Dernière mise à jour le 01/06/2026
IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide
Indications thérapeutiques
IBUPROFENE BIOGARAN CONSEIL est utilisé pour le traitement de courte durée des douleurs légères à modérées telles que maux de tête, douleurs dentaires, règles douloureuses, de la fièvre et des douleurs liées aux rhumes et à la grippe.
En outre, IBUPROFENE BIOGARAN CONSEIL est également utilisé, sur avis médical, pour soulager les douleurs postopératoires.
IBUPROFENE BIOGARAN CONSEIL est destiné aux adultes, aux adolescents et aux enfants à partir de 20 kg (âgés de 6 ans et plus).
Présentations
> 20 sachet(s) polytéréphtalate (PET) aluminium polyéthylène de 10 ml
Code CIP : 34009 301 301 0 0
Déclaration de commercialisation : 11/05/2020
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 02/12/2024
IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Ibuprofène....................................................................................................................... 200,00 mg
Pour un sachet (suspension buvable de 10 mL).
Excipient(s) à effet notoire :
Maltitol liquide (E965): 5 g/10 mL
Sodium: 35,84 mg/10 mL.
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension visqueuse blanche ou presque blanche avec un arôme fraise reconnaissable.
4.1. Indications thérapeutiques
IBUPROFENE BIOGARAN CONSEIL réduit aussi l’inflammation et la fièvre et soulage les douleurs associées à la grippe et aux rhumes.
IBUPROFENE BIOGARAN CONSEIL est destiné aux adultes, aux adolescents et aux enfants à partir de 20 kg (âgés de 6 ans et plus).
4.2. Posologie et mode d'administration
Destiné uniquement à l’administration par voie orale et de courte durée.
Adultes et adolescents à partir de 40 kg (âgés de 12 ans et plus)
Chez les adultes, il est conseillé de consulter un médecin si ce médicament doit être pris pendant plus de 3 jours en cas de fièvre ou si la douleur persiste plus de 4 jours, ou si les symptômes s’aggravent.
Chez les adolescents (âgés de 12 ans et plus), il convient de consulter un médecin si ce médicament doit être pris pendant plus de 3 jours ou si les symptômes s’aggravent.
La dose initiale est de 1 ou 2 sachets de IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide à renouveler, si nécessaire, de 1 ou 2 sachets de IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide toutes les six heures, jusqu’à 1200 mg au maximum (6 sachets de IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide) par période de 24 heures.
L’intervalle entre deux doses ne doit pas être inférieur à 6 heures.
Enfants dont le poids corporel est inférieur ou égal à 39 kg (âgés de 6 ans et plus)
IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ne doit être utilisé que chez les enfants d’au moins 20 kg. La dose maximale totale d’ibuprofène est de 20-30 mg par kg de poids corporel par jour divisés en 3 ou 4 doses avec 6 à 8 heures d’intervalle entre les prises. La dose quotidienne maximale recommandée ne doit pas être dépassée. La dose totale d’ibuprofène de 30 mg/kg sur une période de 24 heures ne doit pas être dépassée.
Les recommandations suivantes doivent être appliquées chez l’enfant :
|
Poids |
Dose par prise en nombre de sachets d’IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable |
Dose quotidienne maximale en nombre de sachets d’IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable |
|
Enfants de 20– 29 kg |
1 sachet (équivalent à 200 mg d’ibuprofène) |
3 sachets (équivalents à 600 mg d’ibuprofène) |
|
Enfants de 30– 39 kg |
1 sachet (équivalent à 200 mg d’ibuprofène) |
4 sachets (équivalents à 800 mg d’ibuprofène) |
Chez les enfants de 6 ans et plus, il est conseillé de consulter un médecin si ce médicament doit être pris pendant plus de 3 jours ou si les symptômes s’aggravent.
Enfants de moins de 20 kg (âgés de moins de 6 ans)
IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide n’est pas destiné aux enfants de moins de 20 kg ou âgés de moins de 6 ans.
Sujets âgés
Chez les patients âgés, la posologie est la même que pour les adultes, mais une attention particulière est nécessaire (voir rubrique 4.4).
Patients souffrant d’une insuffisance hépatique ou rénale
Il n’est pas nécessaire de réduire la dose chez les patients présentant une insuffisance rénale ou hépatique légère à modérée, une attention particulière est toutefois nécessaire (voir rubrique 4.4).
Les effets indésirables peuvent être minimisés en utilisant la dose efficace la plus faible pendant la durée la plus courte nécessaire pour contrôler les symptômes (voir rubrique 4.4).
Mode d’administration
Malaxez le sachet plusieurs fois avant de l’ouvrir.
Le sachet peut être pris avec ou sans aliments. S’il est pris au cours d’un repas ou peu après, le début de l’action peut être retardé. Cependant, la prise pendant les repas améliore la tolérance du produit et la probabilité de troubles gastro-intestinaux est ainsi diminuée. IBUPROFENE BIOGARAN CONSEIL peut être administré directement sans eau ou dilué dans de l’eau.
· Antécédents de réaction d’hypersensibilité (par exemple bronchospasme, asthme, rhinite, angiœdème ou urticaire) associée à l’utilisation d’acide acétylsalicylique ou d’autres médicaments anti-inflammatoires non-stéroïdiens (AINS).
· Antécédents de saignement ou d’ulcère gastrointestinal au cours d’un traitement par AINS antérieur.
· Episode actif ou des antécédents d’ulcération peptique/hémorragie récidivantes (deux ou plusieurs épisodes distincts avérés d’ulcération ou d’hémorragie).
· Hémorragie cérébrovasculaire ou autre hémorragie active.
· Insuffisance hépatique sévère, insuffisance rénale sévère, ou insuffisance cardiaque sévère (Classe IV de la NYHA) (voir rubrique 4.4).
· Déshydratation sévère (causée par des vomissements, diarrhée ou apport hydrique insuffisant).
· Troubles de l’hématopoïèse d’origine inconnue tels que thrombocytopénie.
· Troisième trimestre de grossesse (voir rubrique 4.6).
4.4. Mises en garde spéciales et précautions d'emploi
IBUPROFENE BIOGARAN CONSEIL ne doit être utilisé qu’après un examen rigoureux du rapport bénéfice/risque en cas de :
· lupus érythémateux disséminé (LED) et maladie mixte du tissu conjonctif- en raison du risque accru de méningite aseptique (voir rubrique 4.8),
· trouble congénital du métabolisme de la porphyrine (par exemple porphyrie aiguë intermittente).
Une surveillance médicale particulièrement étroite est requise en cas :
· d’antécédents d’hypertension et/ou d’insuffisance cardiaque car des cas de rétention hydro-sodée et d’œdème ont été rapportés en association avec un traitement par AINS (voir rubriques 4.3 et 4.8),
· d’atteinte de la fonction rénale car une aggravation est possible (voir rubriques 4.3 et 4.8),
· d’atteinte hépatique (voir rubriques 4.3 et 4.8),
· d’intervention chirurgicale importante récente,
· de rhume des foins, polypes nasaux ou bronchopneumopathie obstructive chronique en raison du risque accru de réactions allergiques pouvant se présenter sous la forme de crises d’asthme (appelées asthme analgésique), d’œdème de Quincke ou d’urticaire,
· d’antécédents de réaction d’hypersensibilité à d’autres substances, un risque accru de réactions d’hypersensibilité à ces substances existant également lors de l’utilisation de IBUPROFENE BIOGARAN CONSEIL.
Effets gastro-intestinaux
Des hémorragies, ulcérations ou perforations gastro-intestinales pouvant être fatales ont été rapportées avec tous les AINS à n’importe quel moment du traitement, avec ou sans symptômes d’alerte ou antécédents d’effets indésirables gastro-intestinaux graves.
Le risque d’hémorragie, d’ulcération ou de perforation gastro-intestinale augmente avec des doses d’AINS utilisées et chez les patients présentant des antécédents d’ulcère, en particulier en cas de complication de type hémorragie ou perforation (voir rubrique 4.3) et chez le sujet âgé. Chez ces patients le traitement doit débuter avec la posologie la plus faible possible. Un traitement avec des agents protecteurs de la muqueuse (par exemple misoprostol ou inhibiteurs de la pompe à protons) doit être envisagé chez ces patients, de même que chez les patients recevant un traitement concomitant par de faibles doses d’acide acétylsalicylique ou par d’autres médicaments susceptibles d’augmenter le risque gastro-intestinal (voir ci-dessous et rubrique 4.5).
Les patients ayant des antécédents de toxicité gastro-intestinale, particulièrement s’il s’agit de sujets âgés, doivent signaler tout symptôme abdominal inhabituel (en particulier tout saignement gastro-intestinal), notamment en début de traitement.
Une attention particulière doit être portée aux patients recevant des traitements concomitants susceptibles d’augmenter le risque d’ulcération ou d’hémorragie, tels que les corticostéroïdes oraux, ou les anticoagulants oraux tels que la warfarine, les inhibiteurs sélectifs de la recapture de la sérotonine ou les antiagrégants plaquettaires tels que l’acide acétylsalicylique (voir rubrique 4.5).
En cas d’hémorragie ou d’ulcération gastro-intestinale chez des patients recevant de l’ibuprofène, le traitement doit être interrompu (voir rubrique 4.3).
Les AINS doivent être utilisés avec prudence chez les patients avec des antécédents de maladie gastro-intestinale (rectocolite hémorragique, maladie de Crohn) car ces pathologies peuvent être exacerbées (voir rubrique 4.8).
Des réactions indésirables cutanées sévères (SCAR)
Des réactions indésirables cutanées sévères (SCAR), tels que la dermatite exfoliative, l’érythème polymorphe, le syndrome de Stevens-Johnson (SJS), la nécrolyse épidermique toxique (TEN), la réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS ou syndrome d'hypersensibilité) et la pustulose exanthématique aiguë généralisée (PEAG), qui peuvent engager le pronostic vital ou être fatales ont été rapportées en association avec l’utilisation d'ibuprofène (voir rubrique 4.8). La plupart de ces réactions sont survenues au cours du premier mois de traitement.
En cas d'apparition de signes et de symptômes évocateurs de ces réactions, la prise d'ibuprofène doit être immédiatement interrompue et un autre traitement doit être envisagé (le cas échéant).
Exceptionnellement, de graves complications infectieuses de la peau et des tissus mous peuvent être observées en cas de varicelle. À ce jour le rôle des AINS dans l’aggravation de ces infections ne peut être exclu. Il est donc conseillé d’éviter d’utiliser l’ibuprofène en cas de varicelle.
Effets cardiovasculaires et cérébrovasculaires
La prudence (en discuter avec son médecin ou avec son pharmacien) s’impose avant de débuter un traitement chez des patients présentant des antécédents d’hypertension artérielle et/ou d’insuffisance cardiaque, des cas de rétention hydro-sodée, d’hypertension et d’œdème ayant été rapportés en association au traitement par AINS.
Des études cliniques suggèrent que l’utilisation d’ibuprofène, en particulier à dose élevée (2400 mg/jour), peut être associée à une légère augmentation du risque d’événements thrombotiques artériels (par exemple infarctus du myocarde ou accident vasculaire cérébral). Globalement, les études épidémiologiques ne suggèrent pas que l’ibuprofène à faible dose (par exemple inférieure ou égale à 1200 mg/jour) soit associé à une augmentation du risque d’événements thrombotiques artériels.
Les patients présentant une hypertension artérielle non contrôlée, une insuffisance cardiaque congestive (NYHA II-III), une cardiopathie ischémique démontrée, une artériopathie périphérique et/ou une maladie vasculaire cérébrale ne devront être traités par ibuprofène qu’après un examen attentif, et les doses élevées (2400 mg/jour) devront être évitées.
Une attention similaire doit être portée avant toute instauration d’un traitement de longue durée chez des patients présentant des facteurs de risque cardiovasculaires (par exemple hypertension, hyperlipidémie, diabète sucré, tabagisme), en particulier si des doses élevées (2 400 mg/jour) d’ibuprofène sont nécessaires.
Des cas de syndrome de Kounis ont été rapportés chez des patients traités par IBUPROFENE BIOGARAN CONSEIL. Le syndrome de Kounis a été défini comme des symptômes cardiovasculaires secondaires à une réaction allergique ou hypersensible associée à une constriction des artères coronaires et pouvant conduire à un infarctus du myocarde.
Autres remarques
De réactions d’hypersensibilité aiguë graves (par exemple choc anaphylactique) ont été observées dans de très rares cas. Dès les premiers signes de réaction d’hypersensibilité suivant la prise/l’administration de IBUPROFENE BIOGARAN CONSEIL, il faut interrompre le traitement. Le personnel spécialisé doit instaurer les mesures médicales requises adaptées aux symptômes.
En cas d’administration prolongée de IBUPROFENE BIOGARAN CONSEIL, il est nécessaire de surveiller régulièrement la fonction hépatique, la fonction rénale et la numération sanguine.
Sujets âgés
Les sujets âgés ont un risque élevé de présenter des réactions indésirables aux AINS, en particulier d’hémorragies et de perforations gastro-intestinales pouvant être fatales (voir rubrique 4.8).
Affections respiratoires
Un bronchospasme peut être déclenché chez les patients souffrant ou ayant des antécédents d’asthme ou d’affections allergiques.
Effets rénaux
De manière générale, la prise régulière d’antalgiques, en particulier l’association de plusieurs substances actives antalgiques, peut provoquer des lésions rénales définitives avec un risque d’insuffisance rénale (néphropathie analgésique). Ce risque peut être augmenté en cas d’effort physique associé à une perte de sel et à une déshydratation. Cela doit par conséquent être évité.
Il existe un risque d’insuffisance rénale chez les enfants et les adolescents déshydratés.
Hyperkaliémie
Il existe un risque accru d’hyperkaliémie chez les patients atteints de diabète et lors de l’utilisation concomitante de IBUPROFENE BIOGARAN CONSEIL et de diurétiques épargneurs de potassium.
Effets hématologiques
L’ibuprofène, substance active de IBUPROFENE BIOGARAN CONSEIL peut inhiber temporairement la fonction plaquettaire sanguine (agrégation thrombocytaire). Les patients présentant des troubles de la coagulation doivent donc faire l’objet d’une surveillance particulière.
L’utilisation prolongée de tout type d’antalgiques pour des céphalées peut les aggraver. Dans ces cas ou en cas de suspicion, il faudra demander un avis médical et arrêter le traitement. Le diagnostic de céphalées par abus médicamenteux (CAM) devra être suspecté chez les patients qui ont des céphalées fréquentes ou quotidiennes malgré (ou à cause de) l’utilisation régulière d’antalgiques pour les céphalées.
Autres AINS
L’usage concomitant d’AINS, notamment des inhibiteurs sélectifs de la cyclo-oxygénase 2, doit être évité.
Les effets indésirables liés aux AINS, notamment ceux touchant le tractus gastro-intestinal ou le système nerveux central peuvent être aggravés par la consommation d’alcool.
Dissimulation des symptômes d’une infection sous-jacente
IBUPROFENE BIOGARAN CONSEIL peut masquer les symptômes d’une infection, ce qui peut retarder la mise en place d’un traitement adéquat et ainsi aggraver l’évolution de l’infection. C’est ce qui a été observé dans le cas de la pneumonie communautaire d’origine bactérienne et des complications bactériennes de la varicelle. Lorsque IBUPROFENE BIOGARAN CONSEIL est administré pour soulager la fièvre ou la douleur liée à l’infection, il est conseillé de surveiller l’infection. En milieu non hospitalier, le patient doit consulter un médecin si les symptômes persistent ou s’ils s’aggravent.
Mises en garde relative aux excipients
Ce médicament contient du maltitol liquide. Les patients atteints de troubles héréditaires rares d’intolérance au fructose ne doivent pas prendre ce médicament.
IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide contient 35,84 mg de sodium par dose de 10 mL. Cela est à prendre en considération par les patients qui suivent un régime pauvre en sodium.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
La surveillance des paramètres cliniques et biologiques doit être envisagée chez des patients prenant de l’ibuprofène en association avec les médicaments listés ci-dessous.
Associations déconseillées
Autres AINS, y compris les salicylés et les inhibiteurs sélectifs de la cyclo-oxygénase 2 (COX-2)
L’utilisation concomitante de plusieurs AINS peut augmenter le risque d’ulcères et de saignements gastro-intestinaux dû à un effet synergique. L’utilisation concomitante d’ibuprofène et d’autres AINS doit donc être évitée (voir rubrique 4.4).
Acide acétylsalicylique
L’administration concomitante d’ibuprofène et d’acide acétylsalicylique n’est généralement pas recommandée en raison du risque accru d’effets indésirables.
Des données disponibles suggèrent que l’ibuprofène peut inhiber par compétition l’effet sur l’agrégation plaquettaire de l’acide acétylsalicylique pris à faible dose lorsqu’ils sont administrés en association. Bien qu’il existe des incertitudes quant à l’extrapolation de ces données en situation clinique, la possibilité que l’utilisation régulière à long terme d’ibuprofène puisse réduire l’effet cardioprotecteur de faibles doses d’acide acétylsalicylique ne peut être exclue. Aucun effet cliniquement pertinent n’est considéré comme susceptible de survenir lors d’une utilisation occasionnelle d’ibuprofène (voir rubrique 5.1).
Associations faisant l’objet de précautions d’emploi
Diurétiques, inhibiteurs de l’enzyme de conversion, bêta-bloquants et antagonistes de l’angiotensine II
Les AINS peuvent réduire l’effet des diurétiques et autres médicaments antihypertenseurs. Chez certains patients dont la fonction rénale est altérée (par exemple les patients déshydratés ou les patients âgés dont la fonction rénale est altérée), l’administration concomitante d’un inhibiteur de l’enzyme de conversion, d’un bêta-bloquant ou d’antagonistes de l’angiotensine II et d’inhibiteurs de la cyclooxygénase peut détériorer encore davantage la fonction rénale, jusqu’à provoquer une insuffisance rénale aiguë, en général réversible. Par conséquent une association avec ces médicaments devra être faite avec prudence, notamment chez les sujets âgés. Les patients devront être suffisamment hydratés et une surveillance de la fonction rénale après l’instauration du traitement concomitant doit être envisagée, et régulièrement par la suite.
L’administration concomitante de IBUPROFENE BIOGARAN CONSEIL et de diurétiques épargneurs de potassium peut entraîner une hyperkaliémie.
Digoxine, phénytoïne, lithium
L’usage concomitant de IBUPROFENE BIOGARAN CONSEIL et de préparations à base de digoxine, de phénytoïne ou de lithium peut augmenter la concentration plasmatique de ces médicaments. Il n’est pas nécessaire de contrôler les taux de lithium, de digoxine et de phénytoïne en cas d’utilisation adaptée (de 3 à 4 jours maximum).
Méthotrexate
Un risque d’augmentation des concentrations plasmatiques de méthotrexate a été observé. Les AINS inhibent la sécrétion tubulaire du méthotrexate et la clairance du méthotrexate peut diminuer. En cas de traitement par des doses élevées de méthotrexate, l’ibuprofène (AINS) est à éviter. Le risque d’interaction entre les AINS et le méthotrexate doit aussi être pris en compte en cas de traitement par méthotrexate à faible dose, en particulier chez les patients présentant une insuffisance rénale. En cas d’association de méthotrexate et d’AINS, la fonction rénale doit être surveillée. La prudence est recommandée si des AINS et du méthotrexate sont administrés sur une période de 24 heures, car les concentrations plasmatiques de méthotrexate peuvent augmenter et entraîner une toxicité accrue.
Tacrolimus
Le risque de néphrotoxicité augmente lorsque les deux médicaments sont administrés en association.
Ciclosporine
Des données limitées suggèrent une interaction possible entraînant un risque accru de néphrotoxicité.
Mifépristone
Les AINS ne doivent pas être utilisés dans les 8 à 12 jours suivant l’administration de mifépristone car les AINS peuvent réduire l’effet de la mifépristone.
Corticostéroïdes
Risque accru d’ulcération ou de saignement gastro-intestinal (voir rubrique 4.4).
Anticoagulants
Les AINS peuvent augmenter les effets des anticoagulants tels que la warfarine (voir rubrique 4.4).
Agents antiplaquettaires et inhibiteurs sélectifs de la recapture de la sérotonine (ISRS)
Risque accru de saignement gastro-intestinal (voir rubrique 4.4).
Sulfamides hypoglycémiants
Des études cliniques ont montré des interactions entre les AINS et les hypoglycémiants (sulfamides). Bien que les interactions entre l’ibuprofène et les sulfamides hypoglycémiants n’aient pas été décrites à ce jour, il est recommandé, par précaution, de contrôler le taux de glucose sanguin lors de l’utilisation associée de ces médicaments.
Zidovudine
Un risque accru d’hémarthroses et d’hématomes a été observé chez les patients hémophiles séropositifs recevant un traitement concomitant par zidovudine et ibuprofène.
Probénécide et sulfinpyrazone
Les médicaments contenant du probénécide ou sulfinpyrazone peuvent retarder l’excrétion de l’ibuprofène.
Baclofène
Une toxicité du baclofène peut apparaitre après le début du traitement par ibuprofène.
Ritonavir
Le ritonavir peut augmenter les concentrations plasmatiques d’AINS.
Aminoglycosides
Les AINS peuvent réduire l’excrétion des aminoglycosides.
Antibiotiques de type quinolone
Les données chez l’animal indiquent que les AINS peuvent augmenter le risque de convulsions associé aux antibiotiques de type quinolone. Les patients traités par AINS et quinolones peuvent présenter un risque accru de convulsions.
Voriconazole et fluconazole
Une étude réalisée avec du voriconazole et du fluconazole (inhibiteurs du CYP2C9) a mis en évidence une augmentation de l’exposition à l’ibuprofène S(+) d’environ 80-100 %. Il faut envisager de réduire la dose d’ibuprofène en cas d’administration en association de puissants inhibiteurs du CYP2C9, en particulier en cas d’administration d’ibuprofène à dose élevée avec du voriconazole ou du fluconazole.
Colestyramine
L’administration concomitante d’ibuprofène et de colestyramine peut ralentir et réduire l’absorption de l’ibuprofène (25 %). Ces médicaments doivent être administrés à quelques heures d’intervalle.
4.6. Fertilité, grossesse et allaitement
Grossesse
L’inhibition de la synthèse des prostaglandines peut avoir un effet délétère sur la grossesse et/ou le développement embryonnaire/fœtal. Des données issues d’études épidémiologiques évoquent un risque accru de fausses couches, de malformations cardiaques et de gastroschisis après la prise d’un inhibiteur de la synthèse des prostaglandines en début de grossesse. Le risque semble augmenter en fonction de la dose et la durée du traitement. Chez l’animal, il a été montré que l’administration d’un inhibiteur de la synthèse des prostaglandines entraînait un risque accru de pertes pré- et postimplantatoires et de létalité embryo-fœtale. En outre, une incidence plus élevée de diverses malformations, notamment cardiovasculaires, a été signalée chez les animaux ayant reçu un inhibiteur de la synthèse des prostaglandines pendant la période d’organogénèse.
À partir de la 20e semaine d’aménorrhée, l’utilisation d’ibuprofène peut provoquer un oligoamnios résultant d’une dysfonction rénale fœtale. Cet effet peut survenir peu de temps après le début du traitement et est généralement réversible à l’arrêt de celui-ci. En outre, des cas de constriction du canal artériel ont été signalés à la suite d'un traitement au cours du deuxième trimestre, dont la plupart se sont résorbés après l'arrêt du traitement. Par conséquent, l’ibuprofène ne doit pas être administré pendant le premier et le deuxième trimestre de grossesse, sauf en cas d’absolue nécessité. Si l’ibuprofène est utilisé par une femme qui souhaite concevoir un enfant ou pendant le premier et le deuxième trimestre de grossesse, la dose devra rester la plus faible possible et la durée du traitement la plus brève possible. Une surveillance prénatale de l’oligoamnios et de la constriction du canal artériel doit être envisagée après une exposition à l’ibuprofène pendant plusieurs jours à partir de la 20e semaine d’aménorrhée. Le traitement par ibuprofène doit être interrompu en cas d’oligoamnios ou de constriction du canal artériel.
Pendant le troisième trimestre de grossesse, tous les inhibiteurs de la synthèse des prostaglandines peuvent exposer le fœtus à :
· une toxicité cardiopulmonaire (constriction/fermeture prématurée du canal artériel et hypertension pulmonaire) ;
· une atteinte fonctionnelle rénale (voir ci-dessus), pouvant évoluer vers une insuffisance rénale avec oligo-hydramnios.
En fin de grossesse, la mère et le nouveau-né peuvent être exposés à :
· un allongement du temps de saignement, un effet antiagrégant plaquettaire pouvant survenir même à de très faibles doses ;
· une inhibition des contractions utérines, retardant ou allongeant la durée du travail.
Par conséquent, l’ibuprofène est contre-indiqué pendant le troisième trimestre de la grossesse (voir rubrique 4.3).
Seules de faibles quantités d’ibuprofène et de ses métabolites passent dans le lait maternel. Aucun effet nocif pour les nourrissons n’étant connu à ce jour, il n’est généralement pas nécessaire d’interrompre l’allaitement durant un traitement de courte durée par de l’ibuprofène pris aux doses recommandées.
Fertilité
Les données limitées disponibles indiquent que des substances inhibant la cyclo-oxygénase/synthèse des prostaglandines peuvent altérer la fertilité féminine en raison d’un effet sur l’ovulation. Cela est réversible à l’arrêt du traitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Affections gastro-intestinales
Les effets indésirables les plus fréquemment observés sont de nature gastro-intestinale. Des ulcères peptiques, perforations ou hémorragies gastro-intestinales, parfois fatales, peuvent survenir, en particulier chez le sujet âgé (voir rubrique 4.4). Des nausées, vomissements, diarrhées, flatulences, constipation, dyspepsie, douleurs abdominales, méléna, hématémèse, stomatite ulcérative et exacerbation de colite et de maladie de Crohn (voir rubrique 4.4) ont été rapportés à la suite de l’administration d’ibuprofène. Des cas de gastrite ont été observés moins fréquemment. Le patient doit être prévenu d’arrêter le traitement et de consulter un médecin immédiatement en cas de douleur relativement intense dans le haut de l’abdomen, de méléna ou d’hématémèse.
Affections cardio-vasculaires
Des études cliniques suggèrent que l’utilisation d’ibuprofène, en particulier à dose élevée (2 400 mg/jour), peut être associée à une légère augmentation du risque d’événements thrombotiques artériels tels qu’infarctus du myocarde ou accident vasculaire cérébral (voir rubrique 4.4).
Des cas d’œdème, d’hypertension et d’insuffisance cardiaque ont été rapportés en association avec des traitements par AINS.
La liste des effets indésirables ci-dessous comprend tous les effets indésirables connus pouvant survenir lors d’un traitement par ibuprofène, y compris ceux consécutifs à un traitement à dose élevée de longue durée chez des patients souffrant de rhumatismes. Les fréquences mentionnées, qui vont au-delà de rapports très rares, font référence à l’utilisation à court terme de doses quotidiennes d’un maximum de 1 200 mg d’ibuprofène pour les formes administrées par voie orale et d’un maximum de 1 800 mg pour les suppositoires (= 6 sachets de IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide, dose quotidienne maximale pour les adultes et les enfants de plus de 12 ans).
Les fréquences suivantes sont prises comme point de référence lors de l’évaluation des effets indésirables :
· Très fréquent : ≥ 1/10
· Fréquent : ≥ 1/100, < 1/10
· Peu fréquent : ≥ 1/1 000, < 1/100
· Rare : ≥ 1/10 000, < 1/1000
· Très rare : < 1/10 000
· Fréquence indéterminée : ne peut être estimée sur la base des données disponibles
En ce qui concerne les réactions indésirables au médicament suivantes, il faut prendre en considération le fait qu’elles sont essentiellement dépendantes de la dose et varient d’une personne à l’autre.
|
Classe de systèmes d’organes MedDRA |
Fréquence |
Effet indésirable |
|
Infections et infestations |
Rare |
Cystite, rhinite. |
|
Très rare |
Fasciite nécrosante1), méningite aseptique2) avec symptômes tels que raideur de la nuque, maux de tête, nausée, vomissements, fièvre ou troubles de la conscience. |
|
|
Affections hématologiques et du système lymphatique |
Très rare |
Troubles de l’hématopoïèse3) : anémie, leucopénie, thrombocytopénie, pancytopénie, agranulocytose. Les premiers signes peuvent être : fièvre, mal de gorge, ulcères superficiels de la bouche, symptômes pseudo-grippaux, épuisement sévère, saignements de nez et saignements cutanés. |
|
Affections du système immunitaire |
Peu fréquent |
Réactions d’hypersensibilité4) : rash, démangeaison, crise d’asthme (avec possibilité de chute de la tension artérielle). |
|
Très rare |
Réactions sévères d’hypersensibilité5) : œdème du visage, de la langue, du larynx avec rétrécissement des voies respiratoires, détresse respiratoire, palpitations, chute de la tension artérielle et choc. |
|
|
Troubles du métabolisme et de la nutrition |
Fréquence indéterminée |
Rétention hydro-sodée, diminution de l’appétit. |
|
Affections psychiatriques |
Très rare |
Réaction psychotique, dépression. |
|
Affections du système nerveux |
Peu fréquent |
Maux de tête, étourdissements, insomnie, agitation, irritabilité, fatigue. |
|
Affections oculaires |
Peu fréquent |
Troubles de la vision6). |
|
Rare |
Sécheresse oculaire. |
|
|
Affections de l’oreille et du labyrinthe |
Rare |
Acouphènes. |
|
Fréquence indéterminée |
Déficience auditive. |
|
|
Affections cardiaques |
Très rare |
Palpitations, œdème, insuffisance cardiaque, infarctus du myocarde. |
|
Fréquence indéterminée |
Syndrome de Kounis |
|
|
Affections vasculaires |
Très rare |
Hypertension artérielle, vasculite. |
|
Affections respiratoires, thoraciques et médiastinales |
Fréquence indéterminée |
Asthme, obstruction du larynx, bronchospasme ou apnée, dyspnée. |
|
Affections gastro-intestinales |
Fréquent |
Pyrosis, douleurs abdominales, nausée, vomissements, dyspepsie, flatulences, diarrhée, constipation, saignements gastro-intestinaux légers7). |
|
Peu fréquent |
Ulcères gastro-intestinaux, perforation ou hémorragie gastro-intestinale, méléna, hématémèse, stomatite ulcérative, exacerbation de colite et maladie de Crohn (voir rubrique 4.4), gastrite. |
|
|
Très rare |
Œsophagite, pancréatite, formation de sténoses de l’intestin de type diaphragmatique. |
|
|
Affections hépatobiliaires |
Très rare |
Dysfonction hépatique, lésion hépatique8), jaunisse, hépatite aiguë, nécrose hépatique, insuffisance hépatique. |
|
Affections de la peau et du tissu sous-cutané |
Peu fréquent |
Rash cutané variés. |
|
Rare |
Dermatite exfoliative, photodermatose. |
|
|
Très rare |
Réactions indésirables cutanées sévères (SCAR) (y compris érythème polymorphe, dermatite exfoliative, syndrome de Stevens-Johnson, nécrolyse épidermique toxique, alopécie. Infections cutanées sévères et complications des tissus mous survenant pendant une infection par la varicelle9). |
|
|
Indéterminée |
Réaction d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS ou syndrome d’hypersensibilité), pustulose exanthématique aiguë généralisée (PEAG), réactions de photosensibilité |
|
|
Affections du rein et des voies urinaires10) |
Rare |
Augmentation du taux sanguin de l’urée et de l’acide urique, polyurie, hématurie, lésion du tissu rénal (nécrose papillaire)8). |
|
Très rare |
Diminution de la sécrétion urinaire, formation d’œdèmes11), syndrome néphrotique, néphrite interstitielle, insuffisance rénale. |
|
|
Investigations |
Rare |
Diminution du taux d’hématocrite. |
|
Très rare |
Diminution du taux d’hémoglobine. |
1) Infections et infestations : Très rarement, une exacerbation d’inflammations liées à une infection (par exemple développement d’une fasciite nécrosante) coïncidant avec l’utilisation d’anti-inflammatoires non stéroïdiens a été décrite. Cela peut être associé au mode d’action des médicaments antiinflammatoires non stéroïdiens. Si des signes d’infection apparaissent ou s’aggravent pendant l’utilisation de IBUPROFENE BIOGARAN CONSEIL, il est donc recommandé au patient de consulter immédiatement un médecin. Il faudra déterminer si un traitement anti-infectieux ou antibiotique est nécessaire.
2) Les patients ayant des troubles auto-immuns (LED, maladie mixte du tissu conjonctif) pourraient être plus susceptibles d’être affectés.
3) Dans de tels cas, il faut recommander au patient d’arrêter immédiatement le médicament, d’éviter toute automédication avec des analgésiques ou des antipyrétiques et de consulter un médecin. La numération sanguine doit être vérifiée régulièrement en cas de traitement de longue durée.
4) Dans ce cas, il faut prévenir le patient d’informer immédiatement un médecin et à ne plus prendre IBUPROFENE BIOGARAN CONSEIL.
5) Si l’un de ces symptômes survient, ce qui peut se produire même lors de la première utilisation, une prise en charge médicale urgente est nécessaire.
6) Dans ce cas, il faut prévenir le patient d’informer le médecin immédiatement et à arrêter l’ibuprofène.
7) Peut entraîner une anémie dans des cas exceptionnels.
8) En particulier lors de traitements de longue durée.
9) Dans des cas exceptionnels, des infections cutanées sévères et des complications au niveau des tissus mous peuvent survenir pendant une infection par la varicelle (voir également « Infections et infestations »).
10) La fonction rénale doit être régulièrement surveillée.
11) En particulier chez les patients présentant une hypertension artérielle ou une insuffisance rénale.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
Symptômes
Des perturbations du système nerveux central comme les céphalées, étourdissements, sensations de vertige et pertes de conscience (de même que les convulsions myocloniques chez l’enfant), ainsi que les douleurs abdominales, nausées et vomissements constituent des symptômes possibles de surdosage. En outre, des saignements gastro-intestinaux ainsi que des perturbations des fonctions hépatiques et rénales sont possibles. Une hypotension, une dépression du système respiratoire et une cyanose peuvent également survenir.
Le produit contient du glycérol qui, à doses élevées, peut causer des céphalées, des maux d’estomac (dyspepsie) et la diarrhée, ces symptômes peuvent être exacerbés par un surdosage de IBUPROFENE BIOGARAN CONSEIL.
Prise en charge
Le patient doit être immédiatement transféré à l’hôpital.
Il n’existe pas d’antidote spécifique.
Le traitement d’une intoxication mis en place dépend des symptômes cliniques et de leur gravité selon les pratiques courantes des soins intensifs.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
L’ibuprofène est un médicament anti-inflammatoire non stéroïdien qui, dans les modèles standards d’expérimentation animale visant à évaluer l’inflammation, a montré son efficacité par inhibition de la synthèse des prostaglandines. Chez l’humain, l’ibuprofène a un effet antipyrétique et réduit la douleur et l’œdème d’origine inflammatoire. De plus, l’ibuprofène inhibe de manière réversible l’agrégation plaquettaire induite par l’ADP et le collagène.
Des données expérimentales suggèrent que l’ibuprofène peut inhiber par compétition l’effet sur l’agrégation plaquettaire de l’acide acétylsalicylique pris à faible dose lorsqu’ils sont administrés en association. Certaines études pharmacodynamiques mettent en évidence que l’administration de doses uniques d’ibuprofène 400 mg dans les 8 heures précédant ou dans les 30 minutes suivant la prise de 81 mg d’acide acétylsalicylique à libération immédiate diminue l’effet de l’acide acétylsalicylique sur la formation de thromboxane ou l’agrégation plaquettaire.
Bien qu’il existe des incertitudes quant à l’extrapolation de ces données en situation clinique, la possibilité que l’utilisation régulière à long terme d’ibuprofène puisse réduire l’effet cardioprotecteur de faibles doses d’acide acétylsalicylique ne peut être exclue. Aucun effet cliniquement significatif n’est susceptible de survenir lors d’une utilisation occasionnelle d’ibuprofène (voir rubrique 4.5).
5.2. Propriétés pharmacocinétiques
Après administration orale, l’ibuprofène est partiellement absorbé dans l’estomac, puis complètement dans l’intestin grêle. Après métabolisation hépatique (hydroxylation, carboxylation), les métabolites pharmacologiquement inactifs sont complètement éliminés, principalement par voie rénale (90 %), mais aussi par la bile.
La demi-vie d’élimination chez les volontaires sains et celles atteintes d’affections hépatiques et rénales est de 1,8 à 3,5 heures, le taux de liaison aux protéines plasmatiques est d’environ 99 %. Le pic de concentration plasmatique est atteint 1 à 2 heures après l’administration par voie orale d’une forme pharmaceutique à libération immédiate.
Insuffisance rénale
Les effets suivants ont été rapportés chez des patients présentant une légère insuffisance rénale : augmentation du (S)-ibuprofène non lié, valeurs plus élevées de l’ASC pour le (S)-ibuprofène et taux plus élevés des ASC énantiomériques (S/R) par rapport aux témoins sains.
Chez les patients atteints d’insuffisance rénale terminale qui sont en dialyse, la fraction libre moyenne d’ibuprofène était d’environ 3 % contre environ 1 % chez les volontaires sains. Une insuffisance rénale sévère peut provoquer une accumulation de métabolites de l’ibuprofène. La portée de cet effet est inconnue. Les métabolites peuvent être éliminés par hémodialyse (voir rubriques 4.2, 4.3 et 4.4).
Insuffisance hépatique
Chez les patients cirrhotiques présentant une insuffisance hépatique modérée (score de Child– Pugh de 6-10), qui sont traités par l’ibuprofène racémique, un doublement moyen de la demi-vie a été observé et le taux de l’ASC énantiomérique (S/R) était significativement moins élevé que celui des témoins en bonne santé, ce qui suggère une altération de l’inversion métabolique du (R)-ibuprofène en énantiomère (S) actif (voir rubriques 4.2, 4.3 et 4.4).
5.3. Données de sécurité préclinique
L’ibuprofène a entraîné une inhibition de l’ovulation chez la lapine et une diminution de l’implantation chez plusieurs espèces animales (lapin, rat, souris). Des études expérimentales sur le rat et le lapin ont montré que l’ibuprofène traverse le placenta. Après administration de doses maternotoxiques, une augmentation de l’incidence de malformations (communications interventriculaires) a été observée chez la progéniture du rat.
L’ibuprofène montre un risque pour l’environnement aquatique, en particulier pour les poissons.
* L’arôme de fraise contient : substances identiques aux arômes naturels, préparations aux arômes naturels, maltodextrine de maïs, citrate d’éthyle (E1505), propylèneglycol (E1520) et alcool benzylique
3 ans.
6.4. Précautions particulières de conservation
À conserver à une température ne dépassant pas 30°C.
6.5. Nature et contenu de l'emballage extérieur
Sachets à dose unique, mesurant 25 mm x 150 mm, d’une capacité de 10 mL, composés d’un assemblage de PET/Aluminium/PET/PE (surface en contact avec le produit).
Taille de l’emballage :
IBUPROFENE BIOGARAN CONSEIL : 10, 12, 18, 20, 24, 30 sachets
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Instructions de préparation avant utilisation :
Ce produit est une suspension. Doit être homogénéisé avant utilisation comme indiqué sur la figure suivante :
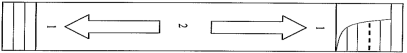
1 – Pressez vos doigts à plusieurs reprises sur le haut et le bas du sachet.
2 – Pressez plusieurs fois de haut en bas et de bas en haut.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 300 7 0 : 10 mL en sachet (PET/Aluminium/PET/PE). Boite de 10.
· 34009 301 300 8 7 : 10 mL en sachet (PET/Aluminium/PET/PE). Boite de 12.
· 34009 301 300 9 4 : 10 mL en sachet (PET/Aluminium/PET/PE). Boite de 18.
· 34009 301 301 0 0 : 10 mL en sachet (PET/Aluminium/PET/PE). Boite de 20.
· 34009 550 483 9 8 : 10 mL en sachet (PET/Aluminium/PET/PE). Boite de 24.
· 34009 550 484 1 1 : 10 mL en sachet (PET/Aluminium/PET/PE). Boite de 30.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[A compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[A compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Médicament non soumis à prescription médicale.
ANSM - Mis à jour le : 02/12/2024
IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide
ibuprofène
Vous devez toujours prendre ce médicament en suivant scrupuleusement les informations fournies dans cette notice ou par votre médecin ou votre pharmacien.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Adressez-vous à votre pharmacien pour tout conseil ou information.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
· Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien après 3 jours pour les enfants et les adolescents ou, pour les adultes, après 4 jours en cas de douleur ou après 3 jours en cas de fièvre.
1. Qu’est-ce que IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ?
3. Comment prendre IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ET DANS QUELS CAS EST-IL UTILISE ?
IBUPROFENE BIOGARAN CONSEIL est utilisé pour le traitement de courte durée des douleurs légères à modérées telles que maux de tête, douleurs dentaires, règles douloureuses, de la fièvre et des douleurs liées aux rhumes et à la grippe.
En outre, IBUPROFENE BIOGARAN CONSEIL est également utilisé, sur avis médical, pour soulager les douleurs postopératoires.
IBUPROFENE BIOGARAN CONSEIL est destiné aux adultes, aux adolescents et aux enfants à partir de 20 kg (âgés de 6 ans et plus).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ?
· vous êtes allergique à l’ibuprofène ou d’autres médicaments anti-inflammatoires non stéroïdiens (AINS) ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· vous avez déjà eu le souffle court, de l’asthme, le nez qui coule, un gonflement du visage, de la langue ou de la gorge, ou de l’urticaire après avoir pris de l’acide acétylsalicylique ou d’autres médicaments anti-inflammatoires non stéroïdiens (AINS) ;
· vous avez déjà eu une perforation ou un saignement au niveau de l’estomac ou des intestins, liés à une précédente utilisation d’AINS ;
· vous avez ou avez déjà eu des ulcères ou des saignements récidivants de l’estomac ou du duodénum (deux ou plusieurs épisodes distincts d’ulcération ou de saignement avérés) ;
· vous souffrez de maladie grave du foie, des reins ou du cœur ;
· vous souffrez de saignement au niveau du cerveau (saignement vasculaire cérébral) ou autre saignement en cours ;
· vous souffrez d’une déshydratation sévère (causée par des vomissements, des diarrhées ou une prise insuffisante de liquide) ;
· vous souffrez de troubles de la formation des cellules sanguines tels qu’une thrombocytopénie ;
· vous êtes une femme pendant le troisième trimestre de grossesse (voir « Grossesse, allaitement et fertilité »).
Avertissements et précautions
Discutez de votre traitement avec votre médecin ou votre pharmacien avant de prendre IBUPROFENE BIOGARAN CONSEIL si :
· vous souffrez d’une maladie héréditaire des cellules sanguines (par exemple porphyrie aiguë intermittente)
· vous avez des antécédents de pression artérielle élevée et/ou insuffisance cardiaque
· vous souffrez de certaines maladies de peau (lupus érythémateux disséminé [LED] ou maladie mixte du collagène)
· vous souffrez ou avez des antécédents de maladies de l’intestin (rectocolite hémorragique ou maladie de Crohn) car ces maladies pourraient s’aggraver (voir rubrique 4 « Quels sont les effets indésirables éventuels ? »)
· vous avez une fonction rénale altérée
· vous avez des problèmes de foie
· vous prenez d’autres médicaments pouvant accroître le risque d’ulcération ou de saignement tels que des corticoïdes par voie orale (comme la prednisolone), des médicaments fluidifiant le sang (comme la warfarine), des inhibiteurs sélectifs de la recapture de la sérotonine (un médicament contre la dépression) ou des agents antiplaquettaires (comme l’acide acétylsalicylique)
· vous prenez un autre AINS (y compris des inhibiteurs de la COX-2 tels que le célécoxib ou l’étoricoxib) étant donné qu’il faut éviter de prendre ces deux médicaments ensemble
· vous avez des problèmes cardiaques, tels qu’une insuffisance cardiaque, une angine de poitrine (douleur de poitrine) ou si vous avez eu une crise cardiaque, un pontage, une maladie artérielle périphérique (mauvaise circulation dans les jambes ou les pieds due à un rétrécissement ou à un blocage des artères) ou tout type d’accident vasculaire cérébral (y compris un « mini-AVC » ou un accident ischémique transitoire « AIT »)
· vous avez une pression artérielle élevée, du diabète, un taux élevé de cholestérol, un membre de votre famille a eu une maladie cardiaque ou un AVC, ou si vous fumez
· vous venez de subir une intervention chirurgicale importante
· vous êtes déshydraté(e), car cela entraîne un risque accru de troubles rénaux
· vous avez des problèmes de coagulation du sang
· vous avez une infection – veuillez consulter le chapitre « Infections » ci-dessous.
Les effets indésirables peuvent être moins importants en utilisant la dose efficace la plus faible pendant la durée la plus courte.
L’utilisation prolongée de tout type d’antalgiques pour des maux de tête peut les aggraver. Dans cette situation, demandez conseil à votre médecin et arrêtez le traitement par IBUPROFENE BIOGARAN CONSEIL.
En général, l’utilisation régulière (de plusieurs sortes) d’analgésiques peut provoquer des problèmes rénaux graves durables. Ce risque peut être augmenté en cas d’effort physique associé à une perte de sel et à une déshydratation. Cette situation doit par conséquent être évitée.
Des difficultés à respirer peuvent apparaître chez les personnes souffrant ou ayant des antécédents d’asthme ou d’allergies.
Si vous souffrez de rhume des foins, de polypose nasale ou d’affections obstructives chroniques des voies respiratoires, il existe un risque accru de réactions allergiques. Ces réactions allergiques peuvent se présenter sous la forme de crises d’asthme (appelé asthme analgésique), d’œdème de Quincke ou d’urticaire.
Dans de très rares cas, de sévères réactions d’hypersensibilité aiguë (par exemple choc anaphylactique) ont été observées. Interrompez immédiatement le traitement aux premiers signes d’une réaction d’hypersensibilité sévère après la prise de IBUPROFENE BIOGARAN CONSEIL. Suivant les symptômes, les procédures médicales requises devront être instaurées par un professionnel de santé qualifié.
Infections
IBUPROFENE BIOGARAN CONSEIL peut masquer des signes d’infections tels que fièvre et douleur. Il est donc possible que IBUPROFENE BIOGARAN CONSEIL retarde la mise en place d’un traitement adéquat de l’infection, ce qui peut accroître les risques de complications. C’est ce que l’on a observé dans le cas de pneumonies d’origine bactérienne et d’infections cutanées bactériennes liées à la varicelle. Si vous prenez ce médicament alors que vous avez une infection et que les symptômes de cette infection persistent ou qu’ils s’aggravent, consultez immédiatement un médecin.
Des réactions cutanées graves, y compris la dermatite exfoliative, l’érythème polymorphe, le syndrome de Stevens-Johnson, la nécrolyse épidermique toxique, la réaction d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS ou syndrome d’hypersensibilité), la pustulose exanthématique aiguë généralisée (PEAG) ont été rapportées en association avec le traitement par l’ibuprofène. Arrêtez de prendre IBUPROFENE BIOGARAN CONSEIL et consultez immédiatement un médecin si vous notez l’un des symptômes liés à ces réactions cutanées graves décrites dans la rubrique 4.
Pendant la varicelle, il est conseillé d’éviter d’utiliser IBUPROFENE BIOGARAN CONSEIL.
Les médicaments anti-inflammatoires/antalgiques tels que l’ibuprofène peuvent être associés à un risque légèrement plus élevé de crise cardiaque ou d’accident vasculaire cérébral, surtout lorsqu’ils sont utilisés à doses élevées. Ne dépassez pas la dose ni la durée du traitement recommandées.
Des signes de réaction allergique à ce médicament, y compris des problèmes respiratoires, un gonflement du visage et du cou (œdème de Quincke), des douleurs thoraciques ont été rapportés avec l’ibuprofène. Arrêtez immédiatement de prendre IBUPROFENE BIOGARAN CONSEIL et contactez immédiatement votre médecin ou allez au service des urgences de l’hôpital le plus proche si vous remarquez l’un de ces signes.
Des hémorragies, ulcérations ou perforations gastro-intestinales parfois fatales ont été rapportées avec tous les AINS à n’importe quel moment du traitement, avec ou sans symptômes annonciateurs ou antécédents d’effets indésirables gastro-intestinaux graves. En cas d’hémorragie ou d’ulcération gastro-intestinale, le traitement doit être immédiatement interrompu. Le risque d’hémorragie, d’ulcération ou de perforation gastro-intestinales augmente avec l’augmentation des doses d’AINS, chez les patients ayant des antécédents d’ulcère, en particulier en cas de complication par une hémorragie ou une perforation (voir rubrique 2 « Ne prenez jamais IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ») et chez les personnes âgées. Ces patients doivent commencer le traitement par la plus faible dose. Un traitement associant des agents protecteurs gastriques (par exemple du misoprostol ou des inhibiteurs de la pompe à protons) doit être envisagé chez ces patients, de même que chez ceux devant prendre simultanément de l’acide acétylsalicylique à faible dose ou d’autres médicaments susceptibles d’augmenter le risque gastro-intestinal.
Consultez un médecin avant d’utiliser IBUPROFENE BIOGARAN CONSEIL si vous avez l’une des affections mentionnées ci-dessus.
En cas d’utilisation prolongée de IBUPROFENE BIOGARAN CONSEIL, il est nécessaire de surveiller régulièrement votre fonction hépatique, votre fonction rénale et votre numération sanguine.
Personnes âgées
Lorsqu’elles prennent des AINS, les personnes âgées présentent un risque plus élevé de développer des effets indésirables, en particulier ceux liés à l’estomac et aux intestins. Voir rubrique 4 « Quels sont les effets indésirables éventuels ? » pour davantage d’informations.
Les patients ayant des antécédents de toxicité gastro-intestinale, surtout s’ils sont âgés, doivent signaler tout symptôme abdominal inhabituel (en particulier un saignement dans l’estomac ou les intestins) surtout en début du traitement.
Enfants et adolescents
Il existe un risque d’insuffisance rénale chez les enfants et les adolescents déshydratés.
IBUPROFENE BIOGARAN CONSEIL n’est pas destiné aux enfants de moins de 20 kg ou âgés de moins de 6 ans en raison de la quantité élevée de substance active contenue dans un sachet.
Autres médicaments et IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
IBUPROFENE BIOGARAN CONSEIL peut affecter ou être affecté par certains autres médicaments. Par exemple :
· les glucocorticoïdes (tels que la prednisolone) car ils peuvent accroître le risque d’ulcération ou de saignement gastro-intestinal ;
· un autre AINS (y compris des inhibiteurs de la COX-2 tels que le célécoxib ou l’étoricoxib), car il pourrait augmenter le risque d’ulcère gastro-intestinal ou de saignement. L’utilisation concomitante d’ibuprofène et d’autres AINS doit être évitée ;
· les médicaments anticoagulants (c’est-à-dire qui fluidifient le sang/empêchent la coagulation, par exemple la warfarine, la ticlopidine) ;
· les agents antiplaquettaires (tels que l’acide acétylsalicylique) et les inhibiteurs sélectifs de la recapture de la sérotonine (des médicaments pour traiter la dépression), car cela peut accroître le risque d’effets indésirables gastro-intestinaux ;
· les médicaments qui réduisent l’hypertension artérielle (inhibiteurs de l’enzyme de conversion tels que le captopril, les bêta-bloquants tels que l’aténolol, les antagonistes du récepteur de l’angiotensine II tels que le losartan) et les diurétiques étant donné que les AINS peuvent diminuer les effets de ces médicaments et potentiellement augmenter le risque de troubles rénaux. Dans ce cas, assurez-vous de boire suffisamment d’eau pendant la journée ;
· le méthotrexate (un médicament pour traiter le cancer ou les rhumatismes) car l’effet du méthotrexate peut être augmenté ;
· le tacrolimus (un médicament pour supprimer les réactions immunitaires) car le risque de toxicité pour les reins est augmenté ;
· la ciclosporine (un médicament pour supprimer les réactions immunitaires) car des données limitées suggèrent une augmentation du risque de toxicité rénale ;
· la mifépristone (un médicament abortif) car l’effet de la mifépristone peut être diminué ;
· la zidovudine (un médicament pour traiter le SIDA) étant donné que l’utilisation de IBUPROFENE BIOGARAN CONSEIL peut accroître le risque de saignement dans une articulation ou de saignement provoquant un gonflement chez les patients hémophiles séropositifs ;
· les sulfamides hypoglycémiants : des investigations cliniques ont montré des interactions entre les AINS et les antidiabétiques (sulfamides hypoglycémiants). Bien que les interactions entre l’ibuprofène et les sulfamides hypoglycémiants n’aient pas été décrites à ce jour, il est recommandé, par précaution, de surveiller les taux de sucre sanguin lors de l’utilisation simultanée de ces médicaments ;
· le probénécide et la sulfinpyrazone : les médicaments contenant du probénécide ou de la sulfinpyrazone peuvent retarder l’excrétion de l’ibuprofène ;
· le baclofène : la toxicité du baclofène peut se développer après le début du traitement par ibuprofène ;
· le ritonavir car la prise de ce médicament peut augmenter les concentrations d’AINS dans le plasma ;
· les aminoglycosides (antibiotiques) car les AINS peuvent réduire l’excrétion des aminoglycosides ;
· la digoxine, la phénytoïne et le lithium : l’ibuprofène peut accroître les concentrations de ces médicaments dans le plasma ;
· les antibiotiques de type quinolone car la prise de ces médicaments en même temps que des AINS peut accroître le risque de convulsions ;
· la cholestyramine car la prise d’ibuprofène avec de la cholestyramine peut retarder et diminuer l’absorption de l’ibuprofène ;
· le voriconazole et le fluconazole car ces médicaments peuvent augmenter les concentrations d’AINS dans le plasma ;
Certains autres médicaments peuvent également affecter ou être affectés par le traitement par IBUPROFENE BIOGARAN CONSEIL. Vous devez donc toujours demander conseil à votre médecin ou à votre pharmacien avant de prendre IBUPROFENE BIOGARAN CONSEIL avec d’autres médicaments.
IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide avec des aliments et de l’alcool
Le sachet peut être pris avec ou sans aliments. S’il est pris au cours d’un repas ou peu après, le début de l’action peut être retardé. Cependant, le produit est mieux toléré s’il est pris avec de la nourriture et la probabilité de problèmes gastro-intestinaux est ainsi diminuée. La consommation de boissons alcoolisées doit être évitée pendant le traitement avec IBUPROFENE BIOGARAN CONSEIL. Certains effets indésirables, comme ceux qui touchent le tractus gastro-intestinal ou le système nerveux central, sont plus susceptibles de se produire en cas de consommation simultanée d’alcool et de IBUPROFENE BIOGARAN CONSEIL.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Grossesse
Prévenez votre médecin si vous débutez une grossesse pendant la prise de IBUPROFENE BIOGARAN CONSEIL.
Ne prenez pas IBUPROFENE BIOGARAN CONSEIL si vous êtes dans les 3 derniers mois de grossesse car cela pourrait nuire à votre bébé ou causer des problèmes à l’accouchement. Il peut causer des problèmes rénaux et cardiaques chez votre bébé. Cela peut avoir des répercussions sur vous et votre bébé en favorisant les saignements et entraîner un accouchement plus tardif ou plus long que prévu. Vous ne devez pas prendre IBUPROFENE BIOGARAN CONSEIL pendant les 6 premiers mois de la grossesse à moins que cela ne soit absolument nécessaire et conseillé par votre médecin. Si un traitement est nécessaire au cours de cette période ou lorsque vous planifiez une grossesse, il est recommandé de prendre la dose la plus faible pendant la durée la plus courte possible. À partir de 20 semaines d’aménorrhée, IBUPROFENE BIOGARAN CONSEIL peut provoquer des problèmes rénaux chez votre bébé, s’il est pris pendant plusieurs jours, ce qui peut entraîner un faible niveau du liquide amniotique dans lequel il se trouve (oligoamnios). Si un traitement de plus de quelques jours est nécessaire, votre médecin peut recommander une surveillance supplémentaire.
Allaitement
Seules de faibles quantités d’ibuprofène et de ses produits de dégradation passent dans le lait maternel. Aucun effet nocif pour les nourrissons n’étant connu à ce jour, il n’est généralement pas nécessaire d’interrompre l’allaitement durant une utilisation à court terme d’ibuprofène aux doses recommandées.
Fertilité
IBUPROFENE BIOGARAN CONSEIL appartient à un groupe de médicaments (AINS) pouvant réduire la fertilité chez les femmes. Cet effet est réversible à l’arrêt du médicament.
Conduite de véhicules et utilisation de machines
Si vous ressentez des étourdissements, des vertiges, des troubles de la vision ou tout autre symptôme pendant la prise de ce médicament, ne conduisez pas de véhicules et n’utilisez pas de machines. Si vous ne prenez qu’une dose d’ibuprofène ou le prenez pendant une courte durée, aucune précaution particulière n’est nécessaire.
IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide contient du maltitol et du sodium
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
Le maltitol peut avoir un léger effet laxatif. Valeur énergétique 2,3 kcal/g de maltitol.
IBUPROFENE BIOGARAN CONSEIL contient 35,84 mg de sodium par dose de 10 mL. Cela est à prendre en considération par les patients qui suivent un régime pauvre en sodium.
3. COMMENT PRENDRE IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ?
Pour atténuer les symptômes, la dose efficace la plus faible devra être utilisée pendant la durée la plus courte possible. Si vous avez une infection et que les symptômes (tels que fièvre et douleur) persistent ou qu’ils s’aggravent, consultez immédiatement un médecin (voir rubrique 2).
Voie orale.
Avant de l’ouvrir, malaxez le sachet pour bien en mélanger le contenu tel qu’indiqué sur la figure suivante :
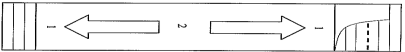
1 – Pressez vos doigts à plusieurs reprises sur le haut et le bas du sachet.
2 – Pressez plusieurs fois de haut en bas et de bas en haut.
IBUPROFENE BIOGARAN CONSEIL peut être administré directement sans eau ou dilué dans de l’eau.
Ce produit est destiné à une utilisation de courte durée uniquement.
La dose recommandée est :
Adultes et adolescents à partir de 40 kg (âgés de 12 ans et plus)
La dose initiale est de 1 ou 2 sachets de IBUPROFENE BIOGARAN CONSEIL et par la suite, si nécessaire, de 1 ou 2 sachets de IBUPROFENE BIOGARAN CONSEIL, toutes les six heures.
Attendez au moins 6 heures entre deux prises.
Ne prenez pas plus de 6 sachets de IBUPROFENE BIOGARAN CONSEIL par jour.
Enfants de 20 à 39 kg (âgés de 6 ans à 11 ans)
IBUPROFENE BIOGARAN CONSEIL ne doit être utilisé que chez les enfants d’au moins 20 kg. La dose quotidienne maximale totale d’ibuprofène est de 20-30 mg par kg de poids corporel divisés en 3 ou 4 doses uniques.
Le dosage pour les enfants de 6 à 11 ans est calculé comme suit en fonction de leur poids corporel :
· Enfants de 20 à 39 kg : prendre 1 sachet de IBUPROFENE BIOGARAN CONSEIL jusqu’à trois fois par jour selon les besoins. Ne prenez pas plus de 3 sachets (jusqu’à 600 mg d’ibuprofène) par jour. Attendez au moins 8 heures entre deux prises.
· Enfants de 30 à 39 kg : prendre 1 sachet d’IBUPROFENE BIOGARAN CONSEIL jusqu’à quatre fois par jour selon les besoins. Ne prenez pas plus de 4 sachets (jusqu’à 800 mg d’ibuprofène) par jour. Attendez au moins 6 à 8 heures entre deux prises.
Chez les enfants âgés de 6 ans et plus et chez les adolescents, il convient de consulter un médecin si ce médicament est nécessaire pendant plus de 3 jours ou si les symptômes s’aggravent.
Enfants de moins de 20 kg (âgés de moins de 6 ans)
IBUPROFENE BIOGARAN CONSEIL n’est pas destiné aux enfants de moins de 20 kg ou âgés de moins de 6 ans.
Personnes âgées
Chez les sujets âgés, la posologie est la même que pour les adultes, mais une attention particulière est nécessaire.
Patients présentant une insuffisance hépatique ou rénale
Si vous souffrez d’une maladie du foie ou des reins, vous devez toujours consulter votre médecin avant de prendre IBUPROFENE BIOGARAN CONSEIL. Votre médecin vous conseillera en conséquence.
Si vous êtes un(e) adulte, vous devez consulter votre médecin si vos symptômes s’aggravent ou ne s’améliorent pas après 4 jours en cas de douleur et après 3 jours en cas de fièvre. Ne prenez pas IBUPROFENE BIOGARAN CONSEIL pendant plus de 7 jours sans avoir consulté un médecin.
Si vous avez pris plus de IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide, que vous n’auriez dû
Demandez immédiatement une assistance médicale en cas de surdosage ou d’ingestion accidentelle de sachets par un enfant.
Si vous avez pris plus de IBUPROFENE BIOGARAN CONSEIL que vous n'auriez dû, ou si des enfants ont pris le médicament accidentellement, contactez toujours un médecin ou l'hôpital le plus proche afin d'obtenir un avis sur le risque et des conseils sur les mesures à prendre.
Les symptômes d’un surdosage peuvent inclure nausées, douleurs abdominales, vomissements (pouvant contenir des traces de sang), maux de tête, bourdonnements dans les oreilles, confusion et mouvements oculaires instables. À fortes doses, les symptômes suivants ont été signalés : somnolence, douleur thoracique, palpitations, perte de conscience, convulsions (principalement chez les enfants), faiblesse et étourdissements, sang dans les urines, faible taux de potassium dans le sang, sensation de froid corporel et problèmes respiratoires.
Si vous oubliez de prendre IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide
Si vous avez oublié de prendre votre dose, ne prenez pas plus que la dose habituelle recommandée lors de votre prochaine dose.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables peuvent être réduits en prenant la dose la plus faible pendant la durée la plus courte nécessaire pour soulager les symptômes. Vous pourriez présenter l’un des effets indésirables connus des AINS. Si c’est le cas, ou si vous avez des inquiétudes, arrêtez de prendre ce médicament et parlez-en à votre médecin dès que possible.
Les personnes âgées utilisant ce produit ont un risque plus important de développer des problèmes associés aux effets indésirables.
En ce qui concerne les effets indésirables suivants, il faut prendre en considération le fait qu’ils sont essentiellement dépendants de la dose et varient d’un patient à l’autre.
Arrêtez la prise de ce médicament et demandez immédiatement une assistance médicale si vous développez :
· des signes de saignement intestinal tels que : douleur sévère de l’abdomen, selles noires et goudronneuses, si vous vomissez du sang ou des particules sombres ressemblant à des grains de café.
· des signes de réaction allergique très rare mais grave, tels que respiration sifflante ou essoufflement inexpliqué, gonflement du visage, de la langue ou de la gorge, difficultés à respirer, accélération des battements de cœur, baisse de la pression artérielle entraînant un choc. Cela peut arriver même lors de la première utilisation de ce médicament. Si vous observez l’un de ces symptômes, appelez immédiatement un médecin.
· des réactions graves de la peau telles que des éruptions cutanées couvrant tout le corps, la peau qui pèle ou qui s’écaille, ou la formation de cloques (par exemple syndrome de Stevens-Johnson, érythème multiforme, nécrolyse épidermique toxique/syndrome de Lyell).
· des troubles ou des modifications de la vision.
· Tâches rougeâtres non surélevées, en forme de cibles ou de cercles, sur le tronc, souvent accompagnées de cloques centrales, d’une desquamation de la peau, d’ulcères de la bouche, de la gorge, du nez, des organes génitaux et des yeux. Ces graves rash cutanés peuvent être précédés par de la fièvre et des symptômes grippaux [dermatite exfoliative, érythème polymorphe, syndrome de Stevens-Johnson, nécrolyse épidermique toxique].
· Éruption cutanée généralisée, température corporelle élevée et gonflement des ganglions lymphatiques (DRESS ou syndrome d’hypersensibilité).
· Éruption cutanée généralisée rouge et squameuse avec des bosses sous la peau et des vésicules s’accompagnant de fièvre. Les symptômes apparaissent généralement dès l’instauration du traitement (pustulose exanthématique aiguë généralisée).
Prévenez votre médecin si vous présentez l’un des effets indésirables suivants, si ces effets s’aggravent ou si vous observez des effets non mentionnés.
Fréquent (peut affecter jusqu’à 1 personne sur 10) :
· Problèmes gastriques, tels que brûlures d’estomac, maux d’estomac, nausées, vomissements, flatulences (gaz), diarrhée, constipation et pertes de sang légères au niveau de l’estomac et/ou des intestins pouvant entraîner de l’anémie dans des cas exceptionnels.
Peu fréquent (peut affecter jusqu’à 1 personne sur 100) :
· Inflammation de l’estomac, exacerbation de colite et maladie de Crohn.
· Perturbations du système nerveux central, telles que maux de tête, étourdissements, insomnie, agitation, irritabilité ou fatigue.
· Ulcères gastro-intestinaux avec possibilité de saignement ou de perforation.
· Ulcères de la bouche et/ou gonflement et irritation de la bouche.
· Réactions d’hypersensibilité avec éruptions cutanées et démangeaisons, et crises d’asthme (pouvant s’accompagner d’une chute de la pression artérielle). Dans ce cas, il faut prévenir un médecin et arrêter de prendre IBUPROFENE BIOGARAN CONSEIL.
· Rashs variés.
Rare (peut affecter jusqu’à 1 personne sur 1000) :
· Acouphènes (bourdonnements dans les oreilles).
· Inflammation de la vessie.
· Inflammation de la muqueuse nasale.
· Sécheresse oculaire.
· Augmentation de la fréquence urinaire, sang dans les urines.
· Rougeur de la peau et peau qui pèle, réactions cutanées à la lumière du soleil.
· Augmentation de la concentration d’acide urique dans le sang, augmentation de la concentration d’urée dans le sang.
· Diminution du nombre de globules rouges.
· Une douleur dans les flancs et/ou l’abdomen, du sang dans les urines et de la fièvre peuvent être des signes de lésion aux reins (nécrose papillaire).
Très rare (peut affecter jusqu’à 1 personne sur 10 000) :
· Inflammation de l’œsophage ou du pancréas, occlusions intestinales.
· Dans des cas exceptionnels, des infections cutanées sévères et des complications des tissus mous peuvent survenir pendant une infection par la varicelle.
· Diminution de l’excrétion d’urine, oedèmes (en particulier chez les patients ayant une pression artérielle élevée ou une atteinte de la fonction rénale) et urine trouble (syndrome néphrotique) ; maladie inflammatoire des reins (néphrite interstitielle) pouvant entraîner une insuffisance rénale aiguë. Si l’un des symptômes susmentionnés survient ou si vous vous sentez vraiment mal, arrêtez de prendre IBUPROFENE BIOGARAN CONSEIL et consultez immédiatement votre médecin car il pourrait s’agir des premiers signes de lésion des reins ou d’insuffisance rénale.
· Problèmes de production des cellules sanguines (les premiers signes sont fièvre, mal de gorge, ulcères buccaux superficiels, symptômes pseudo-grippaux, épuisement sévère, saignement du nez et de la peau, bleus inexpliqués ou inhabituels). Dans de tels cas, arrêtez immédiatement le traitement et consultez un médecin. Ne pas utiliser d’antalgiques ou de médicaments pour diminuer la fièvre (antipyrétiques) en automédication.
· Réactions psychotiques et dépression.
· Aggravation de l’inflammation due à une infection (par exemple fasciite nécrosante). Si des signes d’infection apparaissent ou s’aggravent pendant l’utilisation de IBUPROFENE BIOGARAN CONSEIL, parlez-en à votre médecin sans délai.
· Oedèmes, pression artérielle élevée, palpitations, insuffisance cardiaque, crise cardiaque.
· Problèmes au foie ou inflammation du foie. Insuffisance hépatique ou lésions hépatiques, en particulier lors d’une utilisation de longue durée, se manifestant par un jaunissement de la peau et des yeux, des selles décolorées et des urines foncées.
· Très rarement, des symptômes de méningite aseptique avec raideur de la nuque, maux de tête, nausées, vomissements, fièvre ou altération de la conscience ont été observés lors de l’utilisation d’ibuprofène. Les patients ayant des troubles auto-immuns (LED, connectivite mixte) sont plus susceptibles d’être affectés. Contactez immédiatement un médecin si cela se produit.
· Jaunisse (jaunissement de la peau et/ou du blanc des yeux), lésion hépatique grave.
· Diminution de la quantité d’hémoglobine (pigment transporteur d’oxygène) dans le sang.
· Inflammation des vaisseaux sanguins.
· Perte de cheveux (alopécie).
Fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles) :
· Déficience auditive.
· Rétention d’eau, diminution de l’appétit.
· Asthme, obstruction du larynx, contraction spasmodique du muscle lisse des bronches, comme cela se produit en cas d’asthme ou d’arrêt respiratoire, respiration difficile ou laborieuse.
· Une réaction cutanée sévère appelée syndrome d'hypersensibilité (en anglais : DRESS syndrome) peut survenir. Les symptômes d'hypersensibilité sont : éruption cutanée, fièvre, gonflement des ganglions lymphatiques et augmentation des éosinophiles (un type de globules blancs).
· Douleur thoracique, qui peut être le signe d’une réaction allergique potentiellement grave appelée syndrome de Kounis.
Des médicaments tels que celui-ci peuvent être associés à une légère augmentation du risque de crise cardiaque (infarctus du myocarde) ou d’accident vasculaire cérébral.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER IBUPROFENE BIOGARAN CONSEIL 200 mg, suspension buvable en sachet édulcorée au maltitol liquide ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
À conserver à une température ne dépassant pas 30°C.
Ne jetez aucun médicament au tout-à-l’égout. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Ibuprofène................................................................................................................. 200,00 mg
Pour un sachet (suspension buvable de 10 mL).
· Les autres composants sont :
Benzoate de sodium (E211) ; acide citrique anhydre (E330) ; citrate de sodium dihydraté (E331) ; saccharine de sodium anhydre (E954) ; chlorure de sodium ; hypromellose 15 cP ; gomme xanthane (E415) ; maltitol liquide (E965) ; glycérol 99,8 % (E422) ; arôme de fraise (l’arôme de fraise contient : substances identiques aux arômes naturels, préparations aux arômes naturels, maltodextrine de maïs, citrate d’éthyle (E1505), propylèneglycol (E1520) et alcool benzylique); eau purifiée.
IBUPROFENE BIOGARAN CONSEIL est une suspension visqueuse blanche ou presque blanche avec un arôme de fraise reconnaissable.
Type et taille de l’emballage : sachets à dose unique, mesurant 25 mm x 150 mm, d’une capacité de 10 mL, composés d’un assemblage de PET/Aluminium/PET/PE (surface en contact avec le produit).
IBUPROFENE BIOGARAN CONSEIL : 10, 12, 18, 20, 24, 30 sachets
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
Exploitant de l’autorisation de mise sur le marché
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
A RELVA S/N, O PORRIÑO
36400 PONTEVEDRA
ESPAGNE
OU
LABORATORIOS ALCALA FARMA, S.L.
AVENIDA DE MADRID, 82
ALCALÁ DE HENARES
28802 MADRID
ESPAGNE
OU
FARMALIDER, S.A.
C/ARAGONESES 2, ALCOBENDAS
28108 MADRID
ESPAGNE
OU
EDEFARM, S.L.,
POLÍGONO INDUSTRIAL ENCHILAGAR DEL RULLO, 117,
VILLAMARCHANTE,
46191 VALENCIA,
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l’Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[A compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Sans objet.