Dernière mise à jour le 03/08/2026
PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : psycholeptiques, autres antipsychotiques, code ATC : N05AX13.
PALIPERIDONE BIOGARAN contient la substance active palipéridone qui appartient à la classe des antipsychotiques et est utilisé dans le traitement d’entretien des symptômes de la schizophrénie chez les patients adultes stabilisés par la palipéridone ou la rispéridone.
Si vous avez présenté une réponse à la palipéridone ou à la rispéridone dans le passé et avez des symptômes légers à modérés, votre médecin peut initier le traitement par PALIPERIDONE BIOGARAN sans stabilisation préalable par la palipéridone ou la rispéridone.
La schizophrénie est une maladie avec des symptômes « positifs » et « négatifs ». Les symptômes positifs sont un excès de symptômes qui ne sont normalement pas présents. Par exemple, une personne atteinte de schizophrénie peut entendre des voix ou voir des choses qui ne sont pas là (appelées hallucinations), croire des choses qui ne sont pas vraies (appelées illusions), ou se sentir inhabituellement suspicieuse envers les autres. Les symptômes négatifs représentent une absence de comportements ou de sentiments qui sont normalement présents. Par exemple, une personne atteinte de schizophrénie peut sembler en retrait et peut ne manifester aucune réaction émotionnelle ou peut avoir des difficultés à parler de manière claire et logique. Les personnes atteintes de cette maladie peuvent également se sentir déprimées, anxieuses, coupables ou tendues.
PALIPERIDONE BIOGARAN peut aider à soulager les symptômes de votre maladie et empêcher vos symptômes de revenir.
Présentations
> 1 seringue préremplie polycyclooléfine avec 2 aiguilles
Code CIP : 34009 302 408 9 2
Déclaration d'arrêt de commercialisation : 14/01/2026
Cette présentation n'est pas agréée aux collectivités
- Prix hors honoraire de dispensation : 36,13 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 37,15 €
- Taux de remboursement :65 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : BIOGARAN
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale réservée à certains spécialistes
- prescription réservée aux spécialistes et services PSYCHIATRIE
- renouvellement non restreint
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 262 020 0
ANSM - Mis à jour le : 18/07/2024
PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque seringue préremplie contient 39 mg de palmitate de palipéridone équivalent à 25 mg de palipéridone.
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension injectable à libération prolongée en seringue préremplie.
La suspension est de couleur blanche à blanc cassé. La suspension est de pH neutre (environ 7,0). L’osmolalité de la suspension est de 220-320 mOsm/Kg.
4.1. Indications thérapeutiques
Chez les patients adultes sélectionnés atteints de schizophrénie et ayant précédemment répondu à la palipéridone ou à la rispéridone orale, PALIPERIDONE BIOGARAN peut être utilisé sans stabilisation préalable par un traitement oral si les symptômes psychotiques sont légers à modérés et si un traitement injectable à action prolongée est nécessaire.
4.2. Posologie et mode d'administration
Les doses initiales de Palipéridone Biogaran recommandées sont de 150 mg au jour 1 du traitement et de 100 mg une semaine plus tard (jour 8), les deux doses étant administrées dans le muscle deltoïde afin d’atteindre rapidement des concentrations thérapeutiques (voir rubrique 5.2). La troisième dose doit être administrée un mois après la seconde dose d'initiation. La dose d’entretien mensuelle recommandée est de 75 mg ; certains patients peuvent bénéficier de doses plus faibles ou plus élevées dans l’intervalle recommandé allant de 25 à 150 mg en fonction de la tolérance individuelle du patient et/ou de l’efficacité. Les patients en surpoids ou obèses peuvent avoir besoin de doses comprises dans l’intervalle supérieur (voir rubrique 5.2). Après la seconde dose d’initiation, les doses d’entretien mensuelles peuvent être administrées soit dans le muscle deltoïde soit dans le muscle fessier.
Un ajustement de la dose d’entretien peut être effectué mensuellement. Lors des ajustements de dose, les propriétés de libération prolongée de PALIPERIDONE BIOGARAN doivent être prises en compte (voir rubrique 5.2), car l’effet complet des doses d’entretien peut ne pas être observé avant plusieurs mois.
Substitution de la palipéridone orale à libération prolongée ou de la rispéridone orale par PALIPERIDONE BIOGARAN
PALIPERIDONE BIOGARAN doit être instauré comme décrit au début de la rubrique 4.2 ci-dessus. Durant le traitement par dose d’entretien mensuelle de PALIPERIDONE BIOGARAN, les patients précédemment stabilisés sous différentes doses de palipéridone orale sous forme de comprimés à libération prolongée peuvent atteindre une exposition similaire en palipéridone à l’état d’équilibre avec les injections. Les doses d'entretien de PALIPERIDONE BIOGARAN nécessaires pour atteindre une exposition similaire à l'état d’équilibre sont indiquées dans le tableau suivant :
|
Doses de palipéridone sous forme de comprimés à libération prolongée et de PALIPERIDONE BIOGARAN requises pour atteindre une exposition à la palipéridone similaire à l’état d’équilibre lors d’un traitement d’entretien |
|
|
Dose précédente de palipéridone sous forme de comprimés à libération prolongée |
Injection de PALIPERIDONE BIOGARAN |
|
3 mg par jour 6 mg par jour 9 mg par jour 12 mg par jour |
25-50 mg tous les mois 75 mg tous les mois 100 mg tous les mois 150 mg tous les mois |
La palipéridone orale ou la rispéridone orale précédemment administrée peut être arrêtée au moment de l’instauration du traitement par PALIPERIDONE BIOGARAN. Certains patients peuvent bénéficier d’un arrêt progressif. Certains patients passant d'une dose orale de palipéridone plus élevée (par exemple, 9-12 mg par jour) à des injections dans le muscle fessier avec PALIPERIDONE BIOGARAN peuvent avoir une exposition plasmatique plus faible au cours des 6 premiers mois après le changement. Par conséquent, de façon alternative, il pourrait être envisagé de faire des injections dans le muscle deltoïde pendant les 6 premiers mois.
Substitution de l’injection de rispéridone à action prolongée par PALIPERIDONE BIOGARAN
Lors de la substitution de l’injection de rispéridone à action prolongée, instaurer le traitement par PALIPERIDONE BIOGARAN à la place de l’injection suivante programmée. PALIPERIDONE BIOGARAN doit ensuite être poursuivi à intervalles mensuels. Le schéma posologique d’instauration de la première semaine incluant les injections intramusculaires (jour 1 et 8, respectivement) décrit en rubrique 4.2 ci-dessus n’est pas nécessaire.
Les patients précédemment stabilisés par différentes doses de rispéridone injectable à action prolongée peuvent atteindre une exposition à la palipéridone similaire à l’état d’équilibre lors d’un traitement d’entretien par des doses mensuelles de PALIPERIDONE BIOGARAN comme suit :
|
Doses de rispéridone injectable à action prolongée et de PALIPERIDONE BIOGARAN requises pour atteindre une exposition à la palipéridone similaire à l’état d’équilibre |
|
|
Dose précédente de rispéridone injectable à action prolongée |
Injection de PALIPERIDONE BIOGARAN |
|
25 mg toutes les 2 semaines |
50 mg tous les mois |
|
37,5 mg toutes les 2 semaines |
75 mg tous les mois |
|
50 mg toutes les 2 semaines |
100 mg tous les mois |
L’arrêt des médicaments antipsychotiques doit être effectué conformément aux informations de prescription associées. Si PALIPERIDONE BIOGARAN est interrompu, ses propriétés de libération prolongée doivent être prises en compte. Il est recommandé de réévaluer périodiquement la nécessité de poursuivre un traitement contre les symptômes extrapyramidaux (SE).
Oubli de doses
· Evitez l’oubli de doses
Il est recommandé que la seconde dose d’instauration de PALIPERIDONE BIOGARAN soit administrée une semaine après la première dose. Afin d’éviter un oubli de dose, les patients peuvent recevoir la seconde dose 4 jours avant ou après l’échéance de la première semaine (jour 8). De même, il est recommandé que la troisième injection et les injections suivantes après le schéma d’instauration soient administrées mensuellement. Pour pallier un oubli d’une dose mensuelle, les patients peuvent recevoir l’injection jusqu’à 7 jours avant ou après l’échéance mensuelle.
Si la date de la seconde injection de PALIPERIDONE BIOGARAN (jour 8 ± 4 jours) est oubliée, la recommandation d’instaurer à nouveau le traitement dépend de la durée qui s’est écoulée depuis la première injection reçue par le patient.
· Oubli de la seconde dose d’instauration (< 4 semaines après la première injection)
Si moins de 4 semaines se sont écoulées depuis la première injection, alors le patient devra recevoir la seconde injection de 100 mg dans le muscle deltoïde dès que possible. Une troisième injection de 75 mg de PALIPERIDONE BIOGARAN dans le muscle deltoïde ou le muscle fessier devra être administrée 5 semaines après la première injection (quelle que soit la date de la seconde injection). Par la suite, le cycle mensuel normal des injections dans le muscle deltoïde ou le muscle fessier de doses allant de 25 à 150 mg, en fonction de la tolérance individuelle du patient et/ou de l’efficacité, sera poursuivi.
· Oubli de la seconde dose d’instauration (4 à 7 semaines après la première injection)
Si 4 à 7 semaines se sont écoulées depuis la première injection de PALIPERIDONE BIOGARAN, reprendre le traitement par deux injections de 100 mg comme suit :
1. une injection dans le muscle deltoïde dès que possible
2. une autre injection dans le muscle deltoïde une semaine plus tard
3. reprise du cycle normal mensuel des injections dans le muscle deltoïde ou le muscle fessier de doses allant de 25 à 150 mg, en fonction de la tolérance individuelle du patient et/ou de l’efficacité.
· Oubli de la seconde dose d’instauration (> 7 semaines après la première injection)
Si plus de 7 semaines se sont écoulées depuis la première injection de PALIPERIDONE BIOGARAN, recommencer le traitement comme décrit ci-dessus dans le schéma d’instauration de Palipéridone Biogaran.
· Oubli d’une dose d’entretien mensuelle (1 mois à 6 semaines)
Après instauration, le cycle d’injection de PALIPERIDONE BIOGARAN recommandé est mensuel. Si moins de 6 semaines se sont écoulées depuis la dernière injection, alors la dose précédemment stabilisante devra être administrée dès que possible, suivie par des injections à intervalles mensuels.
· Oubli d’une dose d’entretien mensuelle (> 6 semaines à 6 mois)
Si plus de 6 semaines se sont écoulées depuis la dernière injection de PALIPERIDONE BIOGARAN, la recommandation est la suivante :
Pour les patients stabilisés par des doses allant de 25 à 100 mg
1. une injection dans le muscle deltoïde dès que possible de la même dose que celle par laquelle le patient était précédemment stabilisé.
2. une autre injection dans le muscle deltoïde (même dose) une semaine plus tard (jour 8).
3. reprise du cycle mensuel normal des injections dans le muscle deltoïde ou dans le muscle fessier de doses allant de 25 à 150 mg, en fonction de la tolérance individuelle du patient et/ou de l’efficacité.
Pour les patients stabilisés par 150 mg
1. une injection d’une dose de 100 mg dans le muscle deltoïde dès que possible.
2. une autre injection d’une dose de 100 mg dans le muscle deltoïde une semaine plus tard (jour 8).
3. reprise du cycle normal mensuel des injections dans le muscle deltoïde ou dans le muscle fessier de doses allant de 25 à 150 mg, en fonction de la tolérance individuelle du patient et/ou de l’efficacité.
· Oubli d’une dose d’entretien mensuelle (> 6 mois)
Si plus de 6 mois se sont écoulés depuis la dernière injection de PALIPERIDONE BIOGARAN, recommencez le traitement comme décrit ci-dessus dans le schéma d’instauration de PALIPERIDONE BIOGARAN.
Populations particulières
Patient âgé
L’efficacité et la sécurité chez les patients âgés de plus de 65 ans n’ont pas été établies.
En général, la posologie de PALIPERIDONE BIOGARAN recommandée pour les patients âgés présentant une fonction rénale normale est la même que celle des patients adultes plus jeunes dont la fonction rénale est normale. Toutefois, certains patients âgés pouvant avoir une fonction rénale diminuée, un ajustement posologique peut être nécessaire (voir Insuffisance rénale ci-dessous pour les recommandations posologiques chez les patients présentant une insuffisance rénale).
Insuffisance rénale
PALIPERIDONE BIOGARAN n’a pas été étudié de manière systématique chez les patients présentant une insuffisance rénale (voir rubrique 5.2). Chez les patients présentant une insuffisance rénale légère (clairance de la créatinine ≥ 50 à < 80 mL/min), les doses initiales de PALIPERIDONE BIOGARAN recommandées sont de 100 mg au jour 1 du traitement et 75 mg une semaine plus tard, les deux doses étant administrées dans le muscle deltoïde. La dose d’entretien mensuelle recommandée est de 50 mg dans un intervalle allant de 25 à 100 mg en fonction de la tolérance individuelle du patient et/ou l’efficacité.
PALIPERIDONE BIOGARAN n’est pas recommandé chez les patients présentant une insuffisance rénale modérée ou sévère (clairance de la créatinine < 50 mL/min) (voir rubrique 4.4).
Insuffisance hépatique
D’après l’expérience acquise avec la palipéridone orale, aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée. La palipéridone n’ayant pas été étudiée chez les patients présentant une insuffisance hépatique sévère, la prudence est recommandée chez ces patients (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité de PALIPERIDONE BIOGARAN chez les enfants et les adolescents de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
PALIPERIDONE BIOGARAN est destiné à l’administration intramusculaire uniquement. Il ne doit pas être administré par une autre voie. Il doit être injecté lentement, en profondeur dans le muscle deltoïde ou fessier. Chaque injection doit être administrée par un professionnel de santé. L’administration doit être effectuée par une injection unique. La dose ne doit pas être administrée par des injections séparées.
Les doses d’instauration des jours 1 et 8 doivent chacune être administrées dans le muscle deltoïde afin d’atteindre rapidement des concentrations thérapeutiques (voir rubrique 5.2). Après la seconde dose d’initiation, les doses d’entretien mensuelles peuvent être administrées soit dans le muscle deltoïde soit dans le muscle fessier. Un changement du muscle fessier au muscle deltoïde (et vice versa) doit être envisagé en cas de douleur au site d’injection et si la gêne associée n’est pas bien tolérée (voir rubrique 4.8). Il est également recommandé d’alterner entre les côtés gauche et droit (voir ci-dessous).
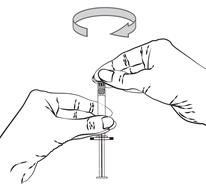
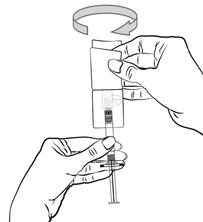
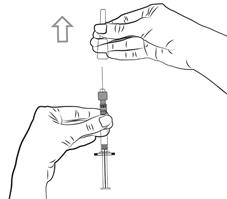
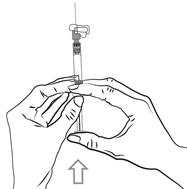
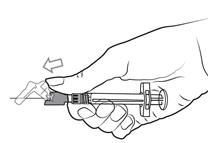
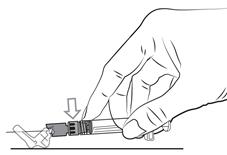
Pour les instructions relatives à l’utilisation et la manipulation de PALIPERIDONE BIOGARAN, voir la notice (information destinée aux professionnels de santé).
Administration dans le muscle deltoïde
La taille de l’aiguille recommandée pour les administrations initiales et d’entretien de PALIPERIDONE BIOGARAN dans le muscle deltoïde est déterminée par le poids du patient. Pour les patients ayant un poids > 90 kg, l’aiguille de 11/2 pouce 22 Gauge (38,1 mm x 0,72 mm) est recommandée. Pour ceux ayant un poids < 90 kg, l’aiguille de 1 pouce 23 Gauge (25,4 mm x 0,64 mm) est recommandée. Les injections dans le muscle deltoïde doivent être alternées entre les deux muscles deltoïdes.
Administration dans le muscle fessier
La taille de l’aiguille recommandée pour l’administration d’entretien de PALIPERIDONE BIOGARAN dans le muscle fessier est l’aiguille de 11/2 pouce 22 Gauge (38,1 mm x 0,72 mm). L’administration doit être faite dans le quadrant supéro-externe de la fesse. Les injections dans le muscle fessier doivent être alternées entre les deux muscles fessiers.
4.4. Mises en garde spéciales et précautions d'emploi
Utilisation chez les patients dans un état d’agitation aiguë ou dans un état psychotique grave
PALIPERIDONE BIOGARAN ne doit pas être utilisé dans la prise en charge des états psychotiques graves ou d’agitation aiguë lorsqu’un contrôle immédiat des symptômes est recherché.
Intervalle QT
La prudence est recommandée lorsque la palipéridone est prescrite à des patients présentant une maladie cardiovasculaire connue ou des antécédents familiaux d’allongement de l’intervalle QT, et en cas d’utilisation concomitante de médicaments suspectés d’allonger l’intervalle QT.
Syndrome malin des neuroleptiques
Le syndrome malin des neuroleptiques (SMN), caractérisé par une hyperthermie, une rigidité musculaire, une instabilité du système nerveux autonome, une altération de la conscience et une augmentation des taux sériques de créatine phosphokinase, a été rapporté avec la palipéridone. Des signes cliniques supplémentaires peuvent inclure une myoglobinurie (rhabdomyolyse) et une insuffisance rénale aiguë. Si un patient développe des signes ou des symptômes indicatifs d’un SMN, la palipéridone doit être interrompue.
Dyskinésie tardive/symptômes extrapyramidaux
Les médicaments qui possèdent des propriétés antagonistes des récepteurs de la dopamine ont été associés à l’induction de dyskinésie tardive caractérisée par des mouvements anormaux involontaires, prédominant au niveau de la langue et/ou du visage. Si les signes et symptômes d’une dyskinésie tardive apparaissent, l’arrêt de tous les antipsychotiques, dont la palipéridone, doit être envisagé.
La prudence est recommandée chez les patients recevant de façon concomitante des psychostimulants (par exemple, méthylphénidate) et la palipéridone, car des symptômes extrapyramidaux peuvent apparaitre lors de l’ajustement de l’un ou des deux médicaments. L’arrêt progressif du traitement stimulant est recommandé (voir rubrique 4.5).
Leucopénie, neutropénie et agranulocytose
Des cas de leucopénie, neutropénie et agranulocytose ont été rapportés avec PALIPERIDONE BIOGARAN. Une agranulocytose a été très rarement rapportée (< 1/10 000 patients) lors de la surveillance après commercialisation. Les patients ayant des antécédents cliniquement significatifs de faible numération des globules blancs (NGB) ou de leucopénie/neutropénie d’origine médicamenteuse doivent être surveillés pendant les tous premiers mois de traitement et l'arrêt de PALIPERIDONE BIOGARAN doit être considéré au premier signe d'une baisse cliniquement significative de la NGB en l'absence d'autres facteurs causaux. Les patients ayant une neutropénie cliniquement significative doivent être attentivement surveillés pour une fièvre ou d'autres symptômes ou signes d'infection et traités rapidement si de tels symptômes ou signes apparaissent. Les patients ayant une neutropénie sévère (numération absolue de neutrophiles < 1 x 109/L) doivent arrêter PALIPERIDONE BIOGARAN et leur NGB doit être suivie jusqu'à rétablissement.
Réactions d'hypersensibilité
Des réactions anaphylactiques chez des patients ayant précédemment toléré la rispéridone orale ou la palipéridone orale ont été rarement rapportées depuis le début de la commercialisation (voir rubriques 4.1 et 4.8).
Si des réactions d'hypersensibilité surviennent : arrêter PALIPERIDONE BIOGARAN ; initier des mesures générales d’accompagnement cliniquement appropriées et surveiller le patient jusqu'à ce que les signes et les symptômes disparaissent (voir rubriques 4.3 et 4.8).
Hyperglycémie et diabète
Hyperglycémie, diabète et exacerbation d’un diabète préexistant, dont coma diabétique et acidocétose, ont été rapportés au cours du traitement par palipéridone. Une surveillance clinique adéquate est recommandée conformément aux recommandations relatives aux antipsychotiques. Les symptômes d'hyperglycémie (tels que polydipsie, polyurie, polyphagie, et fatigue) doivent être recherchés chez les patients traités par PALIPERIDONE BIOGARAN. Une surveillance régulière doit être effectuée chez les patients diabétiques afin de détecter une aggravation de la glycémie.
Prise de poids
Une prise de poids significative a été rapportée avec l’utilisation de PALIPERIDONE BIOGARAN. Le poids doit être contrôlé régulièrement.
Utilisation chez des patients présentant des tumeurs prolactine-dépendantes
Des études sur des cultures de tissus suggèrent que la croissance cellulaire des tumeurs du sein chez l’Homme peut être stimulée par la prolactine. Bien qu’aucune association claire avec l’administration d’antipsychotiques n’ait été démontrée jusqu’à présent dans les études cliniques et épidémiologiques, la prudence est recommandée chez les patients présentant des antécédents médicaux significatifs. La palipéridone doit être utilisée avec précaution chez les patients présentant une tumeur préexistante potentiellement prolactine-dépendante.
Hypotension orthostatique
La palipéridone peut induire une hypotension orthostatique chez certains patients par son activité alpha-bloquante. Sur la base des données poolées de 3 essais contrôlés versus placebo, de 6 semaines, à doses fixes réalisés avec la palipéridone orale sous forme de comprimés à libération prolongée (3, 6, 9 et 12 mg), une hypotension orthostatique a été rapportée chez 2,5 % des patients traités par la palipéridone orale comparé à 0,8 % des patients sous placebo. PALIPERIDONE BIOGARAN doit être utilisé avec précaution chez les patients présentant une maladie cardiovasculaire connue (par exemple, insuffisance cardiaque, infarctus du myocarde ou ischémie, anomalies de la conduction), une maladie cérébrovasculaire ou des situations cliniques prédisposant le patient à l’hypotension (par exemple, déshydratation et hypovolémie).
Convulsions
PALIPERIDONE BIOGARAN doit être utilisé avec précaution chez les patients présentant des antécédents de convulsions ou d’autres situations cliniques pouvant potentiellement abaisser le seuil épileptogène.
Insuffisance rénale
Les concentrations plasmatiques de palipéridone sont augmentées chez les patients présentant une insuffisance rénale et de ce fait, une adaptation posologique est recommandée chez les patients ayant une insuffisance rénale légère. PALIPERIDONE BIOGARAN n’est pas recommandé chez les patients présentant une insuffisance rénale modérée ou sévère (clairance de la créatinine < 50 mL/min) (voir rubriques 4.2 et 5.2).
Insuffisance hépatique
Aucune donnée n’est disponible chez les patients présentant une insuffisance hépatique sévère (score Child-Pugh : classe C). La prudence est recommandée si la palipéridone est utilisée chez ces patients.
Patients âgés déments
PALIPERIDONE BIOGARAN n’a pas été étudié chez les patients âgés déments. PALIPERIDONE BIOGARAN doit être utilisé avec précaution chez les patients âgés déments ayant des facteurs de risque d’accident vasculaire cérébral.
L’expérience acquise avec la rispéridone, citée ci-dessous, est considérée comme également valable pour la palipéridone.
Mortalité globale
Dans une méta-analyse portant sur 17 essais cliniques contrôlés, des patients âgés déments traités par d’autres antipsychotiques atypiques, incluant la rispéridone, l’aripiprazole, l’olanzapine et la quétiapine ont présenté une augmentation du risque de mortalité comparé au placebo. Parmi ceux traités par la rispéridone, le taux de mortalité était de 4 % comparé à 3,1 % avec le placebo.
Effets indésirables cérébrovasculaires
Une augmentation du risque d’effets indésirables cérébrovasculaires d’un facteur 3 environ a été observée dans des essais cliniques randomisés contrôlés versus placebo réalisés chez des patients déments avec certains antipsychotiques atypiques, incluant la rispéridone, l’aripiprazole et l’olanzapine. Le mécanisme de cette augmentation du risque n’est pas connu.
Maladie de Parkinson et démence à corps de Lewy
Les prescripteurs doivent évaluer les risques versus les bénéfices de la prescription de PALIPERIDONE BIOGARAN chez les patients présentant une maladie de Parkinson ou une démence à Corps de Lewy (DCL), ces deux groupes pouvant présenter une augmentation du risque de survenue de syndrome malin des neuroleptiques ainsi qu’une augmentation de la sensibilité aux antipsychotiques. Les manifestations de l’augmentation de sensibilité peuvent inclure une confusion, obnubilation, une instabilité posturale avec des chutes fréquentes, en plus des symptômes extrapyramidaux.
Priapisme
La survenue d’un priapisme a été rapportée avec les médicaments antipsychotiques (dont la rispéridone) ayant des propriétés alpha-bloquantes adrénergiques. Après commercialisation, des cas de priapisme ont également été rapportés avec la palipéridone orale, qui est le métabolite actif de la rispéridone. Les patients doivent être informés d’aller consulter en urgence un médecin si le priapisme n’a pas disparu au bout de 4 heures.
Régulation de la température corporelle
Une altération de la capacité corporelle à diminuer la température corporelle centrale a été rapportée avec les médicaments antipsychotiques. La prudence est recommandée lors de la prescription de PALIPERIDONE BIOGARAN chez des patients susceptibles d’être exposés à certaines situations pouvant contribuer à une augmentation de la température corporelle centrale, par exemple, exercice physique intense, exposition à une température extrême, traitement concomitant par des médicaments ayant une activité anticholinergique ou existence d’une déshydratation.
Thromboembolie veineuse
Des cas de thromboembolie veineuse (TEV) ont été rapportés avec les médicaments antipsychotiques. Les patients traités par antipsychotiques présentant souvent des facteurs de risque acquis de TEV, tout facteur de risque potentiel de TEV doit être identifié avant et pendant le traitement par PALIPERIDONE BIOGARAN et des mesures préventives doivent être mises en œuvre.
Effet antiémétique
Un effet antiémétique a été observé au cours des études précliniques réalisées avec la palipéridone. Cet effet, lorsqu’il survient chez l’Homme, peut masquer les signes et les symptômes de surdosage de certains médicaments ou certaines situations cliniques telles qu’une occlusion intestinale, un syndrome de Reye et une tumeur cérébrale.
Administration
La prudence est recommandée afin d’éviter toute injection accidentelle de PALIPERIDONE BIOGARAN dans un vaisseau sanguin.
Syndrome de l’iris hypotonique peropératoire
Un syndrome de l'iris hypotonique peropératoire (SIHP) a été observé au cours d’interventions chirurgicales de la cataracte chez des patients traités par des médicaments antagonistes des récepteurs alpha 1a-adrénergiques, tels que PALIPERIDONE BIOGARAN (voir rubrique 4.8).
Le SIHP peut augmenter le risque de complications oculaires pendant et après l'opération. L'utilisation actuelle ou antérieure de médicaments ayant un effet antagoniste des récepteurs alpha 1a- adrénergiques doit être portée à la connaissance du chirurgien ophtalmologiste avant l'intervention chirurgicale. Le bénéfice potentiel de l'arrêt du traitement par alpha 1-bloquant avant l’intervention chirurgicale de la cataracte n'a pas été établi et doit être mis en balance avec le risque d'arrêt du traitement antipsychotique.
Excipients
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
La prudence est recommandée lorsque PALIPERIDONE BIOGARAN est prescrit avec des médicaments connus pour allonger l’intervalle QT, par exemple, les antiarythmiques de classe IA (par exemple quinidine, disopyramide) et les antiarythmiques de classe III (par exemple amiodarone, sotalol), certains antihistaminiques, certains autres antipsychotiques et certains antipaludéens (par exemple, méfloquine). Cette liste est indicative et non exhaustive.
Effets potentiels de PALIPERIDONE BIOGARAN sur d’autres médicaments
La palipéridone ne devrait pas entraîner d’interactions pharmacocinétiques cliniquement importantes avec les médicaments métabolisés par les isoenzymes du cytochrome P-450.
Compte tenu des effets primaires de la palipéridone sur le système nerveux central (SNC) (voir rubrique 4.8), PALIPERIDONE BIOGARAN doit être utilisé avec précaution en association avec d’autres médicaments agissant au niveau central, par exemple les anxiolytiques, la majorité des antipsychotiques, les hypnotiques, les opiacés, etc., ou avec l’alcool.
La palipéridone peut antagoniser l’effet de la lévodopa et d’autres agonistes dopaminergiques. Lorsque cette association s’avère nécessaire, en particulier au stade terminal de la maladie de Parkinson, la dose efficace la plus faible de chaque traitement doit être prescrite.
Du fait de son potentiel à induire une hypotension orthostatique (voir rubrique 4.4), un effet additif peut être observé lorsque PALIPERIDONE BIOGARAN est administré avec d’autres médicaments ayant ce potentiel, par exemple d’autres antipsychotiques, antidépresseurs tricycliques.
La prudence est recommandée lorsque la palipéridone est associée à d’autres médicaments connus pour diminuer le seuil épileptogène (par exemple, phénothiazines ou butyrophénones, tricycliques ou IRSSs, tramadol, méfloquine, etc.).
La co-administration de palipéridone orale sous forme de comprimés à libération prolongée à l’état d’équilibre (12 mg une fois par jour) avec du divalproex de sodium sous forme de comprimés à libération prolongée (500 à 2000 mg une fois par jour) n’a pas affecté la pharmacocinétique du valproate à l’état d’équilibre.
Aucune étude d’interaction entre PALIPERIDONE BIOGARAN et le lithium n’a été effectuée, toutefois, une interaction pharmacocinétique est peu probable.
Effets potentiels d’autres médicaments sur PALIPERIDONE BIOGARAN
Les études in vitro indiquent que le CYP2D6 et le CYP3A4 peuvent interférer de façon minime avec le métabolisme de la palipéridone, mais qu’il n’existe pas in vitro ni in vivo de données indiquant que ces isoenzymes jouent un rôle significatif dans le métabolisme de la palipéridone. L’administration concomitante de palipéridone orale avec la paroxétine, un puissant inhibiteur du CYP2D6, n’a pas montré d’effet cliniquement significatif sur la pharmacocinétique de la palipéridone.
La co-administration de la palipéridone orale à libération prolongée une fois par jour avec de la carbamazépine 200 mg administrée deux fois par jour a entraîné une diminution d’environ 37 % de la Cmax et de l’AUC moyennes de la palipéridone à l’état d’équilibre. Cette diminution est due, pour une large part, à une augmentation de 35 % de la clairance rénale de la palipéridone résultant probablement de l’induction de la P-gp rénale par la carbamazépine. Une diminution mineure de la quantité de substance active excrétée sous forme inchangée dans les urines suggère qu’il y a peu d’effet sur le métabolisme via le CYP ou sur la biodisponibilité de la palipéridone au cours de la co- administration avec la carbamazépine. Des diminutions plus importantes des concentrations plasmatiques de palipéridone peuvent survenir avec des doses plus élevées de carbamazépine. Lors de l’initiation de la carbamazépine, la dose de PALIPERIDONE BIOGARAN doit être réévaluée et augmentée si nécessaire. A l’inverse, lors de l’arrêt de la carbamazépine, la dose de PALIPERIDONE BIOGARAN doit être réévaluée et diminuée si nécessaire.
La co-administration d’une dose unique de 12 mg de palipéridone orale sous forme de comprimé à libération prolongée avec le divalproex de sodium sous forme de comprimés à libération prolongée (deux comprimés de 500 mg une fois par jour) a entraîné une augmentation d’environ 50 % de la Cmax et de l’AUC de la palipéridone, résultant probablement d’une augmentation de l’absorption orale.
Aucun effet sur la clairance systémique n’ayant été observé, une interaction cliniquement significative n’est pas attendue entre le divalproex de sodium sous forme de comprimés à libération prolongée et l’injection intramusculaire de PALIPERIDONE BIOGARAN. Cette interaction n’a pas été étudiée avec PALIPERIDONE BIOGARAN.
Utilisation concomitante de PALIPERIDONE BIOGARAN avec la rispéridone ou la palipéridone orale
La palipéridone étant le principal métabolite actif de la rispéridone, une attention particulière est nécessaire lorsque PALIPERIDONE BIOGARAN est coadministré avec la rispéridone ou la palipéridone orale pendant des périodes prolongées. Les données de sécurité concernant l'utilisation concomitante de PALIPERIDONE BIOGARAN avec d'autres antipsychotiques sont limitées.
Utilisation concomitante de PALIPERIDONE BIOGARAN avec des psychostimulants
L’utilisation concomitante de psychostimulants (exemple, méthylphénidate) avec la palipéridone peut entraîner des symptômes extrapyramidaux lors de l’ajustement de l’un ou des deux traitements (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’existe pas de données adéquates sur l’utilisation de la palipéridone pendant la grossesse. Le palmitate de palipéridone injecté par voie intramusculaire et la palipéridone administrée par voie orale n’ont pas montré d’effets tératogènes au cours des études chez l’animal, mais d’autres types de toxicité sur la reproduction ont été observés (voir rubrique 5.3). Les nouveau-nés exposés à la palipéridone pendant le troisième trimestre de la grossesse, présentent un risque de réactions indésirables incluant des symptômes extrapyramidaux et/ou des symptômes de sevrage, pouvant varier en termes de sévérité et de durée après l’accouchement. Les réactions suivantes ont été rapportées : agitation, hypertonie, hypotonie, tremblements, somnolence, détresse respiratoire, trouble de l’alimentation. En conséquence, les nouveau-nés doivent être étroitement surveillés. PALIPERIDONE BIOGARAN ne doit pas être utilisé au cours de la grossesse sauf si manifestement nécessaire.
La palipéridone est excrétée dans le lait maternel en quantités suffisantes pour que des effets sur le nourrisson allaité soient possibles lorsque des doses thérapeutiques sont administrées à la femme allaitante. PALIPERIDONE BIOGARAN ne doit pas être utilisé au cours de l’allaitement.
Fertilité
Aucun effet pertinent n’a été observé dans les études non cliniques.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité d’emploi
Les effets indésirables les plus fréquemment rapportés au cours des essais cliniques ont été insomnie, céphalée, anxiété, infection des voies respiratoires supérieures, réaction au site d’injection, parkinsonisme, prise de poids, akathisie, agitation, sédation/somnolence, nausée, constipation, sensation vertigineuse, douleur musculo-squelettique, tachycardie, tremblement, douleur abdominale, vomissement, diarrhée, fatigue et dystonie.
Parmi ces effets indésirables, l’akathisie et la sédation/somnolence sont apparues dose-dépendantes. Liste récapitulative des effets indésirables
Les effets suivants sont tous les effets indésirables rapportés avec la palipéridone par catégorie de fréquence estimée à partir des essais cliniques menés avec le palmitate de palipéridone. Les termes et fréquences suivants sont utilisés : très fréquent (≥1/10) ; fréquent (≥1/100, <1/10) ; peu fréquent (≥1/1 000, <1/100) ; rare (≥1/10 000, < 1/1 000) ; très rare (<1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes de Systèmes Organes |
Effets indésirables |
||||
|
Fréquence |
|||||
|
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Indéterminée a |
|
|
Infections et infestations |
|
infection des voies respiratoires supérieures, infection des voies urinaires, grippe |
pneumonie, bronchite, infection des voies respiratoires, sinusite, cystite, infection auriculaire, amygdalite, onychomycose, cellulite, abcès sous-cutané |
infection oculaire, acarodermatite |
|
|
Affections hématologiques et du système lymphatique |
|
|
diminution de la numération de globules blancs, anémie |
neutropénie, thrombocytopénie, augmentation de la numération des éosinophiles |
agranulocytose |
|
Affections du système immunitaire |
|
|
hypersensibilité |
|
réaction anaphylactique |
|
Affections endocriniennes |
|
hyperprolactinémieb |
|
sécrétion inappropriée d’hormone antidiurétique, présence de glucose urinaire |
|
|
Troubles du métabolisme et de la nutrition |
|
hyperglycémie, prise de poids, perte de poids, diminution de l’appétit |
diabèted, hyperinsulinémie, augmentation de l’appétit, anorexie, augmentation des triglycérides sanguins, augmentation du cholestérol sanguin |
acidocétose diabétique, hypoglycémie, polydipsie |
intoxication à l’eau |
|
Affections psychiatriques |
insomniee |
agitation, dépression, anxiété |
trouble du sommeil, manie, diminution de la libido, nervosité, cauchemar |
catatonie, état de confusion, somnambulisme, émoussement affectif, anorgasmie |
trouble des conduites alimentaires lié au sommeil |
|
Affections du système nerveux |
|
parkinsonismec, akathisiec, sédation/ somnolence, dystoniec, sensation vertigineuse, dyskinésiec, tremblement, céphalée |
dyskinésie tardive, syncope, hyperactivité psychomotrice, vertige orthostatique, perturbation de l’attention, dysarthrie, dysgueusie, hypoesthésie, paresthésie |
syndrome malin des neuroleptiques, ischémie cérébrale, non réponse aux stimuli, perte de la conscience, diminution du niveau de la conscience, convulsione, trouble de l’équilibre, coordination anormale, titubation céphalique |
coma diabétique |
|
Affections oculaires |
|
|
vision trouble, conjonctivite, sécheresse oculaire |
glaucome, trouble du mouvement oculaire, révulsion oculaire, photophobie, augmentation du larmoiement, hyperémie oculaire |
syndrome de l'iris hypotonique (peropératoire ) |
|
Affections de l’oreille et du labyrinthe |
|
|
vertiges, acouphènes, douleur auriculaire |
|
|
|
Affections cardiaques |
|
tachycardie |
bloc auriculo- ventriculaire, trouble de la conduction, allongement de l’intervalle QT, syndrome de tachycardie orthostatique posturale, bradycardie, électrocardiogramme anormal, palpitations |
fibrillation auriculaire, arythmie sinusale |
|
|
Affections vasculaires |
|
hypertension |
hypotension, hypotension orthostatique |
embolie pulmonaire, thrombose veineuse, bouffées de chaleur |
ischémie |
|
Affections respiratoires, thoraciques et médiastinales |
|
toux, congestion nasale |
dyspnée, douleur pharyngolaryngée, épistaxis |
syndrome d’apnée du sommeil, congestion pulmonaire, congestion de l’appareil respiratoire, râles sibilances |
hyperventilation, pneumonie d’aspiration, dysphonie |
|
Affections gastro- intestinales |
|
douleur abdominale, vomissement, nausée, constipation, diarrhée, dyspepsie, douleur dentaire |
gêne abdominale, gastro-entérite, dysphagie, sécheresse buccale, flatulence |
pancréatite, occlusion intestinale, gonflement de la langue, incontinence fécale, fécalome, chéilite |
iléus |
|
Affections hépatobiliaires |
|
augmentation des transaminases |
augmentation des gamma-glutamyltransférases, augmentation des enzymes hépatiques |
|
jaunisse |
|
Affections de la peau et du tissu sous-cutané |
|
|
urticaire, prurit, rash, alopécie, eczéma, sécheresse cutanée, érythème, acné |
toxidermie, hyperkératose, , dermatite séborrhéique, pellicules |
syndrome de Stevens- Johnson/nécro lyse épidermique toxique, angiœdème, décoloration de la peau |
|
Affections musculo- squelettiques et systémiques |
|
douleur musculo- squelettique, douleur dorsale, arthralgie |
augmentation de la créatine phosphokinase sanguine, spasmes musculaires, raideur articulaire, faiblesse musculaire |
rhabdomyolyse, enflure des articulations |
posture anormale |
|
Affections du rein et des voies urinaires |
|
|
incontinence urinaire, pollakiurie, dysurie |
rétention urinaire |
|
|
Affections gravidiques, puerpérales et périnatales |
|
|
|
|
syndrome de sevrage médicamenteux néonatal (voir rubrique 4.6) |
|
Affections des organes de reproduction et du sein |
|
aménorrhée |
dysfonctionnement érectile, trouble de l’éjaculation, trouble menstruele, gynécomastie, galactorrhée, dysfonctionnement sexuel, douleur mammaire |
priapisme, gêne mammaire, engorgement mammaire, accroissement mammaire, écoulement vaginal |
|
|
Troubles généraux et anomalies au site d’administration |
|
pyrexie, asthénie, fatigue, réaction au site d’injection |
œdème de la face, œdèmee, augmentation de la température corporelle, démarche anormale, douleur thoracique, gêne thoracique, malaise, induration |
hypothermie, frissons, soif, syndrome de sevrage médicamenteux, abcès au site d’injection, cellulite au site d’injection, kyste au site d’injection, hématome au site d’injection |
diminution de la température corporelle, nécrose au site d’injection, ulcère au site d’injection |
|
Lésions, intoxications et complications liées aux procédures |
|
|
chute |
|
|
a La fréquence des effets indésirables est qualifiée comme « indéterminée » car ils n’ont pas été observés lors d’essais cliniques portant sur le palmitate de palipéridone. Ils proviennent soit de rapports spontanés post-commercialisation et la fréquence ne peut être déterminée, soit de données issues d’essais cliniques et/ou de rapports post-commercialisation portant sur la rispéridone (quelle que soit la formulation) ou la palipéridone orale.
b Se référer à « Hyperprolactinémie » ci-dessous.
c Se référer à « Symptômes extrapyramidaux » ci-dessous.
d Dans les essais contrôlés versus placebo, un diabète a été rapporté chez 0,32 % des sujets traités par le palmitate de palipéridone comparé à un taux de 0,39 % dans le groupe placebo. L’incidence globale de tous les essais cliniques était de 0,65 % chez tous les sujets traités par le palmitate de palipéridone.
e L’Insomnie inclut : insomnie initiale, insomnie du milieu de la nuit ; la convulsion inclut : crise de Grand mal ; l’œdème inclut : œdème généralisé, œdème périphérique, œdème qui prend le godet ; les troubles menstruels incluent : menstruation retardée, menstruation irrégulière, oligoménorrhée.
Effets indésirables observés avec les formulations à base de rispéridone
La palipéridone est le métabolite actif de la rispéridone, par conséquent, les profils des effets indésirables de ces composés (incluant les deux formulations orale et injectable) s’appliquent l’un à l’autre.
Description de certains effets indésirables
Réaction anaphylactique
Rarement, des cas de réaction anaphylactique après l'injection de palipéridone ont été rapportés depuis le début de la commercialisation chez les patients ayant précédemment toléré la rispéridone orale ou la palipéridone orale (voir rubrique 4.4).
Réactions au site d’injection
L’effet indésirable lié au site d’injection le plus fréquemment rapporté a été la douleur. La majorité de ces réactions a été rapportée comme étant de sévérité légère à modérée. L’évaluation par les sujets de la douleur au niveau du site d’injection réalisée à l’aide d’une échelle analogique visuelle tendait à diminuer en fréquence et en intensité durant toute la période des études de phases 2 et 3 avec la palipéridone. Les injections dans le muscle deltoïde ont été perçues comme légèrement plus douloureuses que les injections dans le muscle fessier. Les autres réactions au site d’injection ont été principalement d’intensité faible et ont inclus induration (fréquente), prurit (peu fréquent) et nodules (rare).
Symptômes extrapyramidaux
Les symptômes extrapyramidaux incluaient une analyse poolée des termes suivants : parkinsonisme (incluant hypersécrétion salivaire, raideur musculo-squelettique, parkinsonisme, salivation, phénomène de la roue dentée, bradykinésie, hypokinésie, faciès figé, tension musculaire, akinésie, rigidité de la nuque, rigidité musculaire, démarche parkinsonienne, réflexe palpébral anormal et tremblement parkinsonien de repos), akathisie (incluant akathisie, impatience, hyperkinésie et syndrome des jambes sans repos), dyskinésie (dyskinésie, contractions musculaires, choréoathétose, athétose et myoclonie), dystonie (incluant dystonie, hypertonie, torticolis, contractions musculaires involontaires, contracture musculaire, blépharospasme, crise oculogyre, paralysie de la langue, spasme facial, laryngospasme, myotonie, opisthotonus, spasme oropharyngé, pleurothotonus, spasme de la langue et trismus) et tremblement. Il est à noter qu’un spectre plus large de symptômes est inclus, qui n’ont pas nécessairement une origine extrapyramidale.
Prise de poids
Dans l’étude de 13 semaines comprenant la dose d’initiation de 150 mg, la proportion de patients présentant une prise de poids anormale ≥ 7 % a montré une tendance dose-dépendante, avec un taux d’incidence de 5 % dans le groupe placebo comparé à des taux de 6 %, 8 % et 13 % dans les groupes traités par la palipéridone à 25 mg, 100 mg et 150 mg respectivement.
Pendant la période de transition/entretien de 33 semaines en ouvert de l’essai de prévention de rechute à long terme, 12 % des patients traités par la palipéridone répondaient à ce critère (prise de poids ³ 7 % depuis la phase en double aveugle jusqu’à la fin de l’étude) ; le changement de poids moyen (ET) à partir du début de l’étude en ouvert était +0,7 (4,79) kg.
Hyperprolactinémie
Dans les essais cliniques, des augmentations médianes de la prolactine sérique ont été observées chez les patients des deux sexes qui ont reçu la palipéridone. Des effets indésirables pouvant suggérer une augmentation du taux de prolactine (par exemple, aménorrhée, galactorrhée, troubles menstruels, gynécomastie) ont été rapportés au total chez < 1 % des patients.
Effets de classe
Un allongement de l’intervalle QT, des arythmies ventriculaires (fibrillation ventriculaire, tachycardie ventriculaire), une mort subite inexpliquée, un arrêt cardiaque et des torsades de pointes peuvent survenir avec les antipsychotiques.
Des cas de thromboembolies veineuses, incluant des cas d’embolies pulmonaires et de thromboses veineuses profondes, ont été rapportés avec les médicaments antipsychotiques (fréquence indéterminée).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Symptômes
En général, les signes et symptômes attendus sont ceux résultant d’une exacerbation des effets pharmacologiques connus de la palipéridone, à savoir somnolence et sédation, tachycardie et hypotension, allongement de l’intervalle QT et effets extrapyramidaux. Des torsades de pointes et une fibrillation ventriculaire ont été rapportées chez un patient dans le contexte d’un surdosage en palipéridone orale. En cas de surdosage aigu, l’implication possible de plusieurs médicaments doit être prise en compte.
Prise en charge du surdosage
La forme à libération prolongée du médicament et la longue demi-vie d’élimination de la palipéridone doivent être prises en compte dans l’évaluation des besoins en traitement et du rétablissement. Il n’existe pas d’antidote spécifique à la palipéridone. Des mesures générales de maintien des fonctions vitales doivent être mises en place. Etablir et maintenir l’accès aux voies aériennes supérieures et assurer une oxygénation et une ventilation adéquates.
Une surveillance cardiovasculaire doit débuter immédiatement et doit inclure un suivi électrocardiographique en continu pour détecter d’éventuelles arythmies. Une hypotension et un collapsus circulatoire doivent être traités par des mesures appropriées telles que la perfusion intraveineuse de fluides et/ou d’agents sympathomimétiques. En cas de symptômes extrapyramidaux sévères, des agents anticholinergiques doivent être administrés. Une supervision et un suivi rapprochés doivent être poursuivis jusqu’au rétablissement du patient.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : psycholeptiques, autres antipsychotiques, code ATC : N05AX13.
PALIPERIDONE BIOGARAN contient un mélange racémique de palipéridone (+) et (-).
Mécanisme d’action
La palipéridone est un agent sélectif bloquant les effets des monoamines, dont les propriétés pharmacologiques sont différentes de celles des neuroleptiques conventionnels. La palipéridone se lie fortement aux récepteurs sérotoninergiques 5-HT2 et dopaminergiques D2. La palipéridone bloque également les récepteurs alpha 1-adrénergiques et à un moindre degré, les récepteurs histaminergiques
H1 et alpha 2-adrénergiques. L’activité pharmacologique des énantiomères (+) et (-) de la palipéridone est qualitativement et quantitativement comparable.
La palipéridone ne se lie pas aux récepteurs cholinergiques. Bien que la palipéridone soit un puissant antagoniste D2, qui est considéré comme responsable de l’effet bénéfique sur les symptômes positifs de la schizophrénie, elle entraîne moins de catalepsie et diminue moins la motricité que les neuroleptiques conventionnels. L’antagonisme sérotoninergique central dominant peut diminuer la capacité de la palipéridone à induire des effets indésirables extrapyramidaux.
Efficacité clinique
Traitement de la phase aiguë de la schizophrénie
L’efficacité de la palipéridone dans le traitement de la phase aiguë de la schizophrénie a été établie au cours de quatre études à court terme (une étude de 9 semaines et trois études de 13 semaines) en double aveugle, randomisées, contrôlées versus placebo, à doses fixes chez des patients adultes en rechute hospitalisés en phase aiguë et répondant aux critères DSM-IV de la schizophrénie. Les doses fixes de palipéridone dans ces études ont été administrées aux jours 1, 8 et 36 dans l’étude de 9 semaines ainsi qu’au jour 64 dans les études de 13 semaines. Aucune couverture additionnelle avec un antipsychotique oral n’a été requise pendant le traitement de la phase aiguë de la schizophrénie par la palipéridone. Le critère primaire d’efficacité a été défini comme la diminution des scores totaux à l’échelle PANSS (Positive and Negative Syndrome Scale) comme indiqué dans le tableau ci-dessous. L’échelle PANSS est un inventaire validé de plusieurs items portant sur cinq facteurs afin d’évaluer les symptômes positifs, les symptômes négatifs, les pensées désorganisées, l’hostilité/l’excitation incontrôlées et l’anxiété/la dépression. Le fonctionnement a été évalué en utilisant l’échelle PSP (Personal and Social Performance).
L’échelle PSP est une échelle validée notée par les cliniciens qui mesure le fonctionnement personnel et social dans quatre domaines : les activités utiles à la société (travail et études), les relations personnelles et sociales, l’autonomie et les comportements perturbateurs et agressifs.
Dans une étude de 13 semaines (n = 636) comparant trois doses fixes de palipéridone (injection initiale de 150 mg dans le muscle deltoïde suivie de 3 doses dans le muscle fessier ou deltoïde de 25 mg/4 semaines, 100 mg/4 semaines ou 150 mg mg/4 semaines) au placebo, les trois doses de palipéridone ont été supérieures au placebo dans l’amélioration du score total à l’échelle PANSS. Dans cette étude, les groupes de traitement de 100 mg/4 semaines et 150 mg/4 semaines ont montré une supériorité statistique par rapport au placebo sur l’échelle PSP, mais pas celui de 25 mg/4 semaines. Ces résultats soutiennent l’efficacité sur la durée totale du traitement et l’amélioration sur l’échelle PANSS qui a été observée dès le jour 4 avec une différence significative par rapport au placebo dans les groupes palipéridone 25 mg et 150 mg à partir du jour 8.
Les résultats des autres études ont fourni des résultats statistiquement significatifs en faveur de la palipéridone, excepté pour la dose de 50 mg dans une étude (voir tableau ci-dessous).
|
Score total à l’échelle Positive and Negative Syndrome Scale (PANSS) pour la schizophrénie – Variations entre l’état initial et la dernière évaluation – Données en LOCF dans les études R092670-SCH-201, R092670-PSY-3003, R092670-PSY-3004 et R092670-PSY-3007 : Analyse de l’efficacité primaire |
|||||
|
|
Placebo |
25 mg |
50 mg |
100 mg |
150 mg |
|
R092670-PSY-3007* |
n = 160 |
n = 155 |
|
n = 161 |
n = 160 |
|
Moyenne à la ligne de |
|
|
|
|
|
|
base (ET) Variation moyenne (ET) |
86,8 (10,31) -2,9 (19,26) |
86,9 (11,99) -8,0 (19,90) |
-- |
86,2 (10,77) -11,6 (17,63) |
88,4 (11,70) -13,2 (18,48) |
|
Valeur de P (vs placebo) |
-- |
0,034 |
|
< 0,001 |
< 0,001 |
|
R092670-PSY-3003 |
n = 132 |
|
n = 93 |
n = 94 |
n = 30 |
|
Moyenne à la ligne de |
|
|
|
|
|
|
base (ET) |
92,4 (12,55) |
-- |
89,9 (10,78) |
90,1 (11,66) |
92,2 (11,72) |
|
Variation moyenne (ET) |
-4,1 (21,01) |
|
-7,9 (18,71) |
-11,0 (19,06) |
-5,5 (19,78) |
|
Valeur de P (vs placebo) |
-- |
|
0,193 |
0,019 |
-- |
|
R092670-PSY-3004 |
n = 125 |
n = 129 |
n = 128 |
n = 131 |
|
|
Moyenne à la ligne de |
|
|
|
|
|
|
base (ET) Variation moyenne (ET) |
90,7 (12,22) -7,0 (20,07) |
90,7 (12,25) -13,6 (21,45) |
91,2 (12,02) -13,2 (20,14) |
90,8 (11,70) -16,1 (20,36) |
-- |
|
Valeur de P (vs placebo) |
-- |
0,015 |
0,017 |
< 0,001 |
|
|
R092670-SCH-201 |
n = 66 |
|
n = 63 |
n = 68 |
|
|
Moyenne à la ligne de |
|
|
|
|
|
|
base (ET) Variation moyenne (ET) |
87,8 (13,90) 6,2 (18,25) |
-- |
88,0 (12,39) -5,2 (21,52) |
85,2 (11,09) -7,8 (19,40) |
-- |
|
Valeur de P (vs placebo) |
-- |
|
0,001 |
< 0,0001 |
|
* Dans l’étude R092670-PSY-3007, une dose d’initiation de 150 mg a été administrée à tous les sujets des groupes de traitement par la palipéridone au jour 1 suivie par la dose assignée par la suite.
Note : une variation négative du score indique une amélioration.
Maintien du contrôle des symptômes et prévention des rechutes de la schizophrénie
L’efficacité de la palipéridone dans le maintien du contrôle des symptômes et la prévention des rechutes de la schizophrénie a été établie dans une étude à long terme en double aveugle, contrôlée versus placebo, à doses flexibles impliquant 849 patients adultes non âgés répondant aux critères DSM-IV de la schizophrénie. Cette étude incluait une phase de traitement de la phase aigüe et de stabilisation de
33 semaines en ouvert, une phase randomisée, en double aveugle et contrôlée versus placebo pour observer les rechutes et une période d’extension en ouvert de 52 semaines. Dans cette étude, la palipéridone a été administré mensuellement à des doses de 25, 50, 75 et 100 mg ; la dose de 75 mg a été autorisée uniquement dans l’extension ouverte de 52 semaines. Les patients ont reçu initialement des doses flexibles (25-100 mg) de palipéridone pendant une période de transition de 9 semaines, suivie d’une période d’entretien de 24 semaines, au cours de laquelle les patients devaient avoir un score PANSS
≤ 75. Des adaptations posologiques ont été autorisées uniquement pendant les 12 premières semaines de la période d’entretien. Un total de 410 patients stabilisés ont été randomisés sous palipéridone (durée médiane de 171 jours [allant de 1 à 407 jours]) ou sous placebo (durée médiane de 105 jours [allant de 8 à 441 jours]) jusqu’à ce qu’ils présentent une recrudescence des symptômes schizophréniques dans la phase en double aveugle de durée variable. L’essai a été arrêté très tôt pour des raisons d’efficacité car un délai avant rechute significativement plus long (p < 0,0001, Figure 1) a été observé chez les patients traités par la palipéridone comparativement au placebo (hazard ratio= 4,32 ; IC à 95 % : 2,4 à 7,7).
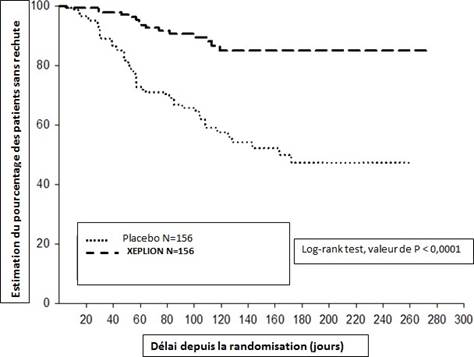
Figure 1 : Courbe de Kaplan-Meier du délai avant rechute – Analyse intermédiaire (Analyse intermédiaire en intention de traiter)
Population pédiatrique
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études avec la palipéridone dans tous les sous-groupes de la population pédiatrique de schizophrénie. Voir rubrique 4.2 pour des informations concernant l’usage pédiatrique.
5.2. Propriétés pharmacocinétiques
Le palmitate de palipéridone est un ester de palmitate, prodrogue de la palipéridone. En raison de sa solubilité extrêmement faible dans l’eau, le palmitate de palipéridone se dissout lentement après une injection intramusculaire avant d’être hydrolysé en palipéridone et absorbé dans la circulation sanguine. Après administration d’une dose unique par voie intramusculaire, les concentrations plasmatiques de palipéridone augmentent graduellement pour atteindre des concentrations plasmatiques maximum à un Tmax médian de 13 jours. La libération de la substance active démarre dès le jour 1 et dure au moins 4 mois.
Après l’injection intramusculaire de doses uniques (25-150 mg) dans le muscle deltoïde, une Cmax, en moyenne 28 % supérieure a été observée par rapport à l’injection dans le muscle fessier. Les deux injections intramusculaires initiales dans le muscle deltoïde de 150 mg le jour 1 et de 100 mg le jour 8 permettent d’atteindre rapidement les concentrations thérapeutiques. Le profil de libération et le schéma posologique du palmitate de palipéridone aboutissent au maintien prolongé des concentrations thérapeutiques. L’exposition totale au palmitate de palipéridone après administration de palmitate de palipéridone a été dose-proportionnelle dans un intervalle de doses allant de 25 à 150 mg et la Cmax a été inférieure à la dose proportionnalité pour les doses dépassant 50 mg. Le ratio moyen pic/vallée à l’état d’équilibre pour une dose de 100 mg de palmitate de palipéridone était de 1,8 après l’administration dans le muscle fessier et de 2,2 après l’administration dans le muscle deltoïde. La demi-vie apparente médiane de la palipéridone après l’administration du palmitate de palipéridone dans l’intervalle de doses 25-150 mg allait de 25 à 49 jours.
La biodisponibilité absolue du palmitate de palipéridone après l’administration de palipéridone est de 100 %.
Après l’administration du palmitate de palipéridone, les énantiomères (+) et (-) de la palipéridone s’interconvertissent atteignant un ratio d’AUC (+) sur (-) d’environ 1,6- 1,8.
La liaison aux protéines plasmatiques de la palipéridone racémique est de 74 %.
Biotransformation et élimination
Une semaine après l’administration d’une dose orale unique de 1 mg de 14C-palipéridone à libération immédiate, 59 % de la dose a été excrétée sous forme inchangée dans les urines, indiquant que la palipéridone n’est pas extensivement métabolisée au niveau hépatique. Environ 80 % de la radioactivité administrée a été retrouvée dans les urines et 11 % dans les fèces. Quatre voies métaboliques ont été identifiées in vivo, aucune d’entre elles ne concerne plus de 6,5 % de la dose : déalkylation, hydroxylation, déshydrogénation et coupure du noyau benzisoxazole. Bien que les études in vitro suggèrent un rôle du CYP2D6 et du CYP3A4 dans le métabolisme de la palipéridone, il n’existe pas de preuve in vivo que ces isoenzymes jouent un rôle significatif dans le métabolisme de la palipéridone. Les analyses de pharmacocinétique de population n’ont indiqué aucune différence notoire sur la clairance apparente de la palipéridone après l’administration de palipéridone orale entre les métaboliseurs rapides et les métaboliseurs lents des substrats du CYP2D6. Les études in vitro sur des microsomes hépatiques humains ont montré que la palipéridone n’inhibe pas de façon substantielle le métabolisme des médicaments métabolisés par les isoenzymes du cytochrome P450, incluant CYP1A2, CYP2A6, CYP2C8/9/10, CYP2D6, CYP2E1, CYP3A4 et CYP3A5.
Des études in vitro ont montré que la palipéridone est un substrat de la P-gp et un inhibiteur faible de la P-gp à des concentrations élevées. Aucune donnée in vivo n’est disponible et la signification clinique n’est pas connue.
Injection de palmitate de palipéridone à action prolongée versus palipéridone orale à libération prolongée
La palipéridone en injection est conçue pour délivrer la palipéridone sur une période mensuelle alors que la palipéridone orale à libération prolongée est administrée quotidiennement. Le schéma initial de palipéridone en injection (150 mg/100 mg dans le muscle deltoïde aux jour 1/jour 8) a été établi afin d’atteindre rapidement des concentrations de palipéridone à l’état d’équilibre lors de l’instauration du traitement sans utiliser de couverture orale supplémentaire.
En général, les concentrations plasmatiques initiales totales de palipéridone étaient comprises dans l’intervalle d’exposition observée avec des doses de palipéridone orale à libération prolongée allant de 6 à 12 mg. L’utilisation du schéma initial de palipéridone a permis aux patients de rester dans la fenêtre d’exposition obtenue avec des doses de 6 à 12 mg de palipéridone orale à libération prolongée même les jours précédant l’administration de la dose (jour 8 et jour 36) où l’exposition est plus faible. En raison de la différence de profils pharmacocinétiques médians entre les deux médicaments, la prudence est recommandée lors d’une comparaison directe de leurs propriétés pharmacocinétiques.
Insuffisance hépatique
La palipéridone n’est pas extensivement métabolisée au niveau hépatique. Bien que la palipéridone n’ait pas été étudiée chez les patients atteints d’insuffisance hépatique, aucune adaptation posologique n’est requise chez les patients atteints d’insuffisance hépatique légère ou modérée. Dans une étude sur la palipéridone orale chez des patients présentant une insuffisance hépatique modérée (score de Child- Pugh : classe B), les concentrations plasmatiques de la palipéridone libre étaient comparables à celles des sujets sains. La palipéridone n’a pas été étudiée chez les patients atteints d’insuffisance hépatique sévère.
Insuffisance rénale
L’élimination d’une dose orale unique de 3 mg de palipéridone sous forme de comprimé à libération prolongée a été étudiée chez des sujets présentant différents degrés de fonction rénale. L’élimination de la palipéridone décroît avec la diminution de la clairance estimée de la créatinine. Chez les sujets avec une fonction rénale altérée, la clairance totale de la palipéridone était réduite de 32 % en moyenne lors d’une insuffisance rénale légère (ClCr = 50 à < 80 mL/min), de 64 % lors d’une insuffisance rénale modérée (ClCr = 30 à < 50 mL/min) et de 71 % lors d’une insuffisance rénale sévère (ClCr = 10 à < 30 mL/min), ce qui correspondait à une augmentation moyenne de l’exposition (ASCinf) de 1,5, 2,6 et 4,8 fois, respectivement, par rapport à des sujets sains. Sur la base d’un nombre limité d’observations de la palipéridone chez les sujets atteint d’insuffisance rénale légère et de simulations pharmacocinétiques, une réduction de dose est recommandée (voir rubrique 4.2).
Sujet âgé
L’analyse pharmacocinétique de population n’a pas mis en évidence de différences pharmacocinétiques liées à l'âge.
Indice de masse corporelle (IMC)/poids corporel
Des études pharmacocinétiques sur le palmitate de palipéridone ont montré des concentrations plasmatiques de palipéridone légèrement plus faibles (10 à 20 %) chez les patients en surpoids ou obèses par rapport aux patients présentant un poids normal (voir rubrique 4.2).
Origine ethnique
Une analyse de pharmacocinétique de population sur les données d’études menées avec la palipéridone orale n’a pas mis en évidence de différences liées à l’origine ethnique sur la pharmacocinétique de la palipéridone après administration de PALIPERIDONE BIOGARAN.
Sexe
Aucune différence cliniquement significative n’a été observée entre les hommes et les femmes. Tabagisme
D’après des études in vitro utilisant des enzymes hépatiques humaines, la palipéridone n’est pas un substrat du CYP1A2 ; le tabagisme ne devrait donc pas avoir d’effet sur la pharmacocinétique de la palipéridone. L’effet du tabagisme sur la pharmacocinétique de la palipéridone n’a pas été étudié avec PALIPERIDONE BIOGARAN. Une analyse de pharmacocinétique de population basée sur des données de la palipéridone orale sous forme de comprimés à libération prolongée a montré une exposition à la palipéridone légèrement plus faible chez les fumeurs que chez les non-fumeurs. Il est peu probable que la différence soit cliniquement significative.
5.3. Données de sécurité préclinique
Au cours des études de reproduction chez le rat avec la rispéridone orale, qui est extensivement convertie en palipéridone chez le rat et l’Homme, des effets indésirables ont été observés sur le poids de naissance et la survie des progénitures. Aucune embryotoxicité ou malformation n’a été observée après l’administration intramusculaire du palmitate de palipéridone à des rats gravides jusqu’à la dose la plus élevée (160 mg/kg/jour), correspondant à 4,1 fois l’exposition chez l’homme à la dose maximale recommandée de 150 mg. D’autres antagonistes dopaminergiques, lorsqu’ils ont été administrés à des animaux gravides, ont entraîné des effets délétères sur les capacités d’apprentissage et le développement moteur des progénitures.
Le palmitate de palipéridone et la palipéridone n'ont pas été génotoxiques. Au cours d’études de cancérogénèse orale réalisées avec la rispéridone chez le rat et la souris, des augmentations de l’incidence des adénomes hypophysaires (souris), des adénomes du pancréas endocrine (rat), et de la glande mammaire (chez les deux espèces) ont été observées. Le potentiel cancérogène du palmitate de palipéridone injecté par voie intramusculaire a été évalué chez le rat. Une augmentation statistiquement significative des adénocarcinomes des glandes mammaires chez les rats femelles à 10, 30 et 60 mg/kg/mois a été observée. Les rats mâles ont montré une augmentation statistiquement significative des adénomes et des carcinomes des glandes mammaires à 30 et 60 mg/kg/mois, ce qui correspond à 1,2 et 2,2 fois l’exposition chez l’Homme à la dose maximale recommandée. Ces tumeurs peuvent être liées à un antagonisme D2 dopaminergique prolongé et à une hyperprolactinémie. La signification de ces données tumorales chez les rongeurs en termes de risque pour l’espèce humaine est inconnue.
Macrogol 4000
Acide citrique monohydraté
Phosphate disodique
Phosphate monosodique monohydraté
Hydroxyde de sodium (pour l’ajustement du pH)
Eau pour préparations injectables
Ce médicament ne doit pas être mélangé avec d’autres médicaments.
36 mois.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30 °C.
6.5. Nature et contenu de l'emballage extérieur
Seringue préremplie (copolymère d’oléfine cyclique) munie d’un bouchon-piston (caoutchouc chlorobutyle), d’une valve antiretour et d’un capuchon (caoutchouc chlorobutyle) avec une aiguille de sécurité de 11/2 pouce 22 Gauge (0,72 mm x 38,1 mm) et une aiguille de sécurité de 1 pouce 23 Gauge (0,64 mm x 25,4 mm).
Présentation :
Le conditionnement contient 1 seringue préremplie et 2 aiguilles.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
A usage unique.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
15 BOULEVARD CHARLES DE GAULLE
92707 COLOMBES CEDEX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 408 9 2 : 1 seringue pré-remplie (polycyclooléfine) avec 2 aiguilles.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Prescription initiale réservée aux spécialistes en psychiatrie. Renouvellement non restreint.
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Informations importantes
Les informations importantes disponibles pour ce médicament sont les suivantes :
- Neuroleptiques et constipation : vigilance indispensable pour limiter le risque de complications
- Rappel des médicaments injectables Palipéridone LP (Biogaran), Rispéridone LP et Octréotide LP (Téva) sans impact sur la couverture des besoins qui est assurée
- Point de situation sur l'approvisionnement en médicaments psychotropes en France au 8 janvier 2026
- Fortes tensions d'approvisionnement en rispéridone injectable : privilégiez le relais de traitement par palipéridone injectable
ANSM - Mis à jour le : 18/07/2024
Palipéridone Biogaran 25 mg, suspension injectable à libération prolongée en seringue préremplie
Palipéridone
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie ?
3. Comment utiliser PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : psycholeptiques, autres antipsychotiques, code ATC : N05AX13.
PALIPERIDONE BIOGARAN contient la substance active palipéridone qui appartient à la classe des antipsychotiques et est utilisé dans le traitement d’entretien des symptômes de la schizophrénie chez les patients adultes stabilisés par la palipéridone ou la rispéridone.
Si vous avez présenté une réponse à la palipéridone ou à la rispéridone dans le passé et avez des symptômes légers à modérés, votre médecin peut initier le traitement par PALIPERIDONE BIOGARAN sans stabilisation préalable par la palipéridone ou la rispéridone.
La schizophrénie est une maladie avec des symptômes « positifs » et « négatifs ». Les symptômes positifs sont un excès de symptômes qui ne sont normalement pas présents. Par exemple, une personne atteinte de schizophrénie peut entendre des voix ou voir des choses qui ne sont pas là (appelées hallucinations), croire des choses qui ne sont pas vraies (appelées illusions), ou se sentir inhabituellement suspicieuse envers les autres. Les symptômes négatifs représentent une absence de comportements ou de sentiments qui sont normalement présents. Par exemple, une personne atteinte de schizophrénie peut sembler en retrait et peut ne manifester aucune réaction émotionnelle ou peut avoir des difficultés à parler de manière claire et logique. Les personnes atteintes de cette maladie peuvent également se sentir déprimées, anxieuses, coupables ou tendues.
PALIPERIDONE BIOGARAN peut aider à soulager les symptômes de votre maladie et empêcher vos symptômes de revenir.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie ?
· Si vous êtes allergique à la palipéridone ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6) ;
· si vous êtes allergique à un autre médicament antipsychotique contenant de la rispéridone.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser PALIPÉRIDONE BIOGARAN.
Ce médicament n’a pas été étudié chez les patients âgés déments. Cependant, les patients âgés déments, qui sont traités par d’autres types de médicaments similaires, peuvent avoir une augmentation du risque d’attaque cérébrale ou de décès (voir rubrique 4).
Tous les médicaments ont des effets indésirables et certains effets indésirables de ce médicament peuvent aggraver les symptômes d’autres affections médicales. Ainsi, il est important de parler avec votre médecin des affections suivantes qui peuvent potentiellement s’aggraver au cours du traitement par ce médicament :
· si vous avez la maladie de Parkinson ;
· si l’on a diagnostiqué précédemment chez vous un état dont les symptômes incluent une température élevée et une raideur musculaire (également connu comme le syndrome malin des neuroleptiques) ;
· si vous avez déjà présenté des mouvements anormaux de la langue ou du visage (dyskinésie tardive) ;
· si vous savez que vous avez eu dans le passé un faible taux de globules blancs (qui peut ou non avoir été causé par d'autres médicaments) ;
· si vous êtes diabétique ou sujet au diabète ;
· si vous avez eu un cancer du sein ou une tumeur de l’hypophyse dans votre cerveau ;
· si vous avez une maladie cardiaque ou un traitement pour une maladie cardiaque qui vous rend sujet à une tension artérielle basse ;
· si vous avez une tension artérielle basse lorsque vous vous mettez debout ou vous redressez soudainement) ;
· si vous êtes épileptique ;
· si vous avez des troubles rénaux ;
· si vous avez des troubles hépatiques ;
· si vous avez une érection prolongée et/ou douloureuse ;
· si vous avez des difficultés à contrôler votre température corporelle centrale ou une température élevée ;
· si vous avez un niveau anormalement élevé d'hormone prolactine dans votre sang ou si vous avez une tumeur, potentiellement liée à la prolactine ;
· si vous ou quelqu’un de votre famille avez des antécédents de formation de caillots sanguins, car la prise d’antipsychotiques a été associée à la formation de caillots sanguins.
Si vous présentez l’un de ces états, parlez-en à votre médecin car il voudra peut-être adapter votre posologie ou vous suivre pendant quelque temps.
Puisqu’un taux dangereusement faible d'un certain type de globules blancs nécessaires pour lutter contre les infections dans votre sang a été très rarement observé chez les patients prenant ce médicament, votre médecin peut vérifier votre numération de globules blancs.
Même si vous avez déjà toléré la rispéridone orale ou la palipéridone orale, des réactions allergiques surviennent rarement après avoir reçu des injections de PALIPÉRIDONE BIOGARAN. Consultez un médecin immédiatement si vous présentez une éruption, un gonflement de la gorge, des démangeaisons ou des problèmes respiratoires qui peuvent être les signes d'une réaction allergique grave.
Ce médicament peut entraîner une prise de poids. Une prise de poids importante peut nuire à votre santé. Votre médecin doit régulièrement mesurer votre poids.
Puisque des diabètes ou l’aggravation de diabètes préexistants ont été observés chez des patients prenant ce médicament, votre médecin doit rechercher des signes d’un taux élevé de sucre dans le sang. Chez les patients ayant un diabète préexistant, le glucose présent dans le sang doit être régulièrement contrôlé.
Ce médicament pouvant réduire votre envie de vomir, il existe un risque qu’il puisse masquer la réaction normale du corps à l’ingestion de substances toxiques ou d’autres états médicaux.
Pendant une opération des yeux pour une nébulosité du cristallin (cataracte), la pupille (le cercle noir au centre de votre œil) peut ne pas se dilater correctement. De même, l’iris (la partie colorée de l’œil) peut devenir flasque pendant l'intervention et ceci pourrait entraîner une lésion oculaire. Si vous devez être opéré des yeux, assurez-vous d’informer votre ophtalmologiste que vous prenez ce médicament.
Enfants et adolescents
Ce médicament n’est pas destiné aux personnes de moins de 18 ans.
Autres médicaments et Palipéridone Biogaran 25 mg, suspension injectable à libération prolongée en seringue préremplie
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Prendre ce médicament avec de la carbamazépine (antiépileptique et régulateur de l'humeur) peut nécessiter une modification de dose de ce médicament.
Comme ce médicament agit principalement au niveau du cerveau, une interférence par d’autres médicaments agissant au niveau du cerveau, tels que d’autres médicaments antipsychotiques, opioïdes, antihistaminiques et médicaments pour dormir, peut provoquer une accentuation des effets indésirables tels que la somnolence ou d’autres effets sur le cerveau.
Ce médicament pouvant diminuer la tension artérielle, la prudence s’impose lorsque ce médicament est utilisé avec d’autres médicaments qui baissent la tension artérielle.
PALIPERIDONE BIOGARAN peut diminuer l’effet de médicaments contre la maladie de Parkinson et le syndrome des jambes sans repos (par exemple, la lévodopa).
Ce médicament peut entraîner une anomalie à électrocardiogramme (ECG) montrant une longue durée pour qu’une impulsion électrique traverse une certaine partie du cœur (connue comme « allongement de l’intervalle QT »). Les autres médicaments qui ont cet effet incluent certains médicaments utilisés pour traiter le rythme du cœur ou pour traiter les infections, et d’autres antipsychotiques.
Si vous êtes sujet aux convulsions, ce médicament peut augmenter votre risque d’en avoir. Les autres médicaments qui ont cet effet incluent certains médicaments utilisés pour traiter la dépression et pour traiter les infections, et d’autres antipsychotiques.
PALIPERIDONE BIOGARAN doit être utilisé avec prudence avec des médicaments augmentant l’activité du système nerveux central (psychostimulants tels que méthylphénidate).
PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie avec de l’alcool
L’alcool doit être évité.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Vous ne devez pas utiliser ce médicament au cours de la grossesse à moins d’en avoir parlé avec votre médecin. Les symptômes suivants peuvent apparaître chez les nouveau-nés dont les mères ont utilisé la palipéridone durant le dernier trimestre (les trois derniers mois de leur grossesse) : tremblement, raideur et/ou faiblesse musculaire, endormissement, agitation, problème de respiration et difficulté à s’alimenter. Si votre bébé développe l’un de ces symptômes, vous devez contacter votre médecin.
Ce médicament peut passer de la mère à l’enfant par le lait maternel et peut être nocif pour le nourrisson. Vous ne devez donc pas allaiter lors du traitement par ce médicament.
Conduite de véhicules et utilisation de machines
Des sensations de vertige, une extrême fatigue et des problèmes de vision peuvent survenir au cours du traitement par ce médicament (voir rubrique 4). Ceci doit être pris en compte lorsqu’une vigilance totale est nécessaire, par exemple, en cas de conduite de véhicules ou d’utilisation de machines.
PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie contient du sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie ?
Vous recevrez la première injection (150 mg) et la seconde injection (100 mg) de ce médicament dans la partie supérieure du bras à environ 1 semaine d’intervalle. Par la suite, vous recevrez une injection (allant de 25 mg à 150 mg) soit dans la partie supérieure du bras soit dans les fesses une fois par mois.
En fonction de vos symptômes, votre médecin peut augmenter ou diminuer la quantité de médicament que vous recevez d’un niveau de dose lors de votre prochaine injection mensuelle prévue.
Patients avec des troubles rénaux
Votre médecin peut ajuster votre dose de ce médicament selon votre fonction rénale. Si vous avez de légers troubles rénaux, votre médecin peut vous donner une dose plus faible. Si vous avez des troubles rénaux modérés ou sévères, ce médicament ne doit pas être utilisé.
Personnes âgées
Votre médecin peut réduire votre dose de ce médicament si votre fonction rénale est réduite.
Si vous avez reçu plus de PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie que vous n’auriez dû
Ce médicament vous sera administré sous surveillance médicale ; il est, par conséquent, peu probable que vous en receviez trop.
Les patients ayant reçu trop de palipéridone peuvent ressentir les symptômes suivants : somnolence ou sédation, fréquence cardiaque rapide, tension artérielle basse, électrocardiogramme anormal (tracé électrique du cœur) ou mouvements lents ou anormaux du visage, du corps, des bras ou des jambes.
Si vous oubliez d’utiliser Palipéridone Biogaran 25 mg, suspension injectable à libération prolongée en seringue préremplie
Si vous cessez de recevoir vos injections, vous perdrez les effets de ce médicament. Vous ne devez pas arrêter d’utiliser ce médicament sans que votre médecin vous ait dit de le faire car vos symptômes peuvent réapparaître.
Si vous arrêtez d’utiliser Palipéridone Biogaran 25 mg, suspension injectable à libération prolongée en seringue préremplie
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Informez immédiatement votre médecin :
· si vous présentez des caillots sanguins veineux, particulièrement au niveau des jambes (les symptômes incluent gonflement, douleur et rougeur au niveau des jambes), qui peuvent se déplacer via les vaisseaux sanguins jusqu’aux poumons et provoquer une douleur dans la poitrine et une difficulté à respirer. Si vous ressentez un de ces symptômes, consultez un médecin immédiatement ;
· si vous avez une démence et présentez un changement soudain de votre état mental ou une soudaine faiblesse ou engourdissement du visage, des bras ou des jambes, surtout d’un côté, ou des troubles de l’élocution, même pour une courte période de temps. Ceux-ci peuvent être des signes d’un accident vasculaire cérébral ;
· si vous présentez de la fièvre, une raideur musculaire, des sueurs ou une baisse du niveau de la conscience (un trouble appelé « syndrome malin des neuroleptiques »). Un traitement médical immédiat peut être nécessaire ;
· si vous êtes un homme et présentez une érection prolongée ou douloureuse. C’est ce que l’on appelle le priapisme. Un traitement médical immédiat peut être nécessaire ;
· si vous présentez des mouvements rythmiques involontaires de la langue, de la bouche et du visage. L’arrêt de la palipéridone peut être nécessaire ;
· si vous présentez une réaction allergique sévère caractérisée par de la fièvre, un gonflement de la bouche, du visage, des lèvres ou de la langue, un essoufflement, des démangeaisons, une éruption cutanée et parfois une baisse de la tension artérielle (constituant une « réaction anaphylactique »). Même si vous avez déjà toléré la rispéridone orale ou la palipéridone orale, des réactions allergiques surviennent rarement après avoir reçu des injections de palipéridone ;
· si vous prévoyez de subir une opération des yeux, assurez-vous d’informer votre ophtalmologiste que vous prenez ce médicament. Pendant une opération des yeux pour une nébulosité du cristallin (cataracte), l’iris (la partie colorée de l’œil) peut devenir flasque pendant l'intervention (connu sous le nom de « syndrome de l'iris hypotonique ») et ceci pourrait entraîner une lésion oculaire ;
· si vous savez que vous avez un nombre dangereusement faible d'un certain type de globules blancs nécessaires pour lutter contre les infections dans votre sang.
Les effets indésirables suivants peuvent survenir :
Effets indésirables très fréquents : peuvent affecter plus de 1 personne sur 10
· Difficulté à s’endormir ou à rester endormi.
Effets indésirables fréquents : peuvent affecter jusqu’à 1 personne sur 10
· Symptôme du rhume, infection urinaire, syndrome grippal ;
· PALIPERIDONE BIOGARAN peut augmenter votre taux d'une hormone appelée « prolactine » retrouvé grâce à un test sanguin (ce qui peut ou non causer des symptômes). Lorsque les symptômes d’un taux élevé de prolactine apparaissent, ils peuvent inclure (chez les hommes) un gonflement des seins, une difficulté à avoir ou maintenir une érection, ou d'autres troubles sexuels ; (chez les femmes) une gêne mammaire, un écoulement de lait au niveau des seins, une absence des règles ou d’autres problèmes avec vos règles ;
· taux élevé de sucre sanguin, prise de poids, perte de poids, diminution de l’appétit ;
· irritabilité, dépression, anxiété ;
· parkinsonisme. Cet état peut inclure des mouvements lents ou altérés, une sensation de raideur ou de crampe musculaire (rendant vos mouvements saccadés), et parfois même une sensation de mouvement « gelés » et qui redémarrent ensuite. D'autres signes de parkinsonisme incluent une marche traînante et lente, un tremblement au repos, une augmentation de la salive et/ou baver et une perte d'expression du visage ;
· impatience, sensation d’être endormi ou moins alerte ;
· dystonie. C’est un état impliquant une contraction involontaire lente ou soutenue des muscles. Bien que n'importe quelle partie du corps peut être touchée (et peut entraîner une posture anormale), la dystonie implique souvent les muscles du visage, y compris des mouvements anormaux des yeux, de la bouche, de la langue ou de la mâchoire ;
· sensation vertigineuse ;
· dyskinésie. C’est un état impliquant des mouvements musculaires involontaires, et peut inclure des mouvements répétitifs, spastiques ou tordus, ou des secousses ;
· tremblement (secousse) ;
· céphalée ;
· rythme cardiaque rapide ;
· hypertension artérielle ;
· toux, nez bouché ;
· douleur abdominale, vomissement, nausée, constipation, diarrhée, indigestion, mal de dent ;
· augmentation des transaminases hépatiques dans votre sang ;
· douleur osseuse ou musculaire, douleur dorsale, douleur articulaire ;
· absence des règles ;
· fièvre, faiblesse, fatigue (épuisement) ;
· réaction au site d’injection, y compris des démangeaisons, des douleurs ou un gonflement.
Effets indésirables peu fréquents : peuvent affecter jusqu’à 1 personne sur 100
· Pneumonie, infection thoracique (bronchite), infection des voies respiratoires, sinusite, infection de la vessie, infection auriculaire, infection fongique des ongles, infection des amygdales, infection cutanée ;
· diminution du nombre de globules blancs, diminution d’un type de globules blancs qui aident à vous protéger contre les infections, anémie ;
· réaction allergique ;
· diabète ou aggravation de diabète, augmentation de l’insuline (hormone qui contrôle le niveau de sucre sanguin) dans votre sang ;
· augmentation de l’appétit ;
· perte d’appétit entraînant malnutrition et faible poids corporel ;
· taux élevé de triglycéride sanguin (un type de graisse), augmentation du cholestérol dans votre sang ;
· trouble du sommeil, humeur exaltée (manie), diminution du désir sexuel, nervosité, cauchemars ;
· dyskinésie tardive (mouvements saccadés ou secousses que vous ne pouvez pas contrôler au niveau de votre visage, de votre langue ou d'autres parties de votre corps). Prévenez immédiatement votre médecin si vous présentez des mouvements rythmiques involontaires de la langue, de la bouche et du visage. L’arrêt de ce médicament peut être nécessaire ;
· évanouissement, besoin impératif de bouger des parties de votre corps, sensation vertigineuse lors du passage à la position debout, troubles de l’attention, problème d’élocution, perte ou altération du goût, sensation de la peau à la douleur et au toucher diminuée, sensation de picotement, de piqûre, ou un engourdissement de la peau ;
· vision trouble, infection oculaire ou conjonctivite, sécheresse oculaire ;
· sensation de tournoiement (vertige), bourdonnement dans les oreilles, douleur auriculaire ;
· interruption de la conduction entre les parties supérieure et inférieure du cœur, conduction électrique anormale du cœur, allongement de l’intervalle QT de votre cœur, accélération du rythme cardiaque lors du passage à la position debout, pouls lent, tracé électrique anormal du cœur (électrocardiogramme ou ECG), sentiment de battements ou pulsations dans votre poitrine (palpitations) ;
· tension artérielle basse, tension artérielle basse en position debout (par conséquent, certaines personnes prenant ce médicament peuvent se sentir faible, mal ou s’évanouir lorsqu’elles se mettent debout ou se redressent soudainement) ;
· essoufflement, maux de gorge, saignements de nez ;
· gêne abdominale, infection de l’estomac ou des intestins, difficultés à avaler, sécheresse buccale ;
· flatulence ;
· augmentation des GGT (une enzyme hépatique appelée gamma-glutamyltransférase) sanguines, augmentation des enzymes du foie de votre sang ;
· urticaire, démangeaison, éruption cutanée, perte de cheveux, eczéma, sécheresse cutanée, rougeurs de la peau, acné, abcès sous-cutané ;
· augmentation des CPK (créatine phosphokinase) dans votre sang, enzyme qui est parfois libérée lors de rupture musculaire ;
· spasmes musculaires, raideur articulaire, faiblesse musculaire ;
· incontinence (perte de contrôle) urinaire, envies fréquentes d'uriner, douleur en urinant ;
· dysfonctionnement érectile, trouble de l’éjaculation, absence de menstruation ou autres problèmes avec les règles chez la femme, développement des seins chez les hommes, dysfonctionnement sexuel, douleur mammaire, écoulement de lait au niveau des seins ;
· gonflement du visage, de la bouche, des yeux ou des lèvres, gonflement du corps, des bras ou des jambes ;
· augmentation de la température corporelle ;
· changement dans votre façon de marcher ;
· douleur à la poitrine, gêne au niveau de la poitrine, sensation de malaise ;
· durcissement de la peau ;
· chute.
Effets indésirables rares : peuvent affecter jusqu’à 1 personne sur 1 000
· Infection oculaire ;
· inflammation cutanée causée par des acariens, démangeaison squameuse du cuir chevelu ou de la peau ;
· augmentation des éosinophiles (un type de globules blancs) dans votre sang ;
· diminution des plaquettes (cellules sanguines qui vous aident à arrêter les saignements) ;
· secousses de la tête ;
· sécrétion inappropriée d’une hormone qui contrôle le volume d’urine ;
· sucre dans les urines ;
· complications des diabètes non contrôlés pouvant engager le pronostic vital ;
· hypoglycémie ;
· consommation excessive d’eau ;
· absence de mouvement ou de réponse pendant l’état éveillé (catatonie) ;
· confusion ;
· somnambulisme ;
· manque d’émotion ;
· incapacité à atteindre l’orgasme ;
· syndrome malin des neuroleptiques (confusion, réduction ou perte de la conscience, forte fièvre, raideur musculaire sévère), problèmes de vaisseaux sanguins dans le cerveau, dont la perte soudaine de l’arrivée de sang au niveau du cerveau (accident vasculaire cérébral ou « mini » attaque cérébrale), non réponse aux stimuli, perte de conscience, faible niveau de conscience, convulsions (crises), trouble de l’équilibre ;
· anomalie de la coordination ;
· glaucome (augmentation de la pression dans le globe oculaire) ;
· problèmes dans les mouvements de vos yeux, yeux roulants, hypersensibilité des yeux à la lumière, augmentation des larmes, rougeur des yeux ;
· fibrillation auriculaire (rythme cardiaque anormal), battement cardiaque irrégulier ;
· caillots sanguins veineux, particulièrement au niveau des jambes (les symptômes incluent gonflement, douleur et rougeur au niveau des jambes). Si vous ressentez un de ces symptômes, consultez un médecin immédiatement ;
· bouffées de chaleur ;
· difficultés à respirer pendant le sommeil (apnée du sommeil) ;
· congestion pulmonaire, encombrement des voies respiratoires ;
· crépitements pulmonaires, respiration sifflante ;
· inflammation du pancréas, gonflement de la langue, incontinence fécale, selles très dures ;
· lèvres gercées ;
· éruption cutanée liée au médicament, épaississement de la peau, pellicules ;
· cassure des fibres musculaires et douleurs musculaires (rhabdomyolyse) ;
· enflure des articulations ;
· incapacité à uriner ;
· gêne mammaire, élargissement des glandes mammaires, élargissement des seins ;
· écoulement vaginal ;
· priapisme (érection prolongée du pénis qui peut nécessiter un traitement chirurgical) ;
· température corporelle très basse, frissons, sensation de soif ;
· symptôme de sevrage médicamenteux ;
· accumulation de pus causé par une infection au niveau du site d’injection, infection de la peau en profondeur, kyste au niveau du site d’injection, bleus au niveau du site d’injection.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
· Nombre dangereusement faible d’un certain type de globules blancs nécessaires pour lutter contre les infections dans votre sang ;
· réaction allergique sévère caractérisée par de la fièvre, la bouche, le visage, les lèvres ou la langue gonflés, un essoufflement, des démangeaisons, une éruption cutanée et parfois une chute de la pression sanguine ;
· prise excessive d'eau pouvant être dangereuse ;
· trouble des conduites alimentaires lié au sommeil ;
· coma dû à un diabète non contrôlé ;
· diminution de l’oxygène dans certaines parties de votre corps (en raison de la diminution du flux sanguin) ;
· respiration rapide et superficielle, pneumonie causée par l'inhalation d’aliments, trouble de la voix ;
· absence de mouvement des muscles de l'intestin provoquant une occlusion ;
· jaunissement de la peau et des yeux (jaunisse) ;
· éruption cutanée grave ou potentiellement mortelle avec des cloques et une peau qui pèle, pouvant commencer dans et autour de la bouche, du nez, des yeux et des organes génitaux et s'étendre à d'autres parties du corps (syndrome de Stevens-Johnson ou nécrolyse épidermique toxique) ;
· réaction allergique grave avec un gonflement qui peut impliquer la gorge et entraîner des difficultés respiratoires ;
· décoloration de la peau ;
· posture anormale ;
· les nouveau-nés dont les mères ont utilisé PALIPERIDONE BIOGARAN durant leur grossesse peuvent ressentir des effets indésirables et/ou des symptômes de sevrage, tel qu’irritabilité, contraction lente ou soutenue des muscles, tremblement, endormissement, problème de respiration et difficulté à s’alimenter ;
· diminution de la température corporelle ;
· cellules mortes de la peau au site d’injection et ulcère au site d’injection.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER PALIPERIDONE BIOGARAN 25 mg, suspension injectable à libération prolongée en seringue préremplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage et la seringue préremplie. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 30°C.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est la palipéridone
Chaque seringue préremplie de PALIPERIDONE BIOGARAN 25 mg contient 39 mg de palmitate de palipéridone.
· Les autres composants sont :
Polysorbate 20, macrogol 4000, acide citrique monohydraté, phosphate disodique, phosphate monosodique monohydraté, hydroxyde de sodium (pour l’ajustement du pH), eau pour préparations injectables.
Ce médicament se présente sous forme d’une suspension injectable à libération prolongée, de couleur blanche à blanc cassé, en seringue préremplie.
Chaque conditionnement contient 1 seringue préremplie et 2 aiguilles.
Titulaire de l’autorisation de mise sur le marché
15 BOULEVARD CHARLES DE GAULLE
92707 COLOMBES CEDEX
Exploitant de l’autorisation de mise sur le marché
15 BOULEVARD CHARLES DE GAULLE
92707 COLOMBES CEDEX
DERVENAKION 6
PALLINI ATTIKI
15351
GRECE
ou
PHARMATHEN INTERNATIONAL S.A
INDUSTRIAL PARK SAPES
RODOPI PREFECTURE
BLOCK No.5
69300 RODOPI
GRECE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé et doivent être lues par le professionnel de santé conjointement avec l’information produit complète (Résumé des Caractéristiques du Produit).
La suspension injectable est à usage unique. Elle doit être contrôlée visuellement pour détecter d’éventuelles substances étrangères avant administration. Elle ne doit pas être utilisée si la seringue contient des substances étrangères.
Le conditionnement contient une seringue préremplie et 2 aiguilles de sécurité (une aiguille de 11/2 pouce 22 Gauge [38,1 mm x 0,72 mm] et une aiguille de 1 pouce 23 Gauge [25,4 mm x 0,64 mm]) pour injection intramusculaire.
|
|
|
(A) 22 G x 11/2 (Embout gris) (B) 23 G x 1 (Embout bleu) (C) Seringue préremplie (D) Embout (E) Capuchon |
|
1. Agiter vigoureusement la seringue pendant au moins 10 secondes pour s’assurer d’obtenir une suspension homogène. |
|
|
|
2. Sélectionner l’aiguille appropriée. La première dose d’initiation de PALIPERIDONE BIOGARAN (150 mg) doit être administrée le Jour 1 dans le muscle DELTOÏDE à l’aide de l’aiguille pour injection DELTOÏDE. La deuxième dose d’initiation de PALIPERIDONE BIOGARAN (100 mg) doit aussi être administrée dans le muscle DELTOÏDE une semaine après (Jour 8) à l’aide de l’aiguille pour injection DELTOÏDE. Si le patient passe de la rispéridone injectable à action prolongée à PALIPERIDONE BIOGARAN, la première injection de PALIPERIDONE BIOGARAN (allant de 25 mg à 150 mg) peut être administrée dans le muscle DELTOÏDE ou FESSIER utilisant l’aiguille appropriée au site d’injection au moment de la prochaine injection prévue. Par la suite, les injections mensuelles d’entretien peuvent être administrées dans le muscle DELTOÏDE ou FESSIER en utilisant l’aiguille appropriée au site d’injection. Pour une injection dans le muscle DELTOÏDE, si le patient pèse < 90 kg, utiliser l’aiguille de 1 pouce 23 Gauge (25,4 mm x 0,64 mm) (aiguille avec l’embout bleu) ; si le patient pèse ³ 90 kg, utiliser l’aiguille de 11/2 pouce 22 Gauge (38,1 mm x 0,72 mm) (avec l’embout gris). Pour une injection dans le muscle FESSIER, utiliser l’aiguille de 11/2 pouce 22 Gauge (38,1 mm x 0,72 mm) (avec l’embout gris). |
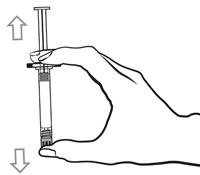
|
3. En tenant la seringue dirigée vers le haut, retirer le capuchon en caoutchouc en le dévissant. |
|
|
|
4. Ouvrir à moitié l’emballage de l’aiguille de sécurité. Saisir le capuchon protecteur de l’aiguille par l’emballage plastique. Fixer l’aiguille de sécurité sur l’embout de la seringue en tournant dans le sens des aiguilles d’une montre. |
|
|
|
5. Retirer le capuchon protecteur de l’aiguille en tirant bien droit. Ne pas tourner le capuchon protecteur, car l’aiguille peut se désolidariser de la seringue. |
|
|
|
6. Diriger la seringue avec l’aiguille fixée vers le haut pour éliminer l’air. Faire sortir l’air de la seringue en enfonçant avec prudence le piston vers le haut. |
|
|
|
7. Injecter la totalité du contenu par voie intramusculaire lentement, profondément dans le muscle deltoïde ou fessier du patient. Ne pas administrer par voie intravasculaire ou sous-cutanée. 8. Une fois l’injection réalisée, utiliser soit le pouce ou un doigt de la main (8a, 8b) ou une surface plane (8c) pour activer le système de protection de l’aiguille. Le système est complètement activé lorsque qu’un « clic » est entendu. Jeter la seringue et l’aiguille dans un endroit prévu à cet effet. |
|
8a |
|
|
|
8b |
|
|
|
8c |
|
|