Dernière mise à jour le 28/04/2026
CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée
Indications thérapeutiques
Qu'est-ce que CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée?
CUVITRU fait partie d'une classe de médicaments appelés « immunoglobulines humaines normales ». Les immunoglobulines sont également connues sous le nom d'anticorps et sont présentes dans le sang des personnes saines. Les anticorps font partie du système immunitaire (défenses naturelles du corps) et aident votre organisme à combattre les infections.
Quel est le mécanisme d'action de CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ?
CUVITRU a été préparé à partir du sang de donneurs sains. Ce médicament agit exactement de la même manière que les immunoglobulines naturellement présentes dans le sang.
Dans quel cas CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée est-il utilisé ?
CUVITRU est utilisé chez les patients présentant un système immunitaire affaibli, qui ne possèdent pas suffisamment d'anticorps dans le sang et qui souffrent d'infections fréquentes. Des doses régulières et suffisantes de CUVITRU peuvent augmenter les taux sanguins d'immunoglobuline anormalement bas afin de leur permettre d'atteindre un niveau normal (traitement substitutif).
CUVITRU est prescrit à
· des patients dont la capacité innée à produire des anticorps est réduite ou inexistante (syndromes d'immunodéficience primaire).
· patients atteints d’infections sévères ou récurrentes dues à un système immunitaire affaibli en raison d’autres pathologies ou traitements.
Présentations
> 1 flacon en verre de 5 mL
Code CIP : 34009 550 524 3 2
Déclaration de commercialisation : 06/04/2021
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon en verre de 10 mL
Code CIP : 34009 550 524 4 9
Déclaration de commercialisation : 06/04/2021
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon en verre de 20 mL
Code CIP : 34009 550 524 5 6
Déclaration de commercialisation : 06/04/2021
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon en verre de 40 mL
Code CIP : 34009 550 524 6 3
Déclaration de commercialisation : 06/04/2021
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre de 50 ml
Code CIP : 34009 550 672 9 0
Déclaration de commercialisation : 08/06/2023
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 07/12/2022 | Inscription (CT) | Le service médical rendu par CUVITRU 200 mg/mL (immunoglobuline humaine normale) est important dans les indications de l’AMM. |
| Important | Avis du 25/07/2018 | Inscription (CT) | Le service médical rendu par CUVITRU est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 07/12/2022 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apportent pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
| V (Inexistant) | Avis du 25/07/2018 | Inscription (CT) | La Commission considère que CUVITRU n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres immunoglobulines humaines normales administrées par voie sous-cutanée ou intraveineuse. |
Autres informations
- Titulaire de l'autorisation : BAXALTA INNOVATIONS GMBH
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- prescription par un médecin exerçant dans un établissement de transfusion sanguine
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 317 262 9
ANSM - Mis à jour le : 04/07/2025
CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Immunoglobuline humaine normale (Ig SC)
Un ml contient :
Immunoglobuline humaine normale ............. 200 mg
(pureté d'au moins 98 % d'IgG)
· Chaque flacon de 5 ml contient : 1 g d'immunoglobuline humaine normale
· Chaque flacon de 10 ml contient : 2 g d'immunoglobuline humaine normale
· Chaque flacon de 20 ml contient : 4 g d'immunoglobuline humaine normale
· Chaque flacon de 40 ml contient : 8 g d'immunoglobuline humaine normale
· Chaque flacon de 50 ml contient : 10 g d’immunoglobuline humaine normale
Distribution des sous-classes d'IgG (valeurs approx.) :
· IgG1 ≥56,9 %
· IgG2 ≥26,6 %
· IgG3 ≥3,4 %
· IgG4 ≥1,7 %
La teneur maximale en IgA est de 280 microgrammes/ml.
Produit à partir du plasma de donneurs humains.
Pour la liste complète des excipients, voir rubrique 6.1.
La solution est limpide et incolore, jaune pâle ou brun clair.
pH de 4,6 à 5,1 (mesuré par une dilution à 1 % dans du sérum physiologique)
4.1. Indications thérapeutiques
Indications pour une administration par voie sous-cutanée (Ig SC)
Traitement substitutif chez l'adulte, l'enfant et l'adolescent (de 0 à 18 ans) atteint de :
· déficits immunitaires primitifs (DIP) avec altération de la production d’anticorps (voir rubrique 4.4) ;
· déficits immunitaires secondaires (DIS) chez les patients souffrant d’infections sévères ou récurrentes, sous traitement antimicrobien inefficace, et présentant soit un déficit avéré des anticorps spécifiques (DAAS)*, soit d’un taux d’IgG sérique de < 4 g/L.
* DAAS : défaut de réponse vaccinale définie par un échec du doublement du titre des anticorps IgG après un vaccin pneumococcique utilisant des antigènes polypeptidiques et polysaccharidiques.
4.2. Posologie et mode d'administration
Posologie
La posologie et le schéma posologique dépendent de l’indication thérapeutique.
Le médicament doit être administré par voie sous-cutanée.
Dans le traitement substitutif, la posologie peut être adaptée à chaque patient en fonction des paramètres pharmacocinétiques et de la réponse clinique. La posologie selon le poids corporel peut nécessiter un ajustement chez les patients maigres ou obèses. Les schémas posologiques suivants sont donnés à titre indicatif.
Traitement substitutif pour les déficits immunitaires primitifs (tels que définis en 4.1)
Le schéma posologique doit assurer un taux résiduel d'IgG (mesuré avant la perfusion suivante) d'au moins 5 à 6 g/l et, si possible, se trouver dans l'intervalle de référence de l'IgG sérique pour l'âge. Une dose de charge d'au moins 0,2 à 0,5 g/kg (1 à 2,5 ml/kg) de poids corporel peut être requise. Il peut être nécessaire de la répartir sur plusieurs jours, avec une dose quotidienne maximale de 0,1 à 0,15 g/kg. Après équilibre des taux d'IgG, des doses d'entretien sont administrées à intervalles réguliers de façon à atteindre une dose cumulative mensuelle de l'ordre de 0,3 à 1,0 g/kg (voir rubrique 5.2 pour plus d'informations). Il peut être nécessaire d'injecter chaque dose dans différents sites anatomiques.
Le taux résiduel doit être mesuré et évalué en association avec l'apparition d'infections. Pour réduire la fréquence des infections, il peut être nécessaire d'augmenter la dose et de viser un taux résiduel plus élevé.
Traitement substitutif pour les déficits immunitaires secondaires (tels que définis en 4.1)
La dose recommandée est administrée à intervalles répétés pour parvenir à une dose mensuelle cumulée de l'ordre de 0,2 à 0,4 g/kg. Il peut être nécessaire d'administrer chaque dose unique sur des sites anatomiques différents.
Les taux résiduels d’IgG doivent être mesurés et évalués conjointement à l'incidence des infections. La dose doit être ajustée si nécessaire pour obtenir une protection optimale contre les infections. Il peut être nécessaire d'augmenter la dose chez les patients présentant une infection persistante ; une diminution de la dose peut être envisagée lorsque le patient reste exempt d'infection.
Population pédiatrique
La posologie chez l'enfant et l'adolescent (de 0 à 18 ans) est identique à celle de l'adulte car la posologie correspondant à chaque indication est mentionnée en fonction du poids corporel et ajustée aux résultats cliniques des indications susmentionnées.
Aucun essai clinique n'a été mené concernant l'utilisation de CUVITRU chez des enfants âgés de 0 à < 2 ans, mais l'expérience en matière d'administration d'immunoglobulines suggère qu'aucun effet néfaste n'est attendu en cas de traitement par CUVITRU dans cette tranche d'âge.
Mode d’administration
Par voie sous-cutanée uniquement.
CUVITRU doit être inspecté visuellement afin de détecter une décoloration ou d’éventuelles particules avant administration. Ne pas utiliser en cas de décoloration et/ou de présence de particules.
La perfusion doit débuter immédiatement après le transfert de CUVITRU dans la seringue. L’administration dure généralement maximum deux heures. S’il n’est pas possible d’administrer la dose complète en moins de deux heures à cause de la quantité nécessaire ou du débit d’administration de CUVITRU, la dose doit être divisée et administrée à différents sites de perfusion. Si CUVITRU reste dans des seringues siliconées pendant plus de deux heures, des particules visibles peuvent se former. Voir rubrique 4.4 pour plus de détails.
CUVITRU ne doit pas être dilué.
La perfusion sous-cutanée réalisée à domicile doit être instaurée et surveillée par un médecin expérimenté dans l'encadrement des patients traités à domicile. Des pompes à perfusion ou une administration manuelle à l’aide d’une seringue qui sont appropriées pour l'administration par voie sous-cutanée d'immunoglobulines peuvent être utilisées. Le patient ou l’aidant doit être formé à l’utilisation d'un pousse-seringue (assisté par un dispositif) ou d’une seringue (administration manuelle), aux techniques de perfusion, à la tenue d’un carnet de traitement, à la reconnaissance des effets indésirables graves et aux mesures à prendre en cas d’apparition de ceux-ci, voir rubrique 4.4.
CUVITRU doit être injecté dans des sites tels que l'abdomen, la cuisse, le haut du bras et la face latérale de la hanche.
L'ajustement de la vitesse et du volume de perfusion selon le site s'effectue en fonction de la tolérance du patient.
Débit de perfusion
CUVITRU peut être perfusé à l’aide :
· d’un dispositif de perfusion, ou
· d’une seringue par administration manuelle.
Le débit de perfusion initial recommandé dépend des besoins individuels du patient. Une augmentation du débit des perfusions successives peut être envisagée selon l’appréciation du patient et sur la base de l’avis des professionnels de santé.
Perfusion assistée par un dispositif :
Il est recommandé d'utiliser une vitesse d'administration initiale de 10 ml/h/site de perfusion. Si elle est bien tolérée (voir rubrique 4.4), le débit d'administration peut être augmenté a minima toutes les 10 minutes jusqu’à 20 ml/heure/site de perfusion au maximum pour les deux premières perfusions. Pour les perfusions suivantes, le débit de perfusion peut être augmenté selon la tolérance.
Plusieurs pompes peuvent être utilisées simultanément. La quantité de produit perfusé varie selon les sites. Chez le nourrisson et l'enfant, le site de perfusion peut être changé tous les 5 à 15 ml. Chez l'adulte, les doses supérieures à 30 ml peuvent être divisées selon les préférences du patient. Il n’y a pas de limites sur le nombre de sites de perfusion.
Perfusion par administration manuelle :
CUVITRU peut être administré à l’aide d’une seringue en un site de perfusion unique. Une nouvelle aiguille d’injection stérile doit être utilisée s’il est nécessaire d’administrer sur d’autres sites.
Le débit de perfusion maximal proposé est d’environ 1 à 2 ml par minute.
Le débit d’administration doit être adapté à la tolérance locale de chaque patient, qui peut dépendre du site de chaque perfusion sous-cutanée et de l’épaisseur de tissu sous-cutané du patient au niveau de ce site.
La quantité de produit perfusé en un site particulier varie. Chez le nourrisson et l’enfant, le site de perfusion peut être changé tous les 5 à 15 ml. Chez l’adulte, les doses supérieures à 30 ml peuvent être divisées selon les préférences du patient.
Déficit sévère en IgA et antécédents d'hypersensibilité à un traitement par immunoglobuline humaine.
CUVITRU ne doit pas être administré par voie intravasculaire ni intramusculaire.
4.4. Mises en garde spéciales et précautions d'emploi
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Si CUVITRU est accidentellement administré dans un vaisseau sanguin, les patients pourraient développer un choc.
Il convient de respecter scrupuleusement le débit de perfusion et les instructions d’administration recommandées indiquées à la rubrique 4.2. Les patients doivent être étroitement surveillés pendant toute la durée de la perfusion afin de détecter tout symptôme éventuel. Si le produit reste dans une seringue siliconée pendant plus de deux heures, des particules visibles peuvent se former.
Certains effets indésirables peuvent survenir plus fréquemment chez les patients recevant pour la première fois une immunoglobuline humaine normale ou, dans de rares cas, lors d'un changement d'immunoglobuline humaine normale ou lorsqu'un long intervalle s'est écoulé depuis la perfusion précédente.
Les complications peuvent souvent être évitées :
· en commençant par injecter le produit lentement (voir rubrique 4.2)
· en s'assurant que les patients sont étroitement suivis pendant toute la durée de la perfusion afin de détecter d'éventuels signes d'intolérance. En particulier, lors de la première administration d'une immunoglobuline humaine normale, lors d'un changement d'immunoglobuline humaine normale ou en cas d'interruption prolongée du traitement, le patient doit être maintenu sous surveillance pendant toute la durée de la première perfusion et pendant l'heure qui suit la fin de l'injection, afin de détecter les effets indésirables éventuels.
Tous les autres patients devront être maintenus en observation pendant au moins 20 minutes après la fin de la perfusion.
En cas d'effet indésirable, le débit d’administration doit être réduit ou la perfusion interrompue. Une suspicion d'hypersensibilité grave ou de réaction de type anaphylactique nécessite l'arrêt immédiat de l'injection. Le traitement requis dépend de la nature et de la sévérité de l'effet indésirable.
En cas de choc, un traitement médical standard du choc doit être instauré.
Une augmentation du nombre et de la gravité des effets indésirables peut survenir lorsque les patients commencent l’administration manuelle. Par conséquent, les patients pour lesquels une administration manuelle est envisagée doivent être stables sur le plan médical et formés de manière adéquate à la reconnaissance des effets indésirables graves et aux mesures à prendre en cas d’apparition de ceux-ci.
Hypersensibilité
Les véritables réactions allergiques sont rares. Elles peuvent survenir en particulier chez les patients avec présence d'anticorps anti‑IgA qui doivent être traités avec une prudence particulière. Les patients avec présence d'anticorps anti-IgA, pour lesquels le traitement par IgG par voie sous-cutanée reste la seule option, doivent être traités avec CUVITRU uniquement sous surveillance médicale étroite. CUVITRU contient des traces d'IgA (280 microgrammes/ml maximum).
Dans de rares cas, l'immunoglobuline humaine normale peut provoquer une chute de la pression artérielle associée à une réaction anaphylactique, même chez les patients ayant toléré de précédentes administrations d'immunoglobuline humaine normale.
Thrombo-embolie
Des événements thromboemboliques artériels et veineux, tels qu’un infarctus du myocarde, un accident vasculaire cérébral, une thrombose veineuse profonde et une embolie pulmonaire, ont été associés à l’utilisation d’immunoglobulines. Il convient d'être particulièrement prudent avec les patients présentant des facteurs de risque pré-existants d'événements thromboemboliques (tels qu'un âge avancé, une hypertension, un diabète ou des antécédents de maladie vasculaire ou d'épisodes thrombotiques, des patients atteints de troubles thrombophiliques acquis ou congénitaux, des patients en immobilisation prolongée, des patients avec une hypovolémie grave, des patients souffrant de maladies augmentant la viscosité du sang). Les patients doivent être informés des premiers symptômes d'événements thromboemboliques, notamment la difficulté respiratoire, la douleur et le gonflement d'un membre, les troubles neurologiques focaux et la douleur thoracique, et ils doivent être avertis de la nécessité de contacter immédiatement leur médecin en cas d'apparition de ces symptômes.
Les patients doivent être suffisamment hydratés avant l'administration. Il convient de surveiller les signes et symptômes de thrombose et d’évaluer la viscosité du sang des patients présentant un risque d’hyperviscosité.
Complications rénales
Des cas d'effets indésirables rénaux sévères ont été rapportés chez des patients traités par immunoglobuline, particulièrement avec des produits contenant du saccharose (CUVITRU ne contient pas de saccharose). Il s'agissait notamment d'insuffisance rénale aiguë, de nécrose tubulaire aiguë, de néphropathie tubulaire proximale et de néphrose osmotique. Les facteurs d'exacerbation du risque de complications rénales englobent, sans s'y limiter, une insuffisance rénale pré-existante, un diabète, une hypovolémie, l'administration concomitante de médicaments néphrotoxiques, un âge supérieur à 65 ans, une septicémie, une hyperviscosité et une paraprotéinémie.
Syndrome de méningite aseptique (SMA)
Le syndrome de méningite aseptique (SMA) a été signalé en association avec l'administration d'immunoglobuline, y compris CUVITRU (voir rubrique 4.8 Effets indésirables – Post commercialisation). Le SMA peut être plus fréquent chez les patients de sexe féminin.
L’interruption du traitement par immunoglobuline peut résulter en une rémission sans séquelle du SMA après plusieurs jours. Les symptômes se déclarent généralement plusieurs heures à 2 jours après le traitement par immunoglobuline. Les analyses du liquide céphalo-rachidien montrent fréquemment une pléocytose pouvant atteindre plusieurs milliers de cellules par mm3, majoritairement de la lignée granulocytaire, ainsi que des taux de protéines élevés, jusqu'à plusieurs centaines de mg/dl.
Les patients doivent être informés des premiers symptômes, à savoir fortes céphalées, raideur de la nuque, endormissement, fièvre, photophobie, nausées et vomissements.
Hémolyse
CUVITRU contient des anticorps anti-érythrocytaires susceptibles d'agir comme des hémolysines et d'induire in vivo le recouvrement des globules rouges par de l'immunoglobuline. Cela peut entraîner un résultat positif au test direct à l'antiglobuline (TDA, test direct de Coombs) et, dans de rares cas, une hémolyse. Une anémie hémolytique différée peut se développer suite à un traitement par immunoglobuline en raison de la séquestration accrue de globules rouges. Des cas d'anémie hémolytique aiguë, correspondant à une hémolyse intravasculaire, ont été rapportés.
Interférence avec les tests sérologiques
Après une injection d'immunoglobulines, l'augmentation transitoire de la concentration des divers anticorps transférés passivement dans le sang des patients peut être responsable de résultats faussement positifs lors de dosages sérologiques, notamment concernant l'hépatite virale A et B, la rougeole et la varicelle. La transmission passive d'anticorps contre les antigènes de surface érythrocytaires (p. ex. A, B, D) peut interférer avec certains tests sérologiques portant sur les anticorps anti-érythrocytaires tels que le test direct à l'antiglobuline (TDA, test direct de Coombs).
L'administration de CUVITRU peut engendrer des résultats faussement positifs dans des tests qui dépendent de la détection de bêta‑D‑glucanes pour diagnostiquer des infections fongiques. Ces données peuvent persister pendant les semaines qui suivent la perfusion du produit.
Agents transmissibles
Les mesures standard de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de plasma, ainsi que la mise en œuvre dans le procédé de fabrication d’étapes efficaces pour l’inactivation/élimination virale. En dépit de cela, le risque de transmission d’agents infectieux par l’administration de médicaments préparés à base de sang ou de plasma humain ne peut être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents et aux autres types d’agents infectieux.
Les mesures prises sont considérées comme efficaces pour les virus enveloppés, tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC) et pour les virus non enveloppés de l’hépatite A et du parvovirus B19.
L'expérience clinique est rassurante, ne rapportant pas de transmission du virus de l’hépatite A ni du parvovirus B19 par les immunoglobulines, les anticorps présents contribuant probablement à la sécurité virale du produit.
Il est fortement recommandé, à chaque administration de CUVITRU à un patient, d’enregistrer le nom et le numéro de lot du médicament afin de maintenir un lien entre le patient et le lot du produit.
Population pédiatrique
Les mises en garde et précautions d'emploi mentionnées s'appliquent aux adultes comme aux enfants.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Vaccins à virus vivant atténué
L'administration d'immunoglobuline peut entraver, pendant une période comprise entre 6 semaines et 3 mois, l'efficacité des vaccins à virus vivant atténué tels que la rougeole, la rubéole, les oreillons et la varicelle. Après administration de CUVITRU, un intervalle de 3 mois doit s'écouler avant une vaccination avec des vaccins constitués de virus vivants atténués. Dans le cas de la rougeole, cette altération de l'efficacité peut persister pendant 1 an. Par conséquent, pour les patients vaccinés contre la rougeole, un contrôle des anticorps protecteurs post-vaccinaux doit être effectué.
Population pédiatrique
Les interactions mentionnées s'appliquent aux adultes comme aux enfants.
4.6. Fertilité, grossesse et allaitement
Les médecins doivent évaluer le rapport risque/bénéfice et ne prescrire CUVITRU qu'en cas de nécessité manifeste.
Grossesse
La sécurité de ce médicament chez les femmes enceintes n’a pas été évaluée au cours d'études cliniques contrôlées. Il doit donc être prescrit avec vigilance aux femmes enceintes et à celles qui allaitent. Il a été démontré que les produits à base d'immunoglobuline pénètrent dans le placenta, et ceci de façon plus importante pendant le troisième trimestre. L’expérience clinique avec les immunoglobulines suggère qu’aucun effet néfaste n'est attendu sur le déroulement de la grossesse, sur le fœtus ni sur le nouveau-né.
Allaitement
Les immunoglobulines sont excrétées dans le lait et peuvent contribuer à protéger le nouveau-né des agents pathogènes qui peuvent pénétrer dans les muqueuses.
Fertilité
L'expérience clinique concernant les immunoglobulines suggère qu'aucun effet délétère sur la fertilité n'est attendu.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Des effets indésirables de type frissons, céphalée, sensation vertigineuse, fièvre, vomissements, réactions allergiques, nausées, arthralgie, chute de la pression artérielle et douleur modérée dans la partie inférieure du dos peuvent survenir occasionnellement.
Dans de rares cas, les immunoglobulines humaines normales peuvent provoquer une chute brutale de la pression artérielle et, dans des cas isolés, un choc anaphylactique, même si le patient n'a pas présenté de réaction d'hypersensibilité lors d'administrations antérieures.
Des réactions locales au site de perfusion, telles que gonflement, endolorissement, rougeur, induration, sensation de chaleur locale, douleur locale, démangeaisons, bleus et rash peuvent survenir fréquemment.
Pour des informations de sécurité en rapport avec les agents transmissibles, voir rubrique 4.4.
Liste des effets indésirables
La sécurité de CUVITRU administré par voie sous-cutanée a été évaluée dans le cadre de deux études prospectives, multicentriques, en ouvert et non contrôlées menées chez 122 patients atteints de déficit immunitaire primitif (DIP).
La majorité (98,8 %) des effets indésirables (EI) locaux étaient d'intensité légère. Un patient a interrompu le traitement en raison d'un EI local (douleur). Sur 122 patients traités par CUVITRU, 112 ont participé jusqu'à la fin de l'étude.
Le tableau ci-dessous utilise la classe de systèmes d'organes MedDRA (SOC et terme préférentiel).
Les fréquences ont été évaluées selon la convention suivante : très fréquent (≥1/10), fréquent (≥1/100, <1/10), peu fréquent (≥1/1 000, <1/100), rare (≥1/10 000, <1/1 000), très rare (<1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Fréquence des effets indésirables (EI) dans les études cliniques menées sur CUVITRU
|
Fréquence des effets indésirables dans les études cliniques menées sur CUVITRU |
|||
|
MedDRA Classes de systèmes d'organes |
Effet indésirable |
Fréquence par patienta |
Fréquence par perfusionb |
|
AFFECTIONS DU SYSTEME NERVEUX |
Céphalée |
Très fréquent |
Fréquent |
|
Sensation vertigineuse |
Fréquent |
Peu fréquent |
|
|
Sensation de brûlure |
Peu fréquent |
Rare |
|
|
Migraine |
Fréquent |
Rare |
|
|
Somnolence |
Fréquent |
Rare |
|
|
AFFECTIONS VASCULAIRES |
Hypotension |
Fréquent |
Rare |
|
AFFECTIONS GASTRO-INTESTINALES |
Diarrhée |
Très fréquent |
Fréquent |
|
Nausées |
Très fréquent |
Peu fréquent |
|
|
Douleur abdominale basse |
Peu fréquent |
Rare |
|
|
Douleur abdominale |
Fréquent |
Peu fréquent |
|
|
AFFECTIONS DE LA PEAU ET DU TISSU SOUS-CUTANE |
Prurit |
Fréquent |
Rare |
|
Urticaire |
Fréquent |
Rare |
|
|
AFFECTIONS MUSCULO-SQUELETTIQUES ET SYSTEMIQUES |
Myalgie |
Fréquent |
Peu fréquent |
|
TROUBLES GENERAUX ET ANOMALIES AU SITE D’ADMINISTRATION |
Réaction locale |
Très fréquent |
Fréquent |
|
Erythème du site de perfusion (y compris érythème du site d'injection) |
Très fréquent |
Fréquent |
|
|
Douleur au niveau du site d'injection (y compris gêne et douleur du site de perfusion) |
Très fréquent |
Fréquent |
|
|
Gonflement au point de perfusion |
Fréquent |
Peu fréquent |
|
|
Prurit au site d'injection (y compris prurit au site de perfusion) |
Fréquent |
Peu fréquent |
|
|
Urticaire au site de perfusion |
Fréquent |
Peu fréquent |
|
|
Contusion au site de perfusion |
Fréquent |
Rare |
|
|
Œdème au site de perfusion |
Peu fréquent |
Rare |
|
|
Fatigue |
Très fréquent |
Peu fréquent |
|
|
Douleur |
Fréquent |
Rare |
|
|
INVESTIGATIONS |
Anticorps anti‑GAD positif |
Peu fréquent |
Rare |
|
Test de Coombs direct positif |
Peu fréquent |
Rare |
|
a La fréquence par patient est calculée à l'aide du nombre de patients associés à l'ensemble des événements indésirables, qu'ils soient liés à CUVITRU ou non.
b La fréquence par perfusion est calculée à l'aide du nombre de perfusions associées à l'ensemble des événements indésirables, qu'ils soient liés à CUVITRU ou non.
Tableau 2 Effets indésirables post-commercialisation
|
Effets indésirables post-commercialisation |
||
|
MedDRA Classes de systèmes d’organes |
Adverse reaction |
Frequency |
|
Infections et infestations |
Méningite aseptique |
Fréquence inconnue |
Les EI supplémentaires suivants ont été identifiés et rapportés pendant l'utilisation post‑commercialisation d'un autre produit à base d'immunoglobuline par voie sous-cutanée : paresthésie, tremblement, tachycardie, dyspnée, laryngospasme et gêne thoracique.
Population pédiatrique
Le profil de sécurité dans la population pédiatrique était semblable à celui des patients adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Les conséquences d'un surdosage sont inconnues.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L'immunoglobuline humaine normale contient essentiellement des immunoglobulines de type G (IgG), qui représentent un large spectre d'anticorps contre les agents infectieux.
L'immunoglobuline humaine normale contient les anticorps IgG présents dans la population normale. En général, elle est préparée à partir de pools de plasma humain provenant d'un minimum de 1 000 dons. La répartition de ses sous-classes d'IgG est proportionnelle à celle du plasma humain natif. Des doses appropriées de ce médicament sont susceptibles de ramener à une valeur normale des taux d'IgG anormalement bas.
Population pédiatrique
Il n'existe aucune différence théorique ou observée entre l'action des immunoglobulines chez l'enfant et chez l'adulte.
5.2. Propriétés pharmacocinétiques
Dans un essai clinique portant sur CUVITRU (n = 48), les patients ont obtenu des taux résiduels constants d'IgG (médiane de 8,26 g/l) sur une période de 52 semaines lors de l'administration de doses hebdomadaires médianes de 0,125 g/kg.
Les résultats de l'essai clinique évaluant CUVITRU démontrent que les taux sériques résiduels d'IgG peuvent être maintenus à l'aide de schémas posologiques de 0,3 à 1,0 g/kg de poids corporel/4 semaines.
Le profil pharmacocinétique de CUVITRU a été évalué lors d'une étude d'efficacité et de sécurité de phase 3 chez 31 patients atteints de DIP et âgés de 12 ans ou plus. Les résultats pharmacocinétiques sont présentés dans le tableau suivant.
|
Paramètres pharmacocinétiques de CUVITRU |
|
|
Paramètre |
CUVITRU Médiane (IC à 95 %), N = 31 |
|
ASC [g*jours/l] |
62,52 (57,16 à 68,86) |
|
ASC / (dose/poids) [(g*jours/l)/(g/kg)] |
589,49 (448,40 à 638,81) |
|
Clairance apparente [mL/kg/jour] |
1,70 (1,57 à 2,23) |
|
Cmax [g/l] |
9,80 (9,31 à 10,62) |
|
Cmin [g/l] |
8,04 (7,30 à 8,99) |
|
Tmax [heures] |
73,92 (69,82 à 120,08) |
Les IgG et les complexes d’IgG sont métabolisés dans les cellules du système réticulo-endothélial.
Posologie hebdomadaire, bimensuelle ou plus fréquente (2‑7 fois par semaine)
La caractérisation pharmacocinétique (PC) d'une posologie bimensuelle ou plus fréquente de CUVITRU a été réalisée à l'aide d'une simulation et d'une modélisation PC de population. Les données de concentration sérique d'IgG sont constituées de 724 échantillons prélevés auprès de 32 patients pédiatriques et adultes atteints de DIP. En comparaison avec une administration hebdomadaire, la modélisation et la simulation PC ont prévu que l'administration bimensuelle d'une dose de CUVITRU correspondant au double de la dose hebdomadaire entraînait un chevauchement de l'exposition à l'IgG sur un intervalle de 2 semaines. En outre, la modélisation et la simulation PC ont permis de prévoir qu'à une dose hebdomadaire totale identique, des perfusions de CUVITRU administrées 2 à 7 fois par semaine (posologie fréquente) entraînaient également un chevauchement de l'exposition à l'IgG sur un intervalle de 2 semaines.
Population pédiatrique
Il n'existe aucune différence théorique ou observée entre le profil pharmacocinétique des immunoglobulines chez l'enfant et chez l'adulte.
5.3. Données de sécurité préclinique
Les immunoglobulines sont des composants normaux du corps humain.
Les données non cliniques ne révèlent pas de risque particulier pour l’homme selon les études conventionnelles de pharmacologie de sécurité et de toxicité relatives aux immunoglobulines. CUVITRU était bien toléré localement en cas de perfusion par voie sous-cutanée chez l'animal. Les études de toxicologie en administration répétée et des fonctions de reproduction chez l'animal sont irréalisables en raison de l'induction du développement d'anticorps aux protéines hétérologues, à l'origine d'interférences.
Aucune étude à long terme sur l’animal pour évaluer le potentiel carcinogène de CUVITRU ou son effet sur la fertilité n’a été réalisée. Un test de mutagénicité in vitro a été réalisé pour l'IGI, 10 % et n'a révélé aucune preuve de mutagénicité.
Eau pour préparations injectables
L'administration de CUVITRU avec d'autres médicaments n'est pas recommandée.
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
CUVITRU ne doit pas être dilué.
Une fois ouvert, utiliser immédiatement.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25 °C.
Ne pas congeler le produit.
Conserver le flacon dans l’emballage extérieur à l’abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
5, 10, 20 ,40 ou 50 ml de solution dans un flacon (verre de type I) muni d'un bouchon (bromobutyl).
Présentation :
1, 10 ou 20 flacon(s) contenant 1 g d'immunoglobuline humaine normale dans 5 ml de solution injectable
1, 10, 20 ou 30 flacon(s) contenant 2 g d'immunoglobuline humaine normale dans 10 ml de solution injectable
1, 10, 20 ou 30 flacon(s) contenant 4 g d'immunoglobuline humaine normale dans 20 ml de solution injectable
1, 5, 10 ou 20 flacon(s) contenant 8 g d'immunoglobuline humaine normale dans 40 ml de solution injectable
1 flacon contenant 10 g d'immunoglobuline humaine normale dans 50 ml de solution injectable
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Si le produit est conservé au réfrigérateur, les flacons non ouverts doivent être placés à température ambiante pendant 90 minutes minimum avant utilisation et conservés à température ambiante pendant l'administration. Ne pas utiliser d’appareil pour réchauffer, dont un four à micro-ondes.
Les solutions troubles ou présentant des dépôts ne doivent pas être utilisées.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
INDUSTRIESTRASSE 67
A-1221 VIENNE
AUTRICHE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 550 524 3 2 : 5 ml de solution en flacon (verre de type I) muni d’un bouchon (bromobutyl) ; boite de 1
· 34009 550 524 4 9 : 10 ml de solution en flacon (verre de type I) muni d’un bouchon (bromobutyl) ; boite de 1
· 34009 550 524 5 6 : 20 ml de solution en flacon (verre de type I) muni d’un bouchon (bromobutyl) ; boite de 1
· 34009 550 524 6 3 : 40 ml de solution en flacon (verre de type I) muni d’un bouchon (bromobutyl) ; boite de 1
· 34009 550 672 9 0 : 50 ml de solution en flacon (verre de type I) muni d’un bouchon (bromobutyl) ; boite de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière. La prescription par un médecin exerçant dans un établissement de transfusion sanguine autorisé à dispenser des médicaments aux malades qui y sont traités est également autorisée.
ANSM - Mis à jour le : 04/07/2025
CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée
Immunoglobuline humaine normale
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que CUVITRU 200 mg/mL, solution injectable par voie sous-cutanée et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ?
3. Comment utiliser CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ET DANS QUELS CAS EST-IL UTILISE ?
Qu'est-ce que CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée?
CUVITRU fait partie d'une classe de médicaments appelés « immunoglobulines humaines normales ». Les immunoglobulines sont également connues sous le nom d'anticorps et sont présentes dans le sang des personnes saines. Les anticorps font partie du système immunitaire (défenses naturelles du corps) et aident votre organisme à combattre les infections.
Quel est le mécanisme d'action de CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ?
CUVITRU a été préparé à partir du sang de donneurs sains. Ce médicament agit exactement de la même manière que les immunoglobulines naturellement présentes dans le sang.
Dans quel cas CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée est-il utilisé ?
CUVITRU est utilisé chez les patients présentant un système immunitaire affaibli, qui ne possèdent pas suffisamment d'anticorps dans le sang et qui souffrent d'infections fréquentes. Des doses régulières et suffisantes de CUVITRU peuvent augmenter les taux sanguins d'immunoglobuline anormalement bas afin de leur permettre d'atteindre un niveau normal (traitement substitutif).
CUVITRU est prescrit à
· des patients dont la capacité innée à produire des anticorps est réduite ou inexistante (syndromes d'immunodéficience primaire).
· patients atteints d’infections sévères ou récurrentes dues à un système immunitaire affaibli en raison d’autres pathologies ou traitements.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ?
N’utilisez jamais CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée :
· si vous êtes allergique aux immunoglobulines ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6.
· si vous avez des anticorps contre l'immunoglobuline de type A (IgA) dans le sang. Cela peut se produire si vous souffrez d'un déficit en IgA. Étant donné que CUVITRU contient des traces d'IgA, vous pouvez présenter une réaction allergique.
· dans un vaisseau sanguin (voie intraveineuse) ou un muscle (voie intramusculaire).
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou infirmier/ère avant d'utiliser CUVITRU.
Si CUVITRU reste dans des seringues siliconées pendant plus de deux heures, des particules visibles peuvent se former. Les instructions détaillées mentionnées sous « Mode et voie d’administration » à la rubrique 3 de cette notice doivent être scrupuleusement respectées.
Réactions allergiques
Vous pouvez être allergique aux immunoglobulines sans le savoir. Des réactions allergiques telles qu'une chute soudaine de la pression artérielle ou un choc anaphylactique (chute brutale de la pression artérielle accompagnée d'autres symptômes tels que le gonflement de la gorge, des difficultés à respirer et un rash cutané) sont rares mais peuvent occasionnellement survenir, même si vous n'avez encore jamais eu de problèmes avec des traitements similaires reçus précédemment. Vous présentez un risque accru de réactions allergiques si vous souffrez de déficit en IgA avec présence d'anticorps anti-IgA. Informez votre médecin ou infirmier/ère avant le traitement si vous souffrez d'un déficit en IgA. CUVITRU contient des quantités résiduelles d'IgA susceptibles d'augmenter le risque de réaction allergique. Les signes ou symptômes de ces réactions allergiques rares englobent :
· des sensations d'étourdissements, un état vertigineux ou une syncope
· un rash cutané et des démangeaisons, un gonflement de la bouche ou de la gorge, des difficultés à respirer, des sibilances
· une fréquence cardiaque anormale, une douleur dans la poitrine, une coloration bleue des lèvres ou des doigts et des orteils
· une vision trouble
Votre médecin ou infirmier/ère perfusera d'abord CUVITRU lentement et vous surveillera attentivement au cours des premières perfusions afin de détecter et de traiter immédiatement toute réaction allergique éventuelle.
o Si vous constatez l'apparition de l'un de ces signes pendant la perfusion, informez immédiatement votre médecin ou infirmier/ère. Il ou elle décidera s'il convient de ralentir la vitesse de la perfusion ou de l'interrompre totalement.
Surveillance pendant la perfusion
Certains effets indésirables risquent d'apparaître plus fréquemment si :
· vous recevez CUVITRU pour la première fois.
· vous avez reçu une autre immunoglobuline auparavant et venez de passer à CUVITRU.
· le traitement a été interrompu pendant une longue période depuis la dernière perfusion de CUVITRU.
o Dans ces circonstances, vous serez surveillé attentivement pendant la première perfusion et l'heure suivant la fin de l'administration.
Dans tous les autres cas, vous devrez être surveillé pendant la perfusion et pendant au moins 20 minutes suivant la fin de l'administration de CUVITRU.
Groupes de patients spécifiques
Votre médecin sera particulièrement attentif si vous souffrez de surcharge pondérale, si vous êtes âgé, diabétique, ou si vous souffrez d'une pression artérielle élevée, d'un faible volume sanguin (hypovolémie), ou de problèmes de vaisseaux sanguins (maladies vasculaires). Dans le cadre de ces pathologies, les immunoglobulines peuvent, dans de très rares cas, augmenter le risque d'infarctus, d’accident vasculaire cérébral, d'embolie pulmonaire ou de thrombose veineuse profonde.
Votre médecin sera également très attentif si vous souffrez ou avez souffert de problèmes de reins ou si vous recevez des médicaments susceptibles d'endommager vos reins (médicaments néphrotoxiques), car il existe un très faible risque d'insuffisance rénale aiguë.
Inflammation des membranes entourant le cerveau (méningite aseptique, SMA)
Les perfusions de produits à base d'immunoglobuline, y compris CUVITRU, peuvent entraîner l'inflammation des membranes entourant le cerveau. L’interruption du traitement par immunoglobuline peut résulter en un soulagement du SMA au bout de plusieurs jours. Le syndrome se déclare généralement plusieurs heures à 2 jours après le traitement par immunoglobuline.
Contactez votre médecin si vous constatez les signes et symptômes suivants : céphalée sévère, raideur de la nuque, endormissement, fièvre, nausées, vomissements et sensibilité ou gêne à la lumière après administration de CUVITRU. Votre médecin déterminera si d'autres examens sont nécessaires et décidera si le traitement par CUVITRU doit être poursuivi.
Destruction des globules rouges (hémolyse)
CUVITRU contient des anticorps anti-érythrocytaires susceptibles d'entraîner la destruction des globules rouges et une anémie hémolytique.
Influence sur les tests sanguins
CUVITRU contient de nombreux anticorps différents, dont certains peuvent altérer les tests sanguins (tests sérologiques).
o Veuillez informer votre médecin de votre traitement par CUVITRU avant tout test sanguin.
Traitement à domicile
Vous et/ou votre soignant serez formé à détecter les premiers signes d'effets indésirables, en particulier de réactions allergiques. Pendant la perfusion, vous ou votre soignant devez être attentif aux premiers signes d'effets indésirables (pour plus de détails, voir rubrique 4, « Quels sont les effets indésirables éventuels ? »).
· Le cas échéant, vous ou votre soignant devez interrompre immédiatement la perfusion et appeler un médecin.
· En présence d'un effet indésirable grave, vous ou votre soignant devez demander immédiatement un traitement d'urgence.
Informations sur la composition de CUVITRU
CUVITRU est fabriqué à base de plasma humain (partie liquide du sang). Lorsque des médicaments sont préparés à partir de sang ou de plasma humain, des mesures de prévention de la transmission d’agents infectieux sont mises en place.
Ces mesures comprennent :
· la sélection soigneuse des donneurs de sang et de plasma de façon à exclure les donneurs risquant d’être porteurs d’infections et
· le contrôle de chaque don et pool de plasma afin de détecter tout signe de virus/d'infection.
· l'inclusion, dans le procédé de traitement du sang ou du plasma, d'étapes d'inactivation/élimination virale.
Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents ou d’autres types d’agents infectieux.
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B, le virus de l’hépatite C, et pour le virus non enveloppé de l’hépatite A et du parvovirus B19.
Les immunoglobulines ne sont pas associées aux infections par le virus de l’hépatite A et le parvovirus B19, probablement grâce aux anticorps protecteurs présents dans CUVITRU.
Il est fortement recommandé de consigner les informations suivantes dans votre carnet de traitement à chaque utilisation de CUVITRU :
· la date d'administration,
· le numéro de lot du médicament, et
· le volume injecté, le débit de perfusion ainsi que les sites de perfusion et leur nombre.
Enfants et adolescents
Les mises en garde et précautions d'emploi mentionnées s'appliquent aux adultes comme aux enfants.
Autres médicaments et CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée
Informez votre médecin, pharmacien ou infirmier/ère si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Vaccinations
CUVITRU peut réduire l'effet de certains vaccins tels que les vaccins contre la rougeole, la rubéole, les oreillons et la varicelle (vaccins à virus vivant). Par conséquent, après l'administration de CUVITRU, vous devez respecter un intervalle de 3 mois avant de pouvoir recevoir certains vaccins. Dans le cas du vaccin contre la rougeole, cette période d'attente peut atteindre un an après l'administration de la dernière dose de CUVITRU.
o Informez le médecin ou l'infirmier/ère administrant votre vaccin de votre traitement par CUVITRU.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
CUVITRU n'a fait l'objet d'aucun essai clinique chez la femme enceinte ou au cours de l'allaitement. L’expérience clinique avec les immunoglobulines suggère toutefois qu’aucun effet néfaste n'est attendu sur le déroulement de la grossesse ni sur l'enfant à naître.
Si vous allaitez et recevez CUVITRU, les anticorps du médicament peuvent se retrouver dans le lait maternel et également protéger votre bébé contre certaines infections.
L'expérience concernant les immunoglobulines suggère qu'aucun effet délétère sur la fertilité n'est attendu.
Conduite de véhicules et utilisation de machines
Pendant le traitement avec CUVITRU, les patients peuvent présenter des effets indésirables (par exemple, sensation vertigineuse ou nausées) susceptibles d'affecter la capacité à conduire des véhicules et à utiliser des machines. Le cas échéant, vous devez attendre que les réactions disparaissent.
3. COMMENT UTILISER CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ?
CUVITRU doit être perfusé sous la peau (par voie sous-cutanée ou SC).
Le traitement par CUVITRU sera démarré par votre médecin ou infirmier/ère, mais vous serez peut-être autorisé à utiliser le médicament à domicile après avoir reçu les premières perfusions sous surveillance médicale et après avoir suivi (vous et/ou votre soignant) une formation adéquate. Vous déciderez, en accord avec votre médecin, si vous pouvez utiliser CUVITRU (par exemple à l’aide d’une pompe à perfusion ou en l’administrant manuellement à l’aide d’une seringue) à domicile. Ne commencez pas le traitement par CUVITRU à domicile avant d'avoir reçu les instructions complètes.
Posologie
Votre médecin calculera la dose correcte pour vous en fonction de votre poids corporel, de vos éventuels traitements antérieurs et de votre réponse au traitement.
Votre médecin déterminera si vous avez besoin d'une dose de charge (pour l'adulte ou l'enfant) d'au moins 1,0 à 2,5 ml/kg de poids corporel, répartie sur plusieurs jours.
Par la suite, vous recevrez une perfusion régulière de CUVITRU, qui peut être quotidienne à bimensuelle ; la dose mensuelle cumulative sera d'environ 1,5 à 5 ml/kg de poids corporel. Votre médecin peut devoir ajuster votre dose en fonction de votre réponse au traitement.
Ne modifiez pas la dose ni l'intervalle de dosage sans consulter votre médecin. Si vous pensez que la fréquence d'administration de CUVITRU doit être modifiée, parlez-en à votre médecin. Si vous pensez avoir oublié une dose, contactez votre médecin dès que possible.
Début du traitement
Votre traitement sera démarré par un médecin ou infirmier/ère expérimenté dans le traitement des patients présentant un système immunitaire affaibli et dans l'encadrement des patients traités à domicile. Vous serez suivi attentivement pendant toute la perfusion et pendant au moins une heure après la fin de la perfusion afin d'évaluer votre tolérance. Au début, le médecin ou l'infirmier/ère utilisera une vitesse de perfusion lente, puis l'augmentera progressivement pendant la première perfusion et les suivantes. Une fois que le médecin ou l'infirmier/ère aura défini la dose et la vitesse de perfusion qui vous conviennent, vous pourrez vous administrer le traitement vous-même, à domicile.
Traitement à domicile
Vous pouvez vous administrer CUVITRU vous-même ou le faire administrer par votre soignant. Vous serez encadré par un médecin ou infirmier/ère expérimenté dans l'encadrement et le traitement de patients tels que vous. Il sera présent avec vous lors des premiers traitements.
Vous ou votre soignant apprendrez :
· les techniques de perfusion stériles (aseptiques),
· l'utilisation d'un dispositif d'administration (si nécessaire),
· la tenue d'un carnet de traitement et
· les mesures à prendre en cas d'effets indésirables graves.
Vous devez respecter scrupuleusement les instructions de votre médecin concernant la dose, la vitesse de perfusion et le calendrier de perfusion de CUVITRU afin d'assurer l'efficacité du traitement.
Mode et voie d’administration
Sélection du ou des sites de perfusion :
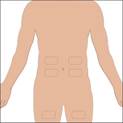
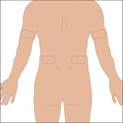
Les zones conseillées pour la perfusion sous-cutanée de CUVITRU sont l'abdomen, les cuisses, le haut des bras ou le bas du dos. CUVITRU peut être perfusé dans plusieurs sites. Les sites de perfusion doivent être distants d'au moins 10 cm. Évitez les zones osseuses, les vaisseaux sanguins visibles, les cicatrices et toute zone enflammée (irritée) ou infectée. Alternez les sites lors de chaque administration, conformément aux instructions de votre médecin ou infirmier/ère.
En cas de perfusion assistée par un dispositif :
Vous pouvez utiliser plusieurs sites de perfusion sous-cutanée en même temps à l'aide d'un kit multi-aiguilles. La quantité de produit perfusée varie selon les sites et les doses supérieures à 30 mL peuvent être réparties selon vos préférences.
En cas d’administration manuelle :
CUVITRU peut être administré à l’aide d’une seringue en un site de perfusion unique. Une nouvelle aiguille d’injection stérile doit être utilisée s’il est nécessaire d’administrer en d’autres sites.
La quantité de produit perfusée varie selon les sites et les doses supérieures à 30 ml peuvent être réparties selon vos préférences.
Débit de perfusion :
Votre médecin déterminera la technique et le débit de perfusion qui vous conviennent en tenant compte de votre dose individuelle, de votre fréquence d’administration et de votre tolérance au produit.
En cas de perfusion assistée par un dispositif :
La vitesse de perfusion initiale recommandée est de 10 ml par heure par site de perfusion. Si elle est bien tolérée, elle peut être augmentée à intervalles de 10 minutes au moins et par paliers de 20 ml par heure par site pendant les deux premières perfusions. Pour les perfusions suivantes, la vitesse peut être augmentée selon la tolérance.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
En cas d’administration manuelle :
Consultez votre médecin. Commencez avec une vitesse de perfusion qui ne provoque pas de gêne. La perfusion ne doit jamais être douloureuse. Le débit de perfusion maximal recommandé est d’environ 1 à 2 ml par minute. Il se peut que certains sites d’injection tolèrent des volumes de perfusion plus importants que d’autres.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
Un mode d'emploi détaillé est fourni à la rubrique suivante :
N'utilisez pas CUVITRU à domicile avant d'avoir reçu des instructions et une formation de votre médecin ou infirmier/ère.
Préparez le ou les flacons de CUVITRU :
· Retirez CUVITRU du carton. Si le produit est conservé au réfrigérateur, laissez les flacons revenir à température ambiante. Cela peut prendre jusqu'à 90 minutes.
· N'utilisez pas d'appareil pour réchauffer ni de four à micro-ondes.
· N'agitez pas le ou les flacons.
|
1. Vérifiez le ou les flacons : · Ne les utilisez pas après la date de péremption. · Ne les utilisez pas si le capuchon protecteur est manquant ou cassé. · Vérifiez la couleur : la solution doit être limpide et incolore à jaune pâle ou brun clair. · Ne les utilisez pas si la solution est trouble ou contient des particules. |
|
|
2. Rassemblez tous les éléments fournis · Rassemblez tous les éléments fournis : Cela inclut : le/les flacon(s) de CUVITRU, et le nécessaire à perfusion : kit d'aiguille sous-cutanée, dispositif(s) de transfert, seringue(s), capuchons protecteurs stériles, pansement stérile transparent, ruban adhésif, gaze, conteneur pour objets pointus, pompe à perfusion (si utilisée), tubulure, carnet de perfusion. · Préparez un espace de travail propre. · En cas d’utilisation d’une pompe à perfusion : programmez la pompe à perfusion selon les débits de perfusion prescrits et les instructions du fabricant. · Lavez-vous soigneusement les mains et laissez-les sécher. · Ouvrez les fournitures en respectant les instructions de votre médecin ou infirmier/ère. |
|
|
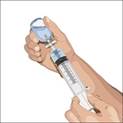
3. Préparez la ou les seringues : · Retirez le capuchon du flacon. · Essuyez chaque bouchon à l'aide d'une lingette stérile imbibée d'alcool et laissez sécher. · Fixez une seringue stérile à un perforateur à évent. · Insérez le perforateur à évent au centre du flacon. · Retournez le flacon et tirez sur le piston pour aspirer la solution dans la ou les seringues. · Répétez ces opérations si vous devez utiliser plusieurs flacons pour atteindre la dose désirée. · La perfusion doit débuter immédiatement après le transfert de CUVITRU dans la seringue. Si l’administration doit prendre plus de deux heures, la dose nécessaire doit être divisée et administrée à différents sites de perfusion. Si CUVITRU reste dans des seringues siliconisées pendant plus de deux heures, des particules visibles peuvent se former. Si vous utilisez une aiguille stérile : Fixez une seringue stérile à l'aiguille stérile et tirez sur le piston de la seringue afin de la remplir d'une quantité d'air égale à la quantité de solution que vous prélèverez du flacon. Insérez l'aiguille au centre du bouchon et injectez l'air. Tirez sur le piston pour aspirer le volume désiré. |
|
|
a) 4. Préparez la perfusion :En cas d’utilisation d’une pompe à perfusion : · Respectez les instructions du fabricant pour le remplissage de la tubulure et l'utilisation de la pompe. · Fixez la seringue remplie de solution au kit d'aiguille. · Dirigez la pointe de la seringue vers le haut et appuyez délicatement sur le piston pour en évacuer l'air et remplir le kit d'aiguille jusqu'à l'embout.
b) En cas d’administration manuelle : · Respectez les instructions de votre infirmier/ère ou de votre professionnel de santé. · Fixez la seringue remplie de solution au kit d’aiguille. Dirigez la pointe de la seringue vers le haut et appuyez délicatement sur le piston pour en évacuer l’air et remplir le kit d’aiguille jusqu’à l’embout. |
|
|
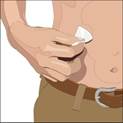
5. Préparez le ou les sites d'injection : · Sélectionnez le nombre de sites de perfusion en fonction du volume de la dose totale. · Choisissez le ou les sites de perfusion : haut des bras, abdomen, cuisses ou bas du dos · Évitez les zones osseuses, les vaisseaux sanguins visibles, les cicatrices et toute zone enflammée (irritée) ou infectée. · Perfusez la solution à partir d'un ou de plusieurs sites de perfusion simultanément. · Choisissez des sites distants d'au moins 10 cm. · Alternez les sites d'une perfusion à l'autre. · Essuyez le ou les sites de perfusion à l'aide d'une lingette stérile imbibée d'alcool, en partant du centre de chaque site et en effectuant des mouvements circulaires vers l'extérieur. Laissez sécher le ou les sites de perfusion (au moins 30 secondes). |
|
|
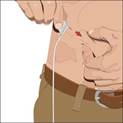
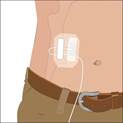
6. Insérez et fixez le kit d'aiguille sous-cutanée : · Retirez le cache-aiguille. Saisissez fermement et pincez au moins 2,5 cm de peau entre deux doigts. · Insérez l'aiguille dans la peau d'un mouvement rapide et rectiligne à un angle de 90 degrés. Fixez l'aiguille en place à l'aide de ruban adhésif stérile (inclus avec le pansement transparent). Si vous utilisez plusieurs sites, répétez les opérations. · Fixez le kit d'aiguille en place en posant un pansement protecteur stérile sur le ou les sites. |
|
|
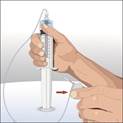
7. Démarrez la perfusion · En cas de perfusion à l’aide d’une pompe : respectez les instructions du fabricant concernant la mise en route de la pompe et le démarrage de la perfusion · En cas de perfusion manuelle : appuyez progressivement sur le piston de la seringue en respectant les instructions de votre médecin ou infirmier/ère, jusqu’à ce que l’intégralité du liquide contenu dans la seringue soit injecté, ou procédez selon les indications fournies par votre médecin ou votre infirmier/ère · Vérifiez occasionnellement le ou les sites de perfusion pendant la durée de la perfusion. |
|
|
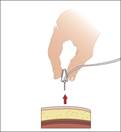
8. Retirez la ou les aiguilles sous-cutanées du ou des sites de perfusion une fois la perfusion terminée : · Retirez le kit d'aiguille en détachant tous les bords du pansement. · Tirez sur les ailettes de l'aiguille d'un mouvement rectiligne. · Pressez doucement une petite compresse de gaze sur le site d'injection et recouvrez-le d'un bandage. · Jetez la ou les aiguilles dans le conteneur pour objets pointus. |
|
|
9. Effectuez la traçabilité de la perfusion : · Décollez l'étiquette détachable du ou des flacons, qui comporte le numéro de lot et la date de péremption du produit, et collez-la dans votre carnet de traitement/de perfusion. · Notez la date, l'heure, la dose, le ou les sites de perfusion (pour vous aider à les alterner) et les réactions éventuelles après chaque perfusion. · Jetez l'éventuelle solution inutilisée restante dans le flacon, les consommables et les flacons conformément aux recommandations de votre médecin ou infirmier/ère. |
|




Utilisation chez les enfants et les adolescents
Les mêmes indications, doses et fréquences de perfusion sont applicables aux adultes, aux enfants et aux adolescents (de 0 à 18 ans).
Si vous avez utilisé plus de CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée que vous n’auriez dû
Si vous pensez avoir utilisé plus de CUVITRU que vous n'auriez dû, contactez votre médecin dès que possible.
Si vous oubliez d’utiliser CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée
N'injectez pas de dose double de CUVITRU pour compenser la dose que vous avez oublié de prendre. Si vous pensez avoir oublié une dose, contactez votre médecin dès que possible.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Certains effets indésirables, tels que céphalée, frissons ou douleurs corporelles, peuvent être réduits en ralentissant la vitesse de perfusion.
Effets indésirables graves
Les perfusions de médicaments tels que CUVITRU peuvent occasionnellement entraîner des réactions allergiques graves, mais rares. Vous pouvez ressentir une chute de pression artérielle soudaine et, dans des cas isolés, un choc anaphylactique. Les médecins sont informés du risque d'apparition de ces effets indésirables et vous surveilleront pendant et après les premières perfusions.
Informez immédiatement votre médecin ou infirmier/ère si vous constatez l'apparition de l'un des signes suivants :
· des sensations d'étourdissement, un état vertigineux ou une syncope,
· un rash cutané et des démangeaisons, un gonflement de la bouche ou de la gorge, des difficultés à respirer, des sibilances,
· une fréquence cardiaque anormale, une douleur dans la poitrine, une coloration bleue des lèvres ou des doigts et des orteils,
· une vision trouble.
En cas d'utilisation de CUVITRU à domicile, vous pouvez effectuer la perfusion en présence de votre soignant, qui vous aidera à détecter l'apparition de réactions allergiques, à arrêter la perfusion et à chercher de l'aide au besoin.
Voir également la rubrique 2 de cette notice concernant le risque de réactions allergiques et l'utilisation de CUVITRU à domicile.
Les effets indésirables suivants sont très fréquents (peuvent toucher plus de 1 personne sur 10) :
· céphalée
· diarrhée et nausées
· rougeur et douleur au site de perfusion
· fatigue
Les effets indésirables suivants sont fréquents (peuvent toucher jusqu’à 1 personne sur 10) :
· sensation vertigineuse, migraine et endormissement
· pression artérielle diminuée
· douleur abdominale
· démangeaisons et rash
· douleur musculaire
· gonflement, démangeaisons, rash et bleus au site de perfusion
· douleur
Les effets indésirables suivants sont peu fréquents (peuvent toucher jusqu’à 1 personne sur 1 00) :
· sensation de brûlure
· douleur abdominale basse
· œdème au site de perfusion
· tests sanguins positifs pour les anticorps
La fréquence des effets indésirables ci-dessous n’est pas connue (ne peut être estimée sur la base des données existantes)
· inflammation des couches du cerveau (méningite aseptique).
Effets indésirables observés avec des médicaments semblables
Les effets indésirables suivants ont été observés lors de la perfusion d'immunoglobuline humaine normale sous la peau (voie sous-cutanée). Bien qu'ils n'aient pas encore été signalés avec CUVITRU, il est possible qu'ils surviennent lors d'un traitement avec CUVITRU.
· picotements
· tremblements
· rythme cardiaque rapide
· difficulté respiratoire
· dysfonctionnement de corde vocale
· douleur dans la poitrine
· durcissement et/ou chaleur au site de perfusion
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance (Site internet : https://signalement.social-sante.gouv.fr/)
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée ?
Tenir hors de la vue et de la portée des enfants.
N'utilisez pas ce médicament après la date de péremption indiquée sur l'étiquette et l'emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
N’utilisez pas ce médicament si vous remarquez que la solution est trouble, contient des particules ou a changé de couleur.
Conserver les flacons dans l’emballage extérieur, à l’abri de la lumière.
A conserver à une température ne dépassant pas 25 °C.
Ne pas congeler.
Si le produit est conservé au réfrigérateur, les flacons non ouverts doivent être retirés du réfrigérateur et placés à température ambiante pendant 90 minutes minimum avant utilisation. Ne pas utiliser d’appareil pour réchauffer, dont un four à micro-ondes.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient CUVITRU 200 mg/ml, solution injectable par voie sous-cutanée
· La substance active est l'immunoglobuline humaine normale.
1 ml de CUVITRU contient 200 mg de protéines humaines, dont au moins 98 % sont des immunoglobulines G (IgG).
· Les autres composants (excipients) sont la glycine et l’eau pour préparations injectables.
Qu’est-ce que CUVITRU et contenu de l’emballage extérieur
· Chaque flacon de 5 ml contient : 1 g d'immunoglobuline humaine normale
· Chaque flacon de 10 ml contient : 2 g d'immunoglobuline humaine normale
· Chaque flacon de 20 ml contient : 4 g d'immunoglobuline humaine normale
· Chaque flacon de 40 ml contient : 8 g d'immunoglobuline humaine normale
· Chaque flacon de 50 ml contient : 10 g d'immunoglobuline humaine normale
Présentations :
1, 10 ou 20 flacon(s) contenant 5 ml de solution injectable
1, 10, 20 ou 30 flacon(s) contenant 10 ml de solution injectable
1, 10, 20 ou 30 flacon(s) contenant 20 ml de solution injectable
1, 5, 10 ou 20 flacon(s) contenant 40 ml de solution injectable
1 flacon contenant 50 ml de solution injectable
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
INDUSTRIESTRASSE 67
A-1221 VIENNE
AUTRICHE
Exploitant de l’autorisation de mise sur le marché
16 PLACE DE L’IRIS
92400 COURBEVOIE
BAXALTA BELGIUM MANUFACTURING SA
BOULEVARD RENE BRANQUART 80
B 7860 - LESSINES
BELGIQUE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).