Dernière mise à jour le 28/04/2026
SUBUTEX 0,4 mg, comprimé sublingual
Indications thérapeutiques
MEDICAMENT UTILISE DANS LA DEPENDANCE AUX OPIOIDES.
SUBUTEX 0,4 mg comprimé sublingual est utilisé pour traiter la dépendance aux opioïdes (stupéfiants). Ce médicament est exclusivement réservé au traitement de substitution des pharmacodépendances, dans le cadre d'un suivi médical et psycho social, chez les patients qui ont accepté d’être traités pour leur dépendance aux opioïdes.
Le traitement par SUBUTEX 0,4 mg comprimé sublingual est réservé aux adultes et adolescents de plus de 15 ans.
Présentations
> plaquette(s) thermoformée(s) nylon aluminium PVC-Aluminium de 7 comprimé(s)
Code CIP : 339 444-2 ou 34009 339 444 2 1
Déclaration de commercialisation : 19/02/1996
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 1,81 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 2,83 €
- Taux de remboursement :65%
Documents de bon usage du médicament
- Prévenir le risque de surdose d’opioïdes
Auteur : Haute autorité de santé
Type : Fiche Bon Usage du Médicament
Date de mise à jour :Avril 2023
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 06/12/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par SUBUTEX reste important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l\'aide)
Autres informations
- Titulaire de l'autorisation : INDIVIOR EUROPE LIMITED
- Conditions de prescription et de délivrance :
- délivrance fractionnée de 7 jours
- liste I
- prescription en toutes lettres sur ordonnance sécurisée
- prescription limitée à 4 semaines
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 302 593 5
ANSM - Mis à jour le : 19/06/2025
SUBUTEX 0,4 mg, comprimé sublingual
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chlorhydrate de buprénorphine.......................................................................................... 0,432 mg
Quantité correspondant à buprénorphine base.................................................................... 0,400 mg
pour un comprimé.
Excipient à effet notoire : lactose
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé blanc à crème, ovale, plat, aux bords biseautés avec la gravure « 04 » sur une face.
4.1. Indications thérapeutiques
Le traitement est réservé aux adultes et adolescents de plus de 15 ans, volontaires pour recevoir un traitement de la dépendance aux opioïdes.
4.2. Posologie et mode d'administration
Le traitement doit se faire sous le contrôle d’un médecin spécialisé dans la prise en charge de la dépendance/addiction aux opiacés.
Il est recommandé de prescrire le traitement par la buprénorphine dans le cadre d’une prise en charge globale de la dépendance aux opioïdes.
Le résultat du traitement dépend, d'une part, de la posologie prescrite et d'autre part, des mesures médico-psychologiques et socio-éducatives associées pour le suivi des patients.
Précautions à prendre avant l’induction du traitement
Avant d'instaurer le traitement, le médecin doit prendre en compte le type de dépendance aux opioïdes (opioïdes à durée d'action longue ou courte), l'intervalle de temps écoulé depuis la dernière prise d'opioïdes et le niveau de dépendance aux opioïdes. Afin d'éviter de précipiter l'apparition d'un syndrome de sevrage, l'induction du traitement par buprénorphine doit être effectuée dès l'apparition des signes objectifs et évidents de sevrage (démontré par ex., par un score indiquant un sevrage léger à modéré sur l'échelle clinique validée des symptômes de sevrage des opioïdes (COWS)).
Chez les patients dépendants à l’héroïne ou aux opioïdes à courte durée d’action, la première dose de buprénorphine doit être prise lors de l’apparition des premiers signes de sevrage mais doit intervenir au moins 6 heures après la dernière prise d’opioïdes.
Chez les patients recevant de la méthadone, la dose de méthadone doit être diminuée à une posologie maximum de 30 mg/jour avant de commencer le traitement par la buprénorphine. La longue demi-vie de la méthadone doit être prise en compte lors de l’instauration du traitement par buprénorphine. La première dose de buprénorphine ne doit être prise que lorsque les premiers signes de sevrage apparaissent et généralement pas moins de 24 heures après la dernière prise de méthadone. La buprénorphine peut précipiter l'apparition de symptômes de sevrage chez les patients dépendants à la méthadone.
Mise en place du traitement (induction) :
La dose initiale recommandée chez l'adulte et l'adolescent âgé de plus de 15 ans est de 2 à 4 mg. Pour minimiser les symptômes de sevrage indésirables et l’usage d’opioïdes conséquents, et maintenir le patient sous traitement, cette dose peut être répétée jusqu’à 12 mg au premier jour. Dans certains cas, selon la réponse du patient, la dose au jour 1 peut être augmentée sans dépasser 24 mg de buprénorphine. Pendant la phase d’instauration du traitement, il est recommandé de contrôler quotidiennement son administration afin de s’assurer que le comprimé est placé correctement sous la langue et d’observer la réponse du patient au traitement, ce qui permettra d’adapter efficacement la dose administrée en fonction de l’effet clinique obtenu chez le patient.
Adaptation posologique et traitement d'entretien :
Suite à l'induction du traitement le jour 1, le patient doit être stabilisé à une dose d'entretien adéquate déterminée individuellement. La titration de la buprénorphine permet d'obtenir une dose qui stabilise le patient et le maintien en traitement, guidé par une évaluation répétée de l'état clinique et psychologique du patient tel que la consommation et le besoin impérieux d'opioïdes, d'autres troubles addictifs associés, des symptômes d'anxiété et de dépression dus à des consommations ou des comorbidités psychiatriques.
Une dose quotidienne d'entretien de 16 mg ou plus est recommandée sans dépasser 24 mg de buprénorphine.
Pendant le traitement d'entretien, il est important de réévaluer périodiquement la dose (augmentation ou diminution) en fonction de l'état clinique et psychologique du patient, comme décrit ci-dessus.
La buprénorphine fait partie d'un traitement intégré décidé avec le patient (décision partagée), impliquant si besoin les pairs, la famille et les proches, et combinant médication, réduction des risques et soutien psychosocial déterminé individuellement comme des interventions motivationnelles, psychoéducation, psychothérapies spécifiques, amélioration des conditions de vie, médicaments pour les comorbidités psychiatriques, etc.
Cette approche centrée sur le patient doit permettre une rémission (disparition des consommations problématiques de substances) et un rétablissement ciblant le développement de nouvelles habitudes de vie et stratégies comportementales et la diminution des vulnérabilités qui contribueront à améliorer l'autonomie fonctionnelle des patients pour leurs projets de vie et leur qualité de vie, et prévenir les rechutes.
Une délivrance quotidienne de la buprénorphine est recommandée, notamment pendant la période d’instauration du traitement. Par la suite et après stabilisation de son état, des quantités de médicament pour plusieurs jours de traitement pourront être remises au patient. Il est recommandé, cependant, de limiter la quantité du médicament délivré en une fois à 7 jours au maximum.
Administration non quotidienne :
Après obtention d'une stabilisation satisfaisante, la fréquence d'administration du traitement peut être réduite à une administration tous les deux jours en doublant la dose quotidienne du patient. Par exemple, un patient stabilisé recevant une dose quotidienne de 8 mg de buprénorphine peut recevoir 16 mg de buprénorphine un jour sur deux, sans traitement les jours intermédiaires. Chez certains patients, après l'obtention d'une stabilisation satisfaisante, la fréquence d'administration du traitement peut être réduite à 3 administrations par semaine (par exemple lundi, mercredi et vendredi). La dose du lundi et du mercredi doit être égale à deux fois la dose quotidienne du patient, et la dose du vendredi doit être égale à trois fois la dose quotidienne du patient, sans traitement les jours intermédiaires. En aucun cas, la dose ne doit dépasser 24 mg de buprénorphine par jour. Cette posologie peut ne pas convenir aux patients nécessitant une dose quotidienne > 8 mg de buprénorphine/jour.
Objectifs du traitement, réduction des doses et arrêt du traitement (arrêt progressif) :
Avant de commencer le traitement par buprénorphine, une stratégie de traitement comprenant la durée du traitement et les objectifs du traitement doit être convenue avec le patient.
Le trouble de l’usage des opioïdes est un trouble chronique, fréquemment associé à des comorbidités psychiatriques (c’est-à-dire des pathologies duelles) et à d’autres vulnérabilités qui contribuent au risque élevé de rechute. Le traitement doit être administré aussi longtemps que nécessaire (plusieurs mois à plusieurs années) pour permettre le processus de rémission et de rétablissement. La durée du traitement d'entretien doit être déterminée individuellement avant d'envisager, par décision partagée, la réduction et l'arrêt de la buprénorphine.
Pendant le traitement, des contacts fréquents doivent être établis entre le médecin et le patient pour évaluer la nécessité de poursuivre le traitement, envisager son arrêt et ajuster les posologies si nécessaire. Lorsqu'un patient n'a plus besoin d'un traitement par buprénorphine, il peut être conseillé de réduire progressivement la dose pour prévenir les symptômes de sevrage (voir rubrique 4.4). La diminution doit être très progressive, au moins sur plusieurs mois, avec une réévaluation périodique comme mentionnée ci-dessus, pour s'assurer que le patient reste stable et éviter les rechutes et/ou le transfert de dépendance.
Lorsque l’évaluation clinique et la volonté du patient conduisent à envisager l’arrêt du traitement, celui-ci doit être effectué avec prudence. La décision d’arrêter le traitement par la buprénorphine après une période d'entretien ou de stabilisation brève doit être prise dans le cadre d’une prise en charge globale. Pour éviter des symptômes de sevrage et une rechute éventuelle, dans les cas favorables, la dose de buprénorphine peut être diminuée progressivement jusqu’à l’arrêt du traitement. Après une période de stabilisation jugée satisfaisante, si le patient l’accepte, le médecin pourra proposer au patient de diminuer progressivement sa dose de buprénorphine, jusqu'à un arrêt total du traitement de substitution dans les cas favorables. La mise à disposition de comprimés sublinguaux dosés respectivement à 0,4 mg, 2 mg et 8 mg permet une diminution progressive de la posologie. Durant la période d'arrêt du traitement, une attention particulière sera portée aux risques de rechute.
Populations particulières
Sujets âgés
La sécurité et l'efficacité de la buprénorphine chez les patients âgés de plus de 65 ans n’ont pas été établies. Aucune recommandation sur la posologie ne peut être donnée.
Insuffisance hépatique
Un bilan hépatique et la recherche d’une hépatite virale sont recommandés avant de commencer le traitement.
L’effet de l’insuffisance hépatique sur la pharmacocinétique de la buprénorphine a été évalué dans une étude réalisée après commercialisation. En raison de la métabolisation importante de la buprénorphine, on retrouve des taux plasmatiques de buprénorphine plus élevés chez les patients atteints d’insuffisance hépatique. L’exposition systémique est légèrement augmentée chez les sujets atteints d’une insuffisance hépatique légère et aucun ajustement de posologie n’est jugé nécessaire.
Après administration d’une dose unique de 2 mg, l’exposition systémique totale est significativement augmentée chez les sujets atteints d’insuffisance hépatique modérée (1,6 fois) et sévère (2,8 fois) comparée aux sujets sains. Les patients doivent être surveillés afin d’éviter les signes et symptômes de toxicité ou de surdosage causés par des taux élevés de buprénorphine. Subutex doit être utilisé avec précaution chez les patients atteints d’insuffisance hépatique modérée et une diminution de la dose initiale et de la dose d’entretien doit être considérée. Compte tenu d’une exposition élevée chez les patients atteints d’insuffisance hépatique sévère et d’une possible accumulation après l’administration de doses répétées, Subutex ne doit pas être utilisé chez les patients atteints d’insuffisance hépatique sévère (voir rubriques 4.3 et 5.2).
Les patients présentant une hépatite virale, sous traitement médical concomitant (voir rubrique 4.5) et/ou souffrant d’un dysfonctionnement hépatique ont un risque plus élevé d’atteinte accélérée du foie. Un bilan hépatique initial et la recherche d'une hépatite virale sont recommandés avant de commencer le traitement. Il est recommandé de contrôler régulièrement la fonction hépatique (voir rubrique 4.4).
Insuffisance rénale
La modification de la posologie de la buprénorphine n’est généralement pas nécessaire chez les patients atteints d’insuffisance rénale. La prudence est recommandée chez les patients présentant une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) (voir rubriques 4.4 et 5.2).
Population pédiatrique
La sécurité et l'efficacité de la buprénorphine chez les enfants âgés de moins de 15 ans n'ont pas été établies. Aucune donnée n'est disponible.
En raison de l'absence de données chez les adolescents (âgés de 15 à 17 ans), ces patients doivent être surveillés plus étroitement pendant le traitement.
Mode d’administration
Administration par voie sublinguale : les médecins doivent informer les patients que la voie sublinguale constitue la seule voie efficace et bien tolérée pour l'administration de ce médicament.
Le comprimé doit être maintenu sous la langue jusqu’à dissolution complète, ce qui intervient habituellement en 5 à 10 minutes. Les patients ne doivent pas avaler ou consommer des aliments ou des boissons avant la dissolution complète du comprimé.
Une dose se compose de comprimés de Subutex de différents dosages, qui peuvent être placés sous la langue simultanément ou en deux parts ; la deuxième part doit être placée sous la langue dès que le ou les comprimés de la première part sont dissous. Pour des instructions spécifiques concernant la posologie pendant le traitement d’induction, de stabilisation et d’entretien, se reporter aux sections ci-dessus intitulées « Mise en place du traitement (induction) » et « Adaptation posologique et traitement d’entretien ».
Hypersensibilité à la buprénorphine ou à l’un des excipients mentionnés à la rubrique 6.1.
Enfants de moins de 15 ans.
Insuffisance respiratoire sévère.
Insuffisance hépatique sévère.
Intoxication alcoolique aiguë ou delirium tremens.
Association à la méthadone, les analgésiques morphiniques de palier III, la naltrexone, et le nalméfène (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Ce médicament est exclusivement réservé au traitement de la pharmacodépendance aux opioïdes.
Utilisation chez l’adolescent- : en raison de l'absence de données chez l’adolescent (âgé de 15 à 17 ans), les patients appartenant à cette tranche d’âge doivent être plus étroitement surveillés pendant le traitement.
Il est recommandé que ce traitement soit prescrit par des médecins assurant une prise en charge thérapeutique globale de la dépendance aux opioïdes.
Mésusage, abus et usage détourné
Tout comme les autres opioïdes, licites ou illicites, la buprénorphine peut être mal utilisée ou utilisée de manière abusive. Parmi les risques de mésusage et d’abus figurent le surdosage, la propagation d’infections virales ou d’infections localisées et systémiques transmises par voie sanguine, la dépression respiratoire et l’atteinte hépatique. Le mauvais usage de la buprénorphine par une personne autre que le patient à qui le produit est destiné risque également de créer une nouvelle catégorie d'individus primodépendants à cette substance ; ce type d’utilisation peut aussi apparaître lorsque le médicament est distribué directement par le patient en vue d’un usage illicite ou lorsque le médicament est volé, n’étant pas conservé en lieu sûr.
En cas de mésusage intentionnel du médicament par voie intraveineuse, des réactions locales, parfois septiques (abcès, cellulite), des hépatites aiguës potentiellement graves et d'autres infections aiguës, telles que des pneumonies ou des endocardites, ont été rapportées.
Un traitement sous-optimal par la buprénorphine peut indiquer un mauvais usage du médicament par le patient, pouvant entraîner un surdosage ou l’abandon du traitement. Un patient sous-dosé en buprénorphine peut continuer à gérer ses symptômes de sevrage et son envie irrépressible de consommer des opioïdes, de l’alcool ou d’autres nooleptiques (par exemple des benzodiazépines).
Afin de réduire le risque de mésusage, d’abus et d’usage détourné, les médecins doivent prendre les mesures qui s’imposent lorsqu’ils prescrivent et administrent la buprénorphine, par exemple éviter de donner des ordonnances pour de multiples renouvellements dès le début de traitement ; d’autre part, ils doivent effectuer des visites de suivi du patient tout en mettant en place un contrôle clinique adapté aux besoins du patient.
Troubles respiratoires du sommeil
Les opioïdes peuvent provoquer des troubles respiratoires du sommeil, notamment l’apnée centrale du sommeil (ACS) et l’hypoxémie du sommeil. Le risque d’ACS augmente en fonction de la dose d’opioïdes utilisée. Chez les patients présentant une ACS, une diminution de la dose totale d’opioïdes doit être envisagée.
Dépression respiratoire
Des cas de décès par dépression respiratoire ont été observés, particulièrement lorsque la buprénorphine avait été utilisée en association avec des benzodiazépines (voir rubrique 4.5) ou lorsque la buprénorphine n’avait pas été utilisée conformément aux informations posologiques. Des décès ont également été rapportés après la prise concomitante de buprénorphine et d’autres dépresseurs tels que l’alcool ou d’autres opioïdes. L’administration de buprénorphine à des personnes non dépendantes aux opioïdes, qui ne sont pas tolérantes aux effets des opioïdes, peut entraîner une dépression respiratoire potentiellement mortelle.
Ce produit doit être utilisé avec précaution chez les patients atteints d’asthme ou d’insuffisance respiratoire (telle qu'une maladie pulmonaire obstructive chronique, un cœur pulmonaire, une diminution de la capacité respiratoire, une hypoxie, une hypercapnie, une dépression respiratoire préexistante ou une cyphoscoliose (déformation de la colonne vertébrale pouvant entraîner une dyspnée).
Les patients présentant les facteurs de risque physiques et/ou pharmacologiques ci-dessus doivent être surveillés et une réduction de la dose peut être envisagée.
La buprénorphine peut provoquer une dépression respiratoire sévère potentiellement mortelle chez les enfants et les personnes non dépendantes qui l’ingèrent accidentellement ou de manière délibérée. Les patients doivent être avertis de conserver les plaquettes en sûreté, de ne jamais sortir à l’avance les comprimés de la plaquette, de tenir les plaquettes hors de portée des enfants et des autres membres de la famille et de ne pas prendre ce médicament devant les enfants. Un service d’urgence doit être immédiatement contacté en cas d’ingestion accidentelle ou de suspicion d’ingestion.
Dépression du SNC
La buprénorphine peut provoquer une somnolence, en particulier lorsqu'il y a prise/administration concomitante avec de l’alcool ou avec des dépresseurs du système nerveux central (tels que benzodiazépines, tranquillisants, sédatifs ou hypnotiques) (voir rubriques 4.5 et 4.7).
L’utilisation concomitante de buprénorphine et de sédatifs tels que les benzodiazépines ou des médicaments apparentés peut entraîner une sédation, une dépression respiratoire, un coma et le décès. En raison de ces risques, la prescription concomitante de ces médicaments sédatifs doit être réservée aux patients pour lesquels il n’existe pas d’alternatives thérapeutiques. S’il est décidé de prescrire la buprénorphine avec des médicaments sédatifs, la dose minimale efficace des médicaments sédatifs doit être utilisée et la durée de traitement doit être la plus courte possible.
Les patients doivent être étroitement surveillés afin que des signes et symptômes de dépression respiratoire et de sédation puissent être détectés. À cet égard, il est fortement recommandé d’informer les patients et leurs soignants qu’ils doivent être vigilants à ces symptômes (voir rubrique 4.5).
Syndrome sérotoninergique
L’administration concomitante de Subutex et d’autres agents sérotoninergiques, tels que les inhibiteurs de la MAO, les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS), les inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSN) ou les antidépresseurs tricycliques, peut engendrer un syndrome sérotoninergique, qui est une maladie potentiellement mortelle (voir rubrique 4.5).
Si un traitement concomitant avec d’autres agents sérotoninergiques est justifié sur le plan clinique, il est conseillé d’observer attentivement le patient, tout particulièrement pendant l’instauration du traitement et les augmentations de dose.
Les symptômes du syndrome sérotoninergique peuvent comprendre des modifications de l’état mental, une instabilité autonome, des anomalies neuromusculaires et/ou des symptômes gastro-intestinaux.
En cas de suspicion de syndrome sérotoninergique, une réduction de dose ou un arrêt du traitement devra être envisagé(e) en fonction de la gravité des symptômes.
Tolérance et trouble de l’usage des opioïdes (abus et dépendance
Des études chez l’animal, ainsi que des données cliniques, ont démontré qu’en cas d’administration chronique, la buprénorphine, un agoniste partiel des récepteurs µ aux opioïdes, peut provoquer une dépendance, celle-ci étant toutefois moindre que celle provoquée par un agoniste complet (tel que la morphine).
Une tolérance, une dépendance physique et psychologique et un trouble de l'usage des opioïdes peuvent se développer lors de l'administration répétée d'opioïdes tels que la buprénorphine. L'abus ou le mésusage de buprénorphine peut entraîner un surdosage et/ou la mort. Le risque de développer un trouble de l'usage des opioïdes est accru chez les patients ayant des antécédents personnels ou familiaux (parents ou fratrie) de troubles liés à l'usage de substances (y compris les troubles liés à la consommation d'alcool), chez les fumeurs ou chez les patients ayant des antécédents personnels d'autres troubles de santé mentale (par ex. dépression majeure, anxiété et troubles de la personnalité).
Avant de commencer le traitement par buprénorphine et pendant le traitement, les objectifs du traitement et un plan d'arrêt doivent être convenus avec le patient (voir rubrique 4.2).
Les patients devront être surveillés pour détecter tout signe de comportement de recherche de médicaments (par exemple, demandes de renouvellement trop précoces). Cela comprend la surveillance de la prise concomitante d’opioïdes et de médicaments psychoactifs (comme les benzodiazépines). Pour les patients présentant des signes et symptômes du trouble de l’usage des opioïdes, une consultation avec un spécialiste en addictologie doit être envisagée.
L'interruption brutale du traitement peut entraîner un syndrome de sevrage, dont les premiers signes peuvent apparaître plus tard.
Hépatite, atteintes hépatiques
Des cas d’hépatite aiguë grave ont été rapportés lors de mésusage, notamment par voie intraveineuse (voir rubrique 4.8). Ces atteintes hépatiques ont surtout été observées à fortes doses, et pourraient être dues à une toxicité mitochondriale. Dans de nombreux cas, la présence d’un dysfonctionnement mitochondrial préexistant (maladie génétique, anomalies enzymatiques hépatiques, infection par le virus de l’hépatite B ou de l’hépatite C, abus d’alcool, anorexie, utilisation concomitante d’autres médicaments potentiellement hépatotoxiques) et la persistance d’injections de drogues peuvent être responsables de l’atteinte hépatique ou y contribuer.
Les patients présentant une hépatite virale, sous traitement médical concomitant (voir rubrique 4.5) et/ou souffrant d’un dysfonctionnement hépatique ont un risque plus élevé d’atteinte du foie et ces facteurs sous-jacents doivent être pris en compte avant la prescription de buprénorphine et au cours du traitement (voir rubrique 4.2).
En cas de suspicion d'atteinte hépatique, un bilan biologique et étiologique approfondi doit être pratiqué. En fonction des résultats obtenus, le traitement peut être interrompu avec prudence afin de prévenir l'apparition de symptômes de sevrage et d'éviter le retour à l'utilisation de drogues illicites. En cas de poursuite du traitement, il faudra étroitement surveiller la fonction hépatique.
Précipitation du syndrome de sevrage aux opioïdes
Lors de l'instauration du traitement par la buprénorphine, le médecin doit prendre en compte le profil agoniste partiel de la buprénorphine et être conscient que le traitement peut précipiter l’apparition d’un syndrome de sevrage chez les patients dépendants aux opioïdes, particulièrement si le traitement est administré moins de 6 heures après la dernière utilisation d’héroïne ou d’un autre opioïde à courte durée d’action, ou s’il est administré moins de 24 heures après la dernière prise de méthadone (conformément à la longue demi-vie de la méthadone). Les patients doivent être surveillés de près lors du passage de la méthadone à la buprénorphine car des symptômes de sevrage ont été signalés. Afin d’éviter de précipiter l’apparition d’un syndrome de sevrage, l’induction du traitement par buprénorphine doit être effectuée dès l’apparition des signes objectifs de sevrage (voir rubrique 4.2).
Les symptômes de sevrage peuvent aussi être associés à un sous-dosage.
Réactions allergiques
Des cas d’hypersensibilité aiguë et chronique à la buprénorphine ont été rapportés dans les études cliniques et après la mise sur le marché. Les signes et symptômes les plus fréquents sont : rash, urticaire et prurit. Des cas de bronchospasme, d’angiœdème et de choc anaphylactique ont été signalés. Des antécédents d’hypersensibilité à la buprénorphine constituent une contre-indication à l’utilisation de la buprénorphine.
Insuffisance hépatique
L’effet de l’insuffisance hépatique sur la pharmacocinétique de la buprénorphine a été évalué dans une étude avec administration d’une dose unique réalisée après commercialisation. En raison de la métabolisation importante de la buprénorphine, on retrouve des taux plasmatiques de buprénorphine plus élevés chez les patients atteints d’insuffisance hépatique modérée et sévère. Les patients doivent être surveillés afin d’éviter les signes et symptômes de toxicité ou de surdosage causés par des taux élevés de buprénorphine. Subutex doit être utilisé avec précaution chez les patients atteints d’insuffisance hépatique modérée. Chez les patients atteints d’insuffisance hépatique sévère, l’utilisation de la buprénorphine est contre-indiquée (voir rubriques 4.3 et 5.2).
Insuffisance rénale
L'élimination rénale peut être prolongée, car 30 % de la dose administrée sont éliminés par la voie rénale. Les métabolites de la buprénorphine s'accumulent chez les patients atteints d'insuffisance rénale. La prudence est recommandée chez les patients présentant une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) (voir rubriques 4.2 et 5.2).
Mises en garde générales relatives à la classe des opioïdes
Les opioïdes peuvent provoquer une hypotension orthostatique.
Les opioïdes peuvent augmenter la pression du liquide céphalo-rachidien, ce qui peut être à l’origine de crises épileptiques. Comme avec les autres opioïdes, la prudence est recommandée chez les patients traités par la buprénorphine qui présentent un traumatisme crânien, des lésions intracrâniennes et une augmentation de la pression intracrânienne ou qui ont des antécédents de crises épileptiques.
Un myosis induit par les opioïdes, des altérations du niveau de conscience ou de la perception de la douleur en tant que symptôme de la maladie peuvent interférer avec l’évaluation du patient ou compliquer le diagnostic ou le traitement clinique d’une maladie concomitante.
Les opioïdes doivent être utilisés avec précaution chez les patients atteints de myxœdème, d’hypothyroïdie ou d’insuffisance corticosurrénale (par exemple maladie d’Addison).
Les opioïdes doivent être utilisés avec précaution chez les patients atteints d’hypotension, d’hypertrophie prostatique ou de sténose urétrale.
Les opioïdes peuvent être responsables d’une augmentation de la pression intra-cholédocienne et doivent donc être utilisés avec précaution chez les patients présentant un dysfonctionnement des voies biliaires.
Les opioïdes doivent être administrés avec précaution chez les patients âgés ou affaiblis.
L’attention des sportifs doit être attirée sur le fait que cette spécialité contient de la buprénorphine et que ce principe actif est inscrit sur la liste des substances dopantes.
Les associations suivantes ne sont pas recommandées avec la buprénorphine : analgésiques de palier II, éthylmorphine et alcool (voir rubrique 4.5).
Excipients
Ce médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
+ Méthadone :
Diminution de l'effet de la méthadone par blocage compétitif des récepteurs, avec risque d’apparition d’un syndrome de sevrage.
+ Analgésiques morphiniques de palier III :
Chez les patients utilisant des analgésiques de palier III, une diminution de l'effet antalgique du morphinique peut être observée, par blocage compétitif des récepteurs, avec risque d'apparition d’un syndrome de sevrage.
La naltrexone et le nalméfène sont des antagonistes des opioïdes susceptibles de bloquer les effets pharmacologiques de la buprénorphine. Pour les patients dépendants aux opioïdes recevant un traitement par buprénorphine, la co-administration de naltrexone et de nalméfène est contre indiquée, la naltrexone et le nalméfène pouvant précipiter l'apparition brutale de symptômes de sevrage aux opioïdes prolongés et intenses.
+ Analgésiques de palier II (tramadol, codéine et dihydrocodéine) : Une diminution de l’effet analgésique du morphinique peut être observée, par blocage compétitif des récepteurs, avec risque d'apparition d’un syndrome de sevrage.
+ Ethylmorphine : Chez les patients utilisant de l’éthylmorphine, une diminution de l’effet analgésique du morphinique peut être observée, par blocage compétitif des récepteurs, avec risque d'apparition d’un syndrome de sevrage.
+ Alcool :
L’alcool augmente l’effet sédatif de la buprénorphine, ce qui peut rendre dangereuses la conduite de véhicules et l’utilisation de machines.
Les patients doivent éviter de prendre la buprénorphine avec des boissons alcoolisées ou des médicaments contenant de l’alcool.
Associations faisant l'objet de précautions d’emploi
+ Médicaments sédatifs tels que les benzodiazépines ou substances apparentées
L’utilisation concomitante d’opioïdes et de sédatifs tels que les benzodiazépines ou substances apparentées majore le risque de sédation, de dépression respiratoire, de coma et de décès en augmentant l’effet dépresseur du SNC. La dose du sédatif et la durée de son utilisation concomitante doivent être limitées Il convient de limiter autant que possible les doses et la durée de l’association (voir rubrique 4.4). Les patients doivent être informés qu’il est extrêmement dangereux de s’administrer soi-même des benzodiazépines qui n’ont pas été prescrites tout en prenant ce produit et doivent également être avertis qu’ils doivent suivre scrupuleusement les indications de leur médecin lorsqu’ils prennent des benzodiazépines (voir rubrique 4.4).
+ Gabapentinoïdes
L'utilisation concomitante de buprénorphine avec des gabapentinoïdes (gabapentine et prégabaline) peut entraîner une dépression respiratoire, une hypotension, une sédation profonde, un coma ou la mort (voir rubrique 4.4).
+ Autres dépresseurs du système nerveux central
D’autres dérivés opioïdes (par exemple les analgésiques et les antitussifs), certains antidépresseurs, antihistaminiques H1 sédatifs, barbituriques, benzodiazépines, anxiolytiques autres que benzodiazépines, neuroleptiques, clonidine et substances apparentées administrés en association avec la buprénorphine majorent la dépression du système nerveux central. L'altération de la vigilance peut rendre dangereuses la conduite de véhicules et l'utilisation de machines.
En outre, les barbituriques augmentent le risque de dépression respiratoire.
+ Médicaments sérotoninergiques, tels que les inhibiteurs de la MAO, les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS), les inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSN) ou les antidépresseurs tricycliques, car le risque de syndrome sérotoninergique, qui est une maladie potentiellement mortelle, est accru (voir rubrique 4.4).
+ Inhibiteurs du CYP3A4
Une étude d’interaction entre la buprénorphine et le kétoconazole (inhibiteur puissant du CYP3A4) a montré une augmentation des Cmax et ASC (aire sous la courbe) de la buprénorphine (d’environ 50 % et 70 % respectivement) et, dans une moindre mesure, de la norbuprénorphine. Les patients traités par la buprénorphine doivent être étroitement surveillés et une diminution de la posologie peut s’avérer nécessaire en cas d’association avec un inhibiteur puissant du CYP3A4 (par exemple les inhibiteurs de la protéase tels que ritonavir, nelfinavir ou indinavir ou les antifongiques azolés tels que kétoconazole, itraconazole, voriconazole ou posaconazole).
+ Inducteurs du CYP3A4
Dans une étude clinique réalisée sur des volontaires sains, l’association de buprénorphine avec la rifampicine ou la rifabutine montre une diminution de 70 % et 35 % respectivement des concentrations plasmatiques de buprénorphine et l’apparition de symptômes de sevrage chez 50 % des 12 volontaires. Une surveillance étroite est recommandée chez les patients traités par buprénorphine si des inducteurs (par exemple phénobarbital, carbamazépine, phénytoïne, rifampicine) sont co-administrés. Le dosage de buprénorphine ou des inducteurs du CYP3A4 peut être ajusté en conséquence.
+ Anticholinergiques
L'administration concomitante de buprénorphine avec des anticholinergiques ou des médicaments ayant une activité anticholinergique (par exemple, antidépresseurs tricycliques, antihistaminiques, antipsychotiques, myorelaxants, médicaments antiparkinsoniens) peut entraîner une augmentation des effets indésirables anticholinergiques.
4.6. Fertilité, grossesse et allaitement
Grossesse
Compte tenu des données disponibles et du bénéfice materno/foetal, la buprénorphine peut être utilisée pendant la grossesse. Cependant, une adaptation de la posologie quotidienne peut être nécessaire afin de maintenir l’efficacité du traitement.
La prise chronique de buprénorphine par la mère, quelle que soit la dose, à la fin de la grossesse, peut entraîner un syndrome de sevrage (cris aigus, mauvaise prise alimentaire, sommeil anormal, irritabilité, tremblement, hypertonie, myoclonie ou convulsions) chez le nouveau-né. Ce syndrome est généralement retardé de plusieurs heures à plusieurs jours après la naissance. Des cas de troubles respiratoires chez les nouveau-nés ont aussi été rapportés. Par conséquent, si la mère est traitée jusqu’à la fin de la grossesse, une surveillance doit être envisagée à la naissance et pendant plusieurs jours après.
De très petites quantités de buprénorphine et de ses métabolites passent dans le lait maternel. Ces quantités ne sont pas suffisantes pour éviter le syndrome de sevrage qui peut être retardé chez les nourrissons allaités. Après une évaluation des facteurs de risque individuels, l'allaitement peut être envisagé chez les patientes traitées par la buprénorphine.
Fertilité
Dans une étude conduite avec des doses pharmacologiques chez la souris, une atrophie testiculaire avec calcification tubulaire a été mise en évidence chez des animaux traités.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés lors de l’étude clinique pivot étaient les effets liés au syndrome de sevrage (par exemple insomnie, céphalées, nausées et hyperhidrose).
Liste tabulée des effets indésirables
Le Tableau 1 présente les effets indésirables les plus fréquemment rapportés lors d’une étude clinique pivot chez les patients traités avec la buprénorphine (n=103) versus placebo (n=107). La fréquence des effets indésirables éventuels répertoriés ci-dessous est définie selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000).
Le Tableau 1 récapitule aussi les effets indésirables les plus fréquemment rapportés dans la base de données de pharmacovigilance globale du titulaire, identifiés dans les études cliniques et la surveillance du produit après commercialisation. La fréquence est dite indéterminée quand l’effet indésirable n’a pas été identifié dans l’étude clinique pivot.
Tableau 1 : Effets indésirables observés dans une étude clinique pivot et/ou notifiés dans le cadre de la pharmacovigilance, présentés par classe de systèmes d’organes
|
Classe de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Fréquence indéterminée |
|
Infections et infestations |
Infection |
Pharyngite |
|
|
|
|
|
Troubles psychiatriques |
Insomnie |
Agitation Anxiété Nervosité |
|
Hallucination |
|
|
|
Affections du système nerveux |
Céphalée |
Migraine Paresthésie Somnolence Syncope Vertige Hyperkinésie |
|
|
|
|
|
Affections vasculaires |
|
Hypotension orthostatique |
|
|
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
Dyspnée |
|
Dépression respiratoire1 |
|
|
|
Affections gastro-intestinales |
Nausées Douleur abdominale |
Constipation Vomissement |
|
|
|
Caries dentaires |
|
Affections de la peau et du tissu sous-cutané |
Hyperhidrose |
|
|
|
|
|
|
Affections musculo-squelettiques et systémiques |
|
Spasmes musculaires |
|
|
|
|
|
Affections des organes reproducteurs et du sein |
|
Dysménorrhée Leucorrhée |
|
|
|
|
|
Troubles généraux et anomalies au site d’administration |
Syndrome de sevrage |
Asthénie |
|
|
|
Syndrome de sevrage néonatal2 |
|
Affections du système immunitaire |
|
|
|
|
|
Réactions d’hypersensibilité3 |
|
Affections hépatiques |
|
|
|
|
|
Augmentation des transaminases, hépatite, ictère4 |
Description de certains effets indésirables
Un résumé des effets indésirables signalés après commercialisation du produit et qui sont considérés comme graves ou présentant un intérêt est présenté ci-dessous :
1 Des cas de dépression respiratoire ont été observés. Des décès dus à une dépression respiratoire ont été rapportés, en particulier lorsque la buprénorphine était utilisée en association avec des benzodiazépines (voir rubrique 4.5) ou n’était pas utilisée conformément aux informations posologiques. Des décès ont également été rapportés lors de l’administration concomitante de buprénorphine et d’autres dépresseurs du SNC tels que l’alcool ou d’autres opioïdes (voir rubriques 4.4 et 4.5).
2 Un syndrome de sevrage néonatal a été signalé chez des nouveau-nés de femmes qui ont reçu de la buprénorphine pendant la grossesse. Le syndrome peut être plus léger et plus long que celui induit par des agonistes complets des récepteurs opioïdes µ à courte durée d’action. La nature du syndrome peut varier en fonction des antécédents d’utilisation de drogues par la mère (voir rubrique 4.6).
3 Les signes et symptômes les plus fréquents d'hypersensibilité comprennent les rash, les urticaires et les prurits. Des cas de bronchospasme, de dépression respiratoire, d'angiœdème et de choc anaphylactique ont été signalés.
4 Des cas d’élévation des transaminases hépatiques et d’hépatite avec ictère généralement d’évolution favorable ont été observés (voir rubrique 4.4).
Dépendance aux drogues
L'utilisation répétée de buprénorphine peut entraîner une pharmacodépendance, même à des doses thérapeutiques. Le risque de dépendance aux médicaments peut varier en fonction des facteurs de risque individuels du patient, de la posologie et de la durée du traitement aux opioïdes (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://www.signalement.social-sante.gouv.fr.
Les propriétés d'agoniste partiel morphinique de la buprénorphine lui confèrent un index thérapeutique élevé.
Symptômes
Le principal symptôme nécessitant une intervention médicale en cas de surdosage est la dépression respiratoire consécutive à une dépression du système nerveux central, car elle peut conduire à un arrêt respiratoire et à la mort (voir rubrique 4.4). Les autres signes d’un surdosage sont notamment la sédation, le myosis, l’hypotension, les nausées et les vomissements.
Traitement / Prise en charge
En cas de surdosage, une prise en charge globale doit être instituée, comprenant une surveillance étroite de l’état respiratoire et cardiaque du patient.
Un traitement symptomatique de la dépression respiratoire et des mesures standard de soins intensifs doivent être mis en place. La libération des voies aériennes supérieures ainsi qu’une ventilation assistée ou contrôlée doivent être assurées si nécessaire. Le patient doit être transféré dans une unité disposant de tous les moyens de réanimation nécessaires.
Si le patient vomit, des précautions doivent être prises afin d’éviter qu’il inhale son vomi.
L’utilisation d’un antagoniste opioïde injectable (à savoir la naloxone) est recommandée, malgré l’effet modeste qu’il peut exercer dans la suppression des symptômes respiratoires induits par la prise de buprénorphine, cette dernière étant fortement liée aux récepteurs morphiniques.
En cas d'utilisation de la naloxone, la longue durée d’action de la buprénorphine doit être prise en compte afin de déterminer la durée de traitement et la surveillance médicale nécessaires pour supprimer les effets du surdosage. La naloxone peut être éliminée plus rapidement que la buprénorphine ; par conséquent, les symptômes de surdosage de la buprénorphine préalablement contrôlés par la naloxone peuvent réapparaître. Une perfusion continue peut s’avérer nécessaire. Les débits de perfusion IV continue doivent être titrés selon la réponse du patient. Si celle-ci est impossible, une dose répétée de naloxone peut être requise.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : médicament utilisé dans la dépendance aux opioïdes, code ATC : N07BC01.
La buprénorphine est un agoniste-antagoniste morphinique et se fixe au niveau des récepteurs cérébraux m et k. Son activité dans le traitement de substitution des opioïdes est attribuée à sa liaison lentement réversible aux récepteurs m qui minimiserait de façon prolongée le besoin des toxicomanes en stupéfiants.
L'activité agoniste partielle de la buprénorphine confère au produit un index thérapeutique élevé en limitant ses effets dépresseurs, notamment sur les fonctions cardio-respiratoires. La marge thérapeutique de la buprénorphine peut être amoindrie en cas d’association à des benzodiazépines ou dans des situations de mésusage de la buprénorphine.
5.2. Propriétés pharmacocinétiques
Par voie orale, la buprénorphine subit une N-désalkylation et une glycuroconjugaison dans l'intestin grêle et dans le foie par un important effet de premier passage. L'administration du médicament par voie orale est donc inappropriée.
Par voie sublinguale, la biodisponibilité absolue de la buprénorphine est mal connue, mais a été estimée entre 15 et 30 %. Le pic de concentration plasmatique est obtenu 90 minutes après administration sublinguale, et la relation dose‑concentration maximale est linéaire entre 2 et 16 mg.
Distribution
L'absorption de la buprénorphine est suivie d'une phase de distribution rapide. La demi‑vie est de 2 à 5 heures.
Métabolisme et élimination
La buprénorphine est métabolisée par 14-N-désalkylation, et glycuroconjugaison de la molécule-mère et du métabolite désalkylé. Des données cliniques confirment que le CYP3A4 est responsable de la N-désalkylation de la buprénorphine.
La N‑désalkylbuprénorphine est un agoniste m de faible activité intrinsèque.
L'élimination de la buprénorphine est bi ou tri-exponentielle, avec une longue phase d'élimination terminale de 20 à 25 heures, due pour partie à une réabsorption de la buprénorphine après hydrolyse intestinale du dérivé conjugué, et pour partie au caractère hautement lipophile de la molécule.
La buprénorphine est essentiellement éliminée dans les fèces par excrétion biliaire des métabolites glycuroconjugués (70 %), le reste étant éliminé par les urines.
Populations particulières
Insuffisance hépatique
L’effet de l’insuffisance hépatique sur la pharmacocinétique de la buprénorphine et de la naloxone a été évalué dans une étude avec administration d’une dose unique réalisée après commercialisation.
Le tableau 2 résume les résultats d’une étude clinique dans laquelle l’exposition après administration de buprénorphine/naloxone 2 mg/0,5 mg comprimé sublingual a été déterminée chez des sujets sains, et chez des sujets présentant différents degrés d’insuffisance hépatique.
|
Tableau 2. Effet de l’insuffisance hépatique sur les paramètres pharmacocinétiques de la buprénorphine après administration de buprénorphine/naloxone (modification par rapport à des sujets sains) |
|||
|
Paramètres PK |
Insuffisance Hépatique Légère (Child-Pugh de Classe A) (n=9) |
Insuffisance Hépatique Modérée (Child-Pugh de Classe B) (n=8) |
Insuffisance Hépatique Sévère (Child-Pugh de Classe C) (n=8) |
|
Buprenorphine |
|||
|
Cmax |
1,2 fois plus |
1,1 fois plus |
1,7 fois plus |
|
ASCdéfini |
Identique |
1,6 fois plus |
2,8 fois plus |
L’exposition plasmatique à la buprénorphine était environ 3 fois plus élevée chez les patients ayant une insuffisance hépatique sévère après administration d’une dose unique de 2 mg.
5.3. Données de sécurité préclinique
Les études de tératogénèse réalisées chez le rat et le lapin permettent de conclure que la buprénorphine n’est ni embryotoxique ni fœtotoxique.
Aucun effet secondaire sur la fertilité n'a été rapporté chez le rat, cependant, une mortalité péri et post-natale élevée a été observée dans cette espèce après administration orale et IM, liée à une difficulté à mettre bas et à une lactation insuffisante.
Aucune preuve d'un potentiel génotoxique n'a été mise en évidence sur une batterie standard de tests.
Les études de cancérogénèse chez la souris et le rat ne montrent pas de différence d'incidence de différents types de tumeurs entre les animaux traités par la buprénorphine et le groupe témoin. Cependant, dans une étude conduite avec des doses pharmacologiques chez la souris, une atrophie testiculaire avec calcification tubulaire a été mise en évidence chez des animaux traités.
3 ans
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30°C.
A conserver dans l’emballage extérieur à l’abri de l’humidité.
6.5. Nature et contenu de l'emballage extérieur
7 comprimés sous plaquettes thermoformées nylon/aluminium/PVC/aluminium.
28 comprimés sous plaquettes thermoformées nylon/aluminium/PVC/aluminium.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
27 WINDSOR PLACE
DUBLIN 2
D02 DK44
IRLANDE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 339 444 2 1 ou 339 444 2 : 7 comprimés sous plaquettes thermoformées nylon/aluminium/PVC/aluminium.
· 34009 345 660 5 9 ou 345 660 5 : 28 comprimés sous plaquettes thermoformées nylon/aluminium/PVC/aluminium.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Prescription sur ordonnance répondant aux spécifications fixées par l’arrêté du 31 mars 1999.
Prescription limitée à 28 jours.
Délivrance fractionnée de 7 jours.
ANSM - Mis à jour le : 19/06/2025
SUBUTEX 0,4 mg, comprimé sublingual
buprénorphine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que Subutex 0,4 mg, comprimé sublingual et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre Subutex 0,4 mg, comprimé sublingual?
3. Comment prendre Subutex 0,4 mg, comprimé sublingual?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver Subutex 0,4 mg, comprimé sublingual ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE SUBUTEX 0,4 mg, comprimé sublingual ET DANS QUELS CAS EST-IL UTILISE ?
MEDICAMENT UTILISE DANS LA DEPENDANCE AUX OPIOIDES.
SUBUTEX 0,4 mg comprimé sublingual est utilisé pour traiter la dépendance aux opioïdes (stupéfiants). Ce médicament est exclusivement réservé au traitement de substitution des pharmacodépendances, dans le cadre d'un suivi médical et psycho social, chez les patients qui ont accepté d’être traités pour leur dépendance aux opioïdes.
Le traitement par SUBUTEX 0,4 mg comprimé sublingual est réservé aux adultes et adolescents de plus de 15 ans.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE SUBUTEX 0,4 mg, comprimé sublingual ?
Ne prenez jamais SUBUTEX 0,4 mg, comprimé sublingual :
· si vous êtes allergique (hypersensible) à la buprénorphine ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6.
· si vous avez moins de 15 ans,
· si vous avez une insuffisance respiratoire grave,
· si vous avez une maladie grave du foie,
· si vous présentez une intoxication alcoolique aiguë ou un delirium tremens (tremblements, sudation, anxiété, confusion ou hallucinations causés par l’alcool),
· si vous prenez de la méthadone,
· si vous prenez des analgésiques morphiniques de palier III,
· si vous prenez de la naltrexone,
· si vous prenez du nalméfène.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre SUBUTEX 0,4 mg comprimé sublingual.
Informez votre médecin en cas de :
· asthme ou autres problèmes respiratoires,
· maladie du foie telle que l’hépatite,
· tension artérielle basse,
· traumatisme crânien récent ou maladie cérébrale,
· affection des voies urinaires (en particulier si elle est liée à une hypertrophie de la prostate chez l’homme),
· néphropathie,
· problèmes thyroïdiens,
· insuffisance surrénalienne (par exemple maladie d’Addison),
· dysfonctionnement des voies biliaires,
· dépression ou autre maladie traitée par des antidépresseurs. L’utilisation concomitante de ces médicaments avec Subutex peut provoquer un syndrome sérotoninergique, une maladie potentiellement mortelle (voir « Autres médicaments et Subutex »).
Informations importantes à prendre en compte
· Mésusage, abus et usage détourné
Ce médicament peut être convoité par les individus qui utilisent de manière abusive des médicaments délivrés sur ordonnance et doit être conservé en un lieu sûr afin d’éviter tout vol. Ne donnez pas ce médicament à d’autres personnes. Il peut entraîner leur décès ou leur être nocif.
· Problèmes respiratoires
Certaines personnes sont décédées suite à une défaillance respiratoire (difficulté à respirer) lors d’une mauvaise utilisation de ce médicament ou lors de la prise concomitante d’autres dépresseurs du système nerveux central tels que l’alcool, les benzodiazépines (tranquillisants) ou d’autres opioïdes.
Troubles respiratoires du sommeil
Subutex peut provoquer des troubles respiratoires du sommeil, tels que l’apnée du sommeil (pause respiratoire pendant le sommeil) et l’hypoxémie du sommeil (faible taux d’oxygène dans le sang). Les symptômes peuvent inclure des pauses respiratoires pendant le sommeil, un réveil nocturne dû à un essoufflement, des difficultés à maintenir le sommeil ou une somnolence excessive pendant la journée. Si vous ou une autre personne observe(z) ces symptômes, contactez votre médecin. Une réduction de dose peut être envisagée par votre médecin.
· Tolérance, dépendance et addiction
Ce produit peut créer une dépendance.
Le risque de dépendance ou d’addiction varie d'une personne à l'autre. Ce risque de dépendance ou d’addiction à la buprénorphine peut être plus élevé si :
· Vous ou un membre de votre famille avez déjà consommé abusivement de l’alcool ou avez été dépendant de l'alcool, de médicaments sur ordonnance ou de drogues illégales (« addiction »).
· Vous êtes fumeur.
· Vous avez déjà eu des problèmes d'humeur (dépression, anxiété ou trouble de la personnalité) ou avez été traité par un psychiatre pour d'autres maladies mentales.
Si vous remarquez l'un des signes suivants pendant que vous prenez la buprénorphine, cela peut être le signe d'une dépendance ou d'une addiction :
· Vous devez prendre le médicament plus longtemps que ce que vous a conseillé votre médecin.
· Vous devez dépasser la dose recommandée
· Vous utilisez le médicament pour des raisons autres que celles prescrites, par exemple « pour rester calme » ou « pour vous aider à dormir ».
· Vous avez fait des tentatives répétées et infructueuses pour arrêter ou contrôler l'utilisation du médicament.
· Lorsque vous arrêtez de prendre le médicament, vous vous sentez mal et vous vous sentez mieux lorsque vous reprenez le médicament (« effets de sevrage »).
Si vous remarquez l'un de ces signes, parlez-en à votre médecin pour discuter de la meilleure voie de traitement pour vous, y compris quand il est approprié d'arrêter et comment arrêter en toute sécurité (voir la section 3, Si vous arrêtez de prendre Subutex).
· Symptômes de sevrage
Ce produit peut entraîner l’apparition de symptômes de sevrage si vous le prenez moins de 6 heures après la prise d’un opioïde à courte durée d’action (morphine, héroïne ou produits apparentés) ou moins de 24 heures après la prise d’un opioïde à longue durée d’action tel que la méthadone.
SUBUTEX 0,4 mg comprimé sublingual peut également provoquer des symptômes de sevrage si vous arrêtez de le prendre brutalement.
· Lésion du foie
Des cas de lésion du foie ont été rapportés suite à la prise de SUBUTEX 0,4 mg comprimé sublingual, notamment lors d’un mauvais usage du médicament par voie intraveineuse et à forte dose. Celles-ci peuvent aussi être favorisées par certaines conditions telles que des infections virales (virus de l’hépatite B ou C), alcoolisme, anorexie ou par l’association avec d’autres médicaments présentant un risque pour votre foie (voir rubrique 4). Votre médecin peut faire pratiquer des tests sanguins réguliers pour surveiller l’état de votre foie. Prévenez votre médecin si vous souffrez de problèmes hépatiques avant de commencer un traitement par SUBUTEX 0,4 mg, comprimé sublingual.
· Somnolence
Ce médicament peut entrainer une somnolence qui peut être augmentée par des produits tels que les boissons alcoolisées ou les médicaments traitant l’anxiété.
· Diagnostic des affections médicales non apparentées
Ce médicament peut masquer les symptômes de douleur susceptibles de contribuer au diagnostic de certaines maladies. N’oubliez pas d’avertir votre médecin si vous prenez ce médicament.
· Tension artérielle
Ce médicament peut provoquer une baisse soudaine de la tension artérielle, provoquant une sensation de vertige lors du passage trop rapide de la position assise ou allongée à la position debout.
Une prescription et une délivrance pour une courte durée sont recommandées, notamment en début de traitement.
Adressez-vous à votre médecin ou pharmacien avant de prendre SUBUTEX 0,4 mg comprimé sublingual.
Autres médicaments et SUBUTEX 0,4 mg, comprimé sublingual
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
N’utilisez pas SUBUTEX 0,4 mg, comprimé sublingual si vous prenez :
· de la méthadone,
· des analgésiques morphiniques (analgésiques de palier III),
· de la naltrexone,
· du nalméfène.
Certaines associations avec SUBUTEX 0,4 mg comprimé sublingual ne sont pas recommandées :
· tramadol, codéine, dihydrocodéine (analgésiques de palier II)
· éthylmorphine,
· alcool ou médicaments contenant de l’alcool.
Certains médicaments peuvent augmenter les effets indésirables de SUBUTEX 0,4 mg comprimé sublingual et risquent d’entraîner des réactions très graves. Ne prenez pas d’autres médicaments en concomitance avec SUBUTEX 0,4 mg comprimé sublingual sans en parler d’abord à votre médecin, notamment :
· Benzodiazépines (utilisées pour traiter l’anxiété ou les troubles du sommeil) telles que le diazépam, le témazépam et l’alprazolam. L’utilisation concomitante de Subutex et de médicaments sédatifs tels que les benzodiazépines et les médicaments apparentés augmente le risque de somnolence, de difficultés pour respirer (dépression respiratoire), de coma et peut engager le pronostic vital. Pour cette raison, l’utilisation concomitante ne doit être envisagée que lorsqu’il n’existe pas d’alternatives thérapeutiques. Cependant, si votre médecin prescrit Subutex avec des sédatifs, la dose et la durée du traitement concomitant doivent être limitées. Informez votre médecin de tous les sédatifs que vous prenez et suivez strictement ses recommandations concernant les doses. Il pourrait être utile d’apprendre à vos amis ou à vos proches à reconnaître les signes et symptômes mentionnés ci-dessus. Contactez votre médecin si vous présentez ces symptômes.
· Gabapentine ou prégabaline pour traiter l'épilepsie ou les douleurs dues à des problèmes nerveux (douleurs neuropathiques)
· Autres médicaments pouvant entraîner une somnolence qui sont utilisés pour traiter des affections telles que l’anxiété, l’insomnie, les convulsions/crises d’épilepsie et la douleur. Ces types de médicaments diminueront votre niveau de vigilance en rendant difficile la conduite de véhicules et l’utilisation de machines. Ils peuvent également provoquer une dépression du système nerveux central, ce qui est très grave : l’utilisation de ces médicaments doit être surveillée étroitement. Une liste d’exemples de ces types de médicaments est présentée ci-dessous :
o les autres opioïdes, certains analgésiques et antitussifs,
o les antidépresseurs (utilisés pour traiter la dépression) tels que l’isocarboxazide et le valproate,
o les antihistaminiques H1 (utilisés pour traiter les réactions allergiques) tels que la diphenhydramine et la chlorphénamine,
o les barbituriques (utilisés pour provoquer le sommeil ou la sédation) tels que le phénobarbital ou l’hydrate de chloral
· Des antidépresseurs tels que le moclobémide, la tranylcypromine, le citalopram, l’escitalopram, la fluoxétine, la fluvoxamine, la paroxétine, la sertraline, la duloxétine, la venlafaxine, l’amitriptyline, la doxépine, ou la trimipramine. Ces médicaments peuvent interagir avec Subutex et vous pouvez présenter des symptômes tels que des contractions rythmiques involontaires des muscles, y compris des muscles qui contrôlent les mouvements de l'œil, une agitation, des hallucinations, un coma, une transpiration excessive, des tremblements, une exagération des réflexes, une augmentation de la tension musculaire, une température corporelle supérieure à 38 °C. Contactez votre médecin si vous ressentez ces symptômes.
· La clonidine (utilisée pour traiter une tension artérielle élevée)
· Les antirétroviraux (utilisés pour traiter le SIDA) tels que le ritonavir, le nelfinavir et l’indinavir,
· Certains agents antifongiques (utilisés pour traiter les infections fongiques) tels que le kétoconazole, l’itraconazole, le voriconazole ou le posaconazole et certains antibiotiques (macrolides)
· Les médicaments utilisés pour traiter le mal des transports ou les nausées (antihistaminiques ou antiémétiques) ;
· Les médicaments destinés au traitement des troubles psychiatriques (antipsychotiques ou neuroleptiques) ;
· Les relaxants musculaires ;
· Les médicaments pour traiter la maladie de Parkinson.
Certains médicaments peuvent diminuer les effets de SUBUTEX 0,4 mg comprimé sublingual et doivent être utilisés avec prudence quand ils sont co-administrés avec SUBUTEX 0,4 mg comprimé sublingual. Parmi ces produits figurent :
· les médicaments utilisés pour traiter l'épilepsie (tels que la carbamazépine, le phénobarbital ou la phénytoïne)
· les médicaments utilisés pour traiter la tuberculose (rifampicine).
La prise concomitante de SUBUTEX 0,4 mg comprimé sublingual avec les médicaments mentionnés ci-dessus doit être étroitement surveillée et peut nécessiter dans certains cas un ajustement des doses par votre médecin.
Vous devez dire à votre médecin ou pharmacien, si vous prenez ou avez pris récemment un autre médicament, y compris un médicament obtenu sans ordonnance.
SUBUTEX 0,4 mg, comprimé sublingual avec des aliments, boissons et de l’alcool
L’alcool peut augmenter la somnolence et le risque de défaillance respiratoire s’il est associé à la prise de SUBUTEX 0,4 mg comprimé sublingual. Vous ne devez pas boire d’alcool ni prendre des médicaments contenant de l’alcool pendant le traitement par SUBUTEX 0,4 mg comprimé sublingual.
Ne pas avaler ou consommer des aliments ou des boissons tant que le comprimé n'est pas complètement dissous.
Grossesse et allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
La buprénorphine peut être utilisée pendant la grossesse.
En cas de prise au cours de la grossesse, plus particulièrement à la fin de la grossesse, les médicaments tels que SUBUTEX 0,4 mg comprimé sublingual peuvent entraîner des symptômes de sevrage et des problèmes respiratoires chez le nouveau-né. Ces symptômes peuvent survenir plusieurs jours après la naissance.
Avant d’allaiter votre enfant, consultez votre médecin : il évaluera vos facteurs de risque personnels et vous dira si vous pouvez allaiter pendant le traitement par ce médicament.
Sportifs
Attention ce médicament contient de la buprénorphine qui est inscrite sur la liste des substances dopantes.
Conduite de véhicules et utilisation de machines
SUBUTEX 0,4 mg, comprimé sublingual peut provoquer une somnolence, des vertiges ou une altération de la pensée. Cela peut surtout se produire pendant les premières semaines de traitement ou lorsque votre dose est modifiée, mais également lorsque vous consommez de l'alcool ou prenez d'autres sédatifs avec SUBUTEX 0,4 mg, comprimé sublingual. Vous ne devez pas conduire ni utiliser des outils ou des machines, ni entreprendre des activités dangereuses tant que vous ne savez pas de quelle manière ce médicament vous affecte.
Demandez conseil à votre médecin ou pharmacien.
SUBUTEX 0,4 mg, comprimé sublingual contient du lactose et du sodium.
Ce médicament contient du lactose. Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE SUBUTEX 0,4 mg, comprimé sublingual ?
Instauration du traitement
La dose initiale recommandée chez l'adulte et l'adolescent âgé de plus de 15 ans est de 2 à 4 mg de Subutex. Cette dose peut être répétée jusqu’à 12 mg le premier jour en fonction de vos besoins. Dans certains cas, selon la réponse du patient, la dose au jour 1 peut être augmentée sans dépasser 24 mg de buprénorphine.
Vous devez présenter des signes manifestes de sevrage avant de prendre la première dose de Subutex. L'évaluation du médecin au regard de votre aptitude à recevoir le traitement permettra de décider du moment où vous prendrez votre première dose de Subutex.
· Instauration du traitement par Subutex lors d’une dépendance à l’héroïne
Si vous êtes dépendant à l'héroïne ou à un opioïde à courte durée d'action, vous devez prendre votre première dose de Subutex lors de l'apparition des premiers signes de sevrage, au moins 6 heures après la dernière prise d'opioïdes.
· Instauration du traitement par Subutex lors d’une dépendance à la méthadone
Si vous prenez de la méthadone ou un opiacé à action prolongée, idéalement la dose de méthadone doit être réduite de manière à être inférieure à 30 mg/jour avant de commencer le traitement par Subutex. La première dose de Subutex doit être prise lorsqu'apparaissent les premiers signes de sevrage, mais doit intervenir au moins 24 heures après la dernière prise de méthadone.
Prise de Subutex
La voie sublinguale constitue la seule voie efficace et bien tolérée pour la prise de ce médicament.
· Prenez la dose en plaçant les comprimés sous la langue.
· Conservez les comprimés sous votre langue jusqu'à leur dissolution complète. Cela peut prendre 5 à 10 minutes.
· Ne mâchez pas et n’avalez pas les comprimés, sous peine d’annuler leur effet et de risquer de provoquer des symptômes de sevrage.
· Ne consommez pas des aliments ou des boissons tant que les comprimés ne sont pas complètement dissous.
Adaptation posologique et dose d'entretien :
Au cours des jours qui suivent le début de votre traitement, votre médecin peut augmenter votre dose de Subutex en fonction de vos besoins. Si vous avez l'impression que l'effet de Subutex est trop fort ou trop faible, parlez-en à votre médecin ou votre pharmacien. La dose quotidienne maximale est de 24 mg.
Après une période pendant laquelle le traitement aura été un succès, vous pourrez vous mettre d'accord avec votre médecin afin de diminuer progressivement la dose jusqu'à parvenir à une dose d'entretien plus faible.
Arrêt du traitement
La durée de traitement doit être déterminée individuellement pour chaque patient (plusieurs mois à plusieurs années) avant d'envisager, par décision partagée avec votre médecin, la réduction et l'arrêt de la buprénorphine. La diminution doit être très progressive, au moins sur plusieurs mois, avec une réévaluation périodique, pour s'assurer que vous restez stable et éviter les rechutes et/ou le transfert de dépendance.
Un arrêt de traitement, par diminution progressive de la posologie, peut être envisagé, avec votre accord, après une stabilisation suffisante de votre état. Les doses doivent être diminuées par paliers successifs sous surveillance de votre médecin.
Ne pas modifier ou arrêter le traitement sans l'accord de votre médecin traitant.
L’efficacité du traitement dépend :
· de la posologie,
· des mesures médico-psychologiques et socio-éducatives associées.
Si vous avez l’impression que l’effet de SUBUTEX 0,4 mg, comprimé sublingual est trop fort ou trop faible, consultez votre médecin ou votre pharmacien.
Si vous avez pris plus de SUBUTEX 0,4 mg, comprimé sublingual que vous n’auriez dû
Un surdosage en buprénorphine nécessite la mise sous surveillance médicale du patient et éventuellement un traitement en urgence à l'hôpital. Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de prendre SUBUTEX 0,4 mg, comprimé sublingual
Si vous avez oublié de prendre une dose, informez votre médecin le plus tôt possible. Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre SUBUTEX 0,4 mg, comprimé sublingual
Ne modifiez pas le traitement de quelque manière que ce soit et n'arrêtez pas le traitement sans l'accord de votre médecin traitant. Un arrêt brutal du traitement peut entraîner l’apparition de symptômes de sevrage.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations <à votre médecin ou à votre pharmacien.
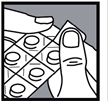
Comment retirer le comprimé de la plaquette thermoformée ?
1- Découper une seule alvéole de la plaquette en suivant les pointillés. 1
2- Décoller le film à partir du coin non scellé dans le sens de la flèche afin de libérer le comprimé. 2
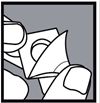
Si la plaquette est endommagée, jetez le comprimé.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Parlez-en immédiatement à votre médecin ou consultez les urgences médicales si vous présentez :
· un gonflement du visage, des lèvres, de la langue ou de la gorge qui peut entraîner une difficulté à avaler ou à respirer, de l'urticaire/rash sévère. Ceci peut être des signes d'une réaction allergique pouvant engager le pronostic vital.
Parlez-en aussi à votre médecin si vous présentez :
· une grande fatigue, des démangeaisons avec jaunissement de la peau ou des yeux. Ceci peut être des symptômes d’une lésion du foie.
Les effets indésirables suivants observés avec la buprénorphine ont été rapportés en utilisant la convention suivante :
· très fréquent (affecte plus de 1 patient sur 10) ;
· fréquent (affecte jusqu’à 1 patient sur 10) ;
· peu fréquent (affecte jusqu’à 1 patient sur 100) ;
· rare (affecte jusqu’à 1 patient sur 1 000) ;
· très rare (affecte jusqu’à 1 patient sur 10 000) ;
· fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Effets indésirables très fréquents :
· Infection,
· Insomnie (incapacité à dormir),
· Céphalée (maux de tête),
· Nausées,
· Douleur abdominale,
· Sueur excessive,
· Syndrome de sevrage.
Effets indésirables fréquents :
· Pharyngite,
· Agitation,
· Anxiété,
· Nervosité,
· Migraine,
· Paresthésie (picotement et engourdissement),
· Somnolence,
· Evanouissement,
· Vertige,
· Hyperkinésie (hyperactivité),
· Chute de la tension artérielle lors du passage de la position assise ou couchée à la position debout,
· Dyspnée (difficulté à respirer),
· Constipation,
· Vomissement,
· Spasme musculaire,
· Règles douloureuses,
· Perte vaginale blanche,
· Fatigue.
Effets indésirables rares :
· Hallucination,
· Dépression respiratoire (difficulté sévère à respirer).
Effets indésirables dont la fréquence est indéterminée :
· Caries dentaires,
· Syndrome de sevrage médicamenteux néonatal,
· Réactions d’hypersensibilité telles que rash, urticaire, démangeaisons,
· Réactions sévères d’hypersensibilité telles que bronchospasme (contraction soudaine des muscles des bronches), dépression respiratoire, gonflement de la face, des lèvres, de la langue et/ou de la gorge, qui peuvent être des signes de réaction allergique pouvant engager le pronostic vital,
· Problèmes de foie avec ou sans jaunisse.
Tous les opioïdes peuvent causer les effets indésirables additionnels suivants : crises d’épilepsie, myosis (contraction de la pupille), altération de la conscience.
Si vous remarquez des effets indésirables non mentionnés dans cette notice, ou si certains effets indésirables deviennent graves, veuillez en informer votre médecin ou votre pharmacien.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://www.signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER SUBUTEX 0,4 mg, comprimé sublingual ?
Tenir ce médicament hors de la vue et de la portée des enfants. Conservez ce médicament dans un endroit sûr et sécurisé, où d'autres personnes ne peuvent pas y accéder. Il peut causer des dommages graves, voire mortels, aux personnes qui pourraient prendre ce médicament par accident ou intentionnellement alors qu'il ne leur a pas été prescrit.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 30ºC.
A conserver dans l’emballage extérieur à l’abri de l'humidité.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient SUBUTEX 0,4 mg, comprimé sublingual
· La substance active est :
Chlorhydrate de buprénorphine..................................................................................... 0,432 mg
Quantité correspondant à buprénorphine base............................................................... 0,400 mg
pour un comprimé sublingual.
· Les autres composants sont :
Lactose monohydraté, mannitol, amidon de maïs, povidone K30, acide citrique anhydre, citrate de sodium, stéarate de magnésium.
Qu’est-ce que SUBUTEX 0,4 mg, comprimé sublingual et contenu de l’emballage extérieur
Ce médicament se présente sous forme de comprimé sublingual.
Boîte de 7 ou 28 comprimés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
27 WINDSOR PLACE
DUBLIN 2
D02 DK44
IRLANDE
Exploitant de l’autorisation de mise sur le marché
INDIVIOR FRANCE
7 AVENUE DE LA CRISTALLERIE
92310 SEVRES
FRANCE
27 WINDSOR PLACE
DUBLIN 2
D02 DK44
IRLANDE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les dernières informations approuvées sur ce médicament sont disponibles en scannant le QR code et sur : [URL à inclure]