Dernière mise à jour le 01/06/2026
ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible
Indications thérapeutiques
ZOLMITRIPTAN VIATRIS est utilisé pour le traitement de la migraine chez les adultes âgés de 18 ans et plus.
Les symptômes de la migraine peuvent être provoqués par une dilatation passagère des vaisseaux sanguins dans la tête. ZOLMITRIPTAN VIATRIS réduit la dilatation de ces vaisseaux sanguins. Cela aide à faire disparaître le mal de tête et les autres symptômes de la crise de migraine, tels que les nausées, les vomissements et la sensibilité à la lumière et au son.
ZOLMITRIPTAN VIATRIS est efficace seulement lorsque la crise de migraine a commencé. ZOLMITRIPTAN VIATRIS ne doit pas être utilisé pour prévenir les crises de migraine.
Présentations
> 6 plaquette(s) polyamide aluminium PVC papier polyester suremballée(s)/surpochée(s) de 1 comprimé(s)
Code CIP : 219 744-9 ou 34009 219 744 9 2
Déclaration de commercialisation : 27/12/2023
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 5,53 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 6,55 €
- Taux de remboursement :65%
> 12 plaquette(s) polyamide aluminium PVC papier polyester suremballée(s)/surpochée(s) de 1 comprimé(s)
Code CIP : 219 747-8 ou 34009 219 747 8 2
Déclaration de commercialisation : 27/12/2023
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 10,98 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 12,00 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 05/10/2023
ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé orodispersible contient 2,5 mg de zolmitriptan.
Excipient à effet notoire : chaque comprimé orodispersible contient 5 mg d'aspartam.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé orodispersible blanc à blanc cassé, rond, plat et biseauté, de 6,5 mm de diamètre, avec « M » gravé sur une face et « ZT1 » gravé sur l'autre face.
4.1. Indications thérapeutiques
ZOLMITRIPTAN VIATRIS n'est pas indiqué dans la prophylaxie de la migraine.
4.2. Posologie et mode d'administration
La dose recommandée de ZOLMITRIPTAN VIATRIS pour le traitement d'une crise de migraine est de 2,5 mg. Il est conseillé de prendre le zolmitriptan le plus tôt possible dès l'apparition de la céphalée migraineuse. Cependant, il est aussi efficace lorsqu'il est administré plus tard.
Si les symptômes de la migraine réapparaissent dans les 24 heures suivant la réponse initiale, une seconde dose peut être prise. En cas de besoin d'une seconde dose, celle-ci ne doit pas être prise dans les 2 heures suivant la prise de la première dose. Si un patient n'est pas soulagé après la première dose, il est peu probable qu'une seconde dose apportera un bénéfice pour la même crise.
Si un patient n'est pas soulagé de façon satisfaisante avec des doses de 2,5 mg, des doses de 5 mg de zolmitriptan pourront être envisagées pour les crises suivantes. La prudence est recommandée en raison d'un risque accru d'effet indésirable. La supériorité de la dose de 5 mg comparée à la dose de 2,5 mg n'a pas pu être démontrée lors d'une étude clinique contrôlée. Néanmoins, une dose de 5 mg peut être bénéfique pour certains patients.
La dose maximale ne doit pas dépasser 10 mg par 24 heures. Ne pas prendre plus de 2 doses de ZOLMITRIPTAN VIATRIS au cours d'une période de 24 heures.
Populations particulières
Utilisation chez les patients âgés de plus de 65 ans
La sécurité d'emploi et l'efficacité du zolmitriptan chez les personnes âgées de plus de 65 ans n'ont pas été établies. Par conséquent, l'utilisation de ZOLMITRIPTAN VIATRIS chez les personnes âgées n'est pas recommandée.
Insuffisants hépatiques
Le métabolisme du zolmitriptan est réduit chez les patients atteints d'insuffisance hépatique (voir rubrique 5.2). Chez les patients atteints d'insuffisance hépatique modérée ou sévère, une dose maximale de 5 mg en 24 heures est recommandée. Cependant, aucun ajustement de dose n'est nécessaire pour les patients atteints d'insuffisance hépatique légère.
Insuffisants rénaux
Aucun ajustement de dose n'est nécessaire chez les patients dont la clairance de la créatinine est supérieure à 15 ml/min (voir rubriques 4.3. et 5.2).
Interactions nécessitant un ajustement de dose (voir rubrique 4.5)
Pour les patients prenant des inhibiteurs de la MAO-A, une dose maximale de 5 mg en 24 heures est recommandée.
Une dose maximale de 5 mg de zolmitriptan en 24 heures est recommandée chez les patients prenant de la cimétidine.
Une dose maximale de 5 mg de zolmitriptan en 24 heures est recommandée chez les patients prenant des inhibiteurs spécifiques du CYP 1A2, comme la fluvoxamine et les quinolones (par exemple, ciprofloxacine).
Population pédiatrique
Utilisation chez les enfants (de moins de 12 ans)
La sécurité d'emploi et l'efficacité des comprimés de zolmitriptan chez les enfants âgés de 0 à < 12 ans n’ont pas été établies. Par conséquent, l'utilisation de ZOLMITRIPTAN VIATRIS n'est pas recommandée chez les enfants.
Adolescents (de 12 à 17 ans)
L'efficacité des comprimés de zolmitriptan chez les enfants âgés de 12 à 17 ans n’a pas été établie. Les données actuellement disponibles sont décrites à la rubrique 5.1 mais aucune recommandation sur la posologie ne peut être donnée. Par conséquent, l'utilisation de ZOLMITRIPTAN VIATRIS n'est pas recommandée chez les adolescents.
Mode d'administration
Voie orale.
Le comprimé n'a pas besoin d'être pris avec une boisson. Il se dissout sur la langue et s'avale avec la salive. Cette formulation peut être utilisée lorsqu'une boisson n'est pas disponible, ou pour éviter les nausées et les vomissements qui peuvent accompagner la prise de comprimés avec une boisson.
Néanmoins, l'apparition de l'effet peut être retardée en raison du délai d'absorption du zolmitriptan contenu dans ZOLMITRIPTAN VIATRIS.
La plaquette thermoformée doit être ouverte comme indiqué sur la feuille d’aluminium (les comprimés ne doivent pas être expulsés à travers cette feuille). Le comprimé de ZOLMITRIPTAN VIATRIS doit être placé sur la langue, où il se dissoudra et sera avalé avec la salive.
· Hypersensibilité à la substance active ou à l'un des excipients mentionnés dans la rubrique 6.1.
· Hypertension modérée ou sévère et hypertension bénigne non contrôlée.
· Cette classe de médicaments (agonistes des récepteurs 5HT1B/1D) a été associée à des vasospasmes coronariens, ce qui a entraîné l'exclusion des patients présentant une cardiopathie ischémique dans les essais cliniques. Le zolmitriptan ne doit donc pas être administré chez les patients ayant un antécédent d'infarctus du myocarde ou une pathologie cardiaque ischémique, un vasospasme coronarien (angor de Prinzmetal), une pathologie vasculaire périphérique ou chez les patients présentant des symptômes ou des signes compatibles avec une pathologie cardiaque ischémique.
· L'association d'ergotamine, de dérivés de l'ergotamine (y compris le méthysergide), sumatriptan, naratriptan et autres agonistes du récepteur 5HT1B/1D est contre-indiquée avec le zolmitriptan (voir rubrique 4.5).
· Le zolmitriptan ne doit pas être administré chez les patients ayant des antécédents d'accident vasculaire cérébral (AVC) ou d'accident ischémique transitoire (AIT).
· Le zolmitriptan est contre-indiqué chez les patients dont la clairance de la créatinine est inférieure à 15 ml/min.
4.4. Mises en garde spéciales et précautions d'emploi
Le zolmitriptan ne doit pas être administré chez les patients présentant un syndrome de Wolff‑Parkinson-White symptomatique ou souffrant d’arythmie associée à une voie de conduction cardiaque accessoire.
Dans de très rares cas, comme avec d'autres agonistes des récepteurs 5HT1B/1D, des cas de vasospasme coronarien, angine de poitrine et infarctus du myocarde ont été rapportés. Le zolmitriptan ne doit pas être administré aux patients ayant des facteurs de risque de maladie cardiaque ischémique (par exemple, tabagisme, hypertension, hyperlipidémie, diabète, antécédents familiaux), sans un bilan cardiovasculaire préalable (voir rubrique 4.3). Une attention particulière doit être portée aux femmes ménopausées et aux hommes de plus de 40 ans présentant ces facteurs de risque. Cependant, ce bilan peut ne pas identifier tous les patients qui ont une maladie cardiovasculaire et, dans de très rares cas, des événements cardiaques graves sont survenus chez des patients sans maladie cardiovasculaire sous-jacente.
Comme avec d'autres agonistes des récepteurs 5HT1B/1D, des sensations de lourdeur, pression ou oppression dans la région précordiale (voir rubrique 4.8) ont été rapportées après l’administration de zolmitriptan. En cas de douleurs thoraciques ou de symptômes évoquant une ischémie cardiaque, il ne faut pas prendre de doses supplémentaires de zolmitriptan jusqu'à ce qu'une évaluation médicale appropriée soit réalisée.
Comme avec d'autres agonistes des récepteurs 5HT1B/1D, des augmentations transitoires de la pression artérielle ont été rapportées chez des patients avec ou sans antécédents d'hypertension. Très rarement, ces augmentations transitoires de la pression artérielle ont été associées à des événements cliniques significatifs. Ne pas dépasser la dose recommandée de zolmitriptan.
Un syndrome sérotoninergique a été signalé lors de l'utilisation combinée de triptans et de médicaments sérotoninergiques, tels que les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) et les inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSN). Le syndrome sérotoninergique est une affection potentiellement mortelle et le diagnostic est probable lorsque (en présence d'un agent sérotoninergique) l'un des éléments suivants est observé :
· Clonus spontané,
· Clonus inductible ou oculaire avec agitation ou diaphorèse,
· Tremblements et hyperréflexie,
· Hypertonie et température corporelle > 38° C et clonus inductible ou oculaire.
Une observation attentive du patient est conseillée si un traitement concomitant par zolmitriptan et un ISRS ou un IRSN est nécessaire, en particulier lors de l'initiation du traitement et des augmentations posologiques (voir rubrique 4.5). L’arrêt des médicaments sérotoninergiques entraîne généralement une amélioration rapide. Le traitement dépend du type et de la gravité des symptômes.
L'utilisation prolongée d'un traitement antalgique pour traiter les céphalées peut entraîner une aggravation de celles-ci. Dans ces cas ou en cas de suspicion, un avis médical est nécessaire et le traitement doit être interrompu. Le diagnostic de céphalée par abus médicamenteux (CAM) doit être suspecté chez les patients présentant des céphalées fréquentes ou quotidiennes malgré (ou à cause) de l'utilisation régulière d'un traitement antimigraineux.
La prévention des crises migraineuses, lorsque le zolmitriptan est administré par voie orale sous forme de comprimés classiques au moment de l'aura, n'a pas été démontrée. ZOLMITRIPTAN VIATRIS doit donc être pris au cours de la céphalée migraineuse.
Ce médicament contient 5 mg d’aspartam par comprimé orodispersible. Ce médicament contient de l’aspartam, une source de phénylalanine, ce qui peut être dangereux pour les personnes atteintes de phénylcétonurie.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Des études d'interaction ont été réalisées avec la caféine, l'ergotamine, la dihydroergotamine, le paracétamol, le métoclopramide, le pizotifène, la fluoxétine, la rifampicine et le propranolol et aucune différence cliniquement significative dans la pharmacocinétique du zolmitriptan ou de son métabolite actif n'a été observée.
Des données chez le sujet sain suggèrent qu'il n'y a pas d'interactions pharmacocinétiques ou cliniques significatives entre le zolmitriptan et l'ergotamine. Toutefois, le risque accru de vasospasme coronarien représente une possibilité théorique et l'administration concomitante est contre-indiquée. Il est conseillé d'attendre au moins 24 heures suite à l'utilisation de préparations à base d'ergotamine avant l'administration de zolmitriptan. Inversement, il est conseillé d'attendre au moins six heures suite à l'utilisation de zolmitriptan avant l'administration d'un produit à base d'ergotamine (voir rubrique 4.3).
Suite à l'administration de moclobémide, un inhibiteur spécifique de la MAO-A, une légère augmentation (26 %) de l'ASC pour le zolmitriptan et une augmentation 3 fois supérieure de l'ASC du métabolite actif ont été observées. C'est pourquoi une prise maximale de 5 mg de zolmitriptan en 24 heures est recommandée chez les patients prenant un inhibiteur de la MAO-A. Les médicaments ne doivent pas être utilisés en association si des doses de moclobémide supérieures à 150 mg deux fois par jour sont administrées.
Suite à l'administration de cimétidine, un inhibiteur du P450 général, la demi-vie du zolmitriptan a été augmentée de 44 % et l'ASC a augmenté de 48 %. De plus, la demi-vie et l'ASC du métabolite actif N-desméthylé (N-desméthylzolmitriptan) ont doublé. Une dose maximale de 5 mg de zolmitriptan en 24 heures est recommandée pour les patients prenant de la cimétidine. En fonction du profil général d'interaction, une interaction avec des inhibiteurs spécifiques du CYP 1A2 ne peut être exclue. La même réduction de posologie est donc recommandée avec des composés de ce type, comme la fluvoxamine et les quinolones (par exemple, la ciprofloxacine).
La sélégiline (un inhibiteur de la MAO-B) et la fluoxétine (un inhibiteur sélectif de la recapture de la sérotonine, ISRS) n'ont pas entraîné d'interaction pharmacocinétique avec le zolmitriptan. Toutefois, des cas isolés décrivant des patients présentant des symptômes compatibles avec un syndrome sérotoninergique (incluant troubles de la conscience, dysautonomie et anomalies neuromusculaires) ont été rapportés suite à l'utilisation d'un inhibiteur sélectif de la recapture de la sérotonine (ISRS) avec des triptans (voir rubrique 4.4).
Les effets indésirables peuvent être plus fréquents lors de l’utilisation concomitante de triptans et de préparations à base de plantes contenant du millepertuis (Hypericum perforatum).
Comme avec d'autres agonistes des récepteurs 5HT1B/1D, le zolmitriptan peut retarder l'absorption d'autres médicaments.
L'administration concomitante d'autres agonistes 5HT1B/1D doit être évitée dans les 24 heures qui suivent un traitement par le zolmitriptan. De même, l'administration de zolmitriptan doit être évitée dans les 24 heures qui suivent l'utilisation d'autres agonistes 5HT1B/1D.
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
La sécurité de ce médicament dans l'utilisation au cours de la grossesse chez l'Homme n'a pas été établie. Les études expérimentales chez l'animal n'ont pas mis en évidence d'effet tératogène direct. Toutefois, certains résultats d'études d'embryotoxicité ont suggéré une viabilité altérée de l'embryon. L'administration de zolmitriptan ne doit être envisagée que si les bénéfices escomptés pour la mère sont supérieurs aux risques éventuels pour le fœtus.
Des études ont montré que le zolmitriptan passe dans le lait des animaux allaitant. Il n'existe aucune donnée chez l'Homme sur le passage de zolmitriptan dans le lait. Il faut donc faire preuve de prudence lors de l'administration de zolmitriptan chez les femmes allaitantes. L'exposition du nouveau-né doit être minimisée en évitant l'allaitement dans les 24 heures suivant le traitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables éventuels sont habituellement transitoires, ont tendance à apparaître dans les quatre heures suivant l'administration, ne sont pas plus fréquents suite à une administration répétée et disparaissent spontanément sans traitement.
Les définitions suivantes s'appliquent à l'incidence des effets indésirables :
Très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000).
Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Les effets indésirables suivants ont été signalés suite à l'administration de zolmitriptan :
Affections du système immunitaire
Rare : réactions d'hypersensibilité, comprenant urticaire, angio-œdème et réactions anaphylactiques.
Affections du système nerveux
Fréquent : anomalies ou troubles de la sensibilité, vertiges, céphalées, hyperesthésie, paresthésie, somnolence, sensation de chaleur.
Affections cardiaques
Fréquent : palpitations.
Peu fréquent : tachycardie.
Très rare : infarctus du myocarde, angine de poitrine, vasospasme coronarien.
Affections vasculaires
Peu fréquent : légère augmentation de la pression artérielle, augmentation transitoire de la pression artérielle.
Affections gastro-intestinales
Fréquent : douleurs abdominales ; nausées ; vomissements ; sècheresse de la bouche, dysphagie.
Très rare : ischémie ou infarctus (par exemple, ischémie intestinale, infarctus intestinal, infarctus splénique) qui peut se manifester sous forme de diarrhées sanglantes ou douleurs abdominales.
Affections musculo-squelettiques et systémiques
Fréquent : faiblesse musculaire, myalgie.
Affections du rein et des voies urinaires
Peu fréquent : polyurie, miction fréquente.
Très rare : miction impérieuse.
Troubles généraux et anomalies au site d'administration
Fréquent : asthénie, lourdeur, oppression, douleur ou pression au niveau de la gorge, du cou, des membres ou de la poitrine.
Certains symptômes peuvent faire partie de la crise de migraine elle-même.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site Internet : https://signalement.social-sante.gouv.fr/.
Les volontaires sains recevant des doses orales uniques de 50 mg présentent fréquemment une sédation.
La demi-vie d'élimination des comprimés de zolmitriptan est de 2,5 à 3 heures (voir rubrique 5.2) et, en cas de surdosage avec le zolmitriptan, la surveillance des patients devra être poursuivie pendant au moins 15 heures ou tant que les symptômes ou signes persistent.
Il n'existe pas d'antidote spécifique du zolmitriptan. En cas d'intoxication grave, une surveillance en soins intensifs est recommandée, celle-ci inclut le maintien de la perméabilité des voies respiratoires, une ventilation et une oxygénation adéquate, le contrôle et le maintien des fonctions cardiovasculaires.
L'effet d'une hémodialyse ou d'une dialyse péritonéale sur les concentrations sériques du zolmitriptan est inconnu.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le zolmitriptan est un agoniste sélectif des récepteurs 5-HT1B/1D qui induisent la contraction vasculaire. Le zolmitriptan présente une forte affinité pour les récepteurs recombinants humains 5-HT1B et 5-HT1D et une faible affinité pour les récepteurs 5-HT1A. Le zolmitriptan ne présente aucune affinité ou activité pharmacologique significative avec les autres sous-types de récepteurs de la 5-HT (5-HT2, 5-HT3, 5‑HT4) ou récepteurs adrénergiques, histaminiques, muscariniques ou dopaminergiques.
Effets pharmacodynamiques
Chez l'animal, l'administration de zolmitriptan induit une vasoconstriction de la circulation artérielle carotidienne. En outre, des données expérimentales chez l'animal suggèrent que le zolmitriptan inhibe l'activité périphérique et centrale du nerf trijumeau en inhibant la libération de neuropeptides (peptide lié au gène de la calcitonine (CGRP), peptide intestinal vaso-actif (VIP) et substance P).
Efficacité clinique et sécurité
Dans les études cliniques avec des comprimés classiques de zolmitriptan, le début de l'efficacité est apparent au bout d'une heure, avec une efficacité accrue entre 2 et 4 heures sur les céphalées et autres symptômes de la migraine, comme les nausées, la photophobie et la phonophobie.
Le zolmitriptan, lorsqu’il est administré par voie orale sous forme de comprimés classiques, est efficace dans la migraine avec ou sans aura et dans la migraine associée aux menstruations. La prévention des crises migraineuses, lorsque le zolmitriptan est administré par voie orale sous forme de comprimés classiques au moment de l'aura, n'a pas été démontrée. ZOLMITRIPTAN VIATRIS doit donc être pris au cours de la phase céphalalgique de la migraine.
Population pédiatrique
Un essai clinique contrôlé mené chez 696 adolescents migraineux n'a pas démontré la supériorité des comprimés de zolmitriptan à des doses de 2,5 mg, 5 mg et 10 mg par rapport au placebo. L'efficacité n'a pas été démontrée.
5.2. Propriétés pharmacocinétiques
Le zolmitriptan sous forme de comprimés classiques est rapidement et efficacement absorbé (au moins 64 %) après l’administration orale chez l'Homme. La biodisponibilité absolue moyenne de la molécule mère est d'environ 40 %.
Chez les volontaires sains, après l’administration d'une dose unique, le zolmitriptan et son métabolite actif, le N-desméthyl, ont une ASC et une Cmax proportionnelles à la dose dans la fourchette de dose de 2,5 à 50 mg. L'absorption du zolmitriptan est rapide. Chez les volontaires sains, 75 % de la Cmax sont atteints en 1 heure, puis la concentration plasmatique du zolmitriptan se maintient approximativement à ce niveau jusqu'à 4 à 5 heures après l'administration.
L'absorption du zolmitriptan n'est pas modifiée par la nourriture. La répétition des prises de zolmitriptan n’a pas mis en évidence d'accumulation du produit.
La concentration plasmatique de zolmitriptan et de ses métabolites est plus basse au cours des 4 premières heures après l'administration du médicament lors d'une crise de migraine comparativement à une période sans migraine, ce qui suggère une absorption retardée compatible avec une vitesse réduite de la vidange gastrique observée lors d'une crise de migraine.
Il a été démontré que les comprimés orodispersibles de zolmitriptan sont bio-équivalents aux comprimés classiques en termes d'ASC et de Cmax pour le zolmitriptan et son métabolite actif N‑desméthylzolmitriptan. Les données pharmacologiques cliniques montrent que le tmax du zolmitriptan peut apparaître plus tard pour les comprimés orodispersibles (entre 0,6 et 5h, médiane 3h) que pour les comprimés classiques (entre 0,5 et 3 h, médiane 1h30). Le tmax pour le métabolite actif était similaire pour les deux formulations (médiane 3h).
Distribution
Le volume de distribution suite à l'administration intraveineuse est de 2,4 l/kg. La liaison aux protéines plasmatiques du zolmitriptan et du métabolite N-desméthyl est faible (approximativement 25 %).
Biotransformation
Le métabolisme du zolmitriptan dépend du CYP1A2 et le métabolisme du métabolite actif N‑desméthylzolmitriptan s'effectue via le système enzymatique de la monoamine oxydase A (MAOA). Les trois métabolites principaux sont : l'acide indole acétique (principal métabolite plasmatique et urinaire), les analogues N-oxyde et N-desméthyl. Le métabolite N-desméthylé est actif contrairement aux autres. Le métabolite N‑desméthyl, est également un agoniste des récepteurs 5HT1B/1D et il est 2 à 6 fois plus puissant, dans les modèles animaux, que le zolmitriptan. Les concentrations plasmatiques du métabolite N-desméthylé représentent environ la moitié de celles de la molécule mère, ce qui permet de penser qu'il contribue à l'action thérapeutique du zolmitriptan.
Elimination
Le zolmitriptan est largement éliminé par biotransformation hépatique suivie d'une excrétion urinaire des métabolites. Plus de 60 % d'une dose orale unique sont éliminés dans l'urine (principalement sous forme de métabolite indole acétique) et environ 30 % dans les selles principalement sous forme de molécule mère inchangée.
Suite à une administration intraveineuse, la clairance plasmatique totale moyenne est d'environ 10 ml/min/kg, dont un quart est une clairance rénale. La clairance rénale est supérieure au taux de filtration glomérulaire, ce qui suggère une sécrétion tubulaire rénale. La demi-vie d'élimination moyenne du zolmitriptan est de 2,5 à 3 heures. Les demi-vies de ses métabolites sont similaires, ce qui suggère que leur élimination est limitée par la vitesse de formation.
Populations particulières
Insuffisance rénale
La clairance rénale du zolmitriptan et de tous ses métabolites est réduite (7 à 8 fois) chez les patients présentant une insuffisance rénale modérée à sévère comparativement aux sujets sains, même si l'ASC de la molécule mère et du métabolite actif est légèrement plus élevée (respectivement de 16 et 35 %) avec une augmentation d'une heure de la demi-vie de 3 à 3,5 heures. Ces paramètres restent dans la gamme des valeurs observées chez les volontaires sains.
Une étude réalisée afin d’évaluer l’effet d’une insuffisance hépatique sur la pharmacocinétique du zolmitriptan a montré que l’ASC et la Cmax étaient respectivement augmentées de 94 % et 50 % chez les patients atteints d’une insuffisance hépatique modérée, et étaient respectivement augmentées de 226 % et 47 % chez les patients atteints d’une insuffisance hépatique sévère, par comparaison aux volontaires sains. L’exposition aux métabolites, y compris le métabolite actif, était diminuée. Pour le métabolite actif N‑desméthylzolmitriptan, l’ASC et la Cmax étaient respectivement réduites de 33 % et 44 % chez les patients atteints d’une insuffisance hépatique modérée, et respectivement de 82 % et 90 % chez les patients atteints d’une insuffisance hépatique sévère.
Sujets âgés
La pharmacocinétique du zolmitriptan chez le sujet âgé sain est similaire à celle du volontaire sain jeune.
5.3. Données de sécurité préclinique
Des effets lors d’études portant sur l’administration d’une dose unique et de doses répétées n’ont été observés qu’à des expositions considérées comme suffisamment supérieures à l’exposition maximale observée chez l’Homme et ont peu de signification clinique.
Les résultats des études de génotoxicité in vitro et in vivo démontrent que les effets génotoxiques du zolmitriptan ne sont pas attendus dans des conditions d'utilisation clinique normale.
Aucune tumeur suite à une utilisation clinique normale n'a été trouvée chez la souris et le rat dans des études de cancérogénicité.
Comme avec d'autres agonistes des récepteurs de la 5HT1B/1D, le zolmitriptan se lie à la mélanine.
2 ans.
Plaquette avec suremballage : Après la première ouverture du suremballage, utiliser dans les 90 jours.
Flacon : Après la première ouverture, utiliser dans les 100 jours.
6.4. Précautions particulières de conservation
À conserver à une température ne dépassant pas 30 °C.
Plaquettes : À conserver dans l'emballage d'origine, à l'abri de l'humidité.
Flacons : Conserver le flacon soigneusement fermé, à l'abri de l'humidité.
6.5. Nature et contenu de l'emballage extérieur
2, 3, 4, 5, 6, 10, 12, 18, 20, 24 ou 48 comprimés orodispersibles sous plaquettes pelables (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC) ou en blisters unitaires perforés en boîte de 6x1, 12x1 ou 24x1 comprimé orodispersible.
2, 3, 4, 5, 6, 10, 12, 18, 20, 24 ou 48 comprimés orodispersibles sous plaquettes pelables (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC) ou en blisters unitaires perforés en boîte de 6x1, 12x1, 24x1 ou 48x1 comprimé orodispersible. Les plaquettes sont placées dans une poche triple laminé contenant des sachets de dessicant à base de gel de silice.
100 comprimés orodispersibles en flacon (PEHD) contenant un dessicant à base de gel de silice, un coton absorbant et fermé par un bouchon en polypropylène (PP) opaque blanc.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1 RUE DE TURIN
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 219 738 9 1 : 2 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 219 739 5 2 : 3 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 219 740 3 4 : 4 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 219 743 2 4 : 5 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 222 526 9 8 : 6 comprimés sous plaquettes (polyamide/aluminium/PVC- papier/polyester/aluminium/PVC).
· 34009 219 746 1 4 : 10 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 222 527 5 9 : 12 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 219 750 9 3 : 18 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 581 568 1 6 : 20 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 581 569 8 4 : 24 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 581 570 6 6 : 48 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 581 571 2 7 : 100 comprimés en flacon (PEHD).
· 34009 219 744 9 2 : 6x1 sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
· 34009 219 747 8 2 : 12x1 comprimés sous plaquettes (polyamide/aluminium/PVC-papier/polyester/aluminium/PVC).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 05/10/2023
ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible
Zolmitriptan
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible ?
3. Comment prendre ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible ET DANS QUELS CAS EST-IL UTILISE ?
ZOLMITRIPTAN VIATRIS est utilisé pour le traitement de la migraine chez les adultes âgés de 18 ans et plus.
Les symptômes de la migraine peuvent être provoqués par une dilatation passagère des vaisseaux sanguins dans la tête. ZOLMITRIPTAN VIATRIS réduit la dilatation de ces vaisseaux sanguins. Cela aide à faire disparaître le mal de tête et les autres symptômes de la crise de migraine, tels que les nausées, les vomissements et la sensibilité à la lumière et au son.
ZOLMITRIPTAN VIATRIS est efficace seulement lorsque la crise de migraine a commencé. ZOLMITRIPTAN VIATRIS ne doit pas être utilisé pour prévenir les crises de migraine.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible ?
Ne prenez jamais ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible :
· si vous êtes allergique au zolmitriptan ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si vous souffrez d'hypertension modérée ou sévère ou d'hypertension légère non contrôlée ;
· si vous avez ou avez eu une maladie cardiaque, une crise cardiaque, des symptômes ou des signes de maladie coronarienne ou si vous souffrez d'angine de poitrine (douleur dans la poitrine par suite d'un manque d'oxygène au niveau du muscle cardiaque) ;
· si vous avez une maladie vasculaire périphérique (rétrécissement des vaisseaux qui amènent le sang aux bras et aux jambes) ;
· si vous avez déjà eu un accident vasculaire cérébral ou si vous avez déjà eu les symptômes d’un accident vasculaire cérébral, qui n’ont duré qu’un temps très court et dont vous vous êtes totalement remis (accident ischémique transitoire) ;
· si vous prenez ou avez pris dans les dernières 24 heures, certains autres médicaments à base d'ergotamine ou des médicaments similaires utilisés dans le traitement de la migraine (y compris le méthysergide). Pour des informations complémentaires, voir « Autres médicaments et ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible » ;
· si vous prenez ou avez pris dans les dernières 24 heures, tous autres « triptans » (tels que naratriptan ou sumatriptan). Pour des informations complémentaires, voir « Autres médicaments et ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible » ;
· si vous souffrez d'une insuffisance rénale sévère ;
· si vous souffrez d’une pathologie appelée « syndrome de Wolff-Parkinson-White ».
Ne pas prendre ce médicament en cas de formes inhabituelles de migraines dues à des problèmes de cerveau ou d’yeux.
Avertissements et précautions
Adressez-vous à votre médecin ou votre pharmacien avant de prendre ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible :
· si vous avez des risques de maladie cardiaque, par exemple vous avez du diabète, si vous fumez beaucoup, souffrez d'hypertension artérielle, d'hypercholestérolémie ou s’il existe des antécédents de maladies cardiaques dans votre famille. Votre médecin pourra vous prescrire des examens complémentaires, en particulier si vous êtes une femme ménopausée ou un homme de plus de 40 ans ;
· si vous avez des problèmes hépatiques ;
· si vous avez des troubles du rythme cardiaque ;
· si vous prenez un médicament pour le traitement de la dépression (voir « Autres médicaments et ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible » plus loin dans cette rubrique).
Pendant le traitement
Durant le traitement, adressez-vous à votre médecin ou votre pharmacien si :
· vous constatez des douleurs ou une sensation d’oppression de la poitrine ou de la gorge. Si ces symptômes ne disparaissent pas rapidement, consultez votre médecin immédiatement ;
· votre pression artérielle augmente. Afin de limiter ce risque, ne prenez pas plus de ZOLMITRIPTAN VIATRIS que recommandé ;
· vous avez de fréquents maux de tête malgré la prise régulière de médicaments contre les céphalées.
Enfants
Sans objet.
Autres médicaments et ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance.
Informez votre médecin, en particulier si vous prenez :
· des médicaments pour la dépression appelés inhibiteurs sélectifs de la recapture de la sérotonine (appelés ISRS) comme la fluoxétine ou des inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSN) comme la venlafaxine ou la duloxétine (qui peut aussi être utilisée contre les problèmes urinaires) ;
· du moclobémide (un médicament utilisé pour traiter la dépression et la phobie sociale) car il peut augmenter l'effet du ZOLMITRIPTAN VIATRIS. Si vous prenez plus de 150 mg de moclobémide deux fois par jour, n'utilisez pas ZOLMITRIPTAN VIATRIS ;
· de la cimétidine, de la fluvoxamine ou des antibiotiques de la famille des quinolones (comme la ciprofloxacine) car ces médicaments peuvent augmenter l'effet de ZOLMITRIPTAN VIATRIS. Par conséquent, la dose maximale recommandée de ZOLMITRIPTAN VIATRIS est de 5 mg par jour ;
· des médicaments tels que la sélégiline (un inhibiteur de la MAO-B) car la prise de ces médicaments avec ZOLMITRIPTAN VIATRIS peut altérer votre état mental ou entraîner des problèmes musculaires. Votre médecin vous indiquera si ZOLMITRIPTAN VIATRIS convient dans votre cas ;
· des médicaments à base de plantes contenant du millepertuis (les effets indésirables peuvent s'aggraver).
Le syndrome sérotoninergique est une maladie rare potentiellement mortelle qui a été rapportée chez certains patients ayant pris du zolmitriptan en association avec des médicaments dits sérotoninergiques (par exemple certains médicaments pour le traitement de la dépression). Les signes du syndrome sérotoninergique peuvent être, par exemple, une agitation, des tremblements, de l'agitation, de la fièvre, une transpiration excessive, des contractions, une rigidité musculaire, des mouvements non coordonnés des membres ou des yeux et des contractions musculaires incontrôlables. Votre médecin peut vous conseiller à ce sujet.
Si vous prenez ou avez pris d’autres médicaments contre la migraine
Si vous prenez d'autres médicaments contre la migraine tels que l'ergotamine ou des dérivés de l'ergotamine (par exemple, la méthysergide) ou tout autre triptan (comme le naratriptan ou le sumatriptan), vous devez attendre au moins 24 heures avant de prendre ZOLMITRIPTAN VIATRIS (voir la rubrique « Ne prenez jamais ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible »).
Après la prise de ZOLMITRIPTAN VIATRIS, attendez au moins 6 heures avant de prendre de l'ergotamine ou des dérivés de l'ergotamine (par exemple, la méthysergide) et attendez au moins 24 heures avant de prendre tout autre médicament de type « triptan ».
ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible avec des aliments, boissons et de l’alcool
Sans objet.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Grossesse
Vous ne devez pas prendre ZOLMITRIPTAN VIATRIS si vous êtes enceinte, sauf si votre médecin vous a indiqué que ce médicament convenait dans votre cas, même si vous êtes enceinte.
Allaitement
Le zolmitriptan peut passer dans le lait maternel. Afin de minimiser le risque d'exposition de votre bébé au médicament, vous ne devez pas allaiter pendant 24 heures après la prise de ZOLMITRIPTAN VIATRIS.
Conduite de véhicules et utilisation de machines
· Il est peu probable que ZOLMITRIPTAN VIATRIS influence la conduite de véhicules ou l’utilisation de certains outils ou machines. Cependant attendez de connaître la façon dont ZOLMITRIPTAN VIATRIS vous affecte avant de reprendre ces activités.
ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible contient de l’aspartam.
Ce médicament contient 5 mg d’aspartam par comprimé orodispersible. L’aspartame est une source de phénylalanine. Il peut être dangereux si vous êtes atteint de phénylcétonurie (PCU), une affection génétique rare caractérisée par une accumulation de la phénylalanine car le corps ne l’élimine pas correctement.
3. COMMENT PRENDRE ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou votre pharmacien en cas de doute.
Posologie
La dose recommandée est de 2,5 mg pour traiter une crise de migraine. Prenez ce médicament dès que possible après le début de votre crise de migraine. Le médicament est également efficace en cas de prise plus tardive.
En cas de réapparition de la douleur dans les 24 heures, un second comprimé de ZOLMITRIPTAN VIATRIS pourra être pris, à condition de respecter un intervalle d'au moins 2 heures entre les 2 prises.
Si une dose de 2,5 mg de ZOLMITRIPTAN VIATRIS ne suffit pas à soulager vos symptômes, prévenez votre médecin. Il peut vous conseiller de prendre une dose plus élevée de 5 mg lors de la prochaine crise de migraine. Vous êtes plus susceptible de présenter des effets indésirables à la dose de 5 mg.
Ne pas prendre plus de 2 doses au cours d'une période de 24 heures. Il est recommandé de ne pas dépasser la dose maximale de 10 mg par 24 heures.
Utilisations chez les patients âgés (âgés de plus de 65 ans)
L'utilisation de ZOLMITRIPTAN VIATRIS n'est pas recommandée.
Patients présentant des problèmes hépatiques
Si vous avez des problèmes modérés ou graves au niveau du foie, vous ne devez pas prendre plus de 5 mg de ZOLMITRIPTAN VIATRIS par 24 heures. En cas de doute, consultez votre médecin qui vous indiquera la dose que vous pouvez prendre.
Patients présentant des problèmes rénaux
Si vous avez des problèmes graves au niveau des reins, votre médecin vous indiquera la dose de ZOLMITRIPTAN VIATRIS que vous devez prendre.
Utilisation chez les enfants et les adolescents (âgés de moins de 17 ans)
ZOLMITRIPTAN VIATRIS ne doit pas être administré aux enfants ou aux adolescents.
Mode et voie d’administration
Voie orale.
Le comprimé peut être pris avec ou sans aliment ou boisson car il se dissout sur la langue et peut être avalé. Néanmoins, ZOLMITRIPTAN VIATRIS agira plus rapidement sur votre migraine si vous prenez le comprimé avec une boisson.
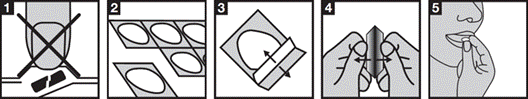
1. Ne poussez pas le comprimé hors de l’alvéole.
2. Tenez la plaquette par les bords et séparez une alvéole du reste de la plaquette en la détachant délicatement en suivant la ligne de prédécoupage.
3. Soulevez le feuillet.
4. Sortez avec précaution le comprimé de son alvéole (ne l'expulsez pas).
5. Placez le comprimé sur votre langue. Laissez-le se dissoudre directement dans votre bouche et avalez-le avec votre salive.
|
|
Si vous avez pris plus de ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible que vous n’auriez dû :
Contactez immédiatement votre médecin ou le service d'urgence de l'hôpital le plus proche de chez vous. Prenez avec vous la boîte et tous les comprimés restants.
Si vous oubliez de prendre ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible :
Si vous arrêtez de prendre ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible :
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Arrêtez de prendre ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible et consultez immédiatement votre médecin ou rendez-vous à l’hôpital le plus proche si vous constatez l’un des effets suivants :
· réactions allergiques graves, pouvant causer un gonflement du visage ou de la gorge, des difficultés à respirer ou des étourdissements ou une éruption cutanée, une urticaire ;
· crise cardiaque (vous pouvez ressentir une forte douleur dans la poitrine, une sensation de moiteur, être essoufflé ou ressentir une douleur dans la mâchoire et les bras) ; douleur à la poitrine (angine) ;
· diminution de la circulation sanguine des intestins ou de la rate (qui peut se manifester par des diarrhées sanglantes ou des douleurs de l'estomac).
Autres effets indésirables possibles :
Effets indésirables fréquents (pouvant affecter 1 personne sur 10) :
· étourdissement, maux de tête, sensibilité accrue au toucher, sensation de fourmillements ou de picotements sur la peau, somnolence, sensation de chaleur, sensations inhabituelles ou altération de la sensibilité ;
· rythme cardiaque irrégulier ou sensation de battements manquants ;
· douleur dans l'abdomen, nausées, vomissements ou bouche sèche, difficulté à avaler (dysphagie) ;
· faiblesse musculaire et douleur musculaire, faiblesse générale, sensation de lourdeur ou d’oppression, douleur ou pression au niveau de la gorge, du cou, des bras et des jambes ou de la poitrine.
Effets indésirables peu fréquents (pouvant affecter 1 personne sur 100) :
· rythme cardiaque accéléré ;
· augmentation de la pression artérielle qui peut aussi apparaître après la prise des comprimés, mais de courte durée ;
· augmentation du volume des urines ; besoin d'uriner plus fréquemment.
Effets indésirables très rares (pouvant affecter 1 personne sur 10 000) :
· envie pressante d'uriner.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site Internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption mentionnée sur l’étiquette, la boîte ou le flacon après EXP. La date de péremption fait référence au dernier jour de ce mois.
À conserver à une température ne dépassant pas 30 °C.
Plaquettes : à conserver dans l'emballage extérieur d'origine, à l'abri de l'humidité.
Plaquettes avec suremballage : à conserver dans l'emballage d'origine, à l'abri de l'humidité. Après la première ouverture du suremballage, utiliser dans les 90 jours.
Flacons : conserver le flacon soigneusement fermé, à l'abri de l'humidité. Après la première ouverture, utiliser dans les 100 jours.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ZOLMITRIPTAN VIATRIS 2,5 mg, comprimé orodispersible
· La substance active est le zolmitriptan.
Chaque comprimé orodispersible contient 2,5 mg de zolmitriptan.
· Les autres composants sont :
Mannitol, silice colloïdale anhydre, crospovidone (type A), crospovidone (type B), aspartam (E951), cellulose microcristalline, gomme guar, stéarate de magnésium, arôme orange (contient : arôme orange, maltodextrine de maïs, alpha tocophérol (E307)).
Ce médicament se présente sous forme de comprimés orodispersibles blancs à blanc cassé, ronds, plats et biseautés, avec « M » marqué sur une face et « ZT1 » sur l’autre face.
Ce médicament est disponible en boîtes de 2, 3, 4, 5, 6, 10, 12, 18, 20, 24 ou 48 comprimés orodispersibles sous plaquettes ou en boîte de 6x1, 12x1, 24x1 ou 48x1 comprimé orodispersible sous plaquettes unitaires prédécoupées. Les plaquettes peuvent être placées dans une poche triple laminé contenant des sachets de dessicant (ne pas avaler le dessicant).
Flacon de 100 comprimés orodispersibles contenant un dessicant (ne pas avaler le dessicant) et un coton absorbant.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
1 RUE DE TURIN
69007 LYON
Exploitant de l’autorisation de mise sur le marché
VIATRIS SANTE
1 RUE DE TURIN
69007 LYON
MCDERMOTT LABORATORIES LIMITED TRADING AS GERARD LABORATORIES
35/36 BALDOYLE INDUSTRIAL ESTATE
GRANGE ROAD
DUBLIN 13
IRLANDE
OU
MYLAN HUNGARY KFT.
H-2900 KOMÁROM
MYLAN UTCA 1
HONGRIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).