Dernière mise à jour le 03/08/2026
MIRTAZAPINE ARROW 15 mg, comprimé orodispersible
Indications thérapeutiques
Classe pharmacothérapeutique : antidépresseurs - code ATC : N06AX11.
MIRTAZAPINE ARROW fait partie d’un groupe de médicaments appelés antidépresseurs.
MIRTAZAPINE ARROW est utilisé pour traiter la maladie dépressive chez les adultes.
MIRTAZAPINE ARROW doit être pris pendant 1 à 2 semaines avant d'agir. Après 2 à 4 semaines, vous pourrez commencer à vous sentir mieux. Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien après 2 à 4 semaines. Vous trouverez plus d'informations dans la rubrique 3 « Quand pouvez-vous espérer commencer à vous sentir mieux ? ».
Présentations
> plaquette(s) polyamide aluminium PVC papier polyester de 30 comprimé(s)
Code CIP : 387 591-1 ou 34009 387 591 1 2
Déclaration de commercialisation : 23/03/2016
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 2,88 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 3,90 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 28/09/2023
MIRTAZAPINE ARROW 15 mg, comprimé orodispersible
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Mirtazapine........................................................................................................................... 15 mg
Pour un comprimé orodispersible
Excipient à effet notoire : chaque comprimé de MIRTAZAPINE ARROW 15 mg, comprimé orodispersible contient 3 mg d’aspartame.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé orodispersible ronds (diamètre 6,5 mm), blancs, à bords chanfreinés, gravés « 36 » sur une face et « A » sur l’autre face.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Adultes
La dose journalière efficace est habituellement comprise entre 15 et 45 mg ; la dose de départ est de 15 ou 30 mg.
En général, les effets de la mirtazapine commencent à apparaître après 1 à 2 semaines de traitement. Un traitement à posologie adaptée devrait en théorie conduire à une réponse positive en 2 à 4 semaines. Si la réponse est insuffisante, la posologie pourra être augmentée jusqu’à la dose maximale. Si aucune réponse n’est constatée au cours des 2 à 4 semaines suivantes, le traitement devra être arrêté.
Les patients présentant une dépression doivent être traités pendant une période suffisante d’au moins 6 mois pour assurer la disparition complète des symptômes.
Il est recommandé d’arrêter le traitement par la mirtazapine progressivement afin d’éviter les symptômes de sevrage (voir rubrique 4.4).
Sujets âgés
La dose recommandée est la même que chez l’adulte. Toute augmentation de posologie chez le sujet âgé impose une surveillance particulière pour obtenir une réponse clinique satisfaisante et bien tolérée.
Insuffisance rénale
La clairance de la mirtazapine peut être diminuée chez les patients atteints d’une insuffisance rénale modérée à sévère (clairance de la créatinine < 40 ml/min). Ceci est à prendre en compte lorsque la mirtazapine est prescrite à cette catégorie de patients (voir rubrique 4.4).
Insuffisance hépatique
La clairance de la mirtazapine peut être diminuée chez les patients atteints d’insuffisance hépatique. Ceci doit être pris en compte lorsque la mirtazapine est prescrit à cette catégorie de patients, en particulier chez les patients présentant une insuffisance hépatique sévère, qui n’ont pas fait l’objet d’études spécifiques (voir rubrique 4.4).
Population pédiatrique
La mirtazapine ne devrait pas être utilisé chez l’enfant et l’adolescent de moins de 18 ans car son efficacité n'a pas été démontrée au cours des deux études cliniques à court terme (voir rubrique 5.1) et pour des raisons de sécurité d'emploi (voir rubriques 4.4, 4.8 et 5.1).
Mode d’administration
La demi-vie d’élimination de la mirtazapine étant de 20 à 40 heures, la mirtazapine peut être administrée en une prise quotidienne unique. Il doit être pris de préférence en une prise unique le soir au coucher. La mirtazapine peut également être administrée en deux prises (une le matin et une au coucher, la dose la plus importante devant être prise au coucher).
Le comprimé doit être pris par voie orale. Le comprimé se désintègre rapidement et peut être avalé sans eau.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Association de la mirtazapine et d’inhibiteurs de la monoamine oxydase (IMAO) (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
L’utilisation de la mirtazapine est déconseillée chez les enfants et les adolescents de moins de 18 ans. Des comportements de type suicidaire (tentatives de suicide et idées suicidaires) et de type hostile (principalement agressivité, comportement d’opposition et colère) ont été plus fréquemment observés au cours des études cliniques chez les enfants et les adolescents traités par antidépresseurs par rapport à ceux traités par placebo. Si, en cas de nécessité clinique, la décision de traiter est néanmoins prise, le patient devra faire l'objet d'une surveillance attentive pour détecter l'apparition de symptômes suicidaires. De plus, on ne dispose d'aucune donnée de tolérance à long terme chez l'enfant et l'adolescent concernant la croissance, la maturation et le développement cognitif et comportemental.
Suicide/idées suicidaires ou aggravation clinique
La dépression est associée à un risque accru d’idées suicidaires, d’auto-agression et de suicide (comportement de type suicidaire). Ce risque persiste jusqu’à l’obtention d’une rémission significative. L’amélioration clinique pouvant ne pas survenir avant plusieurs semaines de traitement, les patients devront être surveillés étroitement jusqu’à obtention de cette amélioration. L’expérience clinique montre que le risque suicidaire peut augmenter en tout début de rétablissement.
Les patients ayant des antécédents de comportement de type suicidaire ou ceux exprimant des idées suicidaires significatives avant de débuter le traitement présentent un risque plus élevé d’idées suicidaires ou de tentatives de suicide, et doivent faire l’objet d’une surveillance étroite pendant le traitement. Une méta-analyse d’essais cliniques contrôlés versus placebo sur l’utilisation d’antidépresseurs chez l’adulte présentant des troubles psychiatriques, a montré une augmentation du risque de comportement de type suicidaire chez les patients de moins de 25 ans traités par antidépresseurs par rapport à ceux recevant un placebo.
Une surveillance étroite des patients, et en particulier de ceux à haut risque, devra accompagner le traitement médicamenteux, particulièrement le début du traitement et lors des changements de dose. Les patients et leur entourage devront être avertis de la nécessité de surveiller la survenue d’une aggravation clinique, l’apparition d’idées ou de comportements suicidaires et tout changement anormal du comportement et de prendre immédiatement un avis médical si ces symptômes survenaient.
Au vu du risque suicidaire, notamment en début de traitement, seule la quantité minimale de comprimés orodispersibles de la mirtazapine permettant une bonne prise en charge du patient devra être donnée à celui-ci afin de réduire le risque de surdosage.
Aplasie médullaire
Des cas d’aplasie médullaire, en général de granulocytopénie ou d’agranulocytose, ont été rapportés au cours d’un traitement par la mirtazapine. De rares cas d’agranulocytose réversibles ont été rapportés au cours d’études cliniques avec la mirtazapine. Depuis la commercialisation de la mirtazapine, de très rares cas d’agranulocytose ont été rapportés, le plus souvent réversibles, mais parfois d’évolution fatale. Les cas ayant entraîné le décès concernaient principalement des patients âgés de plus de 65 ans. Le médecin doit être attentif à l’apparition de symptômes tels que fièvre, maux de gorge, stomatite ou autres signes d’infection ; si de tels symptômes survenaient, le traitement sera arrêté et une numération-formule sanguine sera effectuée.
Ictère
Le traitement devra être arrêté en cas d’apparition d’un ictère.
Cas nécessitant une surveillance
Une adaptation posologique soigneuse ainsi qu’une surveillance étroite et régulière sont nécessaires chez les patients présentant :
· une épilepsie ou un syndrome cérébral organique : bien que l’expérience clinique montre que les crises épileptiques sont rares au cours d’un traitement par la mirtazapine, la mirtazapine doit être, comme les autres antidépresseurs, introduite avec prudence chez les patients présentant des antécédents de convulsions. Le traitement devra être arrêté chez tout patient développant des crises épileptiques, ou présentant une augmentation de la fréquence des crises ;
· une insuffisance hépatique : après administration d’une dose orale unique de 15 mg de mirtazapine, la clairance de la mirtazapine a diminué d’environ 35 % chez les patients présentant une insuffisance hépatique légère à modérée, comparativement aux sujets dont la fonction hépatique était normale. La concentration plasmatique moyenne de mirtazapine a augmenté d’environ 55 % ;
· une insuffisance rénale : après administration d’une dose orale unique de 15 mg de mirtazapine chez des patients atteints d’insuffisance rénale modérée (clairance de la créatinine < 40 ml/min) et sévère (clairance de la créatinine ≤ 10 ml/min), la clairance de la mirtazapine a diminué respectivement d’environ 30 % et 50 %, comparativement aux sujets sains. Les concentrations plasmatiques moyennes de mirtazapine ont respectivement augmenté d’environ 55 % et 115 %. Aucune différence significative n’est apparue entre les patients atteints d’insuffisance rénale légère (clairance de la créatinine < 80 ml/min) et le groupe témoin ;
· une pathologie cardiaque, comme des troubles de la conduction, une angine de poitrine ou un infarctus du myocarde récent : les précautions habituelles doivent être prises et les traitements concomitants administrés avec prudence ;
· une pression artérielle basse ;
· un diabète : chez les patients diabétiques, les antidépresseurs peuvent altérer l’équilibre glycémique. Une adaptation de la posologie d’insuline et/ou d’hypoglycémiant oral peut s’avérer nécessaire et une surveillance étroite est recommandée.
Comme avec les autres antidépresseurs, les situations suivantes doivent être prises en compte :
· une aggravation des symptômes psychotiques peut survenir en cas d’administration d’antidépresseurs à des patients atteints de schizophrénie ou d’autres troubles psychotiques ; les pensées paranoïdes peuvent être majorées ;
· en cas de traitement de la phase dépressive d’un trouble bipolaire, un passage à une phase maniaque est possible. Les patients ayant des antécédents de manie/hypomanie doivent être étroitement surveillés. La mirtazapine doit être arrêtée chez tout patient entrant dans une phase maniaque ;
· bien que la mirtazapine n’entraîne pas de dépendance, l’expérience depuis la commercialisation montre que l’arrêt brutal d’un traitement prolongé peut parfois entraîner des symptômes de sevrage. La plupart des réactions de sevrage sont modérées et spontanément réversibles. Parmi les divers symptômes de sevrage rapportés, les plus fréquents sont : étourdissements, agitation, anxiété, céphalées et nausées. Bien que ces symptômes aient été rapportés comme étant des symptômes de sevrage, il est à noter qu’ils peuvent être dus à la pathologie sous-jacente. Comme précisé en rubrique 4.2, il est recommandé d’arrêter progressivement le traitement par la mirtazapine ;
· des précautions doivent être prises chez les patients présentant des troubles de la miction tels qu’une hypertrophie prostatique et chez les patients présentant un glaucome aigu à angle fermé ou une augmentation de la pression intraoculaire (bien qu’il y ait peu de risque avec la mirtazapine du fait de sa très faible activité anticholinergique) ;
· akathisie/agitation psychomotrice : l’utilisation d’antidépresseurs a été associée avec le développement d’une akathisie, caractérisée par une agitation jugée désagréable ou pénible et un besoin de bouger souvent accompagné d’une incapacité à rester assis ou debout sans bouger. Ces symptômes apparaissent le plus souvent au cours des premières semaines de traitement. Une augmentation de dose peut être préjudiciable chez les patients développant ces symptômes ;
· des cas d’allongement de l’intervalle QT, de torsades de pointe, de tachycardie ventriculaire et de mort subite ont été rapportés depuis la commercialisation de la mirtazapine. La majorité des cas est survenue dans un contexte de surdosage ou chez des patients présentant d’autres facteurs de risque d’allongement de l’intervalle QT, y compris l’utilisation concomitante de médicaments allongeant l’intervalle QTc (voir rubriques 4.5 et 4.9). La prudence est recommandée lorsque la mirtazapine est prescrite chez des patients atteints d’une maladie cardiovasculaire connue ou ayant des antécédents familiaux d’allongement de l’intervalle QT, ainsi qu’en association avec d’autres médicaments susceptibles d’allonger l’intervalle QTc.
Hyponatrémie
Une hyponatrémie, probablement due à une sécrétion inappropriée d’hormone antidiurétique (SIADH), a été très rarement rapportée avec la mirtazapine. Des précautions doivent être prises chez les patients à risque comme les sujets âgés ou les patients déjà traités par des médicaments connus pour provoquer une hyponatrémie.
Syndrome sérotoninergique
Interaction avec des substances à activité sérotoninergique : un syndrome sérotoninergique peut survenir lorsque des inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) sont administrés en association avec d’autres médicaments sérotoninergiques tels que la buprénorphine (voir rubrique 4.5). Les symptômes du syndrome sérotoninergique peuvent être : hyperthermie, rigidité, myoclonies, instabilité du système nerveux autonome, avec possibilité de fluctuations rapides des constantes vitales, modifications de l’état mental incluant confusion, irritabilité et agitation extrême allant jusqu’au délire et au coma. Une prudence particulière et une surveillance clinique plus étroite est requise lorsque ces substances actives sont associées à la mirtazapine. Si des événements de ce type se produisent, le traitement par la mirtazapine devra être interrompu et un traitement symptomatique initié. D’après l’expérience depuis la commercialisation, la survenue d’un syndrome sérotoninergique est très rare chez les patients traités par la mirtazapine seule (voir rubrique 4.8).
Sujets âgés
Les sujets âgés sont souvent plus sensibles, en particulier en ce qui concerne les effets indésirables des antidépresseurs. Au cours des études cliniques avec la mirtazapine, les effets indésirables n’ont pas été rapportés plus fréquemment chez les sujets âgés que dans les autres groupes d’âge.
Réactions indésirables cutanées graves
Des réactions indésirables cutanées graves (SCAR), dont le syndrome de Stevens-Johnson (SSJ), le syndrome de Lyell, le syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), la dermatite bulleuse et l’érythème multiforme, pouvant engager le pronostic vital ou être fatales, ont été signalées dans le cadre de traitements à base de mirtazapine.
Si des signes ou symptômes évoquant l’une de ces réactions apparaissent, la mirtazapine doit être arrêtée immédiatement.
Si le patient a développé l’une de ces réactions en raison de l’utilisation de la mirtazapine, il ne faudra jamais recommencer un traitement à base de mirtazapine chez ce patient.
Aspartame
MIRTAZAPINE ARROW contient de l’aspartame, source de phénylalanine. Chaque comprimé dosé à 15 mg contient 3 mg de phénylalanine. Cela peut être dangereux pour les personnes atteintes de phénylcétonurie.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Interactions pharmacodynamiques
· La mirtazapine ne doit pas être administrée en association avec des IMAO, ni dans les deux semaines qui suivent l’arrêt d’un traitement par IMAO. Inversement, il faut attendre environ deux semaines entre l’arrêt d’un traitement par mirtazapine et le début d’un traitement par IMAO (voir rubrique 4.3). De plus, comme avec les ISRS, l’administration concomitante d’autres substances sérotoninergiques (L-tryptophane, triptans, tramadol, linézolide, bleu de méthylène, ISRS, venlafaxine, lithium, médicaments contenant de la buprénorphine et préparations à base de millerpertuis – Hypericum perforatum) peut entraîner l’apparition d’effets liés à la sérotonine (syndrome sérotoninergique : voir rubrique 4.4). La prudence est recommandée et une surveillance clinique plus étroite est nécessaire quand ces substances sont associées à la mirtazapine.
· La mirtazapine peut augmenter les propriétés sédatives des benzodiazépines et des autres sédatifs (notamment la plupart des antipsychotiques, les antihistaminiques H1 et les opiacés). La prudence s’impose lorsque ces médicaments sont prescrits conjointement à la mirtazapine.
· La mirtazapine peut augmenter les effets dépresseurs du SNC de l’alcool. Il faut donc conseiller aux patients d’éviter la prise de boissons alcoolisées pendant le traitement par mirtazapine.
· La mirtazapine administrée à raison de 30 mg par jour a entraîné une augmentation faible mais statistiquement significative du rapport international normalisé (INR) chez les patients traités par la warfarine. Etant donné qu’avec des doses plus élevées de mirtazapine un effet plus prononcé ne peut pas être exclu, il est recommandé de surveiller l’INR en cas de traitement concomitant par la warfarine et la mirtazapine.
· Le risque d’allongement de l’intervalle QT et/ou d’arythmie ventriculaire (par exemple torsades de pointes) peut être accru en cas d’utilisation concomitante avec des médicaments allongeant l’intervalle QTc (par exemple certains antipsychotiques et antibiotiques).
Interactions pharmacocinétiques
· La carbamazépine et la phénytoïne, inducteurs du CYP3A4, ont entraîné un quasi-doublement de la clairance de la mirtazapine, ainsi qu’une diminution des concentrations plasmatiques moyennes de mirtazapine de respectivement 60 % et 45 %. Quand la carbamazépine ou tout autre inducteur du métabolisme hépatique (comme la rifampicine) est ajouté au traitement par la mirtazapine, il peut être nécessaire d’augmenter la dose de mirtazapine. Si le traitement par un tel médicament est arrêté, il peut s’avérer nécessaire de diminuer la dose de mirtazapine.
· La co-administration de kétoconazole, inhibiteur puissant du CYP3A4, a entraîné une augmentation du pic de concentration plasmatique et de l’AUC de la mirtazapine de respectivement environ 40 % et 50 %.
· Lorsque la cimétidine (faible inhibiteur du CYP1A2, CYP2D6 et CYP3A4) est administrée avec la mirtazapine, les concentrations plasmatiques moyennes de mirtazapine peuvent être augmentées de plus de 50 %. Des précautions doivent être prises et on peut être amené à réduire la dose lors de l’administration concomitante de mirtazapine avec les inhibiteurs puissants du CYP3A4, les inhibiteurs de protéase du VIH, les antifongiques azolés, l’érythromycine, la cimétidine ou la néfazodone.
· Les études d’interactions n’ont mis en évidence aucun effet pharmacocinétique pertinent sur les traitements associant la mirtazapine à la paroxétine, l’amitriptyline, la rispéridone ou le lithium.
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données limitées concernant l’utilisation de la mirtazapine chez la femme enceinte ne montrent pas d’augmentation du risque de malformations congénitales. Des études effectuées chez l’animal n’ont pas mis en évidence d’effet tératogène cliniquement significatif, cependant une toxicité sur le développement a été observée (voir rubrique 5.3).
Des données épidémiologiques suggèrent que l'utilisation d'ISRS pendant la grossesse, en particulier en fin de grossesse, pourrait augmenter le risque d'hypertension artérielle pulmonaire persistante (HTAP) du nouveau-né. Bien qu'aucune étude n'ait étudié l'existence d'une association entre HTAP et traitement par mirtazapine, ce risque potentiel ne peut être exclu, compte tenu du mécanisme d'action impliqué (augmentation des concentrations de sérotonine).
La mirtazapine doit être prescrite avec prudence chez la femme enceinte. Si la mirtazapine est utilisée jusqu’à la naissance ou peu avant, une surveillance post-natale du nouveau-né est recommandée afin de rechercher de possibles réactions de sevrage.
Les études chez l’animal et des données limitées chez l’homme ont montré que la mirtazapine n’était excrétée dans le lait maternel qu’en très faibles quantités. La décision de poursuivre ou non l’allaitement ou le traitement par la mirtazapine doit être prise en tenant compte, d’une part du bénéfice de l’allaitement maternel pour l’enfant, et d’autre part du bénéfice du traitement par la mirtazapine pour la mère.
Fertilité
Des études non cliniques de toxicité sur la reproduction menées chez l'animal n'ont pas montré d'effet sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Des réactions indésirables cutanées graves (SCAR), dont le syndrome de Stevens-Johnson (SSJ), le syndrome de Lyell, le syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), la dermatite bulleuse et l’érythème multiforme, ont été signalées dans le cadre de traitements à base de mirtazapine (voir rubrique 4.4).
Les patients dépressifs présentent un certain nombre de symptômes associés à la pathologie elle-même. Par conséquent, il est parfois difficile de distinguer les symptômes qui résultent de la maladie elle-même de ceux causés par le traitement par mirtazapine.
Les effets indésirables les plus fréquemment rapportés, survenant chez plus de 5 % des patients traités par mirtazapine au cours d’études randomisées versus placebo (voir ci-dessous) sont une somnolence, une sédation, une sécheresse buccale, une prise de poids, une augmentation de l’appétit, un étourdissement et une fatigue.
Tous les essais randomisés versus placebo menés chez des patients (y compris dans des indications autres que l’épisode dépressif majeur) ont évalué les effets indésirables de la mirtazapine. La méta-analyse a étudié 20 essais portant sur une durée prévue de traitement allant jusqu’à 12 semaines et incluant 1 501 patients (134 années-patients) recevant des doses de mirtazapine allant jusqu’à 60 mg, et 850 patients (79 années-patients) recevant un placebo. Les phases d’extension de ces essais ont été exclues de l’analyse afin d’assurer la comparabilité avec le traitement par placebo.
Le tableau 1 présente l’incidence par catégorie des effets indésirables survenus dans les essais cliniques avec une fréquence statistiquement significativement plus élevée sous mirtazapine que sous placebo, ainsi que les effets indésirables spontanément rapportés. Les fréquences des effets indésirables spontanément rapportés se fondent sur le taux de signalement de ces évènements au cours des essais cliniques. La fréquence des effets indésirables spontanément rapportés pour lesquels aucun cas n’a été observé sous mirtazapine au cours des essais cliniques randomisés versus placebo, a été classée comme « indéterminée ».
|
Classe anatomique et fonctionnelle |
Très fréquent (≥ 1/10) |
Fréquent (≥ 1/100, < 1/10) |
Peu fréquent (≥ 1/1 000, < 1/100) |
Rare (≥ 1/10 000, < 1/1 000) |
Fréquence indéterminée (ne peut pas être estimée à partir des données disponibles) |
|
Affections hématologiques et du système lymphatique |
|
|
|
|
Aplasie médullaire (granulocytopénie, agranulocytose, anémie arégénérative, thrombocytopénie) (voir aussi rubrique 4.4) Eosinophilie |
|
Affections endocriniennes |
|
|
|
|
Sécrétion inappropriée d’hormone antidiurétique Hyperprolactinémie (et les symptômes associés galactorrhée et gynécomastie) |
|
Troubles du métabolisme et de la nutrition |
Augmentation de l’appétit 1 Prise de poids 1 |
|
|
|
Hyponatrémie |
|
Affections psychiatriques |
|
Rêves anormaux Confusion Anxiété 2, 5 Insomnie 3, 5 |
Cauchemars 2 Accès maniaques Agitation 2 Hallucinations Agitation psychomotrice (dont akathisie, hyperkinésie) |
Agressivité |
Idées suicidaires 6 Comportement suicidaire 6 Somnambulisme |
|
Affections du système nerveux |
Somnolence 1, 4 Sédation 1, 4 Céphalée 2 |
Léthargie 1 Sensations vertigineuses Tremblements Amnésie7 |
Paresthésies 2 Syndrome des jambes sans repos Syncope |
Myoclonies |
Convulsions (crises) Syndrome sérotoninergique Paresthésies orales Dysarthrie |
|
Affections vasculaires |
|
Hypotension orthostatique |
Hypotension 2 |
|
|
|
Affections gastro-intestinales |
Sécheresse de la bouche |
Constipation 1 Nausées 3 Diarrhée 2 Vomissements 2 |
Hypoesthésie orale |
Pancréatite |
Œdème de la sphère buccale Augmentation de la salivation |
|
Affections hépatobiliaires |
|
|
|
Elévation des transaminases sériques |
|
|
Affections de la peau et du tissu sous-cutané |
|
Exanthème 2 |
|
|
Syndrome de Stevens-Johnson Dermatite bulleuse Erythème polymorphe Syndrome de Lyell Syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) |
|
Affections musculo-squelettiques et systémiques |
|
Arthralgies Myalgies Rachialgies 1 |
|
|
Rhabdomyolyse |
|
Affections du rein et des voies urinaires |
|
|
|
|
Rétention d’urine |
|
Affections des organes de reproduction et du sein |
|
|
|
|
Priapisme |
|
Troubles généraux et anomalies au site d’administration |
|
Œdème périphérique 1 Fatigue |
|
|
Œdème généralisé Œdème localisé |
|
Investigations |
|
|
|
|
Elévation de la créatine-kinase |
1 Au cours des essais cliniques, ces évènements sont survenus avec une fréquence statistiquement significative plus élevée au cours du traitement par mirtazapine qu’avec le placebo.
2 Au cours des essais cliniques, ces évènements sont survenus plus fréquemment pendant le traitement par le placebo qu’avec la mirtazapine, mais sans différence statistiquement significative.
3 Au cours des essais cliniques, ces évènements sont survenus avec une fréquence statistiquement significative plus élevée au cours du traitement par le placebo qu’avec la mirtazapine.
4 N.B. une diminution de dose n’entraîne généralement pas une réduction de la somnolence/sédation, mais peut compromettre l’efficacité antidépressive.
5 Au cours d’un traitement par antidépresseur en général, une anxiété et une insomnie (qui peuvent être des symptômes de la dépression) peuvent apparaître ou s’aggraver. Cela a par ailleurs été rapporté au cours d’un traitement par mirtazapine.
6 Des cas d’idées et de comportements suicidaires ont été rapportés pendant un traitement par la mirtazapine, ou peu après son arrêt (voir rubrique 4.4).
7 Dans la majorité des cas, les patients se sont rétablis après l’arrêt du médicament.
Les analyses de laboratoire effectuées au cours des essais cliniques ont montré des élévations transitoires des transaminases et des gamma-glutamyl-transférases (cependant, aucune augmentation de fréquence statistiquement significative des effets indésirables associés n’a été rapportée sous mirtazapine comparativement au placebo).
Population pédiatrique
Les effets indésirables suivants ont été fréquemment observés au cours des études cliniques chez l'enfant : prise de poids, urticaire et hypertriglycéridémie (voir également la rubrique 5.1).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
L’expérience actuelle sur le surdosage avec la mirtazapine seule indique que les symptômes sont en général légers. Une dépression du système nerveux central, avec désorientation et sédation prolongée, a été rapportée, ainsi qu’une tachycardie et une hyper ou hypotension légère. Cependant, une issue plus sévère (y compris fatale) est possible, à des doses nettement supérieures aux doses thérapeutiques, en particulier en cas de polyintoxication. Dans ces cas, un allongement de l’intervalle QT et des torsades de pointe ont également été rapportés.
En cas de surdosage, un traitement symptomatique approprié et un traitement assurant le maintien des fonctions vitales devront être instaurés. Un contrôle de l’électrocardiogramme (ECG) devra être effectué. L’utilisation de charbon activé ou un lavage gastrique doivent également être envisagés.
Population pédiatrique
Des mesures appropriées telles que décrites chez l'adulte doivent être entreprises en cas de surdosage chez l'enfant.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres antidépresseurs, code ATC : N06AX11.
Mécanisme d’action/effets pharmacodynamiques
La mirtazapine est un antagoniste α2 présynaptique d’action centrale qui augmente la neurotransmission noradrénergique et sérotoninergique centrale.
La stimulation de la neurotransmission sérotoninergique est spécifiquement médiée par les récepteurs 5-HT1, les récepteurs 5-HT2 et 5-HT3 étant bloqués par la mirtazapine. Les deux énantiomères de la mirtazapine semblent intervenir dans l’activité antidépressive, l’énantiomère S(+) en bloquant les récepteurs α2 et 5-HT2 et l’énantiomère R(-) en bloquant les récepteurs 5-HT3.
Efficacité et sécurité clinique
L’activité antagoniste de la mirtazapine sur les récepteurs H1 de l’histamine est associée à ses propriétés sédatives. La mirtazapine n’a pratiquement aucune activité anticholinergique et, aux doses thérapeutiques, a seulement des effets limités (par exemple hypotension orthostatique) sur le système cardiovasculaire.
L'effet de la mirtazapine sur l'intervalle QTc a été évalué dans un essai clinique randomisé et contrôlé versus placebo et moxifloxacine, impliquant 54 volontaires sains utilisant une dose normale de 45 mg et une dose supra-thérapeutique de 75 mg. La modélisation linéaire e-max a suggéré que l’allongement des intervalles QTc est resté inférieur au seuil fixé pour un allongement cliniquement significatif (voir rubrique 4.4).
Population pédiatrique
Deux études randomisées, en double aveugle, contrôlées versus placebo chez des enfants âgés de 7 à 18 ans présentant un trouble dépressif majeur (n = 259) prenant une dose variable pendant les 4 premières semaines (15-45 mg de mirtazapine) suivie d’une dose fixe (15, 30 ou 45 mg de mirtazapine) pendant 4 autres semaines n’ont pas pu démontrer de différence significative entre la mirtazapine et le placebo sur le critère de jugement principal ni sur aucun des critères de jugement secondaires. Une prise de poids significative (≥ 7 %) a été observée chez 48,8 % des patients traités par mirtazapine contre 5,7 % dans le bras placebo. Une urticaire (11,8 % vs 6,8 %) et une hypertriglycéridémie (2,9 % vs 0 %) ont également été observées fréquemment.
5.2. Propriétés pharmacocinétiques
Après administration orale de MIRTAZAPINE ARROW comprimé orodispersible, la mirtazapine, substance active, est rapidement et bien absorbée (biodisponibilité ≈ 50 %), le pic de concentration plasmatique étant atteint en deux heures environ. La prise de nourriture n’a pas d’influence sur la pharmacocinétique de la mirtazapine.
Distribution
La liaison de la mirtazapine aux protéines plasmatiques est d’environ 85 %.
Biotransformation
Les principales voies de biotransformation sont la déméthylation et l’oxydation, suivies de la conjugaison. Les données in vitro provenant de l’étude des microsomes hépatiques humains montrent que les enzymes CYP2D6 et CYP1A2 du cytochrome P450 sont impliqués dans la formation du métabolite 8-hydroxylé, tandis que le CYP3A4 est considéré comme responsable de la formation des métabolites N-déméthylé et N-oxydé. Le métabolite déméthylé est pharmacologiquement actif et semble avoir le même profil pharmacocinétique que la substance mère.
Élimination
La mirtazapine est largement métabolisée et éliminée dans les urines et les fèces en quelques jours.
La demi-vie d'élimination moyenne est comprise entre 20 et 40 heures ; des demi-vies plus longues, pouvant atteindre 65 heures, ont parfois été observées et des demi-vies plus brèves ont été constatées chez des hommes jeunes. La demi-vie d’élimination est suffisante pour justifier une prise quotidienne unique. L'état d'équilibre est atteint en 3 à 4 jours, après lesquels le produit ne s'accumule plus.
Linéarité/non-linéarité
La mirtazapine présente une pharmacocinétique linéaire dans la fourchette des doses recommandées.
Populations particulières
La clairance de la mirtazapine peut être diminuée par une insuffisance rénale ou hépatique.
5.3. Données de sécurité préclinique
Dans des études de toxicité de la reproduction chez le rat et le lapin, aucun effet tératogène n’a été observé. Avec une exposition systémique correspondant à deux fois l’exposition humaine thérapeutique maximale, on a constaté une augmentation des pertes post-implantatoires, une diminution du poids de naissance des petits et une réduction de la survie des petits pendant les trois premiers jours de lactation chez le rat.
La mirtazapine ne s’est pas révélée génotoxique au cours d’une série de tests de mutation génique et de modifications chromosomiques et de l’ADN. Les tumeurs thyroïdiennes observées dans une étude de carcinogénicité chez le rat et les néoplasmes hépatocellulaires observés dans une étude de carcinogénicité chez la souris, sont considérés comme des réponses non génotoxiques, propres à l’espèce et associés à un traitement à long terme par de fortes doses d’inducteurs des enzymes hépatiques.
Arôme fraise et extrait de guarana (maltodextrine, propylène glycol, arômes artificiels, acide acétique (< 1 %)).
Arôme menthe (arômes artificiels, amidon de maïs).
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précaution particulière de conservation.
6.5. Nature et contenu de l'emballage extérieur
Boite de 6, 18, 30, 48, 90 ou 96 comprimés sous plaquette (Polyamide/Aluminium/PVC/Papier/ Polyester/Aluminium).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
26 AVENUE TONY GARNIER
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 387 589 7 9 : 6 comprimés sous plaquette (Polyamide/Aluminium/PVC/Papier/Polyester/ Aluminium).
· 34009 387 590 5 1 : 18 comprimés sous plaquette (Polyamide/Aluminium/PVC/Papier/Polyester/ Aluminium).
· 34009 387 591 1 2 : 30 comprimés sous plaquette (Polyamide/Aluminium/PVC/Papier/Polyester/ Aluminium).
· 34009 573 303 2 3 : 48 comprimés sous plaquette (Polyamide/Aluminium/PVC/Papier/Polyester/ Aluminium).
· 34009 573 304 9 1 : 90 comprimés sous plaquette (Polyamide/Aluminium/PVC/Papier/Polyester/ Aluminium).
· 34009 573 305 5 2 : 96 comprimés sous plaquette (Polyamide/Aluminium/PVC/Papier/Polyester/ Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 28/09/2023
MIRTAZAPINE ARROW 15 mg, comprimé orodispersible
Mirtazapine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que MIRTAZAPINE ARROW 15 mg, comprimé orodispersible et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre MIRTAZAPINE ARROW 15 mg, comprimé orodispersible ?
3. Comment prendre MIRTAZAPINE ARROW 15 mg, comprimé orodispersible ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver MIRTAZAPINE ARROW 15 mg, comprimé orodispersible ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE MIRTAZAPINE ARROW 15 mg, comprimé orodispersible ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : antidépresseurs - code ATC : N06AX11.
MIRTAZAPINE ARROW fait partie d’un groupe de médicaments appelés antidépresseurs.
MIRTAZAPINE ARROW est utilisé pour traiter la maladie dépressive chez les adultes.
MIRTAZAPINE ARROW doit être pris pendant 1 à 2 semaines avant d'agir. Après 2 à 4 semaines, vous pourrez commencer à vous sentir mieux. Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien après 2 à 4 semaines. Vous trouverez plus d'informations dans la rubrique 3 « Quand pouvez-vous espérer commencer à vous sentir mieux ? ».
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE MIRTAZAPINE ARROW 15 mg, comprimé orodispersible ?
· si vous êtes allergique à la mirtazapine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6. Dans ce cas, vous devez contacter votre médecin dès que possible avant de prendre MIRTAZAPINE ARROW ;
· si vous prenez ou avez récemment pris (au cours des deux semaines précédentes) des médicaments appelés inhibiteurs de la monoamine oxydase (IMAO) ;
· si vous avez déjà développé une éruption cutanée ou une desquamation graves, des cloques et/ou des plaies dans la bouche après avoir pris de la mirtazapine ou d’autres médicaments.
Faites particulièrement attention avec la mirtazapine :
Des réactions cutanées graves, dont le syndrome de Stevens-Johnson (SSJ), le syndrome de Lyell et le syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) ont été rapportés dans le cadre de la prise de mirtazapine.
Arrêtez le traitement et consultez immédiatement un médecin si vous remarquez l’un des symptômes décrits dans la rubrique 4 en lien avec ces réactions cutanées graves.
· Si vous avez déjà développé une réaction cutanée grave, il conviendra de ne pas redémarrer un traitement par la mirtazapine.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre MIRTAZAPINE ARROW 15 mg, comprimé orodispersible.
Si vous prenez des médicaments contenant de la buprénorphine. L’utilisation de ces médicaments avec MIRTAZAPINE ARROW peut entraîner un syndrome sérotoninergique, potentiellement mortel (voir « Autres médicaments et MIRTAZAPINE ARROW 15 mg, comprimé orodispersible »)
Enfants et adolescents
MIRTAZAPINE ARROW ne doit habituellement pas être utilisé chez les enfants et les adolescents de moins de 18 ans car son efficacité n'a pas été démontrée. Il est également important de savoir que les patients de moins de 18 ans présentent un risque accru d'effets indésirables tels que tentatives de suicide, pensées suicidaires et comportement hostile (principalement agressivité, comportement d'opposition et colère) lorsqu’ils sont traités par cette classe de médicaments.
Néanmoins, il est possible que votre médecin décide de prescrire MIRTAZAPINE ARROW à des patients de moins de 18 ans, si il/elle décide que c'est dans l’intérêt du patient. Si votre médecin a prescrit MIRTAZAPINE ARROW à un patient de moins de 18 ans et que vous désirez en discuter, adressez-vous à lui. Vous devez informer votre médecin si l'un des symptômes énumérés ci-dessus apparaît ou s'aggrave lors de la prise de MIRTAZAPINE ARROW par un patient de moins de 18 ans. Vous devez également savoir que la sécurité à long terme concernant la croissance, la maturation et le développement cognitif et comportemental de la mirtazapine n'a pas encore été établie dans cette tranche d'âge. De plus, une prise de poids significative a été observée plus souvent que chez l'adulte dans cette tranche d'âge en cas de traitement par mirtazapine.
Idées suicidaires et aggravation de votre dépression
Si vous souffrez de dépression, vous pouvez parfois songer à vous faire du mal ou à vous donner la mort. Ces manifestations peuvent être majorées au début d’un traitement par antidépresseur, car ce type de médicament peut prendre du temps pour agir, généralement 2 semaines mais parfois plus.
Vous êtes plus susceptible de présenter ce type de manifestations dans les cas suivants :
· si vous avez déjà eu des idées suicidaires ou d’auto-agression dans le passé ;
· si vous êtes un jeune adulte. Les études cliniques ont montré que le risque de comportement suicidaire était accru chez les adultes de moins de 25 ans présentant une maladie psychiatrique et traités par antidépresseur.
→ Si vous songez à vous faire du mal ou à vous donner la mort, contactez immédiatement votre médecin ou allez directement à l’hôpital.
Vous pouvez vous faire aider par un ami ou un parent en lui expliquant que vous êtes dépressif et en lui demandant de lire cette notice. Vous pouvez lui demander de vous signaler s’il pense que votre dépression s’aggrave, ou s’il s’inquiète d’un changement dans votre comportement.
Faites également particulièrement attention avec MIRTAZAPINE ARROW 15 mg, comprimé orodispersible :
· si vous avez actuellement ou si vous avez déjà présenté l’une des affections suivantes :
→ si vous ne l’avez pas déjà fait, parlez à votre médecin de ces affections avant de prendre MIRTAZAPINE ARROW.
o convulsions (épilepsie). Si vous développez des crises convulsives ou que leur fréquence augmente, arrêtez de prendre MIRTAZAPINE ARROW et contactez immédiatement votre médecin ;
o affections hépatiques, y compris une jaunisse. Si une jaunisse apparaît, arrêtez de prendre MIRTAZAPINE ARROW et contactez immédiatement votre médecin ;
o maladie du rein ;
o maladie du cœur ou tension artérielle basse ;
o schizophrénie. Si des symptômes psychotiques, tels que des idées paranoïdes, deviennent plus fréquents ou s’aggravent, contactez tout de suite votre médecin ;
o psychose maniaco-dépressive (alternance de périodes d’exaltation/d’hyperactivité et d’humeur dépressive). Si vous commencez à vous sentir exalté(e) ou surexcité(e), arrêtez de prendre MIRTAZAPINE ARROW et contactez immédiatement votre médecin ;
o diabète (il sera peut-être nécessaire d’ajuster votre dose d’insuline ou de vos autres médicaments antidiabétiques) ;
o maladie de l’œil, telle qu’une augmentation de la pression intraoculaire (glaucome) ;
o difficulté à uriner, pouvant être due à une augmentation du volume de la prostate ;
o certains types de maladie cardiaque qui peuvent modifier votre rythme cardiaque, une crise cardiaque récente, une insuffisance cardiaque ou la prise de certains médicaments qui peuvent affecter votre rythme cardiaque ;
· si vous développez des signes d’infection tels qu’une fièvre élevée inexpliquée, des maux de gorge ou des ulcérations de la bouche :
→ Arrêtez de prendre MIRTAZAPINE ARROW et contactez immédiatement votre médecin pour un examen sanguin. Dans de rares cas, ces symptômes peuvent être des signes d’une altération de la production des cellules sanguines par la moelle osseuse. Bien que rares, ces symptômes apparaissent le plus souvent après 4 à 6 semaines de traitement ;
· si vous êtes une personne âgée. Il se peut que vous soyez plus sensible aux effets secondaires des antidépresseurs.
Autres médicaments et MIRTAZAPINE ARROW 15 mg, comprimé orodispersible
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Ne prenez pas MIRTAZAPINE ARROW 15 mg, comprimé orodispersible en association avec :
· des inhibiteurs de la monoamine oxydase (IMAO). Ne prenez pas non plus MIRTAZAPINE ARROW pendant les deux semaines qui suivent l’arrêt d’un traitement par des IMAO. Si vous arrêtez de prendre MIRTAZAPINE ARROW, ne prenez pas non plus d’IMAO pendant les deux semaines qui suivent. Les IMAO comprennent notamment le moclobémide, la tranylcypromine (tous deux des antidépresseurs) et la sélégiline (utilisée dans le traitement de la maladie de Parkinson).
Faites attention si vous prenez MIRTAZAPINE ARROW 15 mg, comprimé orodispersible en association avec :
· des antidépresseurs tels que ISRS, venlafaxine et L-tryptophane ou des triptans (utilisés pour traiter la migraine), du tramadol ou de la buprénorphine (pour traiter des douleurs sévères), du linézolide (un antibiotique), du lithium (utilisé pour traiter certaines affections psychiatriques), du bleu de méthylène (utilisé pour traiter les taux élevés de méthémoglobine dans le sang) et des préparations à base de millepertuis – Hypericum perforatum (remède à base de plante utilisé dans la dépression). Dans de très rares cas, MIRTAZAPINE ARROW seul ou en association avec ces médicaments peut induire ce qu’on appelle un syndrome sérotoninergique. Les symptômes de ce syndrome sont, entre autres, fièvre inexpliquée, sueurs, augmentation de la fréquence cardiaque, diarrhée, contractions musculaires (incontrôlables), frissons, amplification des réflexes, impatience musculaire, sautes d’humeur et perte de connaissance. Si vous ressentez plusieurs de ces symptômes, parlez-en immédiatement à votre médecin ;
· l’antidépresseur néfazodone. Il peut augmenter la quantité de MIRTAZAPINE ARROW dans votre sang. Si vous prenez ce médicament, informez-en votre médecin. Il sera peut-être nécessaire de diminuer la dose de MIRTAZAPINE ARROW, ou, à l’arrêt de la néfazodone, de l’augmenter de nouveau ;
· des médicaments contre l’anxiété ou l’insomnie tels que les benzodiazépines ;
· des médicaments contre la schizophrénie tels que l’olanzapine ;
· des médicaments contre les allergies tels que la cétirizine ;
· des médicaments contre les fortes douleurs tels que la morphine.
Utilisé en association avec ces médicaments, MIRTAZAPINE ARROW peut accentuer les somnolences causées par ces derniers ;
· des médicaments contre les infections : médicaments contre les infections bactériennes (tels que l’érythromycine), contre les infections fongiques (tels que le kétoconazole) et contre les infections par le VIH/SIDA (tels que les inhibiteurs de protéase du VIH) et les médicaments contre les ulcères de l'estomac (tels que la cimétidine).
Utilisés en association avec MIRTAZAPINE ARROW, ces médicaments peuvent augmenter la quantité de MIRTAZAPINE ARROW dans votre sang.
Si vous prenez ces médicaments, informez-en votre médecin. Il sera peut-être nécessaire de diminuer la dose de MIRTAZAPINE ARROW, ou, à l’arrêt de ces médicaments, de l’augmenter de nouveau ;
· des médicaments contre l’épilepsie tels que la carbamazépine et la phénytoïne ;
· des médicaments contre la tuberculose tels que la rifampicine.
Utilisés en association avec MIRTAZAPINE ARROW, ces médicaments peuvent diminuer la quantité de MIRTAZAPINE ARROW dans votre sang. Si vous prenez ces médicaments, informez-en votre médecin. Il sera peut-être nécessaire d’augmenter la dose de MIRTAZAPINE ARROW, ou, à l’arrêt de ces médicaments, de la diminuer de nouveau ;
· des médicaments pour empêcher le sang de coaguler tels que la warfarine.
MIRTAZAPINE ARROW peut augmenter les effets de la warfarine sur le sang. Si vous prenez ce médicament, informez-en votre médecin. En cas d’utilisation concomitante des deux médicaments, une surveillance attentive de votre sang par un médecin est recommandée ;
· des médicaments qui peuvent affecter votre rythme cardiaque tels que certains antibiotiques et certains antipsychotiques.
MIRTAZAPINE ARROW 15 mg, comprimé orodispersible avec des aliments et de l’alcool
Il se peut que vous vous sentiez somnolent si vous consommez de l’alcool pendant votre traitement par MIRTAZAPINE ARROW.
Il vous est recommandé de ne pas boire d’alcool.
Vous pouvez prendre MIRTAZAPINE ARROW accompagné ou non de nourriture.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
L’expérience liée à l’utilisation de MIRTAZAPINE ARROW, bien que limitée chez la femme enceinte, ne montre pas d’augmentation du risque. Cependant, des précautions doivent être prises en cas d’utilisation pendant la grossesse.
Si vous prenez MIRTAZAPINE ARROW jusqu'à la naissance ou peu avant, votre nouveau-né devra être surveillé à la recherche de possibles effets indésirables.
Assurez-vous que votre sage-femme et/ou votre médecin sachent que vous prenez MIRTAZAPINE ARROW. En cas de prise pendant la grossesse, des médicaments similaires (ISRS) peuvent augmenter le risque d'une maladie grave chez le bébé, appelée hypertension artérielle pulmonaire persistante (HTAP) du nouveau-né, qui conduit à une respiration plus rapide et une coloration bleuâtre du bébé. Ces symptômes apparaissent généralement au cours des 24 premières heures après la naissance du bébé. Si cela survient chez votre bébé, contactez immédiatement votre sage-femme et/ou votre médecin.
Conduite de véhicules et utilisation de machines
MIRTAZAPINE ARROW peut affecter votre concentration ou votre vigilance. Assurez-vous que vos capacités ne sont pas altérées avant de conduire ou d’utiliser une machine. Si votre médecin a prescrit MIRTAZAPINE ARROW à un patient de moins de 18 ans, assurez-vous que sa concentration et sa vigilance ne sont pas affectées avant d'emprunter la circulation (par exemple à bicyclette).
MIRTAZAPINE ARROW 15 mg, comprimé orodispersible contient de l’aspartame (E951), une source de phénylalanine
Peut être dangereux pour les personnes atteintes de phénylcétonurie.
3. COMMENT PRENDRE MIRTAZAPINE ARROW 15 mg, comprimé orodispersible ?
Quelle dose de MIRTAZAPINE ARROW 15 mg, comprimé orodispersible prendre ?
La dose de départ recommandée est de 15 ou 30 mg par jour. Votre médecin peut vous conseiller d’augmenter la dose après quelques jours jusqu’à celle qui sera la plus adaptée à votre cas (entre 15 et 45 mg par jour). La posologie est généralement la même quel que soit l’âge. Cependant, si vous êtes une personne âgée ou si vous avez une maladie rénale ou du foie, votre médecin pourra être amené à adapter la dose.
Quand prendre MIRTAZAPINE ARROW 15 mg, comprimé orodispersible ?
→ Prenez MIRTAZAPINE ARROW chaque jour à la même heure.
Il est préférable de prendre MIRTAZAPINE ARROW en une dose unique au coucher. Cependant, votre médecin pourra vous suggérer de diviser la dose de MIRTAZAPINE ARROW – une fois le matin et une fois le soir au coucher.
La dose la plus élevée doit être prise au coucher.
Prendre MIRTAZAPINE ARROW 15 mg, comprimé orodispersible de la manière suivante :
Prenez le comprimé orodispersible par voie orale.
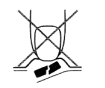
1. N’écrasez pas le comprimé orodispersible
Afin de ne pas écraser le comprimé orodispersible, ne poussez pas à travers l'alvéole (Figure A).
Figure A
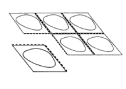
2. Détachez une alvéole
Chaque plaquette contient six alvéoles de comprimés, séparées les unes des autres par une ligne perforée. Détachez une alvéole en suivant les pointillés (Figure 1).
Figure 1
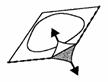
3. Décollez la pellicule protectrice
Décollez la pellicule protectrice avec précaution, en commençant par le coin marqué par une flèche (Figure 2 et 3).
Figure 2 Figure 3
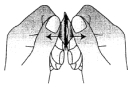
4. Prenez le comprimé orodispersible
Prenez le comprimé orodispersible avec des mains sèches et placez-le sur la langue (Figure 4).
Le comprimé se désagrégera rapidement et pourra être avalé sans eau.
Figure 4
Quand pouvez-vous espérer commencer à vous sentir mieux ?
Habituellement, MIRTAZAPINE ARROW commence à agir après 1 à 2 semaines et vous pourrez commencer à vous sentir mieux après 2 à 4 semaines.
Il est important que, pendant les premières semaines de traitement, vous parliez à votre médecin des effets de MIRTAZAPINE ARROW :
→ 2 à 4 semaines après avoir commencé à prendre MIRTAZAPINE ARROW, parlez à votre médecin de la façon dont le médicament a agi sur vous.
Si vous ne vous sentez toujours pas mieux, votre médecin pourra vous prescrire une dose plus élevée. Dans ce cas, consultez de nouveau votre médecin 2 à 4 semaines plus tard.
Habituellement, vous devrez prendre MIRTAZAPINE ARROW pendant 4 à 6 mois après la disparition de vos symptômes dépressifs.
Si vous avez pris plus de MIRTAZAPINE ARROW 15 mg, comprimé orodispersible que vous n’auriez dû
Si vous ou une autre personne avez pris trop de MIRTAZAPINE ARROW, appelez tout de suite un médecin.
Les effets les plus probables d’un surdosage de MIRTAZAPINE ARROW (quand il n’est pas associé à d’autres médicaments ni à l’alcool) sont une somnolence, une désorientation et une augmentation de la fréquence cardiaque. Les symptômes d’un possible surdosage peuvent inclure des modifications du rythme cardiaque (battements du cœur rapides, irréguliers) et/ou des évanouissements qui peuvent être les signes d’une maladie potentiellement mortelle connue sous le nom de Torsade de Pointes.
Si vous oubliez de prendre MIRTAZAPINE ARROW 15 mg, comprimé orodispersible
Si vous devez prendre votre dose en une prise par jour :
· ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre. Prenez la dose suivante à l’heure habituelle.
Si vous devez prendre votre dose en deux prises par jour :
· si vous avez oublié de prendre votre dose du matin, prenez-la simplement en même temps que votre dose du soir ;
· si vous avez oublié de prendre votre dose du soir, ne la prenez pas avec votre dose du matin suivante ; sautez-la et poursuivez votre traitement en prenant les doses du matin et du soir habituelles ;
· si vous avez oublié de prendre les deux doses, ne tentez pas de rattraper les doses oubliées. Sautez les deux doses et poursuivez votre traitement le lendemain en prenant les doses du matin et du soir habituelles.
Si vous arrêtez de prendre MIRTAZAPINE ARROW 15 mg, comprimé orodispersible
→ N’arrêtez de prendre MIRTAZAPINE ARROW qu’avec l’accord de votre médecin.
Si vous arrêtez trop tôt, votre dépression pourrait réapparaître. Lorsque vous vous sentez mieux, consultez votre médecin. Votre médecin décidera quand le traitement pourra être arrêté.
N’arrêtez pas brutalement de prendre MIRTAZAPINE ARROW, même si votre dépression s’est améliorée. Si vous arrêtez de prendre MIRTAZAPINE ARROW brutalement, vous pourriez vous sentir mal, avec des sensations d’étourdissements, agité(e) ou anxieux(se) et avoir des maux de tête. Ces symptômes peuvent être évités en arrêtant le médicament progressivement. Votre médecin vous expliquera comment diminuer les doses progressivement.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Si vous ressentez un des effets indésirables graves suivants, arrêtez MIRTAZAPINE ARROW 15 mg, comprimé orodispersible et parlez-en immédiatement à votre médecin.
Peu fréquents (pouvant toucher jusqu'à 1 patient sur 100) :
· sentiment d’exaltation ou de surexcitation (manie).
Rares (pouvant toucher jusqu'à 1 patient sur 1 000) :
· coloration jaune des yeux ou de la peau pouvant suggérer une altération de la fonction hépatique (jaunisse).
Fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles) :
· signes d’infection tels que forte fièvre subite inexpliquée, maux de gorge et ulcérations de la bouche (agranulocytose). Dans de rares cas, la mirtazapine peut provoquer une altération de la production des cellules sanguines (aplasie médullaire). Certaines personnes peuvent devenir moins résistantes aux infections dans la mesure où la mirtazapine peut causer une insuffisance temporaire en globules blancs (granulocytopénie). Dans de rares cas, la mirtazapine peut aussi causer une insuffisance en globules rouges et blancs ainsi qu’en plaquettes (anémie aplasique), une insuffisance en plaquettes (thrombocytopénie) ou une augmentation du nombre de globules blancs (éosinophilie) ;
· crises d’épilepsie (convulsions) ;
· combinaison de symptômes tels que fièvre inexpliquée, sueurs, augmentation de la fréquence cardiaque, diarrhée, contractions musculaires (incontrôlables), frissons, augmentation des réflexes, impatience musculaire, sautes d’humeur, perte de connaissance et augmentation de la quantité de salive. Dans de très rares cas, ces symptômes peuvent être les signes d’un syndrome sérotoninergique ;
· envies de se faire du mal ou de se donner la mort ;
· taches rougeâtres sur le tronc qui sont des macules en forme de cibles ou des cercles, souvent avec des cloques centrales, desquamation de la peau, ulcères de la bouche, de la gorge, du nez, des organes génitaux et des yeux. Ces éruptions cutanées graves peuvent être précédées par de la fièvre et des symptômes de type grippal (syndrome de Stevens-Johnson, nécrolyse épidermique toxique) ;
· éruption cutanée étendue, température corporelle élevée et dilatation des ganglions lymphatiques (syndrome DRESS ou syndrome d’hypersensibilité à un médicament).
Autres effets indésirables possibles avec MIRTAZAPINE ARROW 15 mg, comprimé orodispersible :
Très fréquents (pouvant toucher plus de 1 patient sur 10) :
· augmentation de l’appétit et prise de poids ;
· somnolence ou endormissement ;
· maux de tête ;
· sécheresse buccale.
Fréquents (pouvant toucher jusqu'à 1 patient sur 10) :
· léthargie ;
· étourdissements ;
· frissons ou tremblements ;
· nausées ;
· diarrhée ;
· vomissements ;
· constipation ;
· rougeur ou éruption cutanée (exanthème) ;
· douleurs articulaires (arthralgies) ou musculaires (myalgies) ;
· douleur dorsale ;
· sensation de vertiges ou malaise lorsque vous vous levez brutalement (hypotension orthostatique) ;
· gonflement (généralement au niveau des chevilles ou des pieds) dû à une rétention de liquide (œdème) ;
· fatigue ;
· rêves intenses ;
· confusion ;
· anxiété ;
· troubles du sommeil ;
· problèmes de mémoire qui, dans la majorité des cas, disparaissent à l’arrêt du traitement.
Peu fréquents (pouvant toucher jusqu'à 1 patient sur 100) :
· sensations anormales au niveau de la peau : par exemple brûlures, picotements, chatouillements ou fourmillements (paresthésies) ;
· impatiences dans les jambes ;
· évanouissement (syncope) ;
· sensation d’engourdissement dans la bouche (hypoesthésie orale) ;
· tension artérielle basse ;
· cauchemars ;
· agitation ;
· hallucinations ;
· besoin urgent de bouger.
Rares (pouvant toucher jusqu'à 1 patient sur 1 000) :
· secousses ou contractions musculaires (myoclonie) ;
· agressivité ;
· douleurs abdominales et nausées : cela peut évoquer une inflammation du pancréas (pancréatite).
Fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles) :
· sensations anormales dans la bouche (paresthésies orales) ;
· gonflement dans la bouche (œdème buccal) ;
· gonflements sur le corps (œdème généralisé) ;
· gonflements localisés ;
· hyponatrémie ;
· sécrétion inappropriée d’hormone antidiurétique ;
· réactions cutanées sévères (dermatite bulleuse, érythème polymorphe) ;
· somnambulisme ;
· trouble de la parole ;
· élévation du taux de créatine-kinase dans le sang ;
· difficulté à uriner (rétention d’urine) ;
· douleur musculaire, raideur et/ou faiblesse, urine foncée ou décolorée (rhabdomyolyse) ;
· augmentation des taux d'hormone prolactine dans le sang (hyperprolactinémie, y compris les symptômes de gonflement mammaire et/ou d’écoulement laiteux du mamelon) ;
· érection douloureuse et prolongée du pénis.
Effets indésirables supplémentaires chez les enfants et les adolescents
Chez les enfants de moins de 18 ans, les effets indésirables suivants ont été observés fréquemment au cours des études cliniques : prise de poids significative, urticaire et augmentation de la quantité de triglycérides dans le sang.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER MIRTAZAPINE ARROW 15 mg, comprimé orodispersible ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient MIRTAZAPINE ARROW 15 mg, comprimé orodispersible
· La substance active est :
Mirtazapine..................................................................................................................... 15 mg
Pour un comprimé orodispersible
· Les autres composants sont : crospovidone (type B), mannitol (E421), cellulose microcristalline, aspartame (E951), silice colloïdale anhydre, stéarate de magnésium, arôme fraise et extrait de guarana [maltodextrine, propylène glycol, arômes artificiels, acide acétique (< 1 %)], arôme menthe [arômes artificiels, amidon de maïs].
Qu’est-ce que MIRTAZAPINE ARROW 15 mg, comprimé orodispersible et contenu de l’emballage extérieur
MIRTAZAPINE ARROW 15 mg, comprimé orodispersible est disponible en boîte de 6, 18, 30, 48, 90 ou 96 comprimés sous plaquette (Polyamide/Aluminium/PVC/Papier/Polyester/Aluminium).
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
26 AVENUE TONY GARNIER
69007 LYON
Exploitant de l’autorisation de mise sur le marché
ARROW GENERIQUES
26 AVENUE TONY GARNIER
69007 LYON
APL SWIFT SERVICES (MALTA) LTD
HF26, HAL FAR INDUSTRIAL ESTATE, HAL FAR
BIRZEBBUGIA, BBG 3000
MALTE
OU
GENERIS FARMACEUTICA S.A.
RUA JOAO DE DEUS, 19
2700-487 AMADORA
PORTUGAL
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).