Dernière mise à jour le 01/06/2026
GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion
Indications thérapeutiques
Qu’est-ce que GANCICLOVIR HIKMA ?
GANCICLOVIR HIKMA contient la substance active ganciclovir. Elle appartient à un groupe appelé médicaments antiviraux.
Dans quels cas GANCICLOVIR HIKMA est-il utilisé ?
GANCICLOVIR HIKMA est utilisé pour traiter des maladies causées par un virus appelé cytomégalovirus (CMV) chez des patients adultes et adolescents âgés de 12 ans et plus ayant un système immunitaire affaibli. Il est également utilisé pour prévenir l’infection à CMV après une greffe d’organe ou pendant une chimiothérapie chez les adultes et les enfants dès la naissance.
· Le virus peut affecter n’importe quelle partie du corps. Il peut ainsi affecter la rétine au fond de l’œil, ce qui signifie qu’il peut provoquer des troubles de la vision.
· Le virus peut affecter tout le monde, mais il représente un problème particulier pour les personnes dont le système immunitaire est affaibli. Chez ces personnes, le virus CMV peut provoquer une maladie grave. Un affaiblissement du système immunitaire peut être dû à d’autres maladies (telles que le sida) ou à des médicaments (tels qu’une chimiothérapie ou des immunosuppresseurs).
Présentations
> 1 flacon en verre de 20 mL
Code CIP : 34009 302 916 6 5
Déclaration de commercialisation : 01/01/2025
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 01/03/2024
GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Ganciclovir (sous forme de ganciclovir sodique).................................................................... 500 mg
Pour un flacon.
Après reconstitution avec 10 mL d’eau pour préparations injectables, chaque mL contient 50 mg de ganciclovir.
Excipient à effet notoire : environ 43 mg de sodium par flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution à diluer pour perfusion.
Poudre lyophilisée blanche à blanc cassé.
4.1. Indications thérapeutiques
GANCICLOVIR HIKMA est indiqué chez les adultes et les adolescents âgés de ≥ 12 ans pour :
· le traitement des infections à cytomégalovirus (CMV) chez les patients immunodéprimés ;
· la prévention des infections à CMV utilisant un traitement préemptif chez les patients présentant une immunosuppression induite par un traitement médicamenteux (par exemple, à la suite d’une greffe d’organe ou d’une chimiothérapie anticancéreuse).
GANCICLOVIR HIKMA est également indiqué dès la naissance pour :
· la prévention des infections à CMV utilisant une prophylaxie universelle chez les patients présentant une immunosuppression induite par un traitement médicamenteux (par exemple, à la suite d’une greffe d’organe ou d’une chimiothérapie anticancéreuse).
Il convient de prendre en compte les recommandations officielles sur l’utilisation appropriée des agents antiviraux.
4.2. Posologie et mode d'administration
Traitement des infections à CMV
Adultes et population pédiatrique âgée de ≥ 12 ans présentant une fonction rénale normale :
· Traitement d’induction : 5 mg/kg administrés par perfusion intraveineuse d’une heure toutes les 12 heures pendant 14 à 21 jours.
· Traitement d’entretien : un traitement d’entretien peut être administré aux patients immunodéprimés exposés à un risque de rechute. 5 mg/kg administrés par perfusion intraveineuse d’une heure une fois par jour 7 jours par semaine ou 6 mg/kg administrés une fois par jour 5 jours par semaine. La durée du traitement d’entretien doit être déterminée au cas par cas en tenant compte des recommandations thérapeutiques locales.
· Traitement d’une progression de la maladie : le traitement peut être réadministré à la posologie indiquée pour le traitement d’induction chez tout patient dont l’infection à CMV progresse sous traitement d’entretien ou suite à l’arrêt du traitement par ganciclovir.
Population pédiatrique de la naissance à < 12 ans :
Les données pédiatriques actuellement disponibles sont décrites aux rubriques 5.1 et 5.2 mais aucune recommandation sur la posologie ne peut être donnée.
Prophylaxie des infections à CMV utilisant un traitement préemptif
Adultes et population pédiatrique âgée de ≥ 12 ans présentant une fonction rénale normale :
· Traitement d’induction : 5 mg/kg administrés par perfusion intraveineuse d’une heure toutes les 12 heures pendant 7 à 14 jours.
· Traitement d’entretien : 5 mg/kg administrés par perfusion intraveineuse d’une heure une fois par jour 7 jours par semaine ou 6 mg/kg une fois par jour 5 jours par semaine. La durée du traitement d’entretien doit être déterminée selon le risque d’infection à CMV ; les recommandations thérapeutiques locales doivent être consultées.
Population pédiatrique de la naissance à < 12 ans :
Les données pédiatriques actuellement disponibles sont décrites aux rubriques 5.1 et 5.2 mais aucune recommandation sur la posologie ne peut être donnée.
Prévention des infections à CMV utilisant une prophylaxie universelle
Adultes et population pédiatrique âgée de > 16 ans :
5 mg/kg administrés par perfusion intraveineuse d’une heure une fois par jour 7 jours par semaine ou 6 mg/kg une fois par jour 5 jours par semaine. La durée est basée sur le risque de contracter une infection à CMV, tout en tenant compte des recommandations thérapeutiques locales.
Population pédiatrique de la naissance à ≤ 16 ans :
La dose quotidienne unique recommandée de ganciclovir administrée par perfusion intraveineuse pendant une heure est déterminée en fonction de la surface corporelle (SC) à l’aide de la formule de Mosteller et de la clairance de la créatinine dérivée de la formule de Schwartz (ClCrS). Elle est calculée à l’aide des équations ci-dessous. La durée du traitement prophylactique universel est basée sur le risque de contracter une infection à CMV et doit être déterminée au cas par cas.
Dose pédiatrique (mg) = 3 × SC × ClCrS (voir la formule de Mosteller pour le calcul de la SC et la formule de Schwartz pour le calcul de la clairance de la créatinine ci-dessous).
Si la clairance de la créatinine calculée par la formule de Schwartz dépasse 150 mL/min/1,73 m2, une valeur maximale de 150 mL/min/1,73 m2 doit être utilisée dans l’équation :
|
|
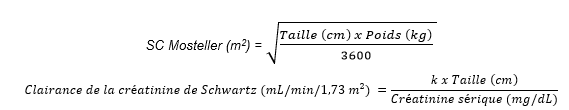
où k = 0,33 pour les patients âgés de < 1 an dont le poids de naissance est bas, 0,45 pour les patients âgés de < 2 ans, 0,55 pour les garçons âgés de 2 à < 13 ans et pour les filles âgées de 2 à 16 ans, et 0,7 pour les garçons âgés de 13 à 16 ans. Pour les patients âgés de plus de 16 ans, se reporter à la posologie pour les adultes.
Les valeurs de k fournies sont basées sur la méthode de Jaffé pour mesurer la créatinine sérique et peuvent nécessiter une correction en cas d’utilisation de méthodes enzymatiques.
Il est recommandé de surveiller régulièrement les taux de créatinine sérique, la taille et le poids, et de modifier la dose le cas échéant.
Instructions posologiques particulières
Insuffisance rénale
Les patients pédiatriques (de la naissance à ≤ 16 ans) présentant une insuffisance rénale et recevant une dose prophylactique de ganciclovir calculée à l’aide de l’algorithme de posologie 3 × SC × ClCrS ne nécessitent pas de modification supplémentaire de la dose car celle-ci est déjà ajustée en fonction de la clairance de la créatinine.
Chez les patients âgés de 12 ans et plus présentant une insuffisance rénale, traités sur la base d’un poids corporel en mg/kg pour un traitement préemptif et un traitement contre les infections à CMV, la dose en mg/kg de ganciclovir doit être modifiée selon la clairance de la créatinine, comme indiqué dans le tableau ci-dessous (voir rubriques 4.4 et 5.2).
Modifications posologiques chez les patients présentant une insuffisance rénale et recevant des doses en mg/kg :
|
ClCr |
Dose d’induction |
Dose d’entretien |
|
> 70 mL/min |
5,0 mg/kg toutes les 12 h |
5,0 mg/kg/jour |
|
50– 69 mL/min |
2,5 mg/kg toutes les 12 h |
2,5 mg/kg/jour |
|
25– 49 mL/min |
2,5 mg/kg/jour |
1,25 mg/kg/jour |
|
10– 24 mL/min |
1,25 mg/kg/jour |
0,625 mg/kg/jour |
|
< 10 mL/min |
1,25 mg/kg 3x/sem après l’hémodialyse |
0,625 mg/kg 3x/sem après l’hémodialyse |
Une estimation de la clairance de la créatinine peut être calculée à partir de la créatininémie à l’aide de la formule suivante :
|
|
Pour une femme : 0,85 × valeur pour un homme.
La créatininémie ou la clairance estimée de la créatinine doit être surveillée car des modifications de posologie sont recommandées chez les patients présentant une insuffisance rénale.
Insuffisance hépatique
La sécurité et l’efficacité du ganciclovir n’ont pas été étudiées chez les patients présentant une insuffisance hépatique (voir rubrique 5.2).
Leucopénie, neutropénie, anémie, thrombopénie et pancytopénie sévères
Voir rubrique 4.4 avant l’instauration du traitement.
Si la numération sanguine diminue significativement au cours du traitement par ganciclovir, un traitement par facteurs de croissance hématopoïétiques et/ou l’arrêt du traitement par ganciclovir doivent être envisagés (voir rubriques 4.4 et 4.8).
Personnes âgées
Aucune étude n’a été menée sur l’efficacité ou la sécurité du ganciclovir chez les personnes âgées. Etant donné que la fonction rénale diminue avec l’âge, le ganciclovir doit être administré aux personnes âgées avec une attention particulière portée à l’état de leur fonction rénale (voir rubrique 5.2).
Mode d’administration
Précautions à prendre avant la manipulation ou l’administration du médicament :
Le ganciclovir étant considéré comme potentiellement tératogène et cancérogène chez l’être humain, des précautions doivent être prises lors de sa manipulation (voir rubrique 6.6).
Mises en garde :
Le ganciclovir doit être administré par perfusion intraveineuse de 1 heure à une concentration ne dépassant pas 10 mg/mL. Ne pas administrer par injection intraveineuse rapide ou en bolus, car les concentrations plasmatiques excessives qui en résulteraient pourraient augmenter la toxicité du ganciclovir.
Ne pas administrer par injection intramusculaire ou sous-cutanée car cela pourrait provoquer une irritation tissulaire sévère due au pH élevé (~11) de la solution de ganciclovir (voir rubrique 4.8).
La posologie, la fréquence et le débit de perfusion recommandés ne doivent pas être dépassés.
GANCICLOVIR HIKMA est une poudre pour solution à diluer pour perfusion. Après reconstitution, GANCICLOVIR HIKMA est une solution incolore à jaune pâle, pratiquement dépourvue de particules visibles.
La perfusion doit être administrée dans une veine dont le débit sanguin est adéquat, de préférence au moyen d’une canule en plastique.
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Allaitement (voir rubrique 4.6)
4.4. Mises en garde spéciales et précautions d'emploi
Hypersensibilité croisée
En raison de la similarité de la structure chimique du ganciclovir et de celle de l’aciclovir et du penciclovir, une réaction d’hypersensibilité croisée entre ces médicaments est possible. Il convient donc d'être prudent lors de la prescription de GANCICLOVIR HIKMA chez des patients présentant une hypersensibilité connue à l’aciclovir ou au penciclovir (ou à leurs prodrogues respectives, le valaciclovir ou le famciclovir).
Mutagenèse, tératogenèse, cancérogenèse, fertilité et contraception
Avant l’instauration du traitement par ganciclovir, les patients doivent être avertis des risques potentiels pour le fœtus. Dans les études menées chez l’animal, le ganciclovir s’est révélé mutagène, tératogène, cancérogène et a altéré la fertilité. D’après des études cliniques et non cliniques, il est probable que le ganciclovir induise une inhibition temporaire ou permanente de la spermatogenèse (voir rubriques 4.6, 4.8 et 5.3).
Le ganciclovir est susceptible d’induire des malformations congénitales et des cancers, il doit donc être considéré comme potentiellement tératogène et cancérogène chez l’être humain. Par conséquent, les femmes en capacité de procréer doivent être informées de la nécessité d’utiliser une méthode de contraception efficace pendant le traitement et pendant au moins 28 semaines par la suite. Les hommes doivent être informés qu’ils doivent utiliser un préservatif et une méthode contraceptive supplémentaire qui, combinés, entraînent un taux d'échec < 1 % par an, pour éviter toute conception pendant le traitement et pendant au moins 95 jours par la suite, sauf s'il est établi que la partenaire féminine n'est pas exposée à un risque de grossesse (voir rubriques 4.6, 4.8 et 5.3).
Ganciclovir doit être utilisé avec une extrême prudence, en particulier dans la population pédiatrique, en raison du risque potentiel d’effets cancérogènes à long terme et de toxicité sur la reproduction. Dans tous les cas, les bénéfices du traitement doivent être soigneusement évalués et l’emporter clairement sur les risques encourus (voir rubrique 4.2). Se référer aux recommandations thérapeutiques.
Myélosuppression
GANCICLOVIR HIKMA doit être utilisé avec prudence chez les patients présentant une cytopénie hématologique préexistante ou des antécédents de cytopénie hématologique d’origine médicamenteuse, ainsi que chez les patients recevant une radiothérapie.
Des cas sévères de leucopénie, de neutropénie, d’anémie, de thrombopénie, de pancytopénie et d’insuffisance médullaire ont été observés chez des patients traités par ganciclovir. Le traitement ne doit pas être instauré si la numération des polynucléaires neutrophiles est inférieure à 500 cellules/μL, si la numération plaquettaire est inférieure à 25 000 cellules/μL ou si l’hémoglobinémie est inférieure à 8 g/dL (voir rubriques 4.2 et 4.8).
Il est recommandé de surveiller la numération formule sanguine complète, y compris la numération plaquettaire, pendant le traitement. Une surveillance hématologique accrue peut être justifiée chez les patients présentant une insuffisance rénale et chez les nouveau-nés et les nourrissons (voir rubrique 4.8). Au cours des 14 premiers jours d’administration, il est recommandé de surveiller la numération des leucocytes (de préférence avec la formule leucocytaire) tous les deux jours ; cette surveillance doit être quotidienne chez les patients dont la numération des polynucléaires neutrophiles est initialement basse (< 1 000 polynucléaires neutrophiles/μL), chez les patients ayant présenté une leucopénie au cours d’un traitement précédent par d’autres substances myélotoxiques et chez ceux présentant une insuffisance rénale.
Chez les patients présentant une leucopénie, une neutropénie, une anémie et/ou une thrombopénie sévères, il est recommandé d’envisager l’utilisation d’un traitement par facteurs de croissance hématopoïétiques et/ou l’interruption du traitement par ganciclovir (voir rubriques 4.2 et 4.8).
Insuffisance rénale
Les patients présentant une altération de la fonction rénale sont exposés à un risque accru de toxicité (particulièrement hématologique). La dose doit être réduite (voir rubriques 4.2 et 5.2).
Utilisation avec d’autres médicaments
Des convulsions ont été rapportées chez des patients traités par imipénème/cilastatine et ganciclovir. Le ganciclovir ne doit pas être utilisé en même temps que l’imipénème/cilastatine sauf si les bénéfices potentiels l’emportent sur les risques encourus (voir rubrique 4.5).
Les patients traités par ganciclovir et didanosine, des médicaments dont l’effet myélosuppresseur est connu ou des médicaments affectant la fonction rénale, doivent faire l’objet d’une surveillance étroite en vue de détecter tout signe de toxicité additive (voir rubrique 4.5).
Excipients
Ce médicament contient environ 43 mg de sodium par flacon, ce qui équivaut à 2 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Interactions pharmacocinétiques
Probénécide
L’administration de probénécide avec du ganciclovir par voie orale a entraîné une diminution statistiquement significative de la clairance rénale du ganciclovir et une augmentation cliniquement significative de l’exposition. Cet effet est également attendu pendant l’administration concomitante de ganciclovir par voie intraveineuse et de probénécide. Les patients traités par probénécide et GANCICLOVIR HIKMA doivent donc faire l’objet d’une surveillance étroite en vue de détecter tout signe de toxicité du ganciclovir.
Didanosine
Les concentrations plasmatiques de didanosine ont systématiquement augmenté en cas d’administration concomitante de ganciclovir. A des doses intraveineuses de 5 et 10 mg/kg/jour, une augmentation de l’ASC de la didanosine allant de 38 % à 67 % a été observée. Aucun effet cliniquement significatif sur les concentrations du ganciclovir n’a été constaté. Les patients doivent faire l’objet d’une surveillance étroite en vue de détecter tout signe de toxicité de la didanosine (voir rubrique 4.4).
Autres antirétroviraux
Les isoenzymes du cytochrome P450 ne jouent aucun rôle dans la pharmacocinétique du ganciclovir. Par conséquent, aucune interaction pharmacocinétique avec les inhibiteurs de protéases et les inhibiteurs non nucléosidiques de la transcriptase inverse n’est attendue.
Interactions pharmacodynamiques
Imipénème/cilastatine
Des convulsions ont été rapportées chez des patients prenant du ganciclovir et de l’imipénème/cilastatine de façon concomitante. Ces médicaments ne doivent pas être administrés de façon concomitante sauf si les bénéfices potentiels l’emportent sur les risques encourus (voir rubrique 4.4).
Zidovudine
La zidovudine et le ganciclovir ont l’un et l’autre la capacité d’induire une neutropénie et une anémie. Une interaction pharmacodynamique peut survenir pendant l’administration concomitante de ces médicaments. Certains patients peuvent ne pas tolérer un traitement concomitant aux posologies maximales autorisées (voir rubrique 4.4).
Autres interactions médicamenteuses potentielles
La toxicité peut être augmentée si le ganciclovir est co-administré avec d’autres médicaments dont l’effet myélosuppresseur est connu ou qui sont associés à une insuffisance rénale. Cela inclut les agents anti-infectieux (tels que dapsone, pentamidine, flucytosine, amphotéricine B, thriméthoprime/sulfaméthoxazole), les immunosuppresseurs (par exemple, ciclosporine, tacrolimus, mycophénolate mofétil), les agents antinéoplasiques (par exemple, vincristine, vinblastine, doxorubicine, et hydroxyurée), ainsi que les analogues nucléosidiques (notamment zidovudine, stavudine et didanosine) et les analogues nucléotidiques (notamment ténofovir et adéfovir). Par conséquent, l’utilisation concomitante de ces médicaments avec le ganciclovir ne doit être envisagée que si les bénéfices potentiels l’emportent sur les risques encourus (voir rubrique 4.4).
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Fertilité
Une étude clinique de faible envergure menée chez des patients transplantés rénaux recevant Valcyte dans le cadre d’une prophylaxie des infections à CMV jusqu’à 200 jours a démontré l’impact du valganciclovir/ganciclovir sur la spermatogenèse avec une diminution de la densité et de la motilité des spermatozoïdes mesurée à la fin du traitement. Cet effet semble être réversible, et environ 6 mois après l’arrêt du traitement par Valcyte, la densité moyenne et la motilité des spermatozoïdes retrouvent des niveaux comparables à ceux observés chez les témoins non traités.
Lors d’études menées chez l’animal, le ganciclovir a altéré la fertilité des souris mâles et femelles et il a été démontré qu’il inhibait la spermatogenèse et induisait une atrophie testiculaire chez la souris, le rat et le chien à des doses considérées comme cliniquement pertinentes.
D’après les études cliniques et non cliniques, il est probable que le ganciclovir puisse inhiber la spermatogenèse humaine de façon temporaire ou permanente (voir rubriques 4.4 et 5.3).
Grossesse
La sécurité du ganciclovir chez la femme enceinte n’a pas été établie. Cependant, le ganciclovir traverse facilement le placenta humain. Lors des études menées chez l’animal, le ganciclovir a été associé à une toxicité pour la reproduction et à une tératogénicité (voir rubriques 4.4 et 5.3). Par conséquent, le ganciclovir ne doit pas être utilisé chez la femme enceinte, sauf si la nécessité clinique du traitement de la femme l'emporte sur le risque tératogène potentiel pour le fœtus.
Contraception chez les hommes et les femmes
En raison du potentiel de toxicité pour la reproduction et de tératogénicité, les femmes en capacité de procréer doivent être informées de la nécessité d’utiliser une méthode de contraception efficace pendant le traitement et pendant au moins 28 semaines après l’arrêt du traitement. Les patients de sexe masculin doivent être informés de la nécessité d’utiliser un préservatif et une méthode contraceptive supplémentaire qui, combinés, entraînent un taux d'échec < 1 % par an, pour éviter toute conception pendant le traitement et pendant au moins 95 jours après l’arrêt du traitement par ganciclovir, sauf s’il est établi que la partenaire n’est pas exposée à un risque de grossesse (voir rubriques 4.4 et 5.3).
Allaitement
On ne sait pas si le ganciclovir est excrété dans le lait maternel humain, mais la possibilité qu’il soit excrété dans le lait maternel et provoque des effets indésirables graves chez le nourrisson allaité ne peut être exclue. Des données chez l’animal indiquent que le ganciclovir est excrété dans le lait de rates allaitantes. L’allaitement doit donc être interrompu durant le traitement par ganciclovir (voir rubrique 4.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Le ganciclovir peut avoir une influence importante sur l’aptitude à conduire des véhicules et à utiliser des machines (voir rubrique 4.8).
Résumé du profil de sécurité
Le valganciclovir est une prodrogue du ganciclovir, et les effets indésirables associés au valganciclovir peuvent être attendus avec le ganciclovir. Le ganciclovir par voie orale n’est plus disponible mais les effets indésirables rapportés lors de son utilisation peuvent être également attendus chez les patients recevant le ganciclovir par voie intraveineuse. Par conséquent, les effets indésirables rapportés avec le ganciclovir par voie intraveineuse ou orale ou avec le valganciclovir sont inclus dans le tableau listant les effets indésirables.
Chez les patients traités par ganciclovir/valganciclovir, les effets indésirables les plus graves et les plus fréquents sont hématologiques, notamment la neutropénie, l’anémie et la thrombopénie (voir rubrique 4.4). D’autres effets indésirables sont présentés dans le tableau ci-dessous.
Les fréquences présentées dans le tableau listant les effets indésirables proviennent d’une population combinée de patients infectés par le VIH (n = 1 704) recevant un traitement d’entretien par ganciclovir ou valganciclovir. Une exception est faite pour l’agranulocytose, la granulocytopénie et la réaction anaphylactique, dont les fréquences proviennent de l’expérience post-commercialisation. Les effets indésirables sont présentés par classe de systèmes d’organes MedDRA. Les catégories de fréquence sont définies à l’aide de la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000) et très rare (< 1/10 000).
Le profil de sécurité global du ganciclovir/valganciclovir est cohérent entre les populations de patients infectés par le VIH et les patients transplantés à l’exception du décollement de la rétine qui a uniquement été rapporté chez les patients infectés par le VIH présentant une rétinite à CMV. Cependant, il existe des différences dans la fréquence de certains effets. Le ganciclovir par voie intraveineuse est associé à un risque plus faible de diarrhée par rapport au valganciclovir par voie orale. La fièvre, les infections à Candida, la dépression, la neutropénie sévère (NAN < 500/μL) et les réactions cutanées sont plus fréquemment rapportées chez les patients infectés par le VIH. Des dysfonctionnements rénaux et hépatiques sont rapportés plus fréquemment chez les receveurs de greffe d’organe.
Tableau listant les effets indésirables
|
Classe de systèmes d’organes (MedDRA) |
Fréquence |
Effets indésirables |
|
Infections et infestations |
Très fréquent |
infections à Candida, y compris candidoses buccales, infection des voies aériennes supérieures |
|
Fréquent |
sepsis, grippe, infection des voies urinaires, cellulite |
|
|
Affections hématologiques et du système lymphatique |
Très fréquent |
neutropénie, anémie |
|
Fréquent |
thrombopénie, leucopénie, pancytopénie |
|
|
Peu fréquent |
insuffisance médullaire |
|
|
Rare |
anémie aplasique, agranulocytose*, granulocytopénie* |
|
|
Affections du système immunitaire |
Fréquent |
hypersensibilité |
|
Rare |
réaction anaphylactique* |
|
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
appétit diminué |
|
Fréquent |
poids diminué |
|
|
Affections psychiatriques |
Fréquent |
dépression, état confusionnel, anxiété |
|
Peu fréquent |
agitation, trouble psychotique, pensée anormale, hallucinations |
|
|
Affections du système nerveux |
Très fréquent |
céphalées |
|
Fréquent |
insomnie, neuropathie périphérique, sensations vertigineuses, paresthésie, hypoesthésie, convulsions, dysgueusie (perturbation du goût) |
|
|
Peu fréquent |
tremblements |
|
|
Affections oculaires |
Fréquent |
altération de la vision, décollement de la rétine, corps flottants du vitré, douleur oculaire, conjonctivite, œdème maculaire |
|
Affections de l’oreille et du labyrinthe |
Fréquent |
douleur auriculaire |
|
Peu fréquent |
surdité |
|
|
Affections cardiaques |
Peu fréquent |
arythmie |
|
Affections vasculaires |
Fréquent |
hypotension |
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent |
toux |
|
Affections gastro-intestinales |
Très fréquent |
diarrhée, nausées, vomissements, douleurs abdominales |
|
Fréquent |
dyspepsie, flatulences, douleurs abdominales, constipation, ulcération buccale, dysphagie, distension abdominale, pancréatite |
|
|
Affections hépatobiliaires |
Fréquent |
phosphatase alcaline sanguine augmentée, fonction hépatique anormale, aspartate aminotransférase augmentée, alanine aminotransférase augmentée |
|
Affections de la peau et du tissu sous-cutané |
Très fréquent |
dermatite |
|
Fréquent |
sueurs nocturnes, prurit, rash, alopécie |
|
|
Peu fréquent |
sécheresse cutanée, urticaire |
|
|
Affections musculosquelettiques et du tissu conjonctif |
Fréquent |
dorsalgies, myalgies, arthralgies, contractures musculaires |
|
Affections du rein et des voies urinaires |
Fréquent |
altération de la fonction rénale, clairance de la créatinine diminuée, créatinine sanguine augmentée |
|
Peu fréquent |
insuffisance rénale, hématurie |
|
|
Affections des organes de reproduction et du sein |
Peu fréquent |
infertilité masculine |
|
Troubles généraux et anomalies au site d’administration |
Très fréquent |
fièvre, fatigue |
|
Fréquent |
réaction au site d’injection, douleur, frissons, malaise, asthénie |
|
|
Peu fréquent |
douleurs thoraciques |
* Les fréquences de ces effets indésirables proviennent de l’expérience post-commercialisation. Toutes les autres catégories de fréquence sont basées sur les fréquences rapportées au cours des essais cliniques.
Description de certains effets indésirables
Neutropénie
Le risque de neutropénie ne peut être anticipé sur la base du nombre de polynucléaires neutrophiles avant traitement. La neutropénie survient habituellement au cours de la première ou de la deuxième semaine du traitement d’induction et à la suite de l’administration d’une dose cumulée ≤ 200 mg/kg. La numération sanguine se normalise habituellement en 2 à 5 jours après l’arrêt du médicament ou une réduction de la dose du médicament (voir rubrique 4.4).
Neutropénie sévère
Des cas de neutropénie sévère ont été plus fréquemment rapportés chez les patients infectés par le VIH (14 %) recevant un traitement d’entretien par valganciclovir ou par ganciclovir par voie orale ou intraveineuse (n = 1 704) que chez les patients ayant reçu une greffe d’organe sous traitement par valganciclovir ou ganciclovir par voie orale. Chez les patients recevant du valganciclovir ou du ganciclovir par voie orale jusqu’au Jour 100 après la greffe, l’incidence de la neutropénie sévère était de 5 % et 3 % respectivement, alors que chez les patients recevant du valganciclovir jusqu’au Jour 200 après la greffe, l’incidence de la neutropénie sévère était de 10 %.
Thrombopénie
Le risque de survenue d’une thrombopénie est plus élevé chez les patients dont la numération plaquettaire est initialement basse (< 100 000/μL). Les patients présentant une immunosuppression iatrogène due à un traitement par des médicaments immunosuppresseurs sont exposés à un risque plus élevé de thrombopénie que les patients atteints du sida (voir rubrique 4.4). Une thrombopénie sévère peut être associée à une hémorragie pouvant engager le pronostic vital.
Convulsions
Des convulsions ont été rapportées chez des patients sous imipénème/cilastatine et ganciclovir (voir rubriques 4.4 et 4.5).
Décollement de la rétine
Cet effet indésirable a uniquement été rapporté au cours d’études menées chez des patients infectés par le VIH et traités par ganciclovir pour une rétinite à CMV.
Réactions au site d’injection
Des réactions au site d’injection surviennent fréquemment chez les patients traités par ganciclovir. GANCICLOVIR HIKMA doit être administré comme indiqué dans la rubrique 4.2 afin de réduire le risque d’irritation tissulaire locale.
Population pédiatrique
Aucune étude sur la sécurité du ganciclovir n’a été menée chez des enfants âgés de < 12 ans mais, sur la base de l’expérience acquise avec le valganciclovir, une prodrogue du ganciclovir, le profil de sécurité global du médicament actif est similaire chez les adultes et les patients pédiatriques. La neutropénie survient plus fréquemment chez les patients pédiatriques, mais aucune corrélation n’a été constatée entre la neutropénie et des effets indésirables infectieux dans la population pédiatrique. Un risque accru de cytopénies chez les nouveau-nés et les nourrissons nécessite une surveillance étroite de la numération sanguine dans ces groupes d’âges (voir rubrique 4.4).
Seules des données limitées sont disponibles chez les nouveau-nés ou les nourrissons infectés par le VIH/atteints de sida ou présentant une infection congénitale symptomatique à CMV traités par valganciclovir ou ganciclovir. Cependant, le profil de sécurité semble être conforme au profil de sécurité connu du valganciclovir/ganciclovir.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Symptômes
Des cas de surdosages de ganciclovir IV, dont certains d’issue fatale, ont été rapportés lors des essais cliniques et au cours de l’expérience post-commercialisation. La majorité de ces cas n’a été associé à aucun effet indésirable, ou a été associé à un ou plusieurs des effets indésirables suivants :
· Toxicité hématologique : myélosuppression incluant pancytopénie, insuffisance médullaire, leucopénie, neutropénie, granulocytopénie
· Hépatotoxicité : hépatite, troubles de la fonction hépatique
· Toxicité rénale : aggravation d’une hématurie chez un patient présentant une insuffisance rénale préexistante, atteinte rénale aiguë, créatininémie élevée
· Toxicité gastro-intestinale : douleurs abdominales, diarrhée, vomissements
· Neurotoxicité : tremblements généralisés, convulsions
Prise en charge
Le ganciclovir est éliminé par hémodialyse. Par conséquent, l’hémodialyse peut être utile pour réduire l’exposition au médicament chez les patients ayant reçu un surdosage de ganciclovir (voir rubrique 5.2).
Informations complémentaires sur les populations particulières
Insuffisance rénale : un surdosage de ganciclovir pourrait entraîner une augmentation de la toxicité rénale chez les patients présentant une insuffisance rénale (voir rubrique 4.4).
Population pédiatrique
Aucune information spécifique n’est disponible.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le ganciclovir est un analogue synthétique de la 2’-désoxyguanosine, qui inhibe la réplication des herpès virus tant in vitro qu’in vivo. Les virus humains sensibles incluent le cytomégalovirus humain (HMCV), les virus herpès simplex 1 et 2 (HSV-1 et HSV-2), les herpès virus humains 6, 7 et 8 (HHV-6, HHV-7, HHV-8), le virus d’Epstein-Barr (EBV), le virus varicelle-zona (VZV) et le virus de l’hépatite B. Les études cliniques se sont limitées à l’évaluation de l’efficacité chez des patients infectés par le CMV.
Dans les cellules infectées par le CMV, le ganciclovir est initialement phosphorylé en ganciclovir monophosphate par la protéine kinase virale UL97. Une phosphorylation ultérieure sous l’effet de plusieurs kinases cellulaires produit du ganciclovir triphosphate, qui subit ensuite un métabolisme intracellulaire lent. Ce mécanisme a été mis en évidence dans des cellules infectées par HSV et HCVM, avec des demi-vies respectives de 18 et 6– 24 heures après l’élimination du ganciclovir extracellulaire. La phosphorylation étant largement dépendante de la kinase virale, la phosphorylation du ganciclovir se produit de préférence dans les cellules infectées par le virus.
L’activité virostatique du ganciclovir résulte de l’inhibition de la synthèse de l’ADN viral par : (1) inhibition compétitive de l’incorporation de la désoxyguanosine triphosphate dans l’ADN par l’ADN polymérase et (2) incorporation de ganciclovir triphosphate dans l’ADN viral provoquant l’arrêt de l’élongation de l’ADN viral ou la limitant très fortement.
Activité antivirale
L’activité antivirale in vitro, mesurée par la CI50 du ganciclovir vis-à-vis du CMV, est comprise entre 0,08 μM (0,02 μg/mL) et 14 μM (3,57 μg/mL).
Efficacité et sécurité clinique
Résistance virale
La possibilité d’une résistance virale doit être envisagée chez les patients dont la réponse clinique est insuffisante de façon répétée, ou chez qui l’excrétion virale persiste pendant le traitement.
Une résistance virale au ganciclovir peut survenir par sélection de mutations au niveau du gène de la kinase virale (UL97) responsable de la monophosphorylation du ganciclovir et/ou du gène de la polymérase virale (UL54). Les virus contenant des mutations au niveau du gène UL97 sont résistants au ganciclovir seul, tandis que les virus contenant des mutations au niveau du gène UL54 sont résistants au ganciclovir, mais peuvent présenter une résistance croisée à d’autres antiviraux qui ciblent également la polymérase virale.
Population pédiatrique
Au cours d’une étude prospective, 36 patients pédiatriques sévèrement immunodéprimés (âgés de 6 mois à 16 ans) infectés par le VIH et le CMV ont reçu du ganciclovir par voie intraveineuse à la dose de 5 mg/kg/jour pendant 2 jours puis du ganciclovir par voie orale pendant une durée médiane de 32 semaines. Le ganciclovir a été efficace, avec un profil de toxicité similaire à celui observé chez les adultes. Le ganciclovir a été associé à une diminution de la détection du CMV par culture ou réaction en chaîne par polymérase (PCR). La neutropénie a été le seul effet indésirable sévère observé pendant l’étude et, bien que l’arrêt du traitement n’ait été nécessaire dans aucun cas, un facteur de croissance granulocytaire (G-CSF) a dû être administré à 4 patients afin de maintenir la numération des polynucléaires neutrophiles > 400 cellules/mm3.
Au cours d’une étude rétrospective, 122 patients pédiatriques (âgés de 16 jours à 18 ans, âge médian : 2,5 ans) ayant reçu une transplantation hépatique, ont été traités pendant au moins 14 jours par 5 mg/kg de ganciclovir par voie intraveineuse deux fois par jour, puis une surveillance préventive de l’infection à CMV a été effectuée par PCR. Le risque d’infection à CMV était jugé élevé chez 43 patients et habituel chez 79 patients. Une infection asymptomatique à CMV a été détectée par PCR chez 34,4 % des patients, et a été plus fréquente chez les receveurs à risque élevé que chez les receveurs à risque habituel (58,1 % vs 21,8 %, p = 0,0001). Une infection à CMV est survenue chez 12 sujets (9,8 % ; 8 à risque élevé vs 4 à risque habituel, p = 0,03). Un rejet aigu est survenu chez 3 sujets dans les 6 mois suivant la détection du CMV, mais la détection du CMV a été précédée d’un rejet chez 13 sujets. Aucun décès secondaire à l’infection à CMV n’est survenu. Au total, 38,5 % des sujets n’ont reçu aucun médicament antiviral en dehors de la prophylaxie postopératoire initiale.
Une analyse rétrospective a comparé la sécurité et l’efficacité du ganciclovir à celles du valganciclovir chez 92 patients pédiatriques (âgés de 7 mois à 18 ans, âge médian : 9 ans) ayant reçu une greffe de rein et/ou de foie. Tous les enfants ont reçu un traitement par ganciclovir par voie intraveineuse 5 mg/kg deux fois par jour pendant 2 semaines à la suite de la greffe. Les enfants traités avant 2004 ont reçu ensuite du ganciclovir par voie orale à raison de 30 mg/kg/dose jusqu’à 1 g/dose trois fois par jour (n = 41), tandis que ceux traités après 2004 ont reçu jusqu’à 900 mg de valganciclovir une fois par jour (n = 51). L’incidence globale de l’infection à CMV a été de 16 % (15/92 patients). Le délai de survenue d’une infection à CMV a été similaire entre les deux groupes.
Lors d’une étude randomisée et contrôlée, 100 nouveau-nés (âgés de ≤ 1 mois) présentant une infection à CMV congénitale symptomatique avec atteinte du SNC ont été traités pendant 6 semaines par 6 mg/kg de ganciclovir par voie intraveineuse toutes les 12 heures ou n’ont reçu aucun traitement. Sur les 100 patients recrutés, 42 répondaient à tous les critères de l’étude et ont fait l’objet d’évaluations audiométriques à l’inclusion et à 6 mois de suivi. Parmi ces patients, 25 ont reçu du ganciclovir et 17 n’ont reçu aucun traitement. L’audition s’est améliorée ou est demeurée normale à 6 mois par rapport à l’inclusion chez 21 des 25 patients du groupe ganciclovir par rapport à 10 des 17 patients du groupe témoin (84 % et 59 %, respectivement ; p = 0,06). Aucun des patients du groupe ganciclovir n’a présenté de dégradation de l’audition à 6 mois par rapport à l’inclusion, comparativement à 7 patients du groupe témoin (p < 0,01). Un an après l’inclusion, l’audition s’était dégradée chez 5/24 patients du groupe ganciclovir et 13/19 patients du groupe témoin (p < 0,01). Au cours de l’étude, 29/46 patients traités par ganciclovir ont présenté une neutropénie, par rapport à 9/43 patients du groupe témoin (p = 0,1). Neuf patients sont décédés au cours de l’étude, soit 3 patients du groupe ganciclovir et 6 patients du groupe témoin. Aucun décès n’a été lié au médicament de l’étude.
Au cours d’une étude randomisée et contrôlée de phase III, 100 nouveau-nés (âgés de 3 à 33 jours, âge médian : 12 jours) présentant une infection à CMV congénitale symptomatique sévère avec atteinte du SNC, ont reçu 6 mg/kg de ganciclovir par voie intraveineuse deux fois par jour pendant 6 semaines (n = 48) ou n’ont reçu aucun traitement antiviral (n = 52). L’évolution neurodéveloppementale à 6 et 12 mois a été meilleure chez les nourrissons qui avaient reçu du ganciclovir par rapport à ceux qui n’avaient pas reçu de traitement antiviral. Bien que les retards aient été moindres et que l’évolution neurologique ait été plus normale chez les nourrissons ayant reçu le ganciclovir, la plupart d’entre eux se situaient encore en deçà d’un développement considéré comme normal aux âges de 6 semaines, 6 mois ou 12 mois. La sécurité n’a pas été évaluée dans cette étude.
Une étude rétrospective a évalué l’effet d’un traitement antiviral sur la perte d’audition d’apparition tardive chez des nourrissons ayant présenté une infection à CMV congénitale (âgés de 4 à 34 mois, âge moyen : 10,3 ± 7,8 mois, âge médian : 8 mois). Cette étude a inclus 21 nourrissons dont l’audition était normale à la naissance, puis s’est dégradée tardivement. Le traitement antiviral a été l’un des suivants :
· ganciclovir par voie intraveineuse 5 mg/kg par jour pendant 6 semaines, suivi d’un traitement par valganciclovir oral 17 mg/kg deux fois par jour pendant 6 semaines puis une fois par jour jusqu’à l’âge de 1 an, ou
· valganciclovir par voie orale 17 mg/kg deux fois par jour pendant 12 semaines puis une fois par jour pendant 9 mois.
Aucun des enfants n’a nécessité d’implant cochléaire, et la perte d’audition s’est améliorée pour 83 % des oreilles affectées par une perte d’audition à l’inclusion. La survenue d’une neutropénie a été le seul effet indésirable rapporté et l’arrêt du traitement n’a été nécessaire chez aucun patient.
5.2. Propriétés pharmacocinétiques
L’exposition systémique (ASC0-∞) rapportée après l’administration d’une dose unique de 5 mg/kg de ganciclovir par perfusion intraveineuse de 1 heure chez des patients adultes transplantés hépatiques était en moyenne de 50,6 μg.h/mL (CV de 40 %). Chez cette population de patients, la concentration plasmatique maximale (Cmax) était en moyenne de 12,2 μg/mL (CV de 24 %).
Distribution
Le volume de distribution du ganciclovir administré par voie intraveineuse est corrélé au poids corporel. Le volume de distribution à l’état d’équilibre était compris entre 0,54 et 0,87 L/kg. La liaison aux protéines plasmatiques était de 1 % à 2 % pour des concentrations de ganciclovir de 0,5 et 51 μg/mL. Le ganciclovir pénètre dans le liquide céphalorachidien, dans lequel les concentrations observées étaient de 24 % à 67 % des concentrations plasmatiques.
Biotransformation
Le ganciclovir n'est pas métabolisé de manière significative.
Elimination
Le ganciclovir est principalement éliminé par excrétion rénale par filtration glomérulaire et sécrétion tubulaire active de ganciclovir sous forme inchangée. Chez les patients dont la fonction rénale est normale, plus de 90 % de la dose de ganciclovir administrée par voie intraveineuse est retrouvée sous forme inchangée dans l’urine au cours des 24 heures suivant l’administration. La clairance systémique moyenne a été de 2,64 ± 0,38 mL/min/kg (N = 15) à 4,52 ± 2,79 mL/min/kg (N = 6) et la clairance rénale de 2,57 ± 0,69 mL/min/kg (N = 15) à 3,48 ± 0,68 mL/min/kg (N = 20), correspondant à 90 % à 101 % du ganciclovir administré. La demi-vie a été de 2,73 ± 1,29 (N = 6) à 3,98 ± 1,78 heures (N = 8) chez des sujets sans insuffisance rénale.
Linéarité/non-linéarité
Les paramètres pharmacocinétiques du ganciclovir par voie intraveineuse sont linéaires de 1,6 à 5,0 mg/kg.
Patients présentant une insuffisance rénale
La clairance corporelle totale du ganciclovir est linéairement corrélée à la clairance de la créatinine. La clairance systémique moyenne a été de 2,1, 1 et 0,3 mL/min/kg chez des patients présentant une insuffisance rénale légère, modérée ou sévère. La demi-vie d’élimination est plus longue chez les patients présentant une insuffisance rénale. Chez les patients présentant une insuffisance rénale sévère, la demi-vie d’élimination était multipliée par 10 (voir rubrique 4.2 pour les modifications posologiques nécessaires chez les patients présentant une insuffisance rénale).
Patients présentant une insuffisance rénale sous hémodialyse
Les concentrations plasmatiques du ganciclovir ont diminué d’environ 50 % après une administration intraveineuse pendant une séance d’hémodialyse de 4 heures.
Durant une hémodialyse intermittente, la clairance estimée du ganciclovir a été de 42 à 92 mL/min, résultant en une demi-vie intra-dialyse de 3,3 à 4,5 heures. La fraction de ganciclovir éliminée durant une seule séance de dialyse a varié de 50 % à 63 %. Les estimations de l’élimination du ganciclovir pour une dialyse continue ont été plus basses (4,0-29,6 mL/min), mais ont indiqué une élimination plus élevée du ganciclovir pendant un intervalle de dose.
Patients présentant une insuffisance hépatique
La sécurité et l’efficacité du ganciclovir n’ont pas été étudiées chez les patients présentant une insuffisance hépatique. L’insuffisance hépatique ne devrait pas affecter la pharmacocinétique du ganciclovir puisqu’il est excrété au niveau rénal et, par conséquent, aucune recommandation particulière de posologie n’est donnée (voir rubrique 4.2).
Population pédiatrique
Les propriétés pharmacocinétiques du ganciclovir par voie IV administré à une dose de 200 mg/m2 ont été étudiées au cours de deux études menées chez des patients pédiatriques transplantés hépatiques (n = 18) et rénaux (n = 25) âgés de 3 mois à 16 ans et évaluées à l’aide d’un modèle pharmacocinétique de population. La clairance de la créatinine (ClCr) a été identifiée comme covariable statistiquement significative pour la clairance du ganciclovir et la taille du patient comme covariable statistiquement significative pour la clairance du ganciclovir, le volume de distribution à l’état d’équilibre et le volume de distribution périphérique. Quand la ClCr et la taille sont incluses dans le modèle, les différences apparentes de la pharmacocinétique du ganciclovir entre différents groupes d’âges ont été prises en compte et ni l’âge, ni le sexe, ni le type de greffe d’organe n’étaient une covariable significative dans ces populations. Le tableau 1 indique les paramètres pharmacocinétiques estimés par groupe d’âges.
Tableau 1 : Paramètres pharmacocinétiques médians (minimum-maximum) après administration de ganciclovir par voie IV basée sur la SC (200 mg/m2) chez des patients ayant subi une greffe d’organe solide rénale et hépatique.
|
|
< 6 ans n = 17 |
6 à < 12 ans n = 9 |
≥ 12 à < 16 ans n = 17 |
|
Cl (L/h) |
4,23 (2,11– 7,92) |
4,03 (1,88– 7,8) |
7,53 (2,89– 16,8) |
|
Vc (L) |
1,83 (0,45– 5,05) |
6,48 (3,34– 9,95) |
12,1 (3,6– 18,4) |
|
Vp (L) |
5,81 (2,9– 11,5) |
16,4 (11,3– 20,1) |
27 (10,6– 39,3) |
|
Vss (L) |
8,06 (3,35– 16,6) |
22,1 (14,6– 30,1) |
37,9 (16,5– 57,2) |
|
ASC0-24h (µg.h/mL) |
24,3 (14,1– 38,9) |
40,4 (17,7– 48,6) |
37,6 (19,2– 80,2) |
|
Cmax (µg/mL) |
12,1 (9,17– 15) |
13,3 (4,73– 15) |
12,4 (4,57– 30,8) |
De plus, les paramètres pharmacocinétiques du ganciclovir par voie intraveineuse d’une dose administrée selon le schéma posologique autorisé chez l’adulte (perfusion IV d’une dose de 5 mg/kg administrée pendant 1 heure) ont été étudiés chez un petit groupe de nourrissons et d’enfants âgés de 9 mois à 12 ans dont la fonction rénale était normale (n = 10, âge moyen : 3,1 ans). L’exposition mesurée par l’ASC0-∞ moyenne au Jour 1 (n = 10) et l’ASC0-12 au Jour 14 (n = 7) ont été de 19,4 ± 7,1 et 24,1 ± 14,6 μg.h/mL avec des valeurs correspondantes de la Cmax de 7,59 ± 3,21 μg/mL (Jour 1) et 8,31 ± 4,9 mg/mL (Jour 14), respectivement. Une tendance vers des expositions plus faibles chez des patients pédiatriques plus jeunes a été observée à une posologie basée sur le poids corporel dans cette étude. Chez les patients pédiatriques âgés de 5 ans maximum, les valeurs moyennes de l’ASC0-∞ au Jour 1 (n = 7) et l’ASC0-12h au Jour 14 (n = 4) étaient de 17,7 ± 5,5 μg.h/mL et de 17,1 ± 7,5 μg.h/mL.
Le schéma posologique du ganciclovir par voie IV basé sur la SC et la fonction rénale (3 × SC × ClCrS), dérivé de l’algorithme de dosage pédiatrique du valganciclovir, permet d’obtenir des expositions similaires au ganciclovir dans la population pédiatrique de la naissance à l’âge de 16 ans (voir tableau 2).
Tableau 2 : Simulation* de l’ASC0-24h du ganciclovir (μg.h/mL) chez les patients pédiatriques traités par une dose (mg) de ganciclovir de 3 × SC × ClCrS administrée par perfusion de 1 heure.
|
|
< 4 mois |
≥ 4 mois à ≤ 2 ans |
> 2 à < 6 ans |
≥ 6 à < 12 ans |
≥ 12 à ≤ 16 ans |
Tous les patients |
|
Nombre de patients (simulation) |
781 |
384 |
86 |
96 |
126 |
1,473 |
|
Médiane |
55,6 |
56,9 |
54,4 |
51,3 |
51,4 |
55,4 |
|
Moyenne |
57,1 |
58,0 |
55,1 |
52,6 |
51,8 |
56,4 |
|
Min |
24,9 |
24,3 |
16,5 |
23,9 |
22,6 |
16,5 |
|
Max |
124,1 |
133,0 |
105,7 |
115,2 |
94,1 |
133,0 |
|
Patients ASC < 40 µg.h/mL |
89 (11 %) |
38 (10 %) |
13 (15 %) |
23 (24 %) |
28 (22 %) |
191 (13 %) |
|
Patients ASC 40– 60 µg.h/mL |
398 (51 %) |
195 (51 %) |
44 (51 %) |
41 (43 %) |
63 (50 %) |
741 (50 %) |
|
Patients ASC > 60 µg.h/mL |
294 (38 %) |
151 (39 %) |
29 (34 %) |
32 (33 %) |
35 (28 %) |
541 (37 %) |
ASC = aire sous la courbe de la concentration plasmatique en fonction du temps ; SC = surface corporelle ; ClCr = clairance de la créatinine ; max = maximum ; min = minimum.
* Les simulations ont été effectuées à l’aide d’un modèle pharmacocinétique de population pédiatrique validée et de données démographiques issues de patients pédiatriques recevant un traitement par valganciclovir ou ganciclovir dans des études cliniques (n = 1 473 données enregistrées)
Personnes âgées
Aucune étude n’a été menée chez des adultes âgés de plus de 65 ans (voir rubrique 4.2).
5.3. Données de sécurité préclinique
Le ganciclovir altère la fertilité et est tératogène chez l’animal. Sur la base des études menées chez l’animal où le ganciclovir a induit une inhibition de la spermatogenèse à des expositions systémiques inférieures aux niveaux thérapeutiques, il est probable que le ganciclovir puisse inhiber la spermatogenèse humaine.
Hydroxyde de sodium (pour ajustement du pH), acide chlorhydrique (pour ajustement du pH).
2 ans
Après reconstitution :
Ne pas mettre au réfrigérateur. Ne pas congeler.
La stabilité physico-chimique en cours d'utilisation a été démontrée pour le produit reconstitué pendant 12 heures à 25 °C après dissolution avec de l'eau pour préparations injectables.
D'un point de vue microbiologique, le produit reconstitué doit être utilisé immédiatement, sauf si la méthode de reconstitution exclut le risque de contamination microbienne. En cas d’utilisation non immédiate, les durées et conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur.
Après dilution :
Ne pas congeler.
La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 24 heures à une température comprise entre 2 et 8 °C.
D'un point de vue microbiologique, le produit dilué doit être utilisé immédiatement, sauf si la méthode de reconstitution exclut tout risque de contamination microbienne. En cas d’utilisation non immédiate, les durées et conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Flacon en verre transparent à usage unique, d'un volume nominal de 20 mL, muni d’un bouchon en caoutchouc et d’une capsule amovible.
Disponible en boîtes de 1 ou 5 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
La prudence est de mise lors de la manipulation de GANCICLOVIR HIKMA.
Le ganciclovir étant considéré comme potentiellement tératogène et cancérogène chez l’être humain, la prudence s’impose lors de sa manipulation. Eviter l’inhalation ou le contact direct de la poudre contenue dans les flacons ou le contact direct de la solution reconstituée avec la peau ou les muqueuses. Les solutions de GANCICLOVIR HIKMA sont alcalines (pH ~11). En cas de contact, laver abondamment à l’eau et au savon ; rincer les yeux abondamment à l’eau claire.
Préparation du concentré reconstitué
La totalité de la reconstitution de GANCICLOVIR HIKMA lyophilisé doit être réalisée dans des conditions aseptiques.
1. La capsule amovible doit être retirée afin d’exposer les parties centrales du bouchon en caoutchouc. Prélever 10 mL d’eau pour préparations injectables dans une seringue, puis injecter lentement dans le flacon à travers le centre du bouchon en caoutchouc en dirigeant l’aiguille vers la paroi du flacon. Ne pas utiliser de l’eau pour préparations injectables à effet bactériostatique contenant des parabènes (para-hydroxybenzoates), car celle-ci est incompatible avec GANCICLOVIR HIKMA.
2. Le flacon doit être agité délicatement afin de mouiller totalement le produit.
3. Faire tourner/agiter délicatement le flacon pendant quelques minutes jusqu’à l’obtention d’une solution reconstituée limpide.
4. La solution reconstituée doit être examinée avec attention pour vérifier que le produit est en solution et pratiquement dépourvu de particules visibles avant dilution dans un solvant compatible. La solution reconstituée de GANCICLOVIR HIKMA est incolore à jaune pâle.
Pour les conditions de conservation du concentré reconstitué, voir rubrique 6.3.
Préparation de la solution diluée pour perfusion finale :
En se basant sur le poids du patient, le volume approprié doit être prélevé du flacon à l’aide d’une seringue puis dilué dans une solution pour perfusion appropriée. Ajouter un volume de 100 mL de diluant à la solution reconstituée. Les concentrations de solution pour perfusion supérieures à 10 mg/mL ne sont pas recommandées.
Les solutions de chlorure de sodium, de glucose à 5 %, de Ringer ou de Ringer lactate sont chimiquement ou physiquement compatibles avec GANCICLOVIR HIKMA.
GANCICLOVIR HIKMA ne doit pas être mélangé avec d’autres produits pour administration intraveineuse.
La solution diluée doit être administrée par perfusion intraveineuse pendant 1 heure comme indiqué dans la rubrique 4.2. Ne pas administrer par injection intramusculaire ou sous-cutanée car cela pourrait provoquer une irritation tissulaire sévère due au pH élevé (~11) de la solution de ganciclovir.
Pour les conditions de conservation de la solution diluée pour perfusion, voir rubrique 6.3.
Elimination
A usage unique exclusivement. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Hikma Farmacêutica (Portugal), S.A.
Estrada do Rio da Mó 8, 8A e 8B
Fervença
2705-906 Terrugem SNT
Portugal
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 916 6 5 : Poudre en flacon (verre). Boîte de 1.
· 34009 302 916 7 2 : Poudre en flacon (verre). Boîte de 5.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation : {JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 01/03/2024
GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion
Ganciclovir
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion ?
3. Comment utiliser GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Qu’est-ce que GANCICLOVIR HIKMA ?
GANCICLOVIR HIKMA contient la substance active ganciclovir. Elle appartient à un groupe appelé médicaments antiviraux.
Dans quels cas GANCICLOVIR HIKMA est-il utilisé ?
GANCICLOVIR HIKMA est utilisé pour traiter des maladies causées par un virus appelé cytomégalovirus (CMV) chez des patients adultes et adolescents âgés de 12 ans et plus ayant un système immunitaire affaibli. Il est également utilisé pour prévenir l’infection à CMV après une greffe d’organe ou pendant une chimiothérapie chez les adultes et les enfants dès la naissance.
· Le virus peut affecter n’importe quelle partie du corps. Il peut ainsi affecter la rétine au fond de l’œil, ce qui signifie qu’il peut provoquer des troubles de la vision.
· Le virus peut affecter tout le monde, mais il représente un problème particulier pour les personnes dont le système immunitaire est affaibli. Chez ces personnes, le virus CMV peut provoquer une maladie grave. Un affaiblissement du système immunitaire peut être dû à d’autres maladies (telles que le sida) ou à des médicaments (tels qu’une chimiothérapie ou des immunosuppresseurs).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion ?
N’utilisez jamais GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion
· si vous êtes allergique au ganciclovir, au valganciclovir ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si vous allaitez (voir sous-rubrique relative à l’allaitement).
N’utilisez pas GANCICLOVIR HIKMA si l’un des cas ci-dessus vous concerne. En cas de doute, adressez-vous à votre médecin, à votre pharmacien ou à votre infirmier/ère avant d’utiliser GANCICLOVIR HIKMA.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou infirmier/ère avant d’utiliser GANCICLOVIR HIKMA :
· si vous êtes allergique à l’aciclovir, au valaciclovir, au penciclovir ou au famciclovir, ce sont d’autres médicaments utilisés pour les infections virales ;
· si la numération de vos globules blancs, de vos globules rouges ou de vos plaquettes est basse votre médecin effectuera des analyses de sang avant le début de votre traitement et pendant celui-ci ;
· si des médicaments ont induit des anomalies du nombre de vos cellules sanguines par le passé ;
· si vous avez des problèmes rénaux votre médecin devra vous prescrire une plus faible dose et vérifiera plus souvent la numération de vos cellules sanguines pendant le traitement ;
· si vous êtes traité(e) par radiothérapie.
Si vous êtes dans l’une des situations ci-dessus (ou en cas de doute), adressez-vous à votre médecin, à votre pharmacien ou à votre infirmier/ère avant d’utiliser GANCICLOVIR HIKMA.
Attention aux effets indésirables
GANCICLOVIR HIKMA peut provoquer certains effets indésirables graves que vous devrez signaler immédiatement à votre médecin. Soyez attentif/ve aux effets indésirables graves répertoriés dans la rubrique 4 et si vous remarquez l’un d’entre eux au cours de votre traitement par GANCICLOVIR HIKMA, informez-en votre médecin. Votre médecin pourra vous demander d’arrêter votre traitement par GANCICLOVIR HIKMA et vous pourrez avoir besoin d’un traitement médical d’urgence.
Analyses et vérifications
Au cours de l’utilisation de GANCICLOVIR HIKMA, votre médecin effectuera des analyses de sang à intervalles réguliers. L’objectif de ces analyses sera de vérifier que la dose que vous recevez est adaptée à votre cas. Ces analyses de sang seront fréquemment effectuées au cours des 2 premières semaines. Elles seront effectuées moins souvent par la suite.
Enfants et adolescents
Les informations sur la sécurité d’emploi et l’efficacité du ganciclovir en tant que traitement dans les infections à CMV chez les enfants âgés de moins de 12 ans sont limitées. Les bébés et les nourrissons ayant reçu GANCICLOVIR HIKMA pour la prévention des infections à CMV devront effectuer régulièrement des bilans sanguins.
Autres médicaments et GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion
Informez votre médecin ou votre pharmacien si vous prenez/utilisez, avez récemment pris/utilisé ou pourriez prendre/utiliser tout autre médicament.
Prévenez particulièrement votre médecin ou votre pharmacien si vous prenez l’un des médicaments suivants :
· imipénème/cilastatine utilisé pour les infections bactériennes ;
· pentamidine utilisée pour les infections par des parasites ou les infections des poumons ;
· flucytosine, amphotéricine B utilisées pour des mycoses ;
· triméthoprime, triméthoprime/sulfaméthoxazole, dapsone utilisés pour les infections bactériennes ;
· probénécide utilisé pour la goutte ;
· mycophénolate mofétil, ciclosporine, tacrolimus utilisés après une greffe d’organe ;
· vincristine, vinblastine, doxorubicine utilisées pour le traitement du cancer ;
· hydroxyurée utilisée pour les troubles appelés « polycythémie », la drépanocytose et le cancer ;
· didanosine, stavudine, zidovudine, ténofovir ou tout autre médicament utilisé pour le traitement de l’infection à VIH ;
· adéfovir ou tout autre médicament utilisé dans le traitement de l’hépatite B.
Si vous êtes dans l’un des cas ci-dessus (ou en cas de doute), adressez-vous à votre médecin ou à votre pharmacien avant d’utiliser GANCICLOVIR HIKMA.
GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant d’utiliser ce médicament.
Grossesse
GANCICLOVIR HIKMA ne doit pas être utilisé chez la femme enceinte, sauf si les bénéfices pour la mère l’emportent sur les risques éventuels pour l’enfant à naître. Si vous êtes enceinte ou si vous pensez l’être, n’utilisez pas ce médicament sans l’avis de votre médecin, car GANCICLOVIR HIKMA pourrait être nocif pour l’enfant à naître.
Si vous débutez une grossesse ou si votre partenaire débute une grossesse au cours du traitement par GANCICLOVIR HIKMA, informez-en immédiatement votre médecin.
Contraception - Femmes
Vous ne devez pas débuter une grossesse au cours de l’utilisation de ce médicament, car il peut être nocif pour l’enfant à naître. Si vous êtes une femme en capacité de procréer, vous devez utiliser une méthode de contraception pendant l’utilisation de GANCICLOVIR HIKMA. Vous devez également utiliser une méthode de contraception pendant au moins 28 semaines après l’arrêt du traitement par GANCICLOVIR HIKMA.
Contraception - Hommes
Si vous êtes un homme et que votre partenaire est en capacité de procréer, vous devez utiliser un préservatif et une méthode de contraception supplémentaire pendant l’utilisation de GANCICLOVIR HIKMA. Faites de même pendant au moins 95 jours après l’arrêt du traitement par GANCICLOVIR HIKMA.
Allaitement
N’utilisez pas GANCICLOVIR HIKMA si vous allaitez. Si votre médecin souhaite que vous commenciez à utiliser GANCICLOVIR HIKMA, vous devez arrêter d’allaiter avant l’utilisation du médicament. Ceci est dû au fait que GANCICLOVIR HIKMA peut passer dans le lait maternel.
Fertilité
GANCICLOVIR HIKMA peut affecter la fertilité. GANCICLOVIR HIKMA peut entraîner un arrêt temporaire ou permanent de la production de sperme. Si vous planifiez d’avoir un enfant, adressez-vous à votre médecin ou pharmacien avant d’utiliser GANCICLOVIR HIKMA.
Conduite de véhicules et utilisation de machines
Une somnolence, des sensations vertigineuses, une confusion, des tremblements, des pertes d’équilibre ou des crises d’épilepsie peuvent se produire au cours de l’utilisation de GANCICLOVIR HIKMA. Si cela se produit, ne conduisez pas de véhicules et n’utilisez ni outils ni machines.
GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion contient du sodium
Ce médicament contient environ 43 mg de sodium (composant principal du sel de cuisine/table) par flacon de 500 mg. Cela équivaut à 2 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
3. COMMENT UTILISER GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion ?
GANCICLOVIR HIKMA vous sera administré par un médecin ou un(e) infirmier/ère. Il sera administré au moyen d’un petit tube inséré dans l’une de vos veines. Cette méthode est appelée « perfusion intraveineuse ». La durée de cette perfusion sera habituellement d’une heure.
La dose de GANCICLOVIR HIKMA varie d’un patient à l’autre. Le médecin déterminera la quantité dont vous avez besoin. Cette dose dépendra :
· de votre poids (chez les enfants, il est également recommandé de tenir compte de la taille) ;
· de votre âge ;
· de l’état de fonctionnement de vos reins ;
· du nombre de vos cellules sanguines ;
· du motif de l’administration du médicament.
La fréquence et la durée du traitement par GANCICLOVIR HIKMA varient également d’un patient à l’autre.
· Votre traitement commencera habituellement par une ou deux perfusions chaque jour.
· Si vous recevez deux perfusions par jour, vous continuerez de les recevoir pendant 21 jours.
· Le médecin pourra ensuite prescrire une perfusion par jour.
Personnes présentant des problèmes rénaux ou sanguins
Si vous avez des problèmes rénaux ou sanguins, votre médecin pourra vous prescrire une dose plus faible de GANCICLOVIR HIKMA et vérifier le nombre de vos cellules sanguines plus souvent pendant le traitement.
Si vous avez utilisé plus de GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion que vous n’auriez dû
Si vous pensez que vous avez reçu trop de GANCICLOVIR HIKMA, informez-en immédiatement votre médecin ou rendez-vous immédiatement à l’hôpital. Si vous avez reçu trop de GANCICLOVIR HIKMA, vous pourriez présenter les symptômes suivants :
· maux d’estomac, diarrhée ou nausées ;
· tremblements ou crises d’épilepsie ;
· sang dans les urines ;
· troubles du foie ou des reins ;
· modification du nombre de cellules sanguines.
Si vous oubliez d’utiliser GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion
Si vous arrêtez d’utiliser GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion
N’arrêtez jamais d’utiliser GANCICLOVIR HIKMA sans en parler à votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables suivants peuvent survenir avec ce médicament :
Effets indésirables graves
Informez immédiatement votre médecin si vous remarquez l’un des effets indésirables graves suivants votre médecin pourra arrêter votre traitement par GANCICLOVIR HIKMA et vous pourrez avoir besoin d’un traitement médical d’urgence :
Très fréquent : pouvant affecter plus de 1 personne sur 10
· faible nombre de globules blancs avec des signes d’infection tels que des maux de gorge, des ulcères buccaux ou de la fièvre ;
· faible nombre de globules rouges les signes comprennent une sensation d’essoufflement ou de fatigue, des palpitations ou une pâleur de la peau.
Fréquent : pouvant affecter jusqu’à 1 personne sur 10
· infection du sang (sepsis) les signes incluent fièvre, frissons, palpitations, confusion et troubles de l’élocution ;
· faible nombre de plaquettes les signes comprennent des saignements ou des ecchymoses (« bleus ») survenant plus facilement que d’habitude, du sang dans les urines ou les selles ou un saignement des gencives ; le saignement peut être sévère ;
· diminution sévère du nombre de cellules sanguines ;
· pancréatite les signes sont des maux d’estomac intenses qui se propagent dans votre dos ;
· crise d’épilepsie.
Peu fréquent : pouvant affecter jusqu’à 1 personne sur 100
· incapacité de la moelle osseuse à produire des cellules sanguines ;
· hallucinations entendre ou voir des choses qui n’existent pas ;
· pensées ou sentiments anormaux, perte de contact avec la réalité ;
· altération de la fonction rénale.
Rare : pouvant affecter jusqu’à 1 personne sur 1 000
· réaction allergique sévère les signes peuvent inclure une rougeur et des démangeaisons de la peau, un gonflement de la gorge, du visage, des lèvres ou de la bouche et des difficultés pour avaler ou respirer ;
Si vous remarquez l’un des effets indésirables ci-dessus, informez-en immédiatement votre médecin.
Autres effets indésirables
Si vous présentez l’un des effets indésirables suivants, informez-en immédiatement votre médecin, votre pharmacien ou votre infirmier/ère :
Très fréquent : pouvant affecter plus de 1 personne sur 10
· muguet et candidose buccale ;
· Infection des voies aériennes supérieures (par exemple, sinusite, angine) ;
· perte d’appétit ;
· maux de tête ;
· toux ;
· sensation d’essoufflement ;
· diarrhée ;
· nausées ou vomissements ;
· douleurs abdominales ;
· eczéma ;
· sensation de fatigue ;
· fièvre.
Fréquent : pouvant affecter jusqu’à 1 personne sur 10
· grippe ;
· infection urinaire les signes incluent de la fièvre, des mictions (action d’uriner) plus fréquentes et des douleurs en urinant ;
· infection de la peau et des tissus sous cutanés ;
· réaction allergique légère les signes peuvent être des rougeurs, des démangeaisons ;
· perte de poids ;
· idées dépressives, anxiété ou confusion ;
· troubles du sommeil ;
· sensation de faiblesse ou d’engourdissement des mains ou des pieds, ce qui peut affecter l’équilibre ;
· modifications du sens du toucher, fourmillements, picotements, sensation de piqûre ou de brûlure ;
· modifications du goût ;
· frissons ;
· inflammation de l’œil (conjonctivite), douleurs oculaires ou troubles de la vision ;
· douleur au niveau de l’oreille ;
· pression artérielle basse, ce qui peut entraîner des sensations vertigineuses ou des évanouissements ;
· difficultés pour avaler ;
· constipation, flatulences, indigestion, maux d’estomac, gonflement de l’abdomen ;
· aphtes ;
· résultats anormaux des bilans biologiques du foie et du rein ;
· sueurs nocturnes ;
· démangeaisons, éruption cutanée ;
· chute de cheveux ;
· douleurs au niveau du dos, des muscles ou des articulations, contractures musculaires ;
· sensations vertigineuses, faiblesse ou malaise général ;
· réaction de la peau à l’endroit où le médicament a été injecté, telle qu’une inflammation, une douleur et un gonflement.
Peu fréquent : pouvant affecter jusqu’à 1 personne sur 100
· sensation d’agitation ;
· tremblements, secousses ;
· surdité ;
· battements cardiaques irréguliers ;
· urticaire, peau sèche ;
· sang dans les urines ;
· infertilité chez les hommes voir rubrique « Fertilité » ;
· douleur thoracique.
Effets indésirables chez les enfants et les adolescents
Un faible nombre de globules blancs est plus probable chez les enfants, en particulier chez les bébés et les nouveau-nés.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette du flacon et l’étui après EXP. La date de péremption fait référence au dernier jour de ce mois.
Flacon de poudre :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Après reconstitution :
Ne pas mettre au réfrigérateur. Ne pas congeler.
La stabilité physico-chimique en cours d'utilisation a été démontrée pour le produit reconstitué pendant 12 heures à 25 °C après dissolution avec de l'eau pour préparations injectables.
D'un point de vue microbiologique, le produit reconstitué doit être utilisé immédiatement, sauf si la méthode de reconstitution exclut le risque de contamination microbienne. En cas d’utilisation non immédiate, les durées et conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur.
Après dilution :
Ne pas congeler.
La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 24 heures à une température comprise entre 2 et 8 °C.
D'un point de vue microbiologique, le produit dilué doit être utilisé immédiatement, sauf si la méthode de reconstitution exclut tout risque de contamination microbienne. En cas d’utilisation non immédiate, les durées et conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient GANCICLOVIR HIKMA 500 mg, poudre pour solution à diluer pour perfusion
· La substance active est :
Ganciclovir (sous forme de ganciclovir sodique).............................................................. 500 mg
Pour un flacon.
Après reconstitution avec 10 mL d’eau pour préparations injectables, chaque mL contient 50 mg de ganciclovir.
· Les autres composants sont : hydroxyde de sodium et/ou acide chlorhydrique pour ajustement du pH.
GANCICLOVIR HIKMA se présente sous forme d’une poudre blanche à blanc cassé pour solution à diluer pour perfusion, présentée dans un flacon unidose en verre muni d’un bouchon en caoutchouc et d’une capsule amovible.
La couleur des solutions reconstituées de GANCICLOVIR HIKMA varie de l'incolore au jaune pâle.
Les flacons de GANCICLOVIR HIKMA sont disponibles en boîtes de 1 ou 5.
Toutes les présentations peuvent ne pas être commercialisées
Titulaire de l’autorisation de mise sur le marché
Hikma Farmacêutica (Portugal), S.A.
Estrada do Rio da Mó 8, 8A e 8B
Fervença
2705-906 Terrugem SNT
Portugal
Exploitant de l’autorisation de mise sur le marché
HIKMA France
105 RUE MARCEL DASSAULT
92100 BOULOGNE-BILLANCOURT
Schiffgraben 23
38690 Goslar
allemagne
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{MM/AAAA}
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
MODE D’EMPLOI ET MANIPULATION
Pour plus d’informations, veuillez-vous reporter au Résumé des Caractéristiques du Produit.
La prudence est de mise lors de la manipulation de GANCICLOVIR HIKMA.
Le ganciclovir étant considéré comme potentiellement tératogène et cancérogène chez l’être humain, la prudence s’impose lors de sa manipulation. Eviter l’inhalation ou le contact direct de la poudre contenue dans les flacons ou le contact direct de la solution reconstituée avec la peau ou les muqueuses. Les solutions de GANCICLOVIR HIKMA sont alcalines (pH ~11). En cas de contact, laver abondamment à l’eau et au savon ; rincer les yeux abondamment à l’eau claire.
Mode d’administration
Mises en garde : le ganciclovir doit être administré par perfusion intraveineuse de 1 heure à une concentration ne dépassant pas 10 mg/mL. Ne pas administrer par injection intraveineuse rapide ou en bolus, car les concentrations plasmatiques excessives qui en résulteraient pourraient augmenter la toxicité du ganciclovir.
Ne pas administrer par injection intramusculaire ou sous-cutanée car cela pourrait provoquer une irritation tissulaire sévère due au pH élevé (~11) de la solution de ganciclovir.
La posologie, la fréquence et le débit de perfusion recommandés ne doivent pas être dépassés.
La perfusion doit être administrée dans une veine dont le débit sanguin est adéquat, de préférence au moyen d’une canule en plastique.
Préparation du concentré reconstitué
La totalité de la reconstitution de GANCICLOVIR HIKMA lyophilisé doit être réalisée dans des conditions aseptiques.
1. La capsule amovible doit être retirée afin d’exposer les parties centrales du bouchon en caoutchouc. Prélever 10 mL d’eau pour préparations injectables dans une seringue, puis injecter lentement dans le flacon à travers le centre du bouchon en caoutchouc en dirigeant l’aiguille vers la paroi du flacon. Ne pas utiliser de l’eau pour préparations injectables à effet bactériostatique contenant des parabènes (para-hydroxybenzoates), car celle-ci est incompatible avec GANCICLOVIR HIKMA.
2. Le flacon doit être agité délicatement afin de mouiller totalement le produit.
3. Faire tourner/agiter délicatement le flacon pendant quelques minutes jusqu’à l’obtention d’une solution reconstituée limpide.
4. La solution reconstituée doit être examinée avec attention pour vérifier que le produit est en solution et pratiquement dépourvu de particules visibles avant dilution dans un solvant compatible. La solution reconstituée de GANCICLOVIR HIKMA est incolore à jaune pâle.
Préparation de la solution diluée pour perfusion finale
En se basant sur le poids du patient, le volume approprié doit être prélevé du flacon à l’aide d’une seringue puis dilué dans une solution pour perfusion appropriée. Ajouter un volume de 100 mL de diluant à la solution reconstituée. Les concentrations de solution pour perfusion supérieures à 10 mg/mL ne sont pas recommandées.
Les solutions de chlorure de sodium, de glucose à 5 %, de Ringer ou de Ringer lactate sont chimiquement ou physiquement compatibles avec GANCICLOVIR HIKMA. GANCICLOVIR HIKMA ne doit pas être mélangé avec d’autres produits pour administration intraveineuse.
Elimination
A usage unique exclusivement. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.