Dernière mise à jour le 01/06/2026
ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique : Autres agents antinéoplasiques, code ATC : L01XX41
ERIBULINE ZENTIVA contient la substance active éribuline et est un médicament anticancéreux qui agit en arrêtant la croissance et la dissémination des cellules cancéreuses.
Il est utilisé chez les adultes pour le cancer du sein localement avancé ou métastatique (le cancer du sein s’est disséminé en dehors de la tumeur d’origine) quand au moins un autre traitement a été essayé et qu’il a perdu son efficacité.
Il est également utilisé chez les adultes pour le liposarcome (un type de cancer qui se développe à partir du tissu adipeux) avancé ou métastatique quand un traitement antérieur a été essayé et qu’il a perdu son efficacité.
Présentations
> 1 flacon en verre de 2 mL
Code CIP : 34009 302 892 6 6
Déclaration de commercialisation : 05/02/2025
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : ZENTIVA France
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription hospitalière
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services CANCEROLOGIE
- prescription réservée aux spécialistes et services HEMATOLOGIE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 243 566 4
ANSM - Mis à jour le : 28/03/2025
ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Eribuline.............................................................................................................................. 0,44 mg
Sous forme de mésilate d’éribuline
Pour un mL.
Chaque flacon de 2 mL contient du mésilate d’éribuline, équivalant à 0,88 mg d’éribuline.
Excipient à effet notoire :
Chaque mL de solution contient 40 mg d’éthanol.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution aqueuse limpide et incolore de pH compris entre 6,0 et 9,0.
4.1. Indications thérapeutiques
L’éribuline est indiquée dans le traitement des patients adultes atteints d’un liposarcome non résécable ayant reçu un protocole de chimiothérapie antérieur comportant une anthracycline (sauf chez les patients ne pouvant pas recevoir ce traitement) pour le traitement d’une maladie avancée ou métastatique (voir rubrique 5.1).
4.2. Posologie et mode d'administration
L’éribuline ne doit être prescrite que par un médecin qualifié, expérimenté dans l’utilisation appropriée de médicaments anticancéreux. Elle ne doit être administrée que par un professionnel de santé disposant des qualifications appropriées.
La dose recommandée d’éribuline sous forme de solution prête à l’emploi est de 1,23 mg/m2 devant être administrée par voie intraveineuse sur 2 à 5 minutes aux Jours 1 et 8 de chaque cycle de 21 jours.
Remarque :
Dans l’Union européenne, la dose recommandée fait référence à la substance active sous forme de base (éribuline). Le calcul de la dose individuelle à administrer à un patient doit être basé sur le dosage de la solution prête à l’emploi qui contient 0,44 mg/mL d’éribuline et sur la recommandation posologique de 1,23 mg/m2. Les recommandations pour la réduction des doses présentées ci-dessous sont également exprimées en dose d’éribuline à administrer sur la base du dosage de la solution prête à l’emploi.
Dans les études pivots, les publications des études et dans certaines autres régions (par exemple les Etats-Unis et la Suisse), la dose recommandée est basée sur la forme sel (mésilate d’éribuline).
Les patients peuvent présenter des nausées ou des vomissements. Un traitement antiémétique prophylactique incluant des corticoïdes doit être envisagé.
Doses différées durant le traitement
Au Jour 1 ou au Jour 8, l’administration d’éribuline doit être différée dans les cas suivants :
· Numération absolue des neutrophiles (NAN) < 1 × 109/L
· Plaquettes < 75 × 109/L
· Toxicités non hématologiques de grade 3 ou 4
Réduction de la dose durant le traitement
Le tableau suivant présente les recommandations concernant la réduction des doses lors de la reprise du traitement.
Recommandations pour la réduction des doses
|
Effet indésirable après une administration antérieure d’éribuline |
Dose d’éribuline recommandée |
|
Hématologique : |
|
|
NAN < 0,5 × 109/L persistant plus de 7 jours |
0,97 mg/m2 |
|
NAN < 1 × 109/L, neutropénie compliquée par de la fièvre ou une infection |
|
|
Plaquettes < 25 × 109/L, thrombopénie |
|
|
Plaquettes < 50 × 109/L, thrombopénie compliquée par une hémorragie ou nécessitant une transfusion de sang ou de plaquettes |
|
|
Non hématologique : |
|
|
Tout événement de grade 3 ou 4 lors du cycle précédent |
|
|
Réapparition de tout effet indésirable hématologique ou non, tel que précisé ci-dessus |
|
|
Malgré la réduction à 0,97 mg/m2 |
0,62 mg/m2 |
|
Malgré la réduction à 0,62 mg/m2 |
Envisager l’arrêt du traitement |
La dose d’éribuline ne doit pas être ré-augmentée après avoir été réduite.
Patients présentant une insuffisance hépatique
Altération de la fonction hépatique en raison de métastases
La dose recommandée d’éribuline chez les patients présentant une insuffisance hépatique légère (Child-Pugh A) est de 0,97 mg/m2 administrée en injection intraveineuse sur 2 à 5 minutes aux Jours 1 et 8 d’un cycle de 21 jours. La dose recommandée d’éribuline chez les patients présentant une insuffisance hépatique modérée (Child-Pugh B) est de 0,62 mg/m2 administrée en injection intraveineuse sur 2 à 5 minutes aux Jours 1 et 8 d’un cycle de 21 jours.
L’insuffisance hépatique sévère (Child-Pugh C) n’a pas été étudiée, mais la réduction de dose devrait être plus importante si l’éribuline est utilisée chez ces patients.
Altération de la fonction hépatique en raison d’une cirrhose
Ce groupe de patients n’a pas été étudié. Les posologies ci-dessus peuvent être utilisées dans l’insuffisance légère et modérée, mais une surveillance étroite est conseillée car les doses pourraient nécessiter un réajustement.
Patients présentant une insuffisance rénale
L’exposition à l’éribuline peut être augmentée chez certains patients présentant une altération de la fonction rénale modérée ou sévère (clairance de la créatinine < 50 mL/min) pour qui une réduction de la dose peut s’avérer nécessaire. La prudence et une surveillance étroite sont recommandées chez tous les patients présentant une insuffisance rénale. (Voir rubrique 5.2).
Patients âgés
Aucune adaptation particulière de la posologie n’est recommandée en fonction de l’âge du patient (voir rubrique 4.8).
Population pédiatrique
Aucune utilisation de l’éribuline n’est justifiée chez les enfants et les adolescents pour l’indication du cancer du sein.
Aucune utilisation de l’éribuline n’est justifiée dans la population pédiatrique pour l’indication du sarcome des tissus mous (voir rubrique 5.1).
Mode d’administration
L’éribuline doit être administrée par voie intraveineuse. La dose peut être diluée dans un volume de solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) pouvant atteindre 100 mL. Elle ne doit pas être diluée dans une solution pour perfusion de glucose à 5 %. Pour les instructions concernant la dilution du médicament avant administration, voir rubrique 6.6. Avant l’administration, il convient de s’assurer de la présence d’un accès veineux périphérique de bonne qualité ou d’une voie veineuse centrale. Rien n’indique que le mésilate d’éribuline puisse être irritant ou vésicant (formation de vésicules). En cas d’extravasation, le traitement doit être symptomatique. Pour des informations relatives à la manipulation des médicaments cytotoxiques, voir rubrique 6.6.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1
· Allaitement
4.4. Mises en garde spéciales et précautions d'emploi
Hématologie
Les effets de myélosuppression sont proportionnels à la dose administrée et se manifestent essentiellement sous forme de neutropénie (rubrique 4.8). Une surveillance de la numération formule sanguine doit être effectuée chez tous les patients avant l'administration de chaque dose d’éribuline. Le traitement par éribuline ne doit être instauré que chez les patients dont la NAN est ≥ 1,5 × 109/L et les plaquettes sont > 100 × 109/L.
Une neutropénie fébrile a été observée chez < 5 % des patients traités par éribuline. Les patients présentant une neutropénie fébrile, une neutropénie sévère ou une thrombopénie doivent être traités selon les recommandations figurant à la rubrique 4.2.
L’incidence des neutropénies de grade 4 et des neutropénies fébriles était plus élevée chez les patients dont les taux d’alanine aminotransférase (ALAT) ou d’aspartate aminotransférase (ASAT) étaient > 3 fois la limite supérieure de la normale (LSN).
Bien que les données soient limitées, l’incidence des neutropénies de grade 4 et des neutropénies fébriles était également plus élevée chez les patients dont la bilirubine était > 1,5 × LSN.
Des cas fatals de neutropénie fébrile, de sepsis neutropénique, de sepsis et de choc septique ont été rapportés.
Une neutropénie sévère peut être corrigée par l’utilisation du facteur stimulant les colonies de granulocytes (G-CSF) ou d’un équivalent à l’appréciation du médecin, conformément aux recommandations appropriées (voir rubrique 5.1).
Neuropathie périphérique
Il convient d’exercer une surveillance étroite des patients en vue de déceler tout signe de neuropathie périphérique motrice et sensitive. La dose doit être différée ou réduite en cas d’apparition d’une neurotoxicité périphérique sévère (voir rubrique 4.2).
Lors des essais cliniques, les patients présentant une neuropathie préexistante, supérieure au grade 2, ont été exclus. Néanmoins, le risque d’apparition de nouveaux symptômes ou d’aggravation des symptômes n’était pas plus élevé chez les patients présentant une neuropathie préexistante de grade 1 ou 2 que chez ceux qui entraient dans l’étude sans cette affection.
Allongement de l’intervalle QT
Au cours d’une étude en ouvert non contrôlée portant sur l’ECG de 26 patients, un allongement de l’intervalle QT a été observé le Jour 8, indépendamment des concentrations d’éribuline, sans allongement de l’intervalle QT le Jour 1. Une surveillance ECG est recommandée chez les patients lorsqu’ils présentent une insuffisance cardiaque congestive, une bradyarythmie, des troubles électrolytiques ou lorsqu’ils sont traités par des médicaments connus pour allonger l’intervalle QT, y compris les antiarythmiques des classes Ia et III. L’hypokaliémie, l’hypocalcémie ou l’hypomagnésémie doivent être corrigées avant le début du traitement par éribuline et ces électrolytes doivent être contrôlés régulièrement pendant le traitement. L’éribuline doit être évitée chez les patients présentant un syndrome du QT long congénital.
Excipients
Ce médicament contient 80 mg d'alcool (éthanol) par flacon. La quantité dans un flacon de ce médicament équivaut à moins de 2 mL de bière ou 1 mL de vin.
La faible quantité d'alcool contenue dans ce médicament n'est pas susceptible d'entraîner d'effet notable.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction médicamenteuse avec les inhibiteurs et les inducteurs du CYP3A4 n’est attendue. L’exposition à l’éribuline (ASC et Cmax) n’a pas été altérée par le kétoconazole, un inhibiteur du CYP3A4 et de la glycoprotéine P (P-gp) et par la rifampicine, un inducteur du CYP3A4.
Effets de l’éribuline sur la pharmacocinétique d’autres médicaments
Les données in vitro indiquent que l’éribuline est un inhibiteur faible du CYP3A4, une enzyme importante pour le métabolisme des médicaments. Aucune donnée in vivo n’est disponible. La prudence et la surveillance des événements indésirables sont recommandées en cas d’utilisation concomitante de substances à marge thérapeutique étroite qui sont éliminées essentiellement par la voie métabolique du CYP3A4 (par exemple, alfentanil, ciclosporine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus).
L’éribuline n’inhibe pas les enzymes CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 ou 2E1 aux concentrations cliniques utilisées.
Aux concentrations cliniques pertinentes, l’éribuline n’a pas inhibé l’activité des transporteurs BCRP, OCT1, OCT2, OAT1, OAT3, OATP1B1 et OATP1B3.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’existe pas de données sur l’utilisation de l’éribuline chez la femme enceinte. L’éribuline est embryotoxique, fœtotoxique et tératogène chez le rat. L’éribuline ne doit pas être utilisée pendant la grossesse, sauf en cas de nécessité absolue et après évaluation minutieuse du bénéfice pour la mère et du risque pour le fœtus.
Les femmes en capacité de procréer doivent être informées d’éviter toute grossesse pendant qu’elles reçoivent l’éribuline et doivent utiliser une méthode de contraception très efficace pendant le traitement par ERIBULINE ZENTIVA et pendant les 7 mois après l’arrêt du traitement.
Les hommes dont la partenaire est en âge de procréer doivent être informés qu'ils ne doivent pas avoir d'enfant pendant le traitement par ERIBULINE ZENTIVA et qu'ils doivent utiliser une méthode de contraception efficace pendant le traitement par ERIBULINE ZENTIVA et pendant les 4 mois après l’arrêt du traitement.
On ne sait pas si l’éribuline/les métabolites sont excrétés dans le lait maternel chez l’être humain ou l’animal. Un risque pour les nouveau-nés/nourrissons ne peut être exclu et, par conséquent, l’éribuline ne doit pas être utilisée pendant l’allaitement (voir rubrique 4.3).
Fertilité
Une toxicité testiculaire a été observée chez le rat et le chien (voir rubrique 5.3). Avant traitement, les patients de sexe masculin doivent se renseigner sur les modalités de conservation du sperme en raison du risque de stérilité irréversible due au traitement par éribuline.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables liés à l’éribuline les plus fréquemment rapportés sont une aplasie médullaire se manifestant par une neutropénie, une leucopénie, une anémie, une thrombopénie et des infections associées. L’apparition ou l’aggravation d’une neuropathie périphérique préexistante a également été rapportée. Les toxicités gastro-intestinales, se manifestant par une anorexie, des nausées, des vomissements, une diarrhée, une constipation et une stomatite, sont des effets indésirables rapportés. Les autres effets indésirables sont notamment : fatigue, alopécie, enzymes hépatiques augmentées, sepsis et syndrome de douleur musculosquelettique.
Tableau listant les effets indésirables
Sauf indication contraire, le tableau indique les taux d’incidence des effets indésirables observés chez les patients atteints d’un cancer du sein ou d’un sarcome des tissus mous et ayant reçu la dose recommandée au cours des études de phase II et de phase III.
Les catégories de fréquence sont définies selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000) et très rare (< 1/10 000).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de fréquence. Lorsque des effets de grade 3 ou 4 se sont produits, les fréquences réelles totales et celles des effets de grade 3 ou 4 sont indiquées.
|
Classe de systèmes d’organes |
Effets indésirables - tous grades |
|||
|
|
Très fréquent (Fréquence en %) |
Fréquent (Fréquence en %) |
Peu fréquent (Fréquence en %) |
Rare ou fréquence indéterminée |
|
Infections et infestations |
|
Infection des voies urinaires (8,5 %) (G3/4 : 0,7 %) Pneumonie (1,6 %) (G3/4 : 1,0 %) Candidose orale Herpès buccal Infection des voies aériennes supérieures Rhinopharyngite Rhinite Zona |
Sepsis (0,5 %) (G3/4 : 0,54 %)a Sepsis neutropénique (0,2 %) (G3/4 : 0,2 %)a Choc septique (0,2 %) (G3/4 : 0,2 %)a |
|
|
Affections hématologiques et du système lymphatique |
Neutropénie (53,6 %) (G3/4 : 46,0 %) Leucopénie (27,9 %) (G3/4 : 17,0 %) Anémie (21,8 %) (G3/4 : 3,0 %) |
Lymphopénie (5,7 %) (G3/4 : 2,1 %) Neutropénie fébrile (4,5 %) (G3/4 : 4,4 %)a Thrombopénie (4,2 %) (G3/4 : 0,7 %) |
|
* Coagulation intravasculaire disséminéeb |
|
Troubles du métabolisme et de la nutrition |
Appétit diminué (22,5 %) (G3/4 : 0,7 %)d |
Hypokaliémie (6,8 %) (G3/4 : 2,0 %) Hypomagnésémie (2,8 %) (G3/4 : 0,3 %) Déshydratation (2,8 %) (G3/4 : 0,5 %)d Hyperglycémie Hypophosphatémie Hypocalcémie |
|
|
|
Affections psychiatriques |
|
Insomnie Dépression |
|
|
|
Affections du système nerveux |
Neuropathie périphériquec (35,9 %) (G3/4 : 7,3 %) Céphalées (17,5 %) (G3/4 : 0,7 %) |
Dysgueusie Sensations vertigineuses (9,0 %) (G3/4 : 0,4 %)d Hypoesthésie Léthargie Neurotoxicité |
|
|
|
Affections oculaires |
|
Augmentation de la sécrétion lacrymale (5,8 %) (G3/4 : 0,1 %)d Conjonctivite |
|
|
|
Affections de l’oreille et du labyrinthe |
|
Vertige Acouphènes |
|
|
|
Affections cardiaques |
|
Tachycardie |
|
|
|
Affections vasculaires |
|
Bouffées de chaleur Embolie pulmonaire (1,3 %) (G3/4 : 1,1 %)a |
Thrombose veineuse profonde |
|
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée (15,2 %)a (G3/4 : 3,5 %)a Toux (15,0 %) G3/4 : 0,5 %)d |
Douleur oropharyngée Epistaxis Rhinorrhée |
Pneumopathie interstitielle diffuse (0,2 %) (G3/4 : 0,1 %) |
|
|
Affections gastro-intestinales |
Nausées (35,7 %) (G3/4 : 1,1 %)d Constipation (22,3 %) (G3/4 : 0,7 %)d Diarrhée (18,7 %) (G3/4 : 0,8 %) Vomissements (18,1 %) (G3/4 : 1,0 %) |
Douleurs abdominales Stomatite (11,1 %) (G3/4 : 1,0 %)d Bouche sèche Dyspepsie (6,5 %) (G3/4 : 0,3 %)d Reflux gastro-œsophagien Distension abdominale |
Ulcération buccale Pancréatite |
|
|
Affections hépatobiliaires |
|
Aspartate aminotransférase augmentée (7.7 %) (G3/4 : 1,4 %)d Alanine aminotransférase augmentée (7,6 %) (G3/4 : 1,9 %)d Gamma-glutamyltransférase augmentée (1,7 %) (G3/4 : 0,9 %)d Hyperbilirubinémie (1,4 %) (G3/4 : 0,4 %) |
Hépatotoxicité (0,8 %) (G3/4 : 0,6 %) |
|
|
Affections de la peau et du tissu sous-cutané |
Alopécie |
Rash (4,9 %) (G3/4 : 0,1 %) Prurit (3,9 %) (G3/4 : 0,1 %)d Trouble unguéal Sueurs nocturnes Sécheresse cutanée Erythème Hyperhidrose Erythrodysesthésie palmo-plantaire (1,0 %) (G3/4 : 0,1 %)d |
Angiœdème |
** Syndrome de Stevens-Johnson/nécrolyse épidermique toxiqueb |
|
Affections musculosquelettiques et du tissu conjonctif |
Arthralgie et myalgie (20,4 %) (G3/4 : 1,0 %) Dorsalgie (12,8 %) (G3/4 : 1,5 %) Extrémités douloureuses (10,0 %) (G3/4 : 0,7 %)d |
Douleur osseuse (6,7 %) (G3/4 : 1,2 %) Contractures musculaires (5,3 %) (G3/4 : 0,1 %)d Douleur musculosquelettique Douleur musculosquelettique du thorax Faiblesse musculaire |
|
|
|
Affections du rein et des voies urinaires |
|
Dysurie |
Hématurie Protéinurie Insuffisance rénale |
|
|
Troubles généraux et anomalies au site d’administration |
Fatigue/asthénie (53,2 %) (G3/4 : 7,7 %) Fièvre (21,8 %) (G3/4 : 0,7 %) |
Inflammation muqueuse (6,4 %) (G3/4 : 0,9 %)d Œdème périphérique Douleur Frissons Douleur thoracique Syndrome grippal |
|
|
|
Investigations |
Poids diminué (11,4 %) (G3/4 : 0,4 %)d |
|
|
|
a Inclut les événements de grade 5.
b Provenant des notifications spontanées
c Comprend les termes préférentiels de neuropathie périphérique, neuropathie motrice périphérique, polyneuropathie, paresthésie, neuropathie périphérique sensitive, neuropathie sensitivomotrice périphérique et polyneuropathie démyélinisante
d Aucun événement de grade 4
* Rare
** Fréquence indéterminée
Globalement, les profils de sécurité dans les populations de patients atteints d’un cancer du sein ou d’un sarcome des tissus mous étaient comparables.
Description de certains effets indésirables
Neutropénie
La neutropénie observée était réversible et non cumulative ; le délai moyen pour atteindre le nadir était de 13 jours et le délai moyen avant résolution d’une neutropénie sévère (< 0,5 × 109/L) était de 8 jours.
Une numération des neutrophiles < 0,5 × 109/L ayant persisté plus de 7 jours s’est produite chez 13 % des patientes atteintes d’un cancer du sein traitées par éribuline dans l’étude EMBRACE.
Une neutropénie a été rapportée comme événement indésirable apparu sous traitement (EIAT) chez 151/404 patients de la population atteinte de sarcome (tous grades : 37,4 %) par rapport à 902/1 559 patientes de la population atteinte d’un cancer du sein (tous grades : 57,9 %). Les fréquences d’EIAT et d’anomalies des taux de neutrophiles combinées étaient de 307/404 (76,0 %) et 1 314/1 559 (84,3 %), respectivement. La durée médiane de traitement était de 12,0 semaines chez les patients atteints d’un sarcome et de 15,9 semaines chez les patientes atteintes d’un cancer du sein.
Des cas fatals de neutropénie fébrile, de sepsis neutropénique, de sepsis et de choc septique ont été rapportés. Chez les 1 963 patients atteints d’un cancer du sein ou d’un sarcome des tissus mous ayant reçu l’éribuline à la dose recommandée dans les études cliniques, il a été rapporté un événement de sepsis neutropénique (0,1 %) et de neutropénie fébrile (0,1 %), tous deux d’issue fatale. Il y a eu de plus 3 événements d’issue fatale de sepsis (0,2 %) et un de choc septique (0,1 %).
Une neutropénie sévère peut être corrigée par l’utilisation de G-CSF ou d’un équivalent à l’appréciation du médecin, conformément aux recommandations. Au cours des deux études de phase III menées dans le cancer du sein (études 305 et 301, respectivement), 18 % et 13 % des patientes traitées par éribuline ont reçu du G-CSF. Au cours de l’étude de phase III menée dans les sarcomes (étude 309), 26 % des patients traités par éribuline ont reçu du G-CSF.
La neutropénie a entraîné l’arrêt du traitement chez < 1 % des patients recevant de l’éribuline.
Coagulation intravasculaire disséminée
Des cas de coagulation intravasculaire disséminée ont été rapportés, généralement en association avec une neutropénie et/ou un sepsis.
Neuropathie périphérique
Parmi les 1 559 patientes atteintes d’un cancer du sein, l’effet indésirable le plus fréquent ayant entraîné l’arrêt du traitement par éribuline était la neuropathie périphérique (3,4 %). Le délai médian d’apparition d’une neuropathie périphérique de grade 2 était de 12,6 semaines (après 4 cycles).
Sur les 404 patients atteints d’un sarcome, deux patients ont arrêté le traitement par éribuline en raison d’une neuropathie périphérique. Le délai médian d’apparition d’une neuropathie périphérique de grade 2 était de 18,4 semaines.
L’apparition d’une neuropathie périphérique de grade 3 ou 4 est survenue chez 7,4 % des patientes atteintes d’un cancer du sein et 3,5 % des patients atteints d’un sarcome. Lors des essais cliniques, le risque d’apparition de nouveaux symptômes ou d’aggravation de ces symptômes était similaire chez les patients présentant une neuropathie préexistante et chez ceux qui entraient dans l’étude sans cette affection.
Chez les patientes atteintes d’un cancer du sein présentant une neuropathie périphérique de grade 1 ou 2 préexistante, la fréquence des neuropathies périphériques de grade 3 survenant pendant le traitement était de 14 %.
Hépatotoxicité
Des augmentations des taux d’enzymes hépatiques ont été rapportées après l’instauration du traitement par éribuline chez certains patients qui présentaient des taux normaux ou anormaux avant le traitement. Ces élévations ont semblé survenir en début de traitement par éribuline, pendant le cycle 1 ou 2 chez la majorité de ces patients, et bien qu’elles soient considérées comme étant probablement un phénomène d’adaptation au traitement par éribuline par le foie et non un signe de toxicité hépatique chez la plupart des patients, une hépatotoxicité a également été rapportée.
Populations particulières
Population âgée
Lors des études, sur les 1 559 patientes atteintes d’un cancer du sein traitées à la dose recommandée d’éribuline, 283 (18,2 %) étaient âgées de ≥ 65 ans. Dans la population de 404 patients atteints d’un sarcome, 90 patients (22,3 %) traités par éribuline étaient âgés de ≥ 65 ans. Le profil de sécurité de l’éribuline chez les patients âgés (≥ 65 ans) était comparable à celui observé chez les patients âgés de < 65 ans, à l’exception de l’asthénie/fatigue, dont l’incidence tendait à augmenter avec l’âge. Aucune adaptation de la posologie n’est recommandée dans la population âgée.
Patients présentant une insuffisance hépatique
L’incidence des neutropénies de grade 4 et des neutropénies fébriles était plus élevée chez les patients dont les taux d’ALAT ou ASAT étaient > 3 × LSN. Bien que les données soient limitées, l’incidence des neutropénies de grade 4 et des neutropénies fébriles était également plus élevée chez les patients dont la bilirubine était > 1,5 × LSN (voir également rubriques 4.2 et 5.2).
Population pédiatrique
Trois études en ouvert, les études 113, 213 et 223, ont été menées chez des patients pédiatriques présentant des tumeurs solides et des lymphomes réfractaires ou récidivants, mais excluant les tumeurs du système nerveux central (SNC) (voir rubrique 5.1).
La sécurité de l’éribuline en monothérapie a été évaluée chez 43 patients pédiatriques ayant reçu jusqu’à 1,58 mg/m2 aux Jours 1 et 8 d’un cycle de 21 jours (études 113 et 223). La sécurité de l’éribuline en association avec l’irinotécan a également été évaluée chez 40 patients pédiatriques ayant reçu 1,23 mg/m2 d’éribuline aux Jours 1 et 8 et 20 ou 40 mg/m2 d’irinotécan des Jours 1 à 5 d’un cycle de 21 jours, ou 100 ou 125 mg/m2 aux Jours 1 et 8 d’un cycle de 21 jours (étude 213).
Dans l’étude 113 (phase I), les effets indésirables les plus fréquemment rapportés étaient la diminution du nombre de globules blancs, la diminution du nombre de lymphocytes, l’anémie et la diminution du nombre de neutrophiles.
Dans l’étude 213 (phase I/II), les effets indésirables les plus fréquemment rapportés étaient la neutropénie (phase I), la diarrhée et la diminution du nombre de neutrophiles (phase II).
Dans l’étude 223 (phase II), les effets indésirables les plus fréquemment rapportés étaient la diminution du nombre de neutrophiles, l’anémie et la diminution du nombre de globules blancs.
Le profil de sécurité de l’éribuline en monothérapie ou en association avec le chlorhydrate d’irinotécan dans cette population pédiatrique était cohérent avec le profil de sécurité connu de chaque médicament à l’étude dans la population adulte.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Dans un cas de surdosage, le patient a reçu par erreur 7,6 mg d’éribuline (environ 4 fois la dose prévue) et a présenté par la suite une réaction d’hypersensibilité (grade 3) au Jour 3 et une neutropénie (grade 3) au Jour 7. Les deux réactions se sont résolues après traitement symptomatique.
Il n’existe pas d’antidote connu en cas de surdosage à l’éribuline. En cas de surdosage, une surveillance étroite du patient s’impose. La prise en charge du surdosage doit comprendre des interventions médicales pour traiter les manifestations cliniques.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Autres agents antinéoplasiques, code ATC : L01XX41
Le mésilate d’éribuline est un inhibiteur de la dynamique des microtubules appartenant à la classe des agents antinéoplasiques de type halichondrine. Le mésilate d’éribuline est un analogue de synthèse à structure simplifiée de l’halichondrine B, une substance naturelle isolée de l’éponge marine Halichondria okadai.
L’éribuline inhibe la phase de croissance des microtubules sans altérer la phase de raccourcissement et piège la tubuline dans des agrégats non productifs. L’éribuline exerce ses effets par un mécanisme antimitotique au niveau de la tubuline, ce qui entraîne le blocage de la phase G2/M du cycle cellulaire, une perturbation des fuseaux mitotiques et finalement la mort cellulaire par apoptose après un blocage mitotique prolongé et irréversible.
Efficacité clinique
Cancer du sein
L’efficacité de l’éribuline dans le cancer du sein est confirmée principalement par deux études de phase III comparatives et randomisées.
Les 762 patientes de l’étude pivot de phase III EMBRACE (étude 305) présentaient un cancer du sein avec récidive locale ou métastatique et avaient reçu au préalable au moins deux et au maximum cinq protocoles de chimiothérapie comprenant une anthracycline et un taxane (sauf en cas de contre- indication). La maladie des patientes devait avoir progressé dans les 6 mois ayant suivi leur dernier protocole de chimiothérapie. Le statut HER2 des patientes était : positif pour 16,1 %, négatif pour 74,2 % et indéterminé pour 9,7 %, tandis que 18,9 % des patientes présentaient un cancer triple négatif. Les patientes ont été randomisées selon un rapport de 2/1 pour recevoir soit l’éribuline, soit un traitement choisi par le médecin (TCM), consistant en une chimiothérapie dans 97 % des cas (26 % vinorelbine, 18 % gemcitabine, 18 % capécitabine, 16 % taxane, 9 % anthracycline, 10 % autre chimiothérapie) ou en une hormonothérapie dans 3 % des cas.
L’étude a atteint le critère d’évaluation principal avec un meilleur résultat de survie globale (SG) dans le groupe éribuline que dans le groupe TCM (différence statistiquement significative) à 55 % des événements.
Ce résultat a été confirmé par une analyse de survie globale actualisée, menée à 77 % des événements.
Etude 305 Analyse de survie globale actualisée (population ITT)
0,805 (0,677 ; 0,958) Valeur de P nominale (log-rank) Eribuline Eribuline TCM Eribuline
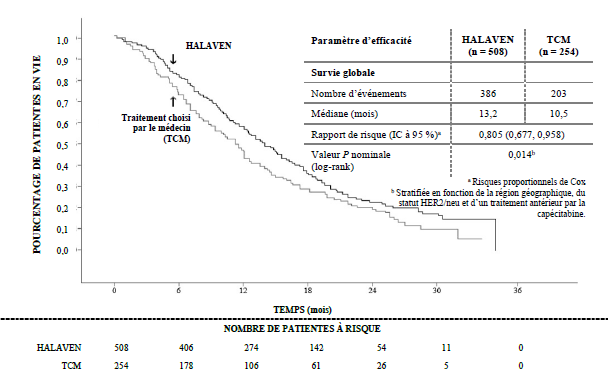
Selon l’évaluation indépendante, la survie sans progression (SSP) médiane était de 3,7 mois dans le bras éribuline versus 2,2 mois dans le bras TCM (RR 0,865, IC à 95 % : 0,714 ; 1,048, p = 0,137). Chez les patientes avec une réponse évaluable, le taux de réponse objective selon les critères RECIST était de 12,2 % (IC à 95 % : 9,4 % ; 15,5 %) selon l’évaluation indépendante pour le bras éribuline versus 4,7 % (IC à 95 % : 2,3 % ; 8,4 %) pour le bras TCM.
L’effet positif sur la SG a été observé dans les groupes de patientes réfractaires et non réfractaires aux taxanes. Dans l’actualisation de la SG, le RR de l’éribuline versus TCM était de 0,90 (IC à 95 % : 0,71 ; 1,14) en faveur de l’éribuline pour les patientes réfractaires aux taxanes et de 0,73 (IC à 95 % : 0,56 ; 0,96) pour les patientes non réfractaires aux taxanes.
L’effet positif sur la SG a été observé à la fois chez les patientes n’ayant jamais reçu de capécitabine et chez celles qui avaient déjà été traitées. L’analyse actualisée de la SG a révélé un bénéfice sur la survie pour le groupe éribuline par rapport au groupe TCM aussi bien chez les patientes ayant été traitées au préalable par la capécitabine avec un RR de 0,787 (IC à 95 % : 0,645 ; 0,961) que pour les patientes n’ayant jamais reçu de capécitabine avec un RR correspondant de 0,865 (IC à 95 % : 0,606 ; 1,233).
La seconde étude de phase III en lignes plus précoces de traitement du cancer du sein métastatique, l’étude 301, était une étude randomisée, en ouvert, menée chez des patientes (n = 1 102) présentant un cancer du sein localement avancé ou métastatique afin d’évaluer l’efficacité de l’éribuline en monothérapie par rapport à la capécitabine en monothérapie en termes de SG et de SSP, les co-critères principaux. Les patientes avaient reçu préalablement jusqu’à trois protocoles de chimiothérapie comportant une anthracycline et un taxane, et deux au maximum en cas de maladie avancée, les pourcentages de patientes ayant reçu 0, 1 ou 2 chimiothérapies antérieures pour un cancer du sein métastatique étant de 20,0 %, 52,0 % et 27,2 %, respectivement. Le statut HER2 des patientes était : positif pour 15,3 %, négatif pour 68,5 % et indéterminé pour 16,2 %, tandis que 25,8 % des patientes présentaient un cancer triple négatif.
Etude 301 Survie globale (population ITT)
a Risques proportionnels de Cox b Stratifiée en fonction de la région géographique et du statut HER2/neu. 0,879 (0,770 ; 1,003) Eribuline Capécitabine Valeur de P (log-rank) Rapport de risque (IC à 95 %)a Médiane (mois) Nombre d’événements Capécitabine Eribuline Paramètre d’efficacité Eribuline Capécitabine NOMBRE DE PATIENTES A RISQUE TEMPS (mois)
![]()
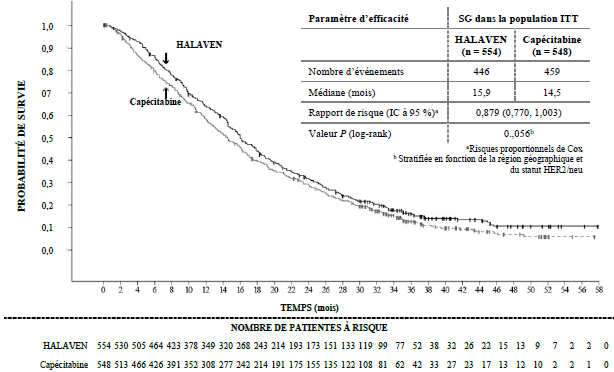
Selon l’évaluation indépendante, la survie sans progression était comparable entre les groupes traités par l’éribuline et la capécitabine, avec des médianes de 4,1 mois versus 4,2 mois respectivement (RR 1,08 ; [IC à 95 % : 0,932 ; 1,250]). Selon l’évaluation indépendante, le taux de réponse objective était également comparable entre l’éribuline et la capécitabine : 11,0 % (IC à 95 % : 8,5 ; 13,9) dans le groupe éribuline et 11,5 % (IC à 95 % : 8,9 ; 14,5) dans le groupe capécitabine.
La survie globale chez les patientes ayant le statut HER2 négatif ou HER2 positif dans les groupes éribuline et témoin des études 305 et 301 est présentée ci-dessous :
|
Paramètre d’efficacité |
Etude 305 - Survie globale actualisée - Population ITT |
|||
|
HER2 négatif |
HER2 positif |
|||
|
|
Eribuline (n = 373) |
TCM (n = 192) |
Eribuline (n = 83) |
TCM (n = 40) |
|
Nombre d’événements |
285 |
151 |
66 |
37 |
|
Médiane (mois) |
13,4 |
10,5 |
11,8 |
8,9 |
|
Rapport de risque (IC à 95 %) |
0,849 (0,695 ; 1,036) |
0,594 (0,389 ; 0,907) |
||
|
Valeur de p (log-rank) |
0,106 |
0,015 |
||
|
Paramètre d’efficacité |
Etude 301 - Survie globale - Population ITT |
|||
|
HER2 négatif |
HER2 positif |
|||
|
|
Eribuline (n = 375) |
Capécitabine (n = 380) |
Eribuline (n = 86) |
Capécitabine (n = 83) |
|
Nombre d’événements |
296 |
316 |
73 |
73 |
|
Médiane (mois) |
15,9 |
13,5 |
14,3 |
17,1 |
|
Rapport de risque (IC à 95 %) |
0,838 (0,715 ; 0,983) |
0,965 (0,688 ; 1,355) |
||
|
Valeur de p (log-rank) |
0,030 |
0,837 |
||
Remarque : aucun traitement anti-HER2 concomitant n’a été administré dans l’étude 305 et l’étude 301.
Liposarcome
L’efficacité de l’éribuline dans le traitement du liposarcome est confirmée par l’étude pivot de phase III menée dans les sarcomes (étude 309). Les patients de cette étude (n = 452) présentaient un sarcome des tissus mous localement récidivant non résécable et/ou métastatique de l’un des deux sous-types, léiomyosarcome ou liposarcome. Les patients avaient reçu antérieurement au moins deux protocoles de chimiothérapie dont l’un devait avoir comporté une anthracycline (sauf en cas de contre-indication).
La maladie des patients devait avoir progressé dans les 6 mois suivant leur dernier protocole de chimiothérapie. Les patients ont été randomisés selon un rapport de 1/1 pour recevoir l’éribuline 1,23 mg/m2 les Jours 1 et 8 d’un cycle de 21 jours ou la dacarbazine 850 mg/m2, 1 000 mg/m2 ou 1 200 mg/m2 (dose déterminée par l’investigateur avant la randomisation) tous les 21 jours.
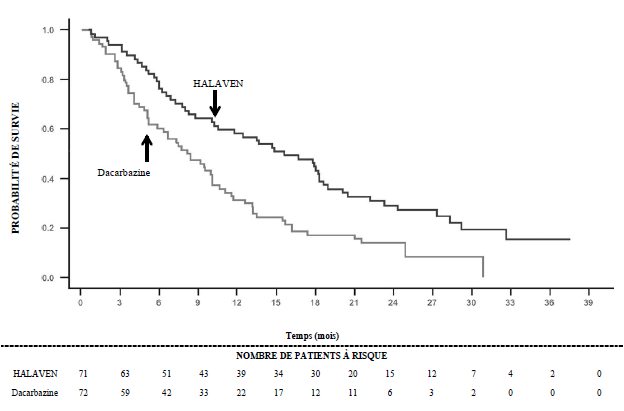
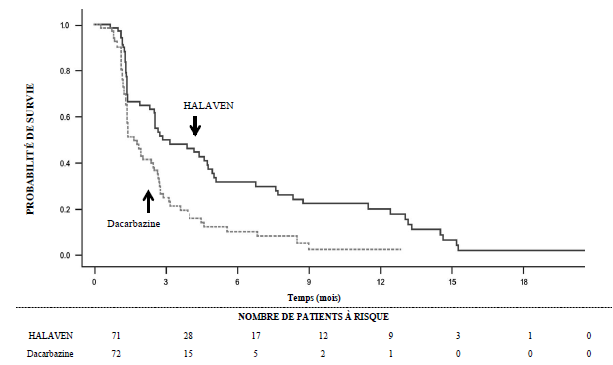
Dans l’étude 309, il a été observé une amélioration statistiquement significative de la SG chez les patients randomisés dans le bras éribuline par rapport au bras témoin. Celle-ci s’est traduite par un allongement de 2 mois de la SG médiane (13,5 mois chez les patients traités par éribuline versus 11,5 mois chez les patients traités par dacarbazine). Il n’a pas été observé de différence significative en termes de survie sans progression ou de taux de réponse globale entre les groupes de traitement dans la population totale.
Selon les analyses en sous-groupes prédéfinies de la SG et de la SSP, les effets du traitement par éribuline étaient limités aux patients présentant un liposarcome (dédifférencié : 45 %, myxoïde/à cellules rondes : 37 % et pléomorphe : 18 % dans l’étude 309). Il n’a pas été observé de différence en termes d’efficacité entre l’éribuline et la dacarbazine chez les patients présentant un léiomyosarcome avancé ou métastatique.
|
|
Etude 309 Sous-groupe liposarcome |
Etude 309 Sous-groupe léiomyosarcome |
Etude 309 Population ITT |
|||
|
Eribuline (n = 71) |
Dacarbazine (n = 72) |
Eribuline (n = 157) |
Dacarbazine (n = 152) |
Eribuline (n = 228) |
Dacarbazine (n = 224) |
|
|
Survie globale |
||||||
|
Nombre d’événements |
52 |
63 |
124 |
118 |
176 |
181 |
|
Médiane (mois) |
15,6 |
8,4 |
12,7 |
13,0 |
13,5 |
11,5 |
|
Rapport de risque (IC à 95 %) |
0,511 (0,346 ; 0,753) |
0,927 (0,714 ; 1,203) |
0,768 (0,618 ; 0,954) |
|||
|
Valeur de p nominale |
0,0006 |
0,5730 |
0,0169 |
|||
|
Survie sans progression |
||||||
|
Nombre d’événements |
57 |
59 |
140 |
129 |
197 |
188 |
|
Médiane (mois) |
2,9 |
1,7 |
2,2 |
2,6 |
2,6 |
2,6 |
|
Rapport de risque (IC à 95 %) |
0,521 (0,346 ; 0,784) |
1,072 (0,835 ; 1,375) |
0,877 (0,710 ; 1,085) |
|||
|
Valeur de p nominale |
0,0015 |
0,5848 |
0,2287 |
|||
Etude 309 Survie globale dans le sous-groupe liposarcome
Dacarbazine Eribuline Dacarbazine Temps (mois) NOMBRE DE PATIENTS A RISQUE Eribuline
![]()
Etude 309 Survie sans progression dans le sous-groupe liposarcome
Eribuline Dacarbazine NOMBRE DE PATIENTS A RISQUE Temps (mois) Dacarbazine Eribuline
![]()
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec l’éribuline dans tous les sous-groupes de la population pédiatrique dans l’indication de cancer du sein (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
L’efficacité de l’éribuline a été évaluée, mais n’a pas été établie, dans trois études en ouvert :
L’étude 113 était une étude de phase I, en ouvert, multicentrique, de détermination de dose, qui visait à évaluer l’éribuline chez des patients pédiatriques présentant des tumeurs solides et des lymphomes réfractaires ou récidivants, mais excluant les tumeurs du SNC. Au total, 22 patients pédiatriques (âgés de 3 à 17 ans) ont été recrutés et traités. Les patients ont reçu de l’éribuline par voie intraveineuse aux Jours 1 et 8 d’un cycle de 21 jours à trois niveaux de dose (0,97, 1,23 et 1,58 mg/m2). La dose maximale tolérée (DMT)/dose recommandée pour la phase II (DRP2) d’éribuline a été établie à 1,23 mg/m2 aux Jours 1 et 8 d’un cycle de 21 jours.
L’étude 223 était une étude de phase II, en ouvert, multicentrique, qui visait à évaluer le profil de sécurité et l’activité préliminaire de l’éribuline chez des patients pédiatriques présentant un rhabdomyosarcome (RMS) réfractaire ou récidivant, un sarcome des tissus mous non-rhabdomyosarcome (STMNR) ou un sarcome d’Ewing (SEW). Vingt-et-un patients pédiatriques (âgés de 2 à 17 ans) ont été recrutés et traités avec de l’éribuline à une dose de 1,23 mg/m2 par voie intraveineuse aux Jours 1 et 8 d’un cycle de 21 jours (soit la DRP2 de l’étude 113). Aucun patient n’a présenté une réponse partielle (RP) ou une réponse complète (RC) confirmée.
L’étude 213 était une étude en ouvert, multicentrique de phase I/II qui visait à évaluer la sécurité et l’efficacité de l’éribuline en association avec le chlorhydrate d’irinotécan chez des patients pédiatriques présentant des tumeurs solides et des lymphomes réfractaires/en rechute, mais excluant les tumeurs du SNC (phase I). Cette étude visait également à évaluer l’efficacité du traitement en association chez des patients pédiatriques présentant un RMS, un STMNR et un SEW (phase II). Au total, 40 patients pédiatriques ont été recrutés et traités dans cette étude. Au cours de la phase I, 13 patients pédiatriques (âgés de 4 à 17 ans) ont été recrutés et traités ; la DRP2 a été établie à 1,23 mg/m2 aux Jours 1 et 8 avec 40 mg/m2 de chlorhydrate d’irinotécan des Jours 1 à 5 d’un cycle de 21 jours. Au cours de la phase II, 27 patients pédiatriques (âgés de 4 à 17 ans) ont été recrutés et traités à la DRP2. Trois patients ont présenté une RP confirmée (1 patient dans chaque cohorte histologique RMS, STMNR et SEW). Le taux de réponse objective (TRO) était de 11,1 %.
Aucun nouveau signal de sécurité n’a été observé dans les trois études pédiatriques (voir rubrique 4.8). Cependant, en raison du faible nombre de patients, aucune conclusion définitive n’a pu être établie.
5.2. Propriétés pharmacocinétiques
La pharmacocinétique de l’éribuline est caractérisée par une phase de distribution rapide suivie d’une phase d’élimination prolongée, avec une demi-vie terminale moyenne d’environ 40 h. Elle a un grand volume de distribution (moyennes comprises entre 43 et 114 L/m2).
L’éribuline est peu liée aux protéines plasmatiques. La liaison de l’éribuline (100– 1 000 ng/mL) aux protéines plasmatiques était comprise entre 49 % et 65 % dans le plasma humain.
Biotransformation
L’éribuline sous forme inchangée constituait la principale espèce circulante dans le plasma après administration de 14C-éribuline à des patients. Les concentrations des métabolites représentaient < 0,6 % de la molécule mère, confirmant qu’il n’existe pas de métabolites humains majeurs de l’éribuline.
Elimination
La clairance de l’éribuline est faible (moyennes comprises entre 1,16 et 2,42 L/h/m2). Aucune accumulation significative d’éribuline n’a été observée avec une administration hebdomadaire. Pour des doses d’éribuline comprises entre 0,22 et 3,53 mg/m2, les propriétés pharmacocinétiques ne sont ni dose-dépendantes ni temps-dépendantes.
L’éribuline est principalement éliminée par excrétion biliaire. La protéine de transport intervenant dans l’excrétion n’est pas connue actuellement. Les études précliniques in vitro indiquent que l’éribuline est transportée par la P-gp. Cependant, aux concentrations cliniquement pertinentes, l’éribuline n’est pas un inhibiteur de la P-gp in vitro.
De plus, l’administration concomitante de kétoconazole, un inhibiteur de la P-gp, n’a pas eu d’effet sur l’exposition à l’éribuline (ASC et Cmax) in vivo. Les études in vitro ont également indiqué que l’éribuline n’était pas un substrat d’OCT1.
Après administration de 14C-éribuline à des patients, environ 82 % de la dose étaient éliminés dans les fèces et 9 % dans l’urine, ce qui indique que la clairance rénale ne constitue pas une voie importante d’élimination de l’éribuline.
L’éribuline sous forme inchangée représentait l’essentiel de la radioactivité totale présente dans les fèces et dans l’urine.
Insuffisance hépatique
Une étude a évalué la pharmacocinétique de l’éribuline chez les patients présentant une insuffisance hépatique légère (Child-Pugh A ; n = 7) et modérée (Child-Pugh B ; n = 4) en raison de métastases hépatiques. Comparée à celle observée chez les patients dont la fonction hépatique est normale (n = 6), l’exposition à l’éribuline a augmenté de 1,8 fois et de 3 fois, respectivement, chez les patients présentant une insuffisance hépatique légère et modérée. L’administration d’éribuline à une dose de 0,97 mg/m2 à des patients présentant une insuffisance hépatique légère et de 0,62 mg/m2 à des patients présentant une insuffisance hépatique modérée a donné lieu à une exposition à l’éribuline quelque peu accrue par rapport à celle observée à la dose de 1,23 mg/m2 administrée à des patients dont la fonction hépatique est normale. L’éribuline n’a pas été étudiée chez les patients présentant une insuffisance hépatique sévère (Child-Pugh C) Aucune étude n’a été menée chez les patients présentant une insuffisance hépatique due à une cirrhose. Voir rubrique 4.2 pour les recommandations posologiques.
Insuffisance rénale
Une augmentation de l’exposition à l’éribuline a été observée chez certains patients présentant une altération de la fonction rénale modérée ou sévère, avec une forte variabilité interindividuelle. La pharmacocinétique de l’éribuline a été évaluée au cours d’une étude de phase I chez des patients présentant une fonction rénale normale (clairance de la créatinine [ClCr] : ≥ 80 mL/min ; n = 6), modérée (30– 50 mL/min ; n = 7) ou sévère (15– < 30 mL/min ; n = 6). La clairance de la créatinine était estimée selon la formule de Cockcroft-Gault. Une augmentation de 1,5 fois (IC à 90 % : 0,9 ; 2,5) de l’ASC(0-inf) normalisée à la dose a été observée chez les patients présentant une insuffisance rénale modérée ou sévère. Voir rubrique 4.2 pour les recommandations thérapeutiques.
Population pédiatrique
Les concentrations plasmatiques d’éribuline ont été recueillies auprès de 83 patients pédiatriques (âgés de 2 à 17 ans) présentant des tumeurs solides et des lymphomes réfractaires/en rechute et récidivants et ayant reçu de l’éribuline dans les études 113, 213 et 223. La pharmacocinétique de l’éribuline chez les patients pédiatriques était comparable à celle chez les patients adultes présentant un STM et les patients présentant d’autres types de tumeurs. L’exposition à l’éribuline chez les patients pédiatriques était similaire à l’exposition chez les patients adultes. L’administration concomitante d’irinotécan n’a pas eu d’effet sur la pharmacocinétique de l’éribuline chez les patients pédiatriques présentant des tumeurs solides réfractaires/en rechute et récidivantes.
5.3. Données de sécurité préclinique
Aucune étude du potentiel cancérogène n’a été menée avec l’éribuline.
Aucune étude de fertilité n’a été menée avec l’éribuline, mais d’après les résultats précliniques obtenus lors d’études à doses répétées où une toxicité pour les testicules a été observée à la fois chez le rat (hypocellularité de l'épithélium séminifère avec hypospermie/aspermie) et chez le chien, le traitement par éribuline pourrait altérer la fertilité masculine. Une étude sur le développement embryo-fœtal menée chez le rat a confirmé la toxicité sur le développement et le potentiel tératogène de l’éribuline. Des rates gravides ont reçu du mésilate d’éribuline, équivalant à 0,009, 0,027, 0,088 et 0,133 mg/kg d’éribuline aux Jours 8, 10 et 12 de la gestation. Une augmentation du nombre de résorptions et une diminution du poids fœtal, proportionnelles à la dose, à des doses ≥ 0,088 mg/kg et une augmentation de l’incidence des malformations (absence de mâchoire inférieure, de langue, d’estomac et de rate) à la dose de 0,133 mg/kg ont été observées.
Flacons avant ouverture
2 ans
Durée de conservation en cours d’utilisation
La stabilité chimique et physique en cours d’utilisation de la solution non diluée dans une seringue a été démontrée pendant 24 heures à une température comprise entre 20 °C et 25 °C et pendant 96 heures à une température comprise entre 2 °C et 8 °C.
La stabilité physico-chimique en cours d’utilisation de la solution diluée (0,018 mg/mL à 0,18 mg/mL d’éribuline dans du chlorure de sodium [0,9 %]) a été démontrée pendant 48 heures à une température comprise entre 2 °C et 8 °C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d’utilisation non immédiate, les conditions et durées de conservation en cours d’utilisation relèvent de la responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C, sauf en cas de dilution réalisée en conditions d’asepsie dûment contrôlées et validées.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après première ouverture ou dilution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon en verre incolore (de type I), muni d’un bouchon en caoutchouc halobutyle et d’un opercule en aluminium avec une capsule « flip-off » en plastique, contenant 2 mL de solution.
Les conditionnements sont des boîtes de 1 ou de 6 flacons. Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
L’éribuline est un médicament anticancéreux cytotoxique et, comme les autres composés toxiques, elle doit être manipulée avec prudence. Il est recommandé de porter des gants, des lunettes de laboratoire et un vêtement protecteur. En cas de contact de la solution avec la peau, laver immédiatement et abondamment la peau à l’eau savonneuse. En cas de contact avec les muqueuses, celles-ci doivent être rincées abondamment à l’eau. L’éribuline ne doit être préparée et administrée que par un personnel convenablement formé à la manipulation des cytotoxiques. Les femmes enceintes ne doivent pas manipuler l’éribuline.
L’éribuline peut être diluée jusqu’à 100 mL avec une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) en respectant les règles d’asepsie. Après l’injection, il est recommandé de rincer la ligne de perfusion intraveineuse avec une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) afin de garantir l’administration de la dose complète. L’éribuline ne doit pas être mélangée avec d’autres médicaments et ne doit pas être diluée dans une solution pour perfusion de glucose à 5 %.
En cas d’utilisation d'un dispositif de prélèvement (spike) pour administrer le produit, se référer aux instructions fournies par le fabricant du dispositif. Les flacons d’ERIBULINE ZENTIVA sont munis d’un bouchon de 20 mm. Le dispositif sélectionné doit être compatible avec les bouchons pour flacons de petite taille.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
35 RUE DU VAL DE MARNE
75013 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 892 6 6 : 1 flacon en verre de 2 mL
· 34009 302 892 7 3 : 6 flacons en verre de 2 mL
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en cancérologie, hématologie ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 28/03/2025
ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable
Eribuline
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre infirmier/ère.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu’est-ce qu’ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d’utiliser ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable ?
3. Comment utiliser ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Autres agents antinéoplasiques, code ATC : L01XX41
ERIBULINE ZENTIVA contient la substance active éribuline et est un médicament anticancéreux qui agit en arrêtant la croissance et la dissémination des cellules cancéreuses.
Il est utilisé chez les adultes pour le cancer du sein localement avancé ou métastatique (le cancer du sein s’est disséminé en dehors de la tumeur d’origine) quand au moins un autre traitement a été essayé et qu’il a perdu son efficacité.
Il est également utilisé chez les adultes pour le liposarcome (un type de cancer qui se développe à partir du tissu adipeux) avancé ou métastatique quand un traitement antérieur a été essayé et qu’il a perdu son efficacité.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable ?
N’utilisez jamais ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable :
· si vous êtes allergique au mésilate d’éribuline ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6) ;
· si vous allaitez.
Avertissements et précautions
Adressez-vous à votre médecin ou infirmier/ère avant d’utiliser ERIBULINE ZENTIVA :
· si vous avez des problèmes au foie ;
· si vous avez de la fièvre ou une infection ;
· si vous présentez des engourdissements, des sensations de fourmillements ou de picotements, une sensibilité au toucher ou une faiblesse musculaire ;
· si vous avez des problèmes cardiaques.
Si vous êtes dans l’un de ces cas, parlez-en à votre médecin qui pourra décider d’arrêter le traitement ou de réduire la dose.
Enfants et adolescents
N’administrez pas ce médicament aux enfants âgés de 0 à 18 ans, car il ne fonctionnera pas.
Autres médicaments et ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable
Informez votre médecin si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Grossesse, allaitement et fertilité
ERIBULINE ZENTIVA peut provoquer de graves malformations congénitales et ne doit pas être utilisé si vous êtes enceinte à moins que cela ne soit considéré comme absolument nécessaire après une évaluation minutieuse de tous les risques pour vous et le bébé. Il peut également entraîner des problèmes ultérieurs de fertilité, irréversibles, chez les hommes qui le prennent et ils doivent en discuter avec leur médecin avant de commencer le traitement. Les femmes en âge de procréer doivent utiliser une méthode de contraception très efficace pendant le traitement par ERIBULINE ZENTIVA et pendant les 7 mois après l’arrêt du traitement par ERIBULINE ZENTIVA.
ERIBULINE ZENTIVA ne doit pas être utilisé pendant l’allaitement à cause des risques possibles pour l’enfant.
Les hommes dont la partenaire est en âge de procréer ne doivent pas avoir d'enfant pendant le traitement par ERIBULINE ZENTIVA. Les hommes doivent utiliser une méthode de contraception efficace pendant le traitement par ERIBULINE ZENTIVA et pendant les 4 mois après l’arrêt du traitement par ERIBULINE ZENTIVA.
Conduite de véhicules et utilisation de machines
ERIBULINE ZENTIVA peut provoquer des effets indésirables tels que fatigue (très fréquent) et sensations vertigineuses (fréquent). Ne conduisez pas de véhicules et n’utilisez pas de machines si vous vous sentez fatigué(e) ou si vous présentez des sensations vertigineuses.
ERIBULINE ZENTIVA contient de l’éthanol (alcool)
Ce médicament contient 80 mg d'alcool (éthanol) par flacon. La quantité dans un flacon de ce médicament équivaut à moins de 2 mL de bière ou 1 mL de vin.
La faible quantité d'alcool contenue dans ce médicament n'est pas susceptible d'entraîner d'effet notable.
3. COMMENT UTILISER ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable ?
A quelle fréquence recevrez-vous ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable ?
ERIBULINE ZENTIVA est généralement administré aux Jours 1 et 8 de chaque cycle de 21 jours. Votre médecin déterminera le nombre de cycles de traitement que vous devez recevoir. En fonction des résultats de vos analyses de sang, le médecin pourra décider de différer l’administration du médicament jusqu’à ce que les analyses de sang soient à nouveau normales. Le médecin pourra alors ensuite décider de réduire la dose que vous recevez.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Si vous présentez l’un des symptômes graves ci-dessous, vous devez arrêter de recevoir ERIBULINE ZENTIVA et consulter immédiatement un médecin :
· Fièvre, avec battements de cœur rapides, respiration rapide et superficielle, peau froide, pâle, moite ou marbrée et/ou confusion. Il peut s’agir de signes d’une affection appelée sepsis, une réaction sévère et grave à une infection. Le sepsis est peu fréquent (pouvant affecter jusqu’à 1 personne sur 100) et peut engager le pronostic vital, voire entraîner le décès.
· Difficultés pour respirer ou gonflement du visage, de la bouche, de la langue ou de la gorge. Il peut s’agir de signes d’une réaction allergique peu fréquente (pouvant affecter jusqu’à 1 personne sur 100).
· Eruptions cutanées graves avec formation de bulles au niveau de la peau, de la bouche, des yeux et des organes génitaux. Il peut s’agir de signes d’une affection appelée syndrome de Stevens-Johnson/nécrolyse épidermique toxique. La fréquence de cette affection est indéterminée, mais elle peut engager le pronostic vital.
Autres effets indésirables :
Effets indésirables très fréquents (pouvant affecter plus de 1 personne sur 10) :
· Diminution du nombre de globules blancs ou de globules rouges
· Fatigue ou faiblesse
· Nausées, vomissements, constipation, diarrhée
· Sensations d’engourdissements, de fourmillements ou de picotements
· Fièvre
· Perte d’appétit, perte de poids
· Difficultés pour respirer, toux
· Douleurs dans les articulations, les muscles et le dos
· Maux de tête
· Chute de cheveux
Effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10) :
· Diminution du nombre de plaquettes (ce qui peut entraîner l’apparition de bleus ou allonger le temps nécessaire pour arrêter les saignements)
· Infection avec fièvre, pneumonie, frissons
· Battements du cœur rapides, bouffées de chaleur
· Vertige, sensations vertigineuses
· Larmoiement excessif, conjonctivite (rougeur et douleur de la surface de l’œil), saignements de nez
· Déshydratation, bouche sèche, boutons de fièvre, muguet, indigestion, brûlures d’estomac, douleurs abdominales ou ballonnements
· Gonflement des tissus mous, douleurs (notamment dans la poitrine, au dos et aux os), spasme ou faiblesse musculaire
· Infections buccales, respiratoires ou urinaires, douleur en urinant
· Angine, nez irrité ou qui coule, symptômes pseudo-grippaux, maux de gorge
· Anomalies des tests de la fonction hépatique, anomalie du taux de sucre, de bilirubine, de phosphate, de potassium, de magnésium ou de calcium dans le sang
· Difficultés d’endormissement, dépression, modification du goût
· Eruption cutanée, démangeaisons, problèmes aux ongles, peau sèche ou rouge
· Transpiration excessive (y compris sueurs nocturnes)
· Bourdonnements dans les oreilles
· Caillots sanguins dans les poumons
· Zona
· Gonflement de la peau et engourdissement des mains et des pieds
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) :
· Caillots sanguins
· Anomalies des tests de la fonction hépatique (hépatotoxicité)
· Insuffisance rénale, sang ou protéines dans les urines
· Inflammation étendue des poumons pouvant entraîner la formation de tissu cicatriciel
· Inflammation du pancréas
· Ulcères dans la bouche
Effets indésirables rares (pouvant affecter jusqu’à 1 personne sur 1 000) :
· Trouble grave de la coagulation sanguine entraînant la formation de caillots sanguins disséminés et des hémorragies internes
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et le flacon après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Durée de conservation en cours d’utilisation
La stabilité physico-chimique en cours d’utilisation de la solution non diluée dans une seringue a été démontrée pendant 24 heures à une température comprise entre 20 °C et 25 °C et pendant 96 heures à une température comprise entre 2 °C et 8 °C.
La stabilité physico-chimique en cours d’utilisation de la solution diluée (0,018 mg/mL à 0,18 mg/mL d’éribuline dans du chlorure de sodium [0,9 %]) a été démontrée pendant 48 heures à une température comprise entre 2 °C et 8 °C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d’utilisation non immédiate, les conditions et durées de conservation en cours d’utilisation relèvent de la responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C, sauf en cas de dilution réalisée en conditions d’asepsie dûment contrôlées et validées.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable
· La substance active est :
Eribuline........................................................................................................................ 0,44 mg
Sous forme de mésilate d’éribuline
Pour un mL.
Chaque flacon de 2 mL contient du mésilate d’éribuline, équivalant à 0,88 mg d’éribuline.
· Les autres composants sont l’éthanol anhydre et l’eau pour préparations injectables, avec de l’acide chlorhydrique (ajustement du pH) et de l’hydroxyde de sodium (ajustement du pH).
Qu’est-ce que ERIBULINE ZENTIVA 0,44 mg/mL, solution injectable et contenu de l’emballage extérieur
ERIBULINE ZENTIVA est une solution injectable aqueuse, limpide et incolore, d’un pH compris entre 6,0 et 9,0, présentée dans des flacons en verre contenant 2 mL de solution. Chaque boîte contient 1 ou 6 flacons.
Titulaire de l’autorisation de mise sur le marché
35 RUE DU VAL DE MARNE
75013 PARIS
Exploitant de l’autorisation de mise sur le marché
ZENTIVA FRANCE
35 RUE DU VAL DE MARNE
75013 PARIS
KAISER-WHILHELM-STR. 89
20355 HAMBURG
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).