Dernière mise à jour le 28/04/2026
GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie
Indications thérapeutiques
GANIRELIX THERAMEX contient la substance active ganirelix et appartient au groupe des médicaments appelés « antagonistes de l’hormone de libération des gonadotrophines » bloquant les effets de l’hormone naturelle de libération des gonadotrophines (GnRH). La GnRH régule la libération des gonadotrophines (hormone lutéinisante (LH) et hormone folliculo-stimulante (FSH)). Les gonadotrophines jouent un rôle important dans la fertilité humaine et la reproduction. Chez les femmes, la FSH est nécessaire pour la croissance et le développement des follicules dans les ovaires. Les follicules sont de petits sacs ronds qui contiennent les ovocytes. La LH est nécessaire pour libérer les ovocytes matures hors des follicules et des ovaires (c’est-à-dire l’ovulation). GANIRELIX THERAMEX inhibe l'action de la GnRH, résultant en une suppression de la libération de LH en particulier.
Utilisation de GANIRELIX THERAMEX
Chez les femmes ayant recours à des techniques médicales d’assistance à la procréation, telles que la fécondation in vitro (FIV) et d’autres méthodes, l’ovulation peut parfois avoir lieu trop tôt et entraîner une diminution significative des chances de devenir enceinte. GANIRELIX THERAMEX est utilisé pour prévenir le pic prématuré de LH qui est susceptible d’entraîner une libération prématurée des ovocytes.
Dans les études cliniques, le ganirelix a été utilisé en association avec l’hormone folliculo-stimulante recombinante (FSH) ou la corifollitropine alfa, un stimulant folliculaire à longue durée d’action.
Présentations
> 1 seringue préremplie en verre de 0,5 mL munie d'une aiguille d'injection
Code CIP : 34009 302 381 5 8
Déclaration de commercialisation : 22/04/2022
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 22,89 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 23,91 €
- Taux de remboursement :100 %
> 5 seringues préremplies en verre de 0,5 mL munies de 5 aiguilles d'injection
Code CIP : 34009 302 381 6 5
Déclaration de commercialisation : 22/04/2022
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 113,12 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 114,14 €
- Taux de remboursement :100 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : THERAMEX IRELAND LIMITED
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription réservée aux spécialistes et services ENDOCRINOLOGIE
- prescription réservée aux spécialistes et services GYNECOLOGIE
- prescription réservée aux spécialistes et services MALADIES METABOLIQUES
- prescription réservée aux spécialistes et services OBSTETRIQUE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 242 848 8
ANSM - Mis à jour le : 15/09/2025
GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
dans 0,5 mL de solution aqueuse
Pour une seringue pré-remplie
Le principe actif, le ganirelix (sous forme d’acétate) (DCI), est un décapeptide synthétique doté d’une haute activité antagoniste de l’hormone naturelle de libération des gonadotrophines (GnRH). Les acides aminés en position 1, 2, 3, 6, 8 et 10 du décapeptide naturel GnRH ont été substitués, donnant le [N-Ac-D-Nal(2)1, D-pClPhe2, D-Pal(3)3, D-hArg(Et2)6, L-hArg(Et2)8, D-Ala10]-GnRH d’un poids moléculaire de 1570,3.
Excipient à effet notoire : Sodium. Chaque seringue pré-remplie contient moins de 1 mmol de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable en seringue pré-remplie.
Solution aqueuse, limpide et incolore avec un pH compris entre 4,5 et 5,5.
4.1. Indications thérapeutiques
GANIRELIX THERAMEX est indiqué dans la prévention des pics prématurés d’hormone lutéinisante (LH) chez les femmes en cours d’hyperstimulation ovarienne contrôlée (HOC) dans le cadre des techniques d’assistance médicale à la procréation (AMP).
Dans les études cliniques, le ganirelix a été utilisé en association avec une hormone folliculo-stimulante humaine recombinante (FSH) ou la corifollitropine alfa, stimulant folliculaire à action prolongée.
4.2. Posologie et mode d'administration
Posologie
Le ganirelix est administré pour prévenir les pics prématurés de LH chez les femmes en cours d’HOC. L’hyperstimulation ovarienne contrôlée par la FSH ou la corifollitropine alfa peut commencer au 2ème ou au 3ème jour des règles. Le ganirelix (0,25 mg) doit être injecté par voie sous-cutanée une fois par jour, en commençant le 5ème ou le 6ème jour de l’administration de FSH ou le 5ème ou le 6ème jour suivant l’administration de la corifollitropine alfa. Le jour d’initiation du traitement par ganirelix est déterminé en fonction de la réponse ovarienne, c’est-à-dire du nombre et de la taille des follicules en croissance et/ou du taux d’estradiol circulant. Le début du traitement par ganirelix peut être retardé en l’absence de croissance folliculaire, bien que l’expérience clinique soit basée sur un début de traitement par ganirelix au 5ème ou au 6ème jour de la stimulation.
Le ganirelix et la FSH doivent être administrés approximativement au même moment. Cependant, les préparations ne doivent pas être mélangées et des sites d’injection différents doivent être utilisés.
Les ajustements de dose de FSH doivent être basés sur le nombre et la taille des follicules en voie de maturation, plutôt que sur le taux d’estradiol circulant (voir rubrique 5.1).
Le traitement quotidien par ganirelix doit être poursuivi jusqu’au jour d’obtention d’un nombre suffisant de follicules de taille adéquate. La maturation finale des follicules peut être induite par l’administration de Gonadotrophine Chorionique Humaine (hCG).
Moment de la dernière injection
En raison de la demi-vie du ganirelix, le délai entre deux injections de ganirelix, ainsi que celui entre la dernière injection de ganirelix et l’injection d’hCG ne doivent pas dépasser 30 heures, dans le cas contraire, un pic prématuré de LH risque de survenir. En conséquence, lorsque le ganirelix est injecté au cours de la matinée, le traitement par ganirelix doit être poursuivi pendant toute la période du traitement par la gonadotrophine, y compris le jour de déclenchement de l’ovulation. Lorsque le ganirelix est injecté au cours de l’après-midi, la dernière injection de ganirelix devra être faite dans l’après-midi, la veille du jour de déclenchement de l’ovulation.
Le ganirelix s’est avéré sûr et efficace chez les femmes ayant eu plusieurs cycles de traitement.
Le besoin d’un support de la phase lutéale pendant les cycles sous ganirelix n’a pas été étudié. Dans les études cliniques, le support de la phase lutéale a été fait selon les pratiques habituelles du centre d’étude ou selon le protocole clinique.
Populations particulières
· Insuffisance rénale
Les sujets présentant une insuffisance rénale n’étant pas inclus dans les études cliniques, il n’y a pas de données sur l’utilisation du ganirelix chez ces sujets. En conséquence, l’utilisation du ganirelix est contre indiquée chez les patientes présentant une insuffisance rénale modérée ou sévère (voir rubrique 4.3).
· Insuffisance hépatique
Les sujets présentant une insuffisance hépatique n’étant pas inclus dans les études cliniques, il n’y a pas de données sur l’utilisation du ganirelix chez ces sujets. En conséquence, l’utilisation du ganirelix est contre indiquée chez les patientes présentant une insuffisance hépatique modérée ou sévère (voir rubrique 4.3).
Population pédiatrique
Il n’y a pas d’utilisation justifiée du ganirelix dans la population pédiatrique.
Mode d’administration
Le ganirelix doit être administré par voie sous-cutanée de préférence dans la cuisse. Pour éviter une lipodystrophie, le point d’injection doit être changé d’une injection à l’autre. La patiente ou son partenaire, peuvent réaliser eux-mêmes les injections de ganirelix, à condition qu’ils aient été convenablement formés et qu’ils aient accès à un avis compétent. Une/des bulle(s) d'air peuvent apparaître dans la seringue pré-remplie. Ceci est attendu, et l'élimination de la/des bulle(s) d'air n'est pas nécessaire
· Hypersensibilité à la substance active ou à l’un des excipients listés à la section 6.1.
· Hypersensibilité à l’hormone de libération des gonadotrophines (GnRH) ou à tout autre analogue de la GnRH.
· Pathologie modérée ou sévère des fonctions rénale ou hépatique.
· Grossesse ou allaitement.
4.4. Mises en garde spéciales et précautions d'emploi
Des précautions particulières doivent être prises chez les femmes présentant des signes et des symptômes de prédisposition allergique. Depuis la commercialisation, des cas de réactions d’hypersensibilité (à la fois généralisées et locales) ont été rapportés avec le ganirelix, dès la première dose. Ces événements comprennent anaphylaxie (y compris choc anaphylactique), angio-œdème et urticaire (voir section 4.8). Si une réaction d’hypersensibilité est suspectée, le ganirelix doit être arrêté et un traitement approprié administré. En l’absence de données cliniques, un traitement par ganirelix n’est pas recommandé chez les femmes présentant des risques importants d’allergie.
Syndrome d'hyperstimulation ovarienne (SHSO)
Le syndrome d’hyperstimulation ovarienne (SHSO) peut survenir pendant ou à la suite d'une stimulation ovarienne. Le SHSO doit être considéré comme un risque intrinsèque de la stimulation par une gonadotrophine. Le SHSO doit être traité de façon symptomatique, par exemple par du repos, une perfusion par voie intraveineuse d’une solution d’électrolytes ou de colloïdes et de l’héparine.
Grossesse extra-utérine
Etant donné que les femmes infertiles suivant des techniques d’assistance à la procréation, notamment des fécondations in vitro (FIV), présentent souvent des anomalies tubaires, l’incidence des grossesses extra-utérines peut être augmentée. Il est important de confirmer par une échographie précoce que la grossesse est intra-utérine.
Malformations congénitales
L'incidence des malformations congénitales après des techniques d’Assistance Médicale à la Procréation (AMP) peut être plus élevée qu'après des conceptions spontanées. Ceci semble dû à des différences au niveau des caractéristiques des parents (par exemple âge de la mère, qualité du sperme) et à une augmentation de l'incidence des grossesses multiples. Dans des études cliniques portant sur plus de 1000 nouveau-nés, il a été démontré que l’incidence des malformations congénitales chez les enfants nés suite à une HOC utilisant le ganirelix était comparable à celle rapportée après HOC utilisant un agoniste de la GnRH.
Femmes pesant moins de 50 kg ou plus de 90 kg
La sécurité et l’efficacité du ganirelix n’ont pas été établies chez les femmes pesant moins de 50 kg ou plus de 90 kg (voir aussi rubriques 5.1 et 5.2).
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par injection, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée.
La possibilité d’interactions avec des médicaments communément utilisés, y compris les médicaments libérant de l’histamine, ne peut pas être exclue.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas de données pertinentes concernant l'utilisation du ganirelix chez la femme enceinte. Chez les animaux, une exposition au ganirelix au moment de l’implantation a abouti à une résorption de la portée (voir rubrique 5.3). On ignore la pertinence de ces données chez l’homme.
On ignore si le ganirelix est excrété dans le lait maternel.
L’utilisation du ganirelix est contre-indiquée pendant la grossesse et l’allaitement (voir rubrique 4.3).
Fertilité
Le ganirelix est utilisé pour traiter les femmes sous hyperstimulation ovarienne contrôlée dans le cadre des programmes de procréation médicalement assistée. Le ganirelix est utilisé pour prévenir les pics prématurés de LH qui pourraient autrement survenir chez ces femmes pendant la stimulation ovarienne. Pour la posologie et la méthode d'administration, voir la rubrique 4.2.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets sur l’aptitude à conduire des véhicules et à utiliser des machines n’ont pas été étudiés.
Résumé du profil de sécurité
Le tableau ci-dessous présente tous les effets indésirables rapportés dans les études cliniques chez les femmes traitées par ganirelix en association avec la FSHrec pour la stimulation ovarienne. Des effets indésirables similaires sont attendus avec ganirelix utilisé en association avec la corifollitropine alfa pour la stimulation ovarienne.
Tableau des effets indésirables
Les effets indésirables sont classés conformément au système classe-organe MedDRA et à la fréquence : très fréquent (³ 1/10) ; fréquent (³ 1/100, < 1/10) ; peu fréquent (³ 1/1000, < 1/100). La fréquence des réactions d’hypersensibilité (très rare, < 1/10 000) est issue de l’expérience post-marketing.
|
Classe de systèmes d’organes |
Fréquence |
Effets indésirables |
|
Affections du système immunitaire |
Très rare |
Réactions d’hypersensibilité (incluant rash, gonflement du visage, dyspnée, anaphylaxie (y compris choc anaphylactique), angio-œdème et urticaire)1 Aggravation d'un eczéma pré-existant2 |
|
Affections du système nerveux |
Peu fréquent |
Céphalées |
|
Affections gastro-intestinales |
Peu fréquent |
Nausées |
|
Troubles généraux et anomalies au site d’administration |
Très fréquent |
Réaction cutanée locale au site d'injection (principalement rougeur, avec ou sans gonflement)3 |
|
Peu fréquent |
Malaises |
|
|
1 Des cas ont été signalés, dès la première dose, chez des patients recevant du ganirelix. 2 Rapporté chez un sujet après la première dose de ganirelix. 3 Dans les études cliniques, une heure après l'injection, 12 % des patientes traitées par ganirelix et 25 % des patientes traitées par un agoniste de la GnRH par voie sous cutanée ont rapporté une réaction cutanée locale modérée ou sévère survenue au moins une fois par cycle de traitement. Les réactions locales disparaissent en général dans les 4 heures suivant l’administration. |
||
Description d’une sélection d’effets indésirables
Les autres effets indésirables rapportés sont liés à l’hyperstimulation ovarienne contrôlée dans le cadre des techniques d’AMP, notamment douleur pelvienne, distension abdominale, SHSO (voir rubrique 4.4), grossesse ectopique et avortement spontané.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Un surdosage peut aboutir à une durée d’action prolongée.
Aucune donnée sur la toxicité aiguë du ganirelix chez l’homme n’est disponible. Les études cliniques avec administration sous-cutanée de ganirelix à des doses uniques allant jusqu’à 12 mg n’ont pas révélé d'effet indésirable systémique. Il a été seulement observé au cours des études de toxicité aiguë chez les rats et les singes, des symptômes de toxicité non spécifique, tels que hypotension et bradycardie, après administration intraveineuse de ganirelix avec, respectivement, plus de 1 et 3 mg/kg.
En cas de surdosage, le traitement par ganirelix doit être (temporairement) arrêté.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le ganirelix est un antagoniste de la GnRH, qui module l’axe hypothalamo-hypophyso-gonadique en se liant de façon compétitive aux récepteurs de la GnRH dans l’hypophyse. De ce fait, une suppression rapide, profonde et réversible des gonadotrophines endogènes survient, sans la stimulation initiale que l’on constate avec les agonistes de la GnRH. A la suite de l’administration de multiples doses de 0,25 mg de ganirelix à des femmes volontaires, les concentrations sériques en LH diminuaient au maximum de 74 %, 4 heures après l’injection et celles en FSH et E2 respectivement de 32 % et 25 %, 16 heures après l’injection. Les taux sériques hormonaux revenaient aux valeurs de pré-traitement dans les 2 jours suivant la dernière injection.
Effets pharmacodynamiques
Chez les patientes en cours de stimulation ovarienne contrôlée, la durée médiane du traitement avec du ganirelix était de 5 jours. Pendant le traitement par ganirelix, l’incidence moyenne des élévations de LH (> 10 UI/L) avec une élévation concomitante de la progestérone (> 1 ng/mL) a été de 0,3 - 1,2 % comparée à 0,8 % pendant le traitement par agoniste de la GnRH. On a observé une tendance à l’augmentation de l’incidence des élévations de LH et de la progestérone chez les femmes présentant un poids corporel élevé (> 80 kg), mais aucun effet sur les résultats cliniques n’a été observé. Cependant, étant donné le faible nombre de patientes traitées à ce jour, un effet ne peut pas être exclu. En cas de réponse ovarienne importante, résultant soit d’une forte exposition aux gonadotrophines au début de la phase folliculaire soit d’une haute réactivité ovarienne, des pics prématurés de LH peuvent survenir plus tôt que le 6ème jour de stimulation. L'initiation du traitement par ganirelix au 5ème jour de la stimulation peut empêcher ces pics prématurés de LH sans compromettre les résultats cliniques.
Efficacité et sécurité clinique
Dans les études contrôlées avec ganirelix associé à la FSH, utilisant un protocole long avec un agoniste de la GnRH comme référence, le traitement par ganirelix a permis une croissance folliculaire plus rapide pendant les premiers jours de stimulation, mais la cohorte de follicules en voie de maturation finale était légèrement plus réduite et a sécrété en moyenne des taux moindres d’estradiol. Ce profil différent de la croissance folliculaire nécessite que les ajustements des doses de FSH soient basés sur le nombre et la taille des follicules en voie de maturation, plutôt que sur le taux d’estradiol circulant. Des études cliniques comparatives similaires avec la corifollitropine alfa utilisant soit un antagoniste de la GnRH soit un protocole agoniste long n’ont pas été réalisées.
5.2. Propriétés pharmacocinétiques
Les paramètres pharmacocinétiques après administrations sous-cutanées répétées de ganirelix (une injection quotidienne) sont comparables à ceux observés après une dose unique sous-cutanée. Des taux plasmatiques à l’équilibre d’environ 0,6 ng/mL sont atteints en 2 à 3 jours, après administration répétée de 0,25 mg/ jour.
L'analyse pharmacocinétique indique une relation inverse entre le poids corporel et les concentrations sériques de ganirelix.
Absorption
Après une administration unique sous-cutanée de 0,25 mg, les taux sériques de ganirelix augmentent rapidement et atteignent leurs niveaux maximaux (Cmax) approximativement de 15 ng/mL en 1 à 2 heures (tmax). La biodisponibilité de ganirelix après administration sous-cutanée est d'environ 91%.
Biotransformation
Le composant principal circulant dans le plasma est le ganirelix. Le ganirelix est aussi le composant principal trouvé dans l’urine. Les fèces ne contiennent que des métabolites. Les métabolites sont des petits fragments peptidiques formés par hydrolyse enzymatique du ganirelix survenant en des sites limités. Le profil métabolique du ganirelix chez l’homme est semblable à celui trouvé chez l’animal.
Élimination
La demi-vie d'élimination (t ½) est d'environ 13 heures et la clairance est d'environ 2,4 L/h. L'excrétion se fait par les fèces (environ 75%) et l'urine (environ 22%).
5.3. Données de sécurité préclinique
Les données précliniques, basées sur des études de sécurité pharmacologique, de toxicité et de génotoxicité à doses répétées, n’ont pas révélé de risque particulier pour l’homme.
Des études de reproduction réalisées avec le ganirelix chez le rat à des doses allant de 0,1 à 10 µg/kg/jour par voie sous-cutanée et chez le lapin à des doses allant de 0,1 à 50 µg/kg/jour par voie sous-cutanée ont montré une augmentation du taux de résorption des portées dans les groupes de dose les plus élevées. Aucun effet tératogène n’a été observé.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Seringues pré-remplies en verre incolore de type I contenant 0,5 mL de solution aqueuse, stérile, prête à l’emploi, fermées par un bouton-piston en caoutchouc et la tige du piston. Chaque seringue pré-remplie est munie d’une aiguille d’injection (27G) protégée par un capuchon en caoutchouc en contact avec l’aiguille. Le capuchon de l’aiguille est protégé par une protection rigide en plastique ou un système de sécurité intégré.
Fourni en boîtes contenant 1 ou 5 seringues pré-remplies.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Examinez la seringue avant utilisation. Utilisez la seringue uniquement si la solution est limpide, ne contient pas de particules et si son conditionnement n’a pas été endommagé. Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
3 RD FLOOR, KILMORE HOUSE
PARK LANE, SPENCER DOCK
D01 YE64 DUBLIN 1
IRLANDE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 381 5 8 : 1 seringue pré-remplie (verre) de 0,5 mL.
· 34009 302 381 6 5 : 5 seringues pré-remplies (verre) de 0,5 mL.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
À compléter ultérieurement par le titulaire
10. DATE DE MISE A JOUR DU TEXTE
À compléter ultérieurement par le titulaire
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à surveillance particulière.
Médicament soumis à une prescription réservée aux spécialistes en gynécologie et/ou en gynécologie-obstétrique et/ou en endocrinologie et métabolisme.
ANSM - Mis à jour le : 15/09/2025
GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie
Ganirelix
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie ?
3. Comment prendre GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie ET DANS QUELS CAS EST-IL UTILISE ?
GANIRELIX THERAMEX contient la substance active ganirelix et appartient au groupe des médicaments appelés « antagonistes de l’hormone de libération des gonadotrophines » bloquant les effets de l’hormone naturelle de libération des gonadotrophines (GnRH). La GnRH régule la libération des gonadotrophines (hormone lutéinisante (LH) et hormone folliculo-stimulante (FSH)). Les gonadotrophines jouent un rôle important dans la fertilité humaine et la reproduction. Chez les femmes, la FSH est nécessaire pour la croissance et le développement des follicules dans les ovaires. Les follicules sont de petits sacs ronds qui contiennent les ovocytes. La LH est nécessaire pour libérer les ovocytes matures hors des follicules et des ovaires (c’est-à-dire l’ovulation). GANIRELIX THERAMEX inhibe l'action de la GnRH, résultant en une suppression de la libération de LH en particulier.
Utilisation de GANIRELIX THERAMEX
Chez les femmes ayant recours à des techniques médicales d’assistance à la procréation, telles que la fécondation in vitro (FIV) et d’autres méthodes, l’ovulation peut parfois avoir lieu trop tôt et entraîner une diminution significative des chances de devenir enceinte. GANIRELIX THERAMEX est utilisé pour prévenir le pic prématuré de LH qui est susceptible d’entraîner une libération prématurée des ovocytes.
Dans les études cliniques, le ganirelix a été utilisé en association avec l’hormone folliculo-stimulante recombinante (FSH) ou la corifollitropine alfa, un stimulant folliculaire à longue durée d’action.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie ?
Ne prenez jamais GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie :
· si vous êtes allergique au ganirelix ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si vous êtes allergique à l’hormone de libération des gonadotrophines (GnRH) ou à un analogue de la GnRH ;
· si vous présentez une maladie modérée ou sévère, du rein ou du foie ;
· si vous êtes enceinte ou si vous allaitez.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant de prendre GANIRELIX THERAMEX.
· Si vous avez des prédispositions allergiques, veuillez-en parler à votre médecin. Votre médecin décidera en fonction de leur sévérité si une surveillance supplémentaire est nécessaire pendant le traitement. Des cas de réactions allergiques ont été rapportés, dès la première dose.
· Des réactions allergiques, à la fois généralisées et locales, incluant urticaire, gonflement du visage, des lèvres, de la langue et/ou de la gorge pouvant provoquer des difficultés à respirer et/ou à avaler (angio-œdème et/ou anaphylaxie) ont été rapportées. (Voir aussi rubrique 4.) Si vous développez une réaction allergique, arrêtez de prendre GANIRELIX THERAMEX et consultez immédiatement un médecin.
· Pendant ou à la suite d’une stimulation hormonale des ovaires, un syndrome d’hyperstimulation ovarienne peut se développer. Ce syndrome est lié au traitement de stimulation par les gonadotrophines. Veuillez-vous rapporter à la notice du médicament contenant de la gonadotrophine qui vous a été prescrit.
· L’incidence des malformations congénitales (anomalies congénitales) après des techniques d’assistance médicale à la procréation peut être légèrement plus élevée qu’après des conceptions spontanées. Cette incidence légèrement plus élevée semble être liée aux caractéristiques des patientes suivant un traitement pour la fertilité (ex : âge de la femme, caractéristiques du sperme) et à une augmentation de l’incidence des grossesses multiples (grossesse avec plus de un bébé à la fois) après des techniques d’assistance médicale à la procréation. L’incidence des malformations congénitales après des techniques d’assistance médicale à la procréation utilisant GANIRELIX THERAMEX est comparable à celle rapportée après utilisation d’autres analogues de la GnRH au cours des techniques d’assistance médicale à la procréation.
· Il y a une légère augmentation du risque de grossesse hors de l’utérus (grossesse ectopique) chez les femmes présentant une anomalie des trompes de Fallope.
· L’efficacité et la sécurité de GANIRELIX THERAMEX n’ont été établies que chez les femmes pesant entre 50 kg et 90 kg. Pour plus d’informations, adressez-vous à votre médecin.
Enfants et adolescents
GANIRELIX THERAMEX n’est pas utilisé chez les enfants ou les adolescents.
Autres médicaments et GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Ganiremix Theramex est indiqué pour une stimulation ovarienne contrôlée dans le cadre d’une assistance médicale à la procréation (AMP).
N’utilisez pas GANIRELIX THERAMEX pendant la grossesse et l’allaitement.
Conduite de véhicules et utilisation de machines
Les effets de GANIRELIX THERAMEX sur l’aptitude à conduire des véhicules et à utiliser des machines n’ont pas été étudiés.
GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie contient du sodium
GANIRELIX THERAMEX contient moins de 1 mmol (23 mg) de sodium par injection, c'est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie ?
Vous allez vous injecter ce médicament par vous même et votre médecin vous expliquera comment faire.
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Si vous n’avez pas compris les indications, vérifiez auprès de votre médecin ou pharmacien.
Etape 1
La stimulation ovarienne par l’Hormone Folliculo-Stimulante (FSH) ou la corifollitropine peut commencer au 2ème ou au 3ème jour de vos règles.
Etape 2
Le contenu de la seringue de GANIRELIX THERAMEX (0,25 mg) doit être injecté juste sous la peau une fois par jour, en commençant le 5ème ou le 6ème jour de la stimulation. En fonction de votre réponse ovarienne, votre médecin peut décider de commencer le traitement un autre jour.
GANIRELIX THERAMEX et la FSH doivent être administrés approximativement au même moment. Toutefois, les préparations ne doivent pas être mélangées et des sites d’injection différents doivent être utilisés.
Le traitement quotidien par GANIRELIX THERAMEX doit être poursuivi jusqu'au jour où un nombre suffisant de follicules de taille adéquate sont présents.
Etape 3
La maturation finale des ovocytes contenus dans les follicules peut être induite par administration de gonadotrophine chorionique humaine (hCG). Le délai entre deux injections de Ganirelix Theramex, ainsi que celui entre la dernière injection de GANIRELIX THERAMEX et l'injection d'hCG ne doit pas dépasser 30 heures car, dans le cas contraire, une ovulation prématurée (libération des ovocytes) peut survenir. Par conséquent, si GANIRELIX THERAMEX est injecté au cours de la matinée, le traitement par GANIRELIX THERAMEX doit être poursuivi pendant toute la période du traitement par la gonadotrophine, y compris le jour de déclenchement de l’ovulation. Si GANIRELIX THERAMEX est injecté au cours de l’après-midi, la dernière injection de GANIRELIX THERAMEX devra être faite dans l’après-midi, la veille du jour de déclenchement de l’ovulation.
Mode d’emploi
· Zone d’injection
GANIRELIX THERAMEX est fourni en seringues pré-remplies qui contiennent une dose chacune. Le contenu doit être injecté lentement, juste sous la peau, de préférence dans la cuisse. Examinez la solution avant utilisation. Ne pas l’utiliser si elle contient des particules ou si elle n’est pas limpide. Vous pouvez remarquer une ou plusieurs bulles d'air dans la seringue pré-remplie. Ceci est attendu, et l'élimination de la/des bulle(s) d'air n'est pas nécessaire. Si vous pratiquez vous-même les injections ou si elles sont faites par votre partenaire, respectez strictement le mode d'emploi ci-dessous. Ne mélangez GANIRELIX THERAMEX avec aucun autre médicament.
· Préparation de la zone d'injection
Lavez-vous minutieusement les mains à l'eau et au savon. Nettoyez la zone d'injection avec un désinfectant (par exemple : alcool) afin d'éliminer toutes les bactéries cutanées. Désinfectez sur une zone d’environ 5 cm (2 pouces) autour du point d'injection et laissez sécher le désinfectant pendant au moins une minute avant de pratiquer l’injection.
· Introduction de l'aiguille
Retirez le capuchon de l'aiguille. Pincez doucement une zone de peau propre afin de former un pli de peau. Introduisez l'aiguille à la base de la peau pincée. Introduisez l'aiguille selon un angle de 45°C par rapport à la surface de la peau.
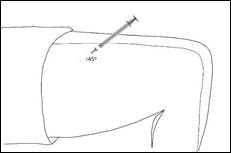
Utiliser une zone différente pour chaque injection.
· Vérification de la position correcte de l'aiguille
Tirez doucement le piston de la seringue pour vérifier que l’aiguille est correctement placée. Si du sang est présent dans la seringue, cela signifie que l’extrémité de l'aiguille a pénétré un vaisseau sanguin. Dans ce cas, ne continuez pas l’injection de Ganirelix Theramex. Retirez la seringue, couvrez le site d'injection d'un coton imbibé de désinfectant et exercez une pression ; le saignement doit cesser en une ou deux minutes. N’utilisez plus cette seringue, et jetez-la soigneusement. Recommencez avec une nouvelle seringue.
· Injection de la solution
Une fois l'aiguille correctement positionnée, poussez le piston lentement et régulièrement, la solution sera ainsi correctement injectée et les tissus cutanés ne seront pas abimés.
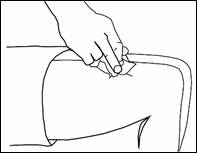
· Retrait de la seringue
Retirez rapidement la seringue.
Exercez une pression sur le point d’injection avec un coton imbibé de désinfectant. N’utilisez la seringue pré-remplie qu’une seule fois.
Si vous avez pris plus de GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie que vous n’auriez dû
Contactez votre médecin.
Si vous oubliez de prendre GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie
Si vous vous rendez compte que vous avez oublié d'injecter une dose, faites l’injection dès que possible. N’injectez pas de dose double pour compenser la dose que vous avez oubliée d’injecter. Si le retard est supérieur à 6 heures (de sorte que l'intervalle entre deux injections dépasse les 30 heures), injectez la dose aussitôt que possible, et contactez votre médecin pour qu’il vous conseille.
Si vous arrêtez de prendre GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie
N’arrêtez pas le traitement par GANIRELIX THERAMEX à moins que cela ne soit conseillé par votre médecin, car cela peut affecter le résultat de votre traitement.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Très fréquent (pouvant affecter plus de 1 femme sur 10)
· une réaction cutanée locale (essentiellement une rougeur, accompagnée ou non de gonflement) au point d'injection. La réaction locale disparaît normalement dans les 4 heures suivant l’administration.
Peu fréquent (pouvant affecter jusqu’à 1 femme sur 100)
· maux de tête
· nausées
· malaises
Très rare (pouvant affecter jusqu’à 1 femme sur 10 000)
· des réactions allergiques ont été observées, dès la première dose.
· éruption cutanée
· gonflement du visage
· difficulté à respirer (dyspnée)
· gonflement du visage, des lèvres, de la langue et/ou de la gorge pouvant provoquer des difficultés à respirer et/ou à avaler (angio-œdème et/ou anaphylaxie)
· urticaire
De plus, des effets indésirables, connus pour survenir pendant un traitement par hyperstimulation ovarienne contrôlée, ont été rapportés, tels que :
· douleurs abdominales
· syndrome d'hyperstimulation ovarienne (SHSO). (SHSO apparait quand vos ovaires réagissent de manière excessive au traitement de la fertilité que vous prenez).
· grossesse extra-utérine (lorsque l’embryon se développe en dehors de l’utérus)
· fausse-couche (voir la notice patient de la préparation contenant de la FSH avec laquelle vous êtes traitée).
Une aggravation d'une éruption de la peau pré-existante (eczéma) a été rapportée chez un sujet après la première dose de ganirelix.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étui et sur l'étiquette après EXP. La date d’expiration fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Examinez la seringue avant utilisation. N’utilisez la seringue que si la solution est limpide, ne contient pas de particules et que si son conditionnement n’a pas été endommagé.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient GANIRELIX THERAMEX 0,25 mg/0,5 mL, solution injectable en seringue pré-remplie
· La substance active est :
Ganirelix (sous forme d’acétate) .................................................................................... 0,25 mg
dans 0,5 mL de solution aqueuse
Pour une seringue pré-remplie
· Les autres composants sont :
Acide acétique glacial, mannitol, et eau pour préparations injectables. Le pH (un indicateur de l’acidité) peut avoir été ajusté avec de l'hydroxyde de sodium et de l’acide acétique.
GANIRELIX THERAMEX est une solution injectable, aqueuse, limpide et incolore. La solution est prête à l’emploi et destinée à une administration sous-cutanée.
GANIRELIX THERAMEX est disponible en boîtes contenant 1 ou 5 seringues pré-remplies avec des aiguilles d’injection (27G).
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
3 RD FLOOR, KILMORE HOUSE
PARK LANE, SPENCER DOCK
D01 YE64 DUBLIN 1
IRLANDE
Exploitant de l’autorisation de mise sur le marché
83 AVENUE CHARLES DE GAULLE
92200 NEUILLY-SUR-SEINE
POLIGONO INDUSTRIAL ELS VINYETS-ELS FOGARS, SECTOR 2
CARRETERA COMARCAL C-244, KM 22
08777 SANT QUINTÍ DE MEDIONA
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
À compléter ultérieurement par le titulaire
La dernière date à laquelle cette notice a été révisée est :
À compléter ultérieurement par le titulaire
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).