Dernière mise à jour le 01/06/2026
VIRPAX 50 mg, comprimé buccogingival muco adhésif
Indications thérapeutiques
Virpax est utilisé dans le traitement des boutons de fièvre récidivants (l'herpès labial récurrent), dus au virus Herpès simplex chez les adultes dont le système immunitaire fonctionne correctement. Virpax doit être appliqué directement sur la gencive (pas à l'endroit même du bouton) dès l’apparition des premiers symptômes (brûlures, picotements, démangeaisons) ou signes (rougeur, gonflement).
Le virus provoque des vésicules ou des inflammations principalement sur les lèvres mais parfois aussi sur d'autres parties du visage. Cette infection virale peut être activée quand le système immunitaire est affaibli, par exemple par des rhumes ou d'autres infections. Une exposition solaire importante, le stress ou les menstruations peuvent également déclencher les symptômes.
Virpax inhibe la capacité du virus à se reproduire et permet ainsi à l'infection de régresser.
Vous devez vous adresser à votre médecin si les symptômes s’aggravent ou ne s’améliorent pas après 5 jours.
Présentations
> 2 plaquette(s) unitaires aluminium de 1 comprimé(s)
Code CIP : 278 274-5 ou 34009 278 274 5 7
Déclaration de commercialisation : 13/05/2025
Cette présentation n'est pas agréée aux collectivités
> 1 plaquette(s) unitaire aluminium de 1 comprimé(s)
Code CIP : 34009 302 533 5 9
Déclaration de commercialisation : 10/02/2024
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 19/09/2025
VIRPAX 50 mg, comprimé buccogingival muco adhésif
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé contient 50 mg d'aciclovir.
Excipient à effet notoire : traces de lactose dérivé de concentré de protéines de lait, laurilsulfate de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé buccogingival muco-adhésif.
Comprimé blanc à jaunâtre de 8 mm avec une face arrondie et une face plane avec la mention gravée «AL21».
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Dose unique. Administration gingivale.
Adultes
Virpax 50 mg comprimé buccogingival muco-adhésif ne doit être appliqué qu'une seule fois par épisode sur la gencive supérieure.
Virpax doit être appliqué dès l’apparition des premiers symptômes ou signes prodromaux d’herpès labial (voir la rubrique 5.1).
Utilisation dans la population pédiatrique
Virpax est uniquement indiqué chez les adultes. La sécurité et l’efficacité chez les enfants n’ont pas été établies. Aucune donnée n'est disponible.
Mode d’administration
Précautions à prendre avant la manipulation ou l'administration du médicament
Virpax doit être appliqué dès l’apparition des premiers symptômes et signes prodromaux d’herpès labial. Le comprimé doit être appliqué avec un doigt sec immédiatement après avoir été sorti de la plaquette. Le comprimé doit être placé contre la gencive supérieure juste au-dessus de la deuxième incisive et maintenu en place avec une légère pression sur la lèvre supérieure pendant 30 secondes pour garantir l'adhérence. Pour des questions de confort, la face arrondie doit être placée contre la gencive supérieure, mais les deux côtés du comprimé peuvent être appliqués. Virpax peut aussi être appliqué à l'intérieur de la lèvre s'il colle à cet endroit plutôt que contre la gencive. Les patients souffrant de sècheresse buccale doivent boire un verre d'eau avant d'appliquer le comprimé afin d'humidifier la muqueuse buccale et ainsi favoriser l'adhérence du comprimé.
Une fois appliqué, Virpax reste en position et se dissout progressivement au cours de la journée.
Les aliments et les boissons peuvent être consommés normalement quand Virpax est en place. Le comprimé ne doit pas être sucé, mâché ou avalé.
Toutes les situations pouvant interférer avec l'adhérence du comprimé doivent être évitées :
· Toucher ou appuyer sur le comprimé déjà en place.
· Mâcher un chewing-gum.
· Brosser les dents pendant la journée du traitement.
Si Virpax n'adhère pas ou tombe dans les 6 premières heures suivant l'application, le même comprimé doit être remis en place immédiatement. Si le comprimé ne peut être remis en place, un nouveau comprimé doit être appliqué.
Si Virpax est avalé dans les 6 premières heures, le patient doit boire un verre d'eau et un nouveau comprimé doit être appliqué. Le comprimé ne doit être remplacé qu’une seule fois.
Si le comprimé Virpax tombe ou est avalé accidentellement au bout de 6 heures, il ne doit pas être remplacé.
· Hypersensibilité à l'aciclovir ou à l'un des excipients mentionnés à la rubrique 6.1.
· Allergie au lait ou produits dérivés.
4.4. Mises en garde spéciales et précautions d'emploi
Il n'y a aucune donnée sur l'utilisation de Virpax chez des patients immunodéprimés. Virpax ne doit pas être utilisé chez des patients immunodéprimés, car un risque d'augmentation de la résistance à l'aciclovir ne peut être exclu.
L’efficacité de Virpax lorsqu’il est appliqué après la formation des lésions vésiculaires n’a pas été démontrée. En conséquence, Virpax doit uniquement être utilisé dès l’apparition des symptômes ou signes prodromaux.
Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose ne doivent pas prendre ce médicament. Ce médicament contient des protéines de lait, qui peuvent provoquer des réactions allergiques chez les personnes présentant une hypersensibilité ou une allergie sévères aux protéines de lait.
Ce médicament contient 5,2 mg de laurilsulfate de sodium par comprimé. Le laurilsulfate de sodium peut provoquer des réactions cutanées locales (comme une sensation de piqûre ou de brûlure) ou augmenter les réactions cutanées causées par d'autres produits lorsqu'ils sont appliqués sur la même zone.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est à dire qu'il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée avec le Virpax. L'aciclovir est principalement éliminé sous forme inchangée dans l'urine par sécrétion tubulaire active. Bien que les concentrations plasmatiques d'aciclovir après application de Virpax soient faibles, tout médicament administré concomitamment qui inhibe ce mécanisme d'élimination risque d'augmenter les concentrations plasmatiques d'aciclovir. Cependant, étant donné la faible dose et la faible exposition systémique d'aciclovir obtenue après l'application de Virpax, il est peu probable que les interactions soient cliniquement significatives.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas de données ou il existe des données limitées, sur l'utilisation de Virpax chez la femme enceinte. Les données post-commercialisation sur l'utilisation locale ou systémique de formulations d'aciclovir pendant la grossesse n'ont pas révélé d'augmentation du nombre de malformations par rapport à la population générale. De plus, les malformations observées ne présentaient aucune caractéristique unique ni aucun profil cohérent suggérant une cause commune. Cependant, aucune donnée épidémiologique ne permet actuellement d'exclure un risque.
Les études effectuées chez l’animal n’ont pas mis en évidence d’effets délétères directs ou indirects en termes de toxicité pour la reproduction à des doses supérieures à la dose thérapeutique chez l’homme (voir rubrique 5.3).
L'utilisation de Virpax chez la femme enceinte ne doit être envisagée que lorsque les bénéfices potentiels l'emportent sur d'éventuels risques inconnus.
Allaitement
D'après des données limitées chez l'Homme, l'aciclovir administré par voie orale est excrété dans le lait maternel en très faibles quantités (dose infantile relative d'environ 1 %). La Cmax et l'ASC plasmatiques après application d'un comprimé muco-adhésif étant environ 8 à 10 fois inférieures à celles obtenues par voie orale, l'utilisation de Virpax pendant l'allaitement ne devrait pas affecter le nourrisson allaité. De plus, la biodisponibilité gastro-intestinale chez le nourrisson est également faible.
Bien que la dose reçue par le nourrisson après administration de Virpax à sa mère soit négligeable, l'utilisation de Virpax pendant l'allaitement ne doit être envisagée que lorsque les bénéfices potentiels l'emportent sur d'éventuels risques inconnus.
Fertilité
Il n'existe aucune donnée concernant l'effet de Virpax comprimé buccogingival muco-adhésif sur la fertilité de la femme. Au cours d'une étude menée sur 20 patients masculins ayant une numération de spermatozoïdes normale, l'administration d'aciclovir par voie orale à des doses allant jusqu'à 1 g par jour pendant une durée de six mois maximum n'a montré aucun effet cliniquement significatif sur la numération de spermatozoïdes, leur motilité ou leur morphologie.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Le profil de sécurité de Virpax a été établi sur la base d'un essai clinique de 775 patients dont 378 avaient reçu Virpax. Les effets indésirables sont listés ci-dessous par système organe classe et par fréquence (très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Les effets indésirables les plus fréquemment signalés sont des troubles généraux et des anomalies au site d'administration.
|
Effet indésirable par système organe classe |
Fréquence |
|
Patients présentant une réaction indésirable pendant l’étude clinique |
|
|
Affections du système nerveux |
|
|
Maux de tête |
Fréquent |
|
Vertiges |
Peu fréquent |
|
Troubles généraux et anomalies au site d'administration |
|
|
Douleur au point d'application |
Fréquent |
|
Irritation au point d'application |
Peu fréquent |
|
Affections gastro-intestinales |
|
|
Nausée |
Peu fréquent |
|
Stomatite aphteuse |
Peu fréquent |
|
Douleur gingivale |
Peu fréquent |
|
Affections de la peau et du tissu sous-cutané |
|
|
Érythème |
Peu fréquent |
Les effets indésirables suspectés d'être liés à l'application de Virpax sont peu fréquents (< 1 %) et incluent la douleur au point d'application, l'irritation au point d'application, la stomatite aphteuse et la douleur gingivale. L'application de Virpax n’a pas été interrompue en raison d’effets indésirables.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
L'absorption d'aciclovir et l'exposition systémique après l'application de Virpax sont minimes. Par conséquent, le risque de surdosage est improbable.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L'aciclovir est un antiviral qui est très actif in vitro contre le virus Herpès Simplex (HSV) types 1 et 2. L'activité d'inhibition de l'aciclovir pour HSV1 et HSV2 est hautement sélective.
Après avoir pénétré dans les cellules infectées par l'herpès, l'aciclovir est phosphorylé pour devenir le composé actif aciclovir triphosphate. La première étape de ce processus dépend de la présence de la thymidine kinase de l'HSV. L'enzyme thymidine kinase (TK) de cellules normales, non infectées, n'utilise pas l'aciclovir comme substrat, par conséquent la toxicité vis-à-vis des cellules hôtes de mammifères est faible. L'aciclovir triphosphate agit comme inhibiteur et comme substrat de l'ADN polymérase de l'herpès, bloquant la synthèse ultérieure de l'ADN viral sans affecter les processus cellulaires normaux. La diminution de la sensibilité à l'aciclovir est très rare chez le patient immunocompétent.
Efficacité et sécurité clinique
Dans un essai de phase 3 randomisé (Virpax 50 mg comparé à un placebo), en double insu, ont été inclus 775 patients adultes (378 dans le groupe Virpax contre 397 dans le groupe placebo) randomisés et traités (population ITT = 771) ayant au moins 4 épisodes d'herpès au cours de l'année précédente (dont 68,4 % avec ≥ 5 épisodes), avec des symptômes prodromaux dans au moins 50 % des épisodes récurrents. Les patients devaient appliquer Virpax dès la survenue des premiers symptômes ou signes prodromaux. Les résultats ont indiqué que l'administration d'une dose unique de Virpax 50 mg comprimé buccogingival muco-adhésif réduisait de façon significative la durée de guérison de la lésion vésiculaire primaire : la durée moyenne était de 5,03 jours dans le groupe Virpax contre 5,95 jours dans le groupe en ITT (p = 0,002) et 7,0 jours contre 7,6 jours en mITT (n = 521, p = 0,015).
Les critères secondaires sont présentés dans le tableau ci-dessous :
|
|
Aciclovir 50 mg (N=376) |
Placebo (N=395) |
Valeur P |
|
Episodes abortifs : n (%) |
130 (34,9 %) |
109 (28,1 %) |
0,0419 |
|
Durée de l’épisode (jours) : médiane (IC 95%) |
5,57 (5,03; 6,01) |
6,38 (5,93; 6,97) |
0,0033 |
|
Lésions non primaires : n (%) |
39 (10,4 %) |
62 (15,7 %) |
0,037 |
|
Durée des symptômes (jours) : médiane (IC 95%) |
3,57 (3,04; 4,01) |
4,16 (3,75; 4,89) |
0,0098 |
|
Intensité des symptômes (jour 5) : médiane (fourchette) |
5,0 (0; 100) |
9,0 (0; 100) |
0,0078 |
Au cours de l'étude pivot, 85 % des patients ont appliqué Virpax dans l'heure suivant le début des symptômes prodromaux. Il n'y a pas de donnée qui valide l'efficacité de Virpax s'il est appliqué une fois les lésions vésiculaires apparues.
Au cours de cette étude, la durée d'adhérence du comprimé était supérieure à 6 heures chez 88,5 % des patients.
La tolérance n'était pas différente dans le groupe Virpax par rapport au groupe témoin.
La satisfaction des patients était significativement supérieure dans le groupe Virpax (81,8 %) par rapport au groupe placebo (72,4 %, p = 0,002).
Population pédiatrique
L’Agence européenne du médicament a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec Virpax dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement de l’herpès simplex labial (voir rubrique 4.2 pour les informations sur l’utilisation pédiatrique).
5.2. Propriétés pharmacocinétiques
La biodisponibilité de l'aciclovir administré par voie orale est variable, de 15 à 30 %. Après l'administration de comprimés d'aciclovir à 200 mg, les pics moyens de concentrations plasmatiques (Cmax) sont de 0,350 ± 0,100 µg/mL et la Tmax est observée entre 1 et 3 heures. La liaison aux protéines plasmatiques varie de 9 à 33 %. La majeure partie de l'aciclovir est éliminée sous forme inchangée dans l'urine.
Après l'application d’une dose unique de Virpax 50 mg comprimé buccogingival muco-adhésif chez des volontaires sains (n = 12), la Cmax plasmatique moyenne de l'aciclovir était d'environ 28 ng/ml. La Cmax et l'ASC dans le plasma étaient respectivement de 10 et 8 fois inférieures après l'application de Virpax 50mg comprimé buccogingival par rapport à l'administration orale d'un comprimé de 200 mg d'aciclovir. La Cmax et la Tmax obtenues dans la salive étaient respectivement de 440 000 ng/ml et 7 heures.
Les concentrations d’aciclovir dans la salive observées chez 56 patients de l’étude de phase 3 sont cohérentes avec celles obtenues chez les volontaires sains.
5.3. Données de sécurité préclinique
Des effets indésirables réversibles pour la plupart sur la spermatogenèse liés à une toxicité globale chez les rats et les chiens ont été rapportés uniquement pour des doses d'aciclovir très largement supérieures à celles utilisées en thérapeutique. Des études sur deux générations de souris n'ont pas mis en évidence d'effet de l'aciclovir (administré par voie orale) sur la fertilité.
Une administration systémique d'aciclovir lors de tests standardisés selon la norme internationale n'a pas produit d'effets embryotoxiques ou tératogènes chez le lapin, le rat et la souris. Lors d'un test non standardisé chez les rats, des anomalies fœtales ont été observées seulement après des doses sous-cutanées tellement élevées qu'une toxicité maternelle est apparue. Les conséquences cliniques de ces découvertes sont incertaines.
Des études de tolérance locale (sur la muqueuse jugale du hamster) n'ont pas indiqué de toxicité.
3 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30°C.
6.5. Nature et contenu de l'emballage extérieur
1 comprimé (1 x 1 comprimé) ou 2 comprimés (2 x 1 comprimé) sous plaquettes unitaires (Alu/Alu).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d'exigences particulières pour l’élimination.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
230 BUREAUX DE LA COLLINE
92213 SAINT-CLOUD CEDEX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
34009 278 274 5 7 : 2 comprimés (2 x 1 comprimé) sous plaquettes unitaires (Alu/Alu).
34009 302 533 5 9 : 1 comprimé (1 x 1 comprimé) sous plaquettes unitaires (Alu/Alu).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Médicament non soumis à prescription médicale.
ANSM - Mis à jour le : 19/09/2025
VIRPAX 50 mg, comprimé buccogingival muco-adhésif
aciclovir
Vous devez toujours utiliser ce médicament en suivant scrupuleusement les informations fournies dans cette notice ou par votre médecin, ou votre pharmacien ou votre infirmier/ère.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Adressez-vous à votre pharmacien pour tout conseil ou information.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien ou votre infirmier/infirmière. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
· Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien après 5 jours.
1. Qu'est-ce que VIRPAX 50 mg, comprimé buccogingival muco-adhésif et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser VIRPAX 50 mg, comprimé buccogingival muco-adhésif?
3. Comment utiliser VIRPAX 50 mg, comprimé buccogingival muco-adhésif ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver VIRPAX 50 mg, comprimé buccogingival muco-adhésif ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE VIRPAX 50 mg, comprimé buccogingival muco-adhésif ET DANS QUELS CAS EST-IL UTILISE ?
Virpax est utilisé dans le traitement des boutons de fièvre récidivants (l'herpès labial récurrent), dus au virus Herpès simplex chez les adultes dont le système immunitaire fonctionne correctement. Virpax doit être appliqué directement sur la gencive (pas à l'endroit même du bouton) dès l’apparition des premiers symptômes (brûlures, picotements, démangeaisons) ou signes (rougeur, gonflement).
Le virus provoque des vésicules ou des inflammations principalement sur les lèvres mais parfois aussi sur d'autres parties du visage. Cette infection virale peut être activée quand le système immunitaire est affaibli, par exemple par des rhumes ou d'autres infections. Une exposition solaire importante, le stress ou les menstruations peuvent également déclencher les symptômes.
Virpax inhibe la capacité du virus à se reproduire et permet ainsi à l'infection de régresser.
Vous devez vous adresser à votre médecin si les symptômes s’aggravent ou ne s’améliorent pas après 5 jours.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER VIRPAX 50 mg, comprimé buccogingival muco-adhésif ?
N’utilisez jamais VIRPAX 50 mg, comprimé buccogingival muco-adhésif :
· si vous êtes allergique à l'aciclovir ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6) ;
· si vous êtes allergique au lait ou aux dérivés du lait.
Avertissements et précautions
· L'ingestion accidentelle de Virpax peut survenir. Si Virpax est avalé accidentellement, il est recommandé de boire un verre d'eau.
· Adressez-vous à votre médecin avant d'utiliser ce médicament si votre système immunitaire est affaibli (en lien avec une autre maladie qui affecte le système immunitaire, par exemple le SIDA) ou en raison d’un traitement qui affecte votre système immunitaire (par exemple des médicaments immunosuppresseurs). Virpax ne doit pas être pris dans ce cas.
· On ignore si Virpax fonctionne en cas d’utilisation après l’apparition des vésicules. Par conséquent, utilisez Virpax uniquement lorsque vous ressentez les premiers symptômes de boutons de fièvre (démangeaisons, rougeur ou picotements).
· Vous devez également contacter un médecin en cas d’épisodes récurrents fréquents de boutons de fièvre (plus de 4 par an), ou d’augmentation récente de l’incidence des épisodes ou d’extension à d’autres parties du corps
Enfants et adolescents
Ce médicament ne doit pas être utilisé chez les enfants et les adolescents de moins de 18 ans.
Autres médicaments et VIRPAX 50 mg, comprimé buccogingival muco-adhésif
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
VIRPAX 50 mg, comprimé buccogingival muco-adhésif avec des aliments et boissons
Vous pouvez manger et boire normalement une fois que VIRPAX est en place.
Grossesse et allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou à votre pharmacien avant de prendre ce médicament.
Grossesse
Il n’est pas attendu que Virpax ait un effet sur le fœtus. Cependant, par mesure de précaution, il est recommandé d'utiliser Virpax pendant la grossesse uniquement si nécessaire.
Allaitement
Il n’est pas attendu que Virpax ait un effet sur le nourrisson allaité. Cependant, par mesure de précaution, il est recommandé de l'utiliser pendant l'allaitement uniquement si nécessaire.
Conduite de véhicules et utilisation de machines
Ce médicament n’a aucun effet ou qu’un effet négligeable sur votre aptitude à conduire des véhicules ou à utiliser des machines.
VIRPAX 50 mg, comprimé buccogingival muco-adhésif contient un concentré de protéines de lait avec des traces de lactose et du laurilsulfate de sodium.
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament. Ce médicament contient des protéines de lait qui peuvent provoquer des réactions allergiques chez les personnes présentant une hypersensibilité ou une allergie sévères aux protéines de lait.
Ce médicament contient 5.2 mg de laurilsulfate de sodium par comprimé.
Le laurilsulfate de sodium peut provoquer des réactions cutanées locales (comme une sensation de piqûre ou de brûlure) ou augmenter les réactions cutanées causées par d’autres produits lorsqu’ils sont appliqués sur la même zone.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est à dire qu'il est essentiellement « sans sodium ».
3. COMMENT UTILISER VIRPAX 50 mg, comprimé buccogingival muco-adhésif ?
La dose recommandée est d'un seul comprimé de Virpax par épisode de bouton de fièvre, à coller sur la gencive dès l'apparition des premiers symptômes ou signes de bouton de fièvre.
Vous devez consulter un médecin si les symptômes s'aggravent ou ne s'améliorent pas après 5 jours.
NE PAS AVALER LE COMPRIME
Si vous l’avalez accidentellement, buvez un verre d’eau.
Instructions pour un bon usage
Si la bouche est sèche, il est recommandé de boire de l'eau afin de l’eau afin d’humidifier votre gencive avant d’appliquer le comprimé.
|
|
Avant d'appliquer le comprimé, repérez l’endroit où le comprimé doit être appliqué sur la gencive supérieure, juste au-dessus de la deuxième incisive.
Retirez le comprimé de sa plaquette.
Le comprimé peut être appliqué sur le côté droit ou gauche, quel que soit le côté où apparaissent les premiers signes ou symptômes de bouton de fièvre. Ce médicament doit être appliqué sur la gencive supérieure (pas à l'endroit même du bouton), juste au-dessus de la deuxième incisive (voie gingivale). Appliquez, avec un doigt sec, le comprimé immédiatement après l’avoir sorti de sa plaquette. Vous remarquerez que le comprimé a une face arrondie et une face plane gravée « AL21 ». Placez le côté plat du comprimé sur le bout de votre doigt sec. Placez la face arrondie du comprimé contre la gencive supérieure. Si le comprimé reste collé à l’intérieur de la lèvre plutôt que de la gencive, le médicament agira quand même.
Maintenez le comprimé en place en appliquant une légère pression avec votre doigt sur l'extérieur de votre lèvre supérieure pendant 30 secondes pour assurer l'adhérence.
Une fois appliqué, le comprimé reste en place et se dissout progressivement au cours de la journée. |

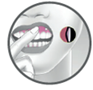


Recommandations particulières
Vous pouvez manger et boire normalement lorsque Virpax est en place. Le comprimé ne doit pas être sucé, mâché ou avalé.
Toutes les situations pouvant interférer avec l'adhérence du comprimé doivent être évitées, notamment :
· Toucher ou appuyer sur le comprimé déjà en place.
· Mâcher un chewing-gum.
· Se brosser les dents alors que le comprimé est en place.
Si vous avalez accidentellement le comprimé de Virpax :
· buvez un verre d'eau ;
· si le comprimé a été avalé dans les 6 premières heures suivant l’application, il ne doit être remplacé qu’une seule fois.
Si Virpax n'adhère pas ou tombe dans les 6 premières heures suivant l’application :
· le même comprimé doit être remis en place immédiatement ;
· si le comprimé ne peut pas être remis en place, un nouveau comprimé doit être appliqué.
Si Virpax tombe ou est avalé accidentellement après 6 heures d’adhérence, il ne doit pas être remplacé.
La forme du comprimé peut changer en raison de l'absorption de la salive afin de s'adapter à la forme de vos gencives.
Utilisation chez les enfants et les adolescents
Ce médicament ne doit pas être utilisé chez les enfants et les adolescents de moins de 18 ans.
Si vous avez pris plus de VIRPAX 50 mg, comprimé buccogingival muco-adhésif, que vous n’auriez dû : Consultez votre médecin ou votre pharmacien immédiatement, même si le risque de surdosage est faible compte tenu de la faible absorption dans le sang après la prise de Virpax.
Visualisez le mode d’administration de Virpax et les recommandations particulières en scannant ce QR code ou en suivant le lien : [à compléter].
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10) :
· Maux de tête
· Douleur au point d'application
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) :
· Vertiges
· Nausées
· Irritation au point d'application
· Aphte
· Douleur gingivale
· Rougeur
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER VIRPAX 50 mg, comprimé buccogingival muco-adhésif?
Tenir ce médicament hors de la vue et de la portée des enfants.
A conserver à une température ne dépassant pas 30°C.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage et la plaquette après "EXP". La date de péremption fait référence au dernier jour de ce mois.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient VIRPAX 50 mg, comprimé buccogingival muco-adhésif
· La substance active est l’aciclovir. Chaque comprimé buccogingival muco-adhésif contient 50 mg d’aciclovir
· Les autres composants sont : hypromellose, concentré de protéines de lait avec des traces de lactose, laurilsulfate de sodium, stéarate de magnésium, cellulose microcristalline, povidone, silice colloïdale anhydre.
Qu’est-ce que VIRPAX 50 mg, comprimé buccogingival muco-adhésif et contenu de l’emballage extérieur
Ce médicament est un comprimé buccogingival muco-adhésif de 8 mm, blanc à jaunâtre avec une face arrondie et une face plane marquée de « AL21 »
Chaque boîte de Virpax contient 1 ou 2 plaquette(s) unitaire(s) de 1 comprimé.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
230 BUREAUX DE LA COLLINE
92213 SAINT-CLOUD CEDEX
Exploitant de l’autorisation de mise sur le marché
ZAMBON FRANCE S.A.
13 RUE RENE JACQUES,
92130 ISSY-LES-MOULINEAUX
10 RUE BOUCHE THOMAS
ZAC D’ORGEMONT
49000 ANGERS CEDEX
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Conseil d’éducation sanitaire :
Les boutons de fièvre ou herpès labial sont dus à une infection provoquée par un virus appelé Herpès simplex. Après une primo-infection, le virus persiste dans l'organisme sans entraîner de symptômes.
A l'occasion d'un événement comme la fatigue, les règles, la fièvre, une émotion ou une exposition solaire, le virus peut parfois être réactivé et provoquer l'apparition de vésicules (cloques) en bouquet autour des lèvres, appelées boutons de fièvre. Ces vésicules sont contagieuses jusqu'à l'apparition des croûtes, signe de cicatrisation.
Il existe des signes avant-coureurs localisés au lieu habituel de la lésion, qui se traduisent généralement par une sensation de brûlure et/ou de picotements.
Ces signes sont importants à connaître :
· c'est dès leur apparition qu'il faut débuter le traitement qui empêche la multiplication virale et permet de prévenir ou de limiter l'évolution du bouton de fièvre,
· c'est aussi dès leur apparition qu'il faut respecter les règles d'hygiène suivantes afin de diminuer le risque de transmission du virus à vous-même et votre entourage.
· Evitez le contact rapproché, un baiser par exemple, avec votre entourage.
· N'EMBRASSEZ JAMAIS UN ENFANT EN BAS AGE.
· Lavez-vous soigneusement les mains en cas de contact avec la lésion.
· Au cours du traitement, afin de ne pas propager les lésions, ne frottez ni vos yeux ni vos paupières avec vos mains et si vous portez des lentilles de contact, ne les humidifiez jamais avec votre salive.
· Evitez de toucher ou de gratter les lésions.
En cas d'une autre localisation de votre crise d'herpès, n'utilisez jamais VIRPAX, PRENEZ L'AVIS DE VOTRE MEDECIN.
En revanche, l'aciclovir ne peut agir sur les virus qui persistent à l'état latent dans l'organisme et l'aciclovir n'empêche pas la survenue de récidives après l'interruption du traitement