèche, peau sèche ou œil sec,
· diminution ou augmentation de la sensibilité cutanée,
· bouffées de chaleur, frissons ou sueurs nocturnes.
Si l’un de ces effets vous affecte sévèrement, informez votre médecin.
Peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) :
· toux, écoulement nasal ou nez bouché, sensation de lourdeur ou de douleur en appuyant sur la zone au-dessus des yeux ou sur les côtés du nez, congestion nasale, éternuements, mal de gorge, avec ou sans maux de tête (signes d’infection des voies respiratoires hautes),
· maux de tête sévères ressentis comme une douleur lancinante ou une sensation de pulsation, généralement d’un côté de la tête et souvent accompagnés de nausées, de vomissements et d’une sensibilité à la lumière ou au bruit (signes de migraine),
· symptômes pseudo-grippaux (grippe),
· douleur ou sensation de brûlure en urinant, augmentation de la température corporelle, douleur dans l’aine ou dans la région pelvienne, urine de couleur rouge ou brune ou urine trouble (signes d’infection des voies urinaires),
· douleur et gonflement au niveau des articulations (signes d’arthralgie),
· un sentiment constant de tristesse et de perte d’intérêt, qui vous empêche de réaliser vos activités normales (signes de dépression),
· un sentiment d’appréhension et d’inquiétude accompagné de symptômes physiques tels que des palpitations, des sueurs, des tremblements, une bouche sèche (signes d’anxiété),
· somnolence, endormissement, sommeil excessif,
· mouvements tremblants (tremblement),
· troubles de la mémoire,
· besoin irrépressible de bouger les jambes (syndrome des jambes sans repos),
· perception de bruits (par exemple : sifflements, bourdonnements) dans les oreilles qui n’ont pas de source extérieure (acouphène),
· pression artérielle élevée (hypertension),
· éructations,
· inflammation des lèvres,
· difficulté à avaler,
· augmentation de la transpiration,
· décoloration de la peau,
· ongles cassants,
· boutons rouges ou blancs autour de la racine des cheveux, éventuellement douloureux, démangeaison ou sensation de brûlure (signes d’une inflammation des follicules pileux, également appelée folliculite),
· éruption cutanée avec desquamation ou peau qui pèle (dermatite exfoliative),
· grossissement des seins (peut survenir chez les hommes ou les femmes),
· douleur diffuse et/ou sensation de lourdeur dans les testicules ou le bas-ventre, douleur en urinant, au cours des rapports sexuels ou pendant l’éjaculation, sang dans les urines (signes d’œdème des testicules),
· incapacité d’obtenir ou de maintenir une érection (dysfonction érectile),
· menstruations abondantes ou irrégulières,
· difficulté à atteindre/maintenir l’excitation sexuelle,
· diminution du désir sexuel,
· douleur aux mamelons,
· sensation générale de malaise,
· infection virale telle que le bouton de fièvre,
· douleur dans le bas du dos résultant d’un trouble rénal,
· augmentation de la fréquence des mictions,
· augmentation de l’appétit,
· douleur ou sensation de brûlure dans la partie supérieure de l’abdomen et/ou de la poitrine (brûlures d’estomac), nausées, vomissements, reflux acides, sensation de satiété et de ballonnement, selles de couleur noire (signes d’un ulcère d’estomac),
· raideur articulaire et musculaire,
· résultats anormaux aux analyses de laboratoire.
Si l’un de ces effets vous affecte sévèrement, informez votre médecin.
Rares (pouvant affecter jusqu’à 1 personne sur 1000) :
· confusion,
· décoloration des ongles.
Fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles) :
· rougeur et/ou gonflement de la paume des mains et de la plante des pieds qui peuvent être accompagnés de sensations de picotements et de brûlures douloureuses,
· lésions cutanées douloureuses et/ou bulleuses,
· retard de croissance chez l’enfant et l’adolescent.
Si l’un de ces effets vous affecte sévèrement, informez votre médecin.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte ou la plaquette après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 30°C.
A conserver dans l’emballage extérieur d'origine à l’abri de l’humidité.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable
· La substance active est :
Imatinib (sous forme de mésilate).................................................................................... 100 mg
Pour un comprimé pelliculé sécable.
· Les autres composants sont :
Noyau du comprimé : cellulose microcristalline, crospovidone (type A), hypromellose, stéarate de magnésium, silice colloïdale anhydre.
Pelliculage du comprimé : oxyde de fer rouge (E172), oxyde de fer jaune (E172), macrogol 4000, talc, hypromellose.
Qu’est-ce que IMATINIB SANDOZ 100 mg, comprimé pelliculé sécable et contenu de l’emballage extérieur
Ce médicament se présente sous forme d’un comprimé pelliculé sécable, de diamètre d’environ 9,2 mm, de couleur jaune très foncé à brun orangé, rond, biconvexe, à bords biseautés, portant l’inscription « NVR » gravée sur une face et « SA » sur l’autre face avec une barre de sécabilité séparant les 2 lettres.
Les comprimés sont disponibles en boîtes de 20, 30, 50, 60, 80, 90 ou 120 comprimés pelliculés sécables.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
49 AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
Exploitant de l’autorisation de mise sur le marché
49 AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
VEROVSKOVA ULICA 57
1526 LJUBLJANA
SLOVENIE
OU
SALUTAS PHARMA GMBH
OTTO-VON-GUERICKE-ALLEE 1
39179 BARLEBEN
ALLEMAGNE
OU
NOVARTIS PHARMA GMBH
ROONSTRASSE 25
90429 NÜRNBERG
ALLEMAGNE
OU
LEK PHARMACEUTICALS D.D.
TRIMLINI 2D
9220 LENDAVA
SLOVENIE
OU
NOVARTIS PHARMACEUTICAL MANUFACTURING LLC
VEROVSKOVA ULICA 57
1000 LJUBLJANA
SLOVENIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< [MM/AAAA] >< [mois AAAA] >
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).