Dernière mise à jour le 01/06/2026
TALOXA 600 mg/5 ml suspension buvable
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : N03AX10
Qu’est-ce que TALOXA 600 mg/5 ml, suspension buvable ?
TALOXA est utilisé comme un médicament antiépileptique. Les crises d'épilepsie sont également connues sous le nom d'épilepsie ou de crises de convulsions.
Dans quels cas TALOXA 600 mg/5 ml, suspension buvable est-il utilisé ?
TALOXA n’est pas destiné à être utilisé chez l’enfant de moins de 4 ans.
TALOXA est utilisé en association avec d’autres médicaments pour contrôler les convulsions chez l'adulte et l'enfant à partir de 4 ans ayant un syndrome de Lennox-Gastaut non contrôlé par les autres antiépileptiques.
Présentations
> 1 flacon(s) en verre de 230 ml avec seringue(s) doseuse(s)
Code CIP : 559 158-9 ou 34009 559 158 9 8
Déclaration de commercialisation : 19/07/1996
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
Autres informations
- Titulaire de l'autorisation : ORGANON France
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription initiale hospitalière semestrielle
- prescription initiale réservée à certains spécialistes
- renouvellement de la prescription réservé aux spécialistes en NEUROLOGIE
- renouvellement de la prescription réservé aux spécialistes en PEDIATRIE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 231 346 8
ANSM - Mis à jour le : 14/04/2025
TALOXA 600 mg/5 ml, suspension buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
5 ml de TALOXA 600 mg/5 ml, suspension buvable contient 600 mg de felbamate.
Excipients à effet notoire :
1,05 g de sorbitol (E420) par 5 ml de TALOXA 600 mg/5 ml, suspension buvable.
6,5 mg de parahydroxybenzoate de méthyle (E218) par 5 ml de TALOXA 600 mg/5 ml, suspension buvable.
1 mg de parahydroxybenzoate de propyle (E216) par 5 ml de TALOXA 600 mg/5 ml, suspension buvable.
10 mg de benzoate de sodium (E211) par 5 ml de TALOXA 600 mg/5 ml, suspension buvable.
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension blanche à blanchâtre, opaque et visqueuse.
4.1. Indications thérapeutiques
Syndrome de Lennox-Gastaut : en complément du traitement antérieur, traitement du syndrome de Lennox-Gastaut chez les patients à partir de 4 ans, non contrôlé par les autres médicaments antiépileptiques appropriés disponibles.
Une évaluation rigoureuse de l'efficacité du felbamate doit être effectuée après 2 à 3 mois de traitement. Seuls les patients pour lesquels une amélioration clinique majeure de leur syndrome épileptique (i.e. réduction marquée de la fréquence des crises ou de leur sévérité) aura été constatée dans ce laps de temps devront continuer le traitement par le felbamate (voir rubrique 4.4).
Les patients doivent être informés, avant l'instauration du traitement, du risque potentiel lié à l'utilisation du felbamate (voir rubrique 4.4).
Les patients doivent être avertis que l'utilisation du felbamate a été associée à la survenue d'aplasie médullaire et d'insuffisance hépatique, toutes deux potentiellement mortelles (voir rubrique 4.4).
4.2. Posologie et mode d'administration
Posologie
SYNDROME DE LENNOX-GASTAUT
Posologie chez l'adulte et l'adolescent à partir de 14 ans
En association avec d'autres antiépileptiques : le felbamate administré en association avec la carbamazépine, la phénytoïne, le phénobarbital ou l'acide valproïque peut augmenter l'incidence de leurs effets indésirables respectifs (voir rubrique 4.5). La posologie initiale recommandée pour le felbamate est de 600 mg à 1 200 mg/jour, administrée en 2 ou 3 prises. Chez les patients déjà traités par carbamazépine, phénytoïne, phénobarbital, et/ou acide valproïque, la posologie de ces produits doit être réduite de 20 % à 30 % lors de l'introduction du felbamate. La posologie quotidienne du felbamate peut alors être augmentée par paliers de 600 à 1 200 mg à intervalles d'environ une semaine, jusqu'à un maximum de 3 600 mg/jour administrés en 3 à 4 prises. Une adaptation des doses de carbamazépine, de phénytoïne, de phénobarbital, et d'acide valproïque peut être nécessaire en fonction de l'augmentation de la posologie du felbamate. Cependant, les interactions médicamenteuses sont dose-dépendantes et sont sujettes à variabilité inter-individuelle. Par conséquent, tout ajustement de posologie des antiépileptiques associés doit être basé non seulement sur les taux plasmatiques à l'état d’équilibre, mais aussi sur l'observation clinique.
Population pédiatrique
Posologie pédiatrique : Enfant de 4 à 11 ans et adolescent de 12 à 14 ans
En association avec d'autres antiépileptiques : le felbamate administré en association avec la carbamazépine, la phénytoïne, le phénobarbital, ou l'acide valproïque peut augmenter l'incidence de leurs effets indésirables respectifs (voir rubrique 4.5). La posologie initiale recommandée pour le felbamate est de 7,5 mg/kg/jour à 15 mg/kg/jour, administrés en 2 ou 3 prises. Chez les patients déjà traités par carbamazépine, phénytoïne, phénobarbital et/ou acide valproïque, la posologie de ces produits doit être réduite de 20 % à 30 % lors de l'introduction du felbamate.
La posologie quotidienne du felbamate peut alors être augmentée par paliers de 7,5 mg/kg à 15 mg/kg à intervalles d'une semaine au moins, jusqu'à un maximum de 45 mg/kg/jour (sans dépasser 3 600 mg/jour) administrés en 3 à 4 prises. Une adaptation des doses de carbamazépine, de phénytoïne, de phénobarbital et/ou d'acide valproïque peut être nécessaire en fonction de l'augmentation de la posologie du felbamate.
Cependant, les interactions médicamenteuses sont dose-dépendantes et sont sujettes à variabilité inter-individuelle. Par conséquent, tout ajustement de posologie des antiépileptiques associés doit être basé non seulement sur les taux plasmatiques à l'état d’équilibre, mais aussi sur l'observation clinique.
La dose de Taloxa suspension buvable doit être prescrite en millilitres (voir tableau de conversion ci-dessous qui donne en millilitre l'équivalence de la dose en milligramme).
Les doses indiquées dans le tableau ci-dessous ne sont applicables que pour les patients âgés de 4 ans et plus. Ces doses quotidiennes doivent être réparties en plusieurs prises dans la journée.
Tableau 1 : Doses quotidiennes totales en ml pour différentes posologies et différents poids de l'enfant.
La dose quotidienne à administrer doit être répartie en deux, trois ou quatre prises par jour (voir le paragraphe ci-dessus).
|
Tableau 1 |
Dose totale quotidienne à administrer en ml |
|||||
|
Poids |
7,5 |
15 |
22,5 |
30 |
37,5 |
45 |
|
en kg |
mg/kg |
mg/kg |
mg/kg |
mg/kg |
mg/kg |
mg/kg |
|
12 |
0,8 |
1,5 |
2,3 |
3,0 |
3,8 |
4,5 |
|
14 |
0,9 |
1,8 |
2,6 |
3,5 |
4,4 |
5,3 |
|
16 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
6,0 |
|
18 |
1,1 |
2,3 |
3,4 |
4,5 |
5,6 |
6,8 |
|
20 |
1,3 |
2,5 |
3,8 |
5,0 |
6,3 |
7,5 |
|
22 |
1,4 |
2,8 |
4,1 |
5,5 |
6,9 |
8,3 |
|
24 |
1,5 |
3,0 |
4,5 |
6,0 |
7,5 |
9,0 |
|
26 |
1,6 |
3,3 |
4,9 |
6,5 |
8,1 |
9,8 |
|
28 |
1,8 |
3,5 |
5,3 |
7,0 |
8,8 |
10,5 |
|
30 |
1,9 |
3,8 |
5,6 |
7,5 |
9,4 |
11,3 |
|
32 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
12,0 |
|
34 |
2,1 |
4,3 |
6,4 |
8,5 |
10,6 |
12,8 |
|
36 |
2,3 |
4,5 |
6,8 |
9,0 |
11,3 |
13,5 |
|
38 |
2,4 |
4,8 |
7,1 |
9,5 |
11,9 |
14,3 |
|
40 |
2,5 |
5,0 |
7,5 |
10,0 |
12,5 |
15,0 |
Utilisation pédiatrique : la sécurité et l’efficacité du felbamate chez l’enfant de moins de 4 ans n’ont pas été établies.
Population gériatrique
Utilisation chez le sujet âgé : Les données cliniques limitées sur l'administration du felbamate à des patients de plus de 65 ans n'indiquent aucune nécessité de restriction d'utilisation du produit dans cette population. Cependant, d’une manière générale, l'adaptation de la posologie doit être déterminée avec prudence chez le sujet âgé.
Patient avec une insuffisance rénale
Posologie chez les patients présentant une insuffisance rénale : chez les patients avec une clairance de la créatinine < 50 ml/min, les doses initiales du felbamate doivent être diminuées de moitié, puis augmentées avec précaution.
Patient avec une insuffisance hépatique
Posologie chez les patients présentant un dysfonctionnement hépatique : le felbamate ne doit pas être utilisé chez les patients ayant un (antécédent de) dysfonctionnement hépatique en raison du risque d’hépatotoxicité (voir rubriques 4.3 et 4.4).
La prise d’un repas n’affecte pas la vitesse et le degré d’absorption du felbamate.
Mode d’administration
Bien agiter le flacon avant utilisation.
Une seringue doseuse de 5,0 ml avec marquage CE avec des graduations de 0,1 ml pour administration orale est fournie avec TALOXA, suspension buvable. La graduation 0,1 ml permet de mesurer une dose de 12 mg ; celle de 0,5 ml, une dose de 60 mg ; et celle de 5,0 ml, une dose de 600 mg de felbamate.
Le felbamate est contre-indiqué en cas :
· d’antécédent de troubles hématologiques ou hépatiques,
· d’hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
|
Information pour les patients : les patients doivent être informés avant le début du traitement que l'utilisation du felbamate a été associée à la survenue d'aplasie médullaire et d'insuffisance hépatique, toutes deux potentiellement mortelles. |
|
Troubles hématologiques : plusieurs cas d'effets indésirables graves de type hématologique, tels que thrombopénie, leucopénie, pancytopénie, anémie et aplasie médullaire, ont été rapportés lors de l'utilisation du felbamate. |
L'aplasie médullaire représente l'effet le plus grave, mortel dans 30 % des cas. L'incidence a été estimée à un cas pour 4 000 patients traités environ, ce qui correspond à une forte augmentation (100 fois) de l'incidence dite normale (2 à 5 par million de personnes par an). En conséquence, le felbamate ne doit être prescrit que chez les patients souffrant d'un syndrome de Lennox-Gastaut réfractaire, lorsqu'aucune autre alternative médicamenteuse n'est disponible.
Les cas d'aplasie médullaire sont survenus 2 à 12 mois après le début du traitement par le felbamate. Cependant, l'atteinte des cellules de la moelle osseuse conduisant finalement à l'aplasie peut se produire plusieurs semaines à plusieurs mois plus tôt. De même, le risque de survenue d'une aplasie médullaire peut persister pendant plusieurs mois après l'arrêt du traitement par le felbamate. On ne sait pas s'il existe une corrélation entre le risque de survenue d'aplasie médullaire et la durée du traitement. On ne peut donc être certain qu'un patient traité par le felbamate pendant plusieurs mois sans signe d'anomalies hématologiques soit exempté de risque.
· Une numération formule sanguine/plaquettes doit être pratiquée avant le début du traitement par le felbamate et toutes les 2 semaines pendant le traitement.
· En présence d'une neutropénie (polynucléaires neutrophiles < 1 500/mm3) et/ou d'une thrombopénie (plaquettes < 150 000/mm3), le traitement par le felbamate doit être arrêté et des investigations doivent être menées afin de rechercher une aplasie médullaire.
· Une surveillance clinique soigneuse à la recherche des symptômes tels qu’ecchymoses, pétéchies, saignement ou signes d'infection et/ou d'anémie (fatigue, faiblesse, etc.) doit être effectuée. Si ces symptômes sont retrouvés, une numération formule sanguine/plaquettes doit être pratiquée immédiatement.
|
Hépatotoxicité : Des cas sévères d'insuffisance hépatique aiguë (mortels dans 30 % des cas) ont été rapportés chez des patients traités par le felbamate. |
· Un bilan hépatique (ASAT, ALAT, bilirubine) doit être pratiqué avant le début du traitement par le felbamate. Les patients avec une fonction hépatique anormale ne doivent pas être traités par le felbamate.
· Pendant toute la durée du traitement par le felbamate, un bilan hépatique doit être effectué toutes les 2 semaines. Le traitement par le felbamate doit être interrompu chez les patients présentant des anomalies cliniquement significatives de la fonction hépatique.
· L'apparition de signes cliniques tels qu’ictère, anorexie, nausées, vomissements et douleurs abdominales devra conduire à un bilan hépatique immédiat.
Ce médicament contient 1,05 g de sorbitol (E 420) par 5 ml de suspension buvable. Le sorbitol dans les médicaments à usage oral peut affecter la biodisponibilité d’autres médicaments à usage oral administrés de façon concomitante. Le sorbitol est une source de fructose ; les patients présentant une intolérance héréditaire au fructose (IHF) ne doivent pas prendre/recevoir ce médicament. Le sorbitol peut causer une gêne gastro-intestinale et un effet laxatif léger.
TALOXA suspension buvable contient des parahydroxybenzoates de méthyle et de propyle et peut provoquer des réactions allergiques (éventuellement retardées).
Ce médicament contient moins de 1 mmol (23 mg) de sodium par 5 ml, c’est-à-dire qu’il est essentiellement « sans sodium ».
Ce médicament contient 10 mg de benzoate de sodium par 5 ml.
Des cas de cristallurie ont été rapportés très rarement. Pour éviter leur survenue, un apport liquidien adéquat est nécessaire pendant le traitement par le felbamate.
Hypersensibilité
· Le felbamate doit être utilisé avec prudence en cas d'hypersensibilité connue à d'autres carbamates.
· Des réactions sévères d'hypersensibilité, y compris choc anaphylactique, syndrome de Stevens-Johnson, éruption bulleuse et épidermolyse nécrosante, ont été rapportées avec le felbamate. Elles sont survenues généralement 2 à 3 semaines après le début du traitement. Les symptômes décrits incluaient rash, fièvre, œdème des muqueuses et anaphylaxie, leucopénie, thrombopénie, élévation des valeurs biologiques hépatiques, arthralgie, myalgie et angine. En cas d'hypersensibilité au felbamate, arrêter le traitement et débuter un traitement symptomatique approprié.
Arrêt du felbamate
Les antiépileptiques, notamment le felbamate, ne doivent pas, en général, être interrompus brutalement, en raison d'une possible augmentation de la fréquence des crises convulsives. Cependant, si la gravité du ou des effet(s) indésirable(s) justifie un arrêt immédiat, une surveillance médicale attentive est obligatoire.
Les patients chez qui le felbamate a été arrêté à cause d'effets indésirables graves ne doivent pas reprendre le traitement.
Augmentation de la fréquence des crises
Comme rapporté avec les autres antiépileptiques, certains patients peuvent présenter une augmentation de la fréquence des crises ou l’apparition de nouveaux types de crises (voir rubrique 4.8). Ces phénomènes peuvent résulter d’un surdosage, d’une diminution des concentrations plasmatiques du traitement antiépileptique administré parallèlement, ou d’un effet paradoxal.
Idées et comportements suicidaires
Des idées et comportements suicidaires ont été rapportés chez des patients traités par des antiépileptiques dans plusieurs indications. Une méta-analyse d’essais randomisés, contrôlés versus placebo portant sur des antiépileptiques a également montré une légère augmentation du risque d’idées et de comportements suicidaires. Les causes de ce risque ne sont pas connues et les données disponibles n’excluent pas la possibilité d’une augmentation de ce risque pour le felbamate.
Par conséquent, les patients doivent être étroitement surveillés pour tout signe d’idées et de comportements suicidaires et un traitement approprié doit être envisagé. Il doit être recommandé aux patients (et leur entourage) de demander un avis médical en cas de survenue de signes d’idées et de comportements suicidaires.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Effets du felbamate sur les autres agents antiépileptiques
Carbamazépine : Le felbamate diminue d'environ 25 % les concentrations plasmatiques de la carbamazépine à l'état d'équilibre, et augmente d'environ 50 % celles de son époxyde.
Phénytoïne : Le felbamate inhibe la clairance de la phénytoïne de manière dose-dépendante, d'où une élévation possible de sa concentration plasmatique de 20 % à 60 %.
Phénobarbital : Le felbamate à la dose de 1 200 mg deux fois par jour augmente l’ASC du phénobarbital d’environ 25 %.
Acide valproïque : Administré à raison de 600 mg ou 1 200 mg, deux fois par jour, le felbamate élève les concentrations plasmatiques du valproate à l'état d'équilibre de façon linéaire et dose-dépendante. Pour la dose la plus faible de felbamate, l'ASC moyenne du valproate et le nadir des concentrations sont augmentés respectivement de 28 % et 18 % ; une élévation proportionnelle de ces valeurs est observée pour la dose la plus forte.
Clonazépam, oxcarbazépine, vigabatrine et lamotrigine : Bien que le felbamate administré à la dose de 1 200 mg toutes les 12 heures produise des modifications statistiquement significatives des paramètres pharmacocinétiques du clonazépam, de la lamotrigine et du vigabatrine, ces modifications sont minimes et ne semblent pas avoir de conséquence clinique. Aucun changement n’a été observé parmi les paramètres pharmacocinétiques du métabolite actif monohydroxy de l’oxcarbazépine. Etant donné qu'une interaction pharmacodynamique du felbamate avec l'un de ces produits ne peut être exclue, l'ajustement des posologies devra toujours être basé sur la réponse clinique et la tolérance.
Effets des autres médicaments antiépileptiques sur le felbamate
Carbamazépine, phénytoïne, phénobarbital : Lorsque la carbamazépine ou la phénytoïne sont associées avec le felbamate, la diminution des taux plasmatiques du felbamate à l'état d'équilibre peut être proche de 20 %. L’association de phénobarbital engendre une diminution de 35 % des taux plasmatiques du felbamate à l'état d'équilibre.
Acide valproïque : L’acide valproïque semble avoir un effet minime sur la clairance du felbamate ; dans une étude, cependant, le nadir des concentrations du felbamate était d’environ 50 % supérieur à celui du felbamate administré seul.
Autres interactions médicamenteuses du felbamate
Contraceptifs oraux : Le felbamate réduit l’ASC du gestodène et de l’éthinylestradiol de 42 % et de 13 % respectivement chez les femmes traitées par un contraceptif oral combiné faiblement dosé. L’efficacité et la tolérance des contraceptifs oraux peuvent être altérées. Les autres associations n’ont pas été étudiées.
Effets du felbamate sur le cytochrome P450
Il a été montré que le felbamate était un substrat du CYP3A4 et du CYP2E1, mais l’inhibition de ces voies mineures n’a pas de conséquence pharmacocinétique attendue.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace durant le traitement et jusqu’à 1 mois après la fin du traitement (voir rubrique 4.5).
Grossesse
L'innocuité de ce médicament pendant la grossesse n'a pas été établie. Les études de reproduction conduites chez le rat et le lapin n'ont pas mis en évidence d’effets nocifs du felbamate sur le fœtus. Cependant, un passage transplacentaire du felbamate a été observé (voir rubrique 5.3). Comme les études de reproduction conduites chez l'animal ne permettent pas toujours de prédire la réponse chez l'homme, et étant donné le risque potentiel d'atteinte de la moelle osseuse du fœtus et d’hépatotoxicité, le felbamate ne doit pas être utilisé chez les femmes en âge de procréer n’utilisant pas de méthode contraception efficace ni chez les femmes enceintes, sauf en cas de nécessité absolue.
Risque lié à l’épilepsie et aux médicaments antiépileptiques en général :
Un avis médical spécialisé doit être donné aux femmes en âge de procréer. Lorsqu’une femme envisage une grossesse, la nécessité du traitement antiépileptique doit être réévaluée. Chez les femmes traitées pour de l’épilepsie, un arrêt brutal du traitement antiépileptique doit être évité car cela pourrait entraîner la réapparition de crises dont les conséquences pour la mère et l’enfant à naître peuvent être graves.
Une monothérapie utilisant la dose minimale efficace doit être préférée autant que possible car une polythérapie avec plusieurs antiépileptiques peut être associée à un risque plus élevé de malformations congénitales qu’une monothérapie, selon les antiépileptiques co-administrés.
Le felbamate est excrété dans le lait maternel. Etant donné le risque potentiel lié au felbamate d’hépatotoxicité et d'atteinte de la moelle osseuse de l’enfant recevant du lait maternel, le felbamate ne doit pas être administré chez la femme allaitante.
Fertilité
Les études de reproduction conduites chez le rat et le lapin n’ont mis en évidence aucune altération de la fertilité due au felbamate.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les patients peuvent ressentir des vertiges ou une somnolence et doivent être mis en garde avant de s’engager dans des activités potentiellement dangereuses.
Le felbamate est associé à une augmentation de l'incidence de troubles hématologiques (voir rubrique 4.4), y compris d'aplasie médullaire. Les autres effets indésirables graves hématologiques comprennent de rares cas de thrombocytopénie, leucopénie, neutropénie, anémie, ou des associations de ces effets, y compris la pancytopénie. Certains apparaissent lors d’une réaction d’hypersensibilité aiguë (voir rubrique 4.4). Des cas d’hépatite sévère, allant jusqu’à l’insuffisance hépatique aiguë mortelle ont été rapportés avec le felbamate (voir rubrique 4.4).
Les effets indésirables rapportés chez les patients adultes traités par le felbamate en association à d’autres antiépileptiques lors des essais cliniques et considérés comme étant liés au traitement sont listés dans le tableau suivant, par système classe-organe et par fréquence.
|
Table 1 : Effets indésirables rapportés lors du traitement Très fréquent (≥ 1/10) ; Fréquent (≥ 1/100, < 1/10) ; Peu fréquent (≥ 1/1 000, < 1/100) ; Rare (≥ 1/10 000, < 1/1 000) ; Très rare (< 1/10 000) ; Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) (CIOMS III) |
|
|
Affections hématologiques et du système lymphatique |
|
|
Rare : |
Thrombocytopénie, leucopénie, neutropénie, anémie ou associations de ces effets y compris la pancytopénie, troubles hématologiques (voir rubrique 4.4) y compris aplasie médullaire |
|
Affections du système immunitaire |
|
|
Rare : |
Choc anaphylactique (voir rubrique 4.4) |
|
Troubles du métabolisme et de la nutrition |
|
|
Fréquent : |
Perte de poids, anorexie |
|
Peu fréquent : |
Hypophosphatémie |
|
Affections psychiatriques |
|
|
Peu fréquent : |
Troubles de l’élocution, dépression, stupeur, anxiété |
|
Affections du système nerveux |
|
|
Fréquent : |
Insomnie, somnolence, ataxie, vertiges, céphalées |
|
Rare : |
Augmentation de la fréquence des crises convulsives (voir rubrique 4.4) |
|
Affections oculaires |
|
|
Fréquent : |
Diplopie, vision anormale |
|
Affections gastro-intestinales |
|
|
Fréquent : |
Nausées, vomissements, dyspepsie, douleur abdominale, diarrhée |
|
Très rare : |
Constipation |
|
Affections hépato-biliaires |
|
|
Très rare : |
Hépatite sévère, insuffisance hépatique aiguë (parfois fatale) (voir rubrique 4.4) |
|
Affections de la peau et du tissu sous-cutané |
|
|
Peu fréquent : |
Rash |
|
Rare : |
Réactions d’hypersensibilité (incluant syndrome de Stevens-Johnson, éruption bulleuse, nécrolyse épidermique toxique) (voir rubrique 4.4) |
|
Affections du rein et des voies urinaires |
|
|
Très rare : |
Cristallurie |
|
Troubles généraux et anomalies du site d’administration |
|
|
Fréquent : |
Démarche anormale |
|
Peu fréquent : |
Fatigue |
Population pédiatrique
Le profil d’effets indésirables chez les enfants était identique. En plus, des infections des voies respiratoires hautes ont été fréquemment observées chez les enfants ; cependant, le lien avec le traitement n’est pas probable.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Lors des études cliniques, des doses allant de 4 000 à 12 000 mg/jour furent involontairement absorbées par des patients traités en poly- ou en monothérapie. La sévérité des effets indésirables observés fut faible à modérée. Ils comprenaient vertige, constipation, purpura, céphalées, nausées, vomissements, perte de poids, fièvre, otite, somnolence et légère tachycardie (100 bpm).
Après commercialisation du produit, des surdosages allant jusqu'à 40 000 mg de felbamate ont été rapportés. La grande majorité des patients s'est rétablie sans incident. Les effets indésirables comprenaient ataxie, nystagmus, diplopie, agitation, cristallurie ou coma. Des décès ont été rapportés chez des patients qui avaient pris de multiples médicaments y compris le felbamate.
En cas de surdosage, une thérapeutique symptomatique globale doit être instituée. On ignore si le felbamate est dialysable.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Système nerveux, antiépileptique, code ATC : N03AX10.
Le felbamate est un nouvel antiépileptique, tant du point de vue chimique que pharmacologique. Il s'agit d'un dicarbamate dont la structure diffère de celle des autres carbamates connus. Son mécanisme d'action précis n'est pas élucidé.
Mécanisme d’action
Les études de liaison aux récepteurs conduites in vitro ont montré un effet inhibiteur faible ou nul du felbamate sur les systèmes de liaison aux récepteurs du GABA et des benzodiazépines. De plus, le felbamate est dénué de propriétés excito-toxiques et n'antagonise pas les effets neuro-toxiques du NMDA, du kaïnate ou du quisqualate in vitro ; ce n'est donc pas un antagoniste du NMDA.
Lors des études précliniques pharmacologiques, le profil anti-convulsivant du felbamate a été démontré grâce à de nombreux modèles animaux. Chez la souris, le felbamate est efficace tant sur les crises induites par électrochoc maximal que par injection sous-cutanée de pentylène-tétrazole. Il est également efficace sur les crises déclenchées par la picrotoxine et la bicuculline. L'efficacité du felbamate sur les modèles de crises déclenchées chimiquement ou par électrochoc maximal suggère que le produit exerce son activité antiépileptique en élevant le seuil épileptogène et en prévenant la généralisation des crises.
Efficacité et sécurité clinique
Le felbamate a montré son efficacité thérapeutique grâce à cinq essais contrôlés menés chez des patients souffrant de crises partielles se généralisant secondairement ou non, et un essai mené chez des patients souffrant du syndrome de Lennox-Gastaut. Dans ce dernier essai, ont été traités des patients présentant des crises atoniques, des absences atypiques ou des crises tonico-cloniques généralisées. Les résultats de cet essai, pendant lequel des doses pouvant aller jusqu'à 45 mg/kg/jour ou 3 600 mg/jour ont été administrées, ont mis en évidence une relation entre les concentrations plasmatiques du produit et le contrôle des crises.
Le felbamate a été administré à des doses uniques allant jusqu'à 1 200 mg ou des doses multiples de 200 mg à 600 mg, deux fois par jour pendant 28 jours, chez des volontaires sains. Des sujets épileptiques (avec crises partielles) ont été traités jusqu'à 28 jours et 6 semaines par des doses de 800 mg et 1 200 mg 2 fois par jour, respectivement. Ces essais n'ont montré aucun effet indésirable significatif au niveau des principaux systèmes de l'organisme : SNC, appareil cardio-vasculaire, système hématopoïétique, rénal, hépatique ou respiratoire.
5.2. Propriétés pharmacocinétiques
Après administration orale de felbamate 14C à des volontaires sains, 90 % environ de chaque dose sont retrouvés dans l'urine, et moins de 5 % dans les fèces. La biodisponibilité systémique absolue n'a pas été étudiée. Le felbamate est donc bien absorbé. La prise d’aliments n’affecte pas la vitesse et le degré d’absorption du felbamate.
Biotransformation et élimination
La comparaison des ASC a montré que plus de 85 % de la radioactivité plasmatique correspondait à du felbamate sous forme inchangée.
Outre le felbamate inchangé, les métabolites suivants ont pu être identifiés dans l'urine : le p-hydroxy felbamate, le 2-hydroxy felbamate, des dérivés monocarbamates du felbamate et des métabolites polaires du felbamate (notamment des dérivés conjugués).
Des études à doses uniques et multiples conduites chez des sujets sains et chez des patients épileptiques ont montré que le Tmax apparaissait 2 à 6 heures après la prise. La demi-vie d'élimination terminale du felbamate est comprise entre 15 et 23 heures.
Après administration en doses uniques ou multiples par voie orale à des sujets sains de sexe masculin, le felbamate a montré une cinétique linéaire pour des doses allant jusqu'à 3 600 mg/jour, la Cmax et l'ASC augmentant linéairement avec la dose.
Liaison aux protéines plasmatiques
Chez l'homme, 22 à 25 % du felbamate sont liés aux protéines plasmatiques, essentiellement à l'albumine.
Taux plasmatiques
Des essais cliniques contrôlés ont montré que les concentrations plasmatiques moyennes efficaces du produit étaient comprises entre 32 et 82 microgrammes/ml. Dans un essai sur le syndrome de Lennox-Gastaut, un effet sur les crises atoniques a pu être observé dès la concentration plasmatique de 18 microgrammes/ml. Des taux de felbamate atteignant 137 microgrammes/ml ont été observés chez certains patients traités dans la gamme posologique recommandée, sans problème de tolérance.
Distribution
Le felbamate et ses métabolites traversent la barrière hémato-encéphalique.
Patients ayant une insuffisance rénale
Lors d’un essai de pharmacocinétique à dose unique, la clairance et l’excrétion du felbamate ont été diminuées et la demi-vie augmentée en fonction du degré d’insuffisance rénale. L’ASC du felbamate chez les patients ayant une clairance de la créatinine < 50 ml/min a été augmentée d’environ 100 % par rapport à celle des sujets du groupe témoin (voir rubrique 4.2).
5.3. Données de sécurité préclinique
Des études de toxicité aiguë ont été conduites chez la souris (p.o. ; i.p.), chez le rat (p.o. ; i.p.) et chez le chien (p.o.). La DL50 orale fut supérieure à 5 g/kg tant chez la souris que chez le rat et supérieure à 2 g/kg chez le chien. La toxicité s'est manifestée essentiellement par un ptosis, une ataxie, des tremblements, une diminution de l'activité et du tonus musculaire.
Lors d'études de toxicité conduites chez l'animal sur une durée maximale de un an, des modifications hépatiques suggérant des phénomènes d'induction enzymatique sont apparues chez le rat. Chez le rat et la souris, des études conduites sur deux ans n'ont montré aucune élévation de l'incidence des tumeurs bénignes et malignes ni de l'incidence totale des tumeurs. Une augmentation d'incidence des tumeurs testiculaires interstitielles, observée chez des rats traités à dose élevée, n'a pas été retrouvée avec la dose faible ni lors d'études conduites pendant un an chez le rat et le chien, et pendant deux ans chez la souris. L'observation de ce type de tumeur n'est pas rare chez le rat. Cependant, chez l'homme, les tumeurs des cellules interstitielles des testicules sont rares. La pertinence de ces données pour l'évaluation du risque chez l'homme est incertaine.
*Composition du prosweet G#859 : glycérol (E422), vanilline, éthylmaltol.
Avant ouverture : 5 ans.
Après première ouverture du flacon : 1 mois.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
Conserver le flacon soigneusement fermé afin de le protéger de l’humidité.
6.5. Nature et contenu de l'emballage extérieur
flacon de 230 ml (verre)
flacon de 250 ml (verre)
flacon de 450 ml (verre)
flacon de 230 ml (PEHD)
flacon de 250 ml (PEHD)
flacon de 450 ml (PEHD)
Une seringue doseuse de 5,0 ml avec marquage CE est fournie, pour administration orale (voir rubrique 6.6).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Bien agiter avant l’emploi.
Pas d'exigences particulières pour l'élimination de ce produit.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
176 RUE MONTMARTRE
75002 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 559 158 9 8 : 230 ml en flacon (verre) avec seringue doseuse
· 34009 559 160 3 1 : 250 ml en flacon (verre) avec seringue doseuse
· 34009 559 163 2 1 : 450 ml en flacon (verre) avec seringue doseuse
· 34009 559 159 5 9 : 230 ml en flacon (PEHD) avec seringue doseuse
· 34009 559 162 6 0 : 250 ml en flacon (PEHD) avec seringue doseuse
· 34009 559 165 5 0 : 450 ml en flacon (PEHD) avec seringue doseuse
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation : {JJ mois AAAA}
Date de dernier renouvellement : {JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription initiale hospitalière de 6 mois, réservée aux spécialistes en neurologie ou en pédiatrie.
Renouvellement réservé aux spécialistes en neurologie ou en pédiatrie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 14/04/2025
TALOXA 600 mg/5 ml, suspension buvable
Felbamate
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que TALOXA 600 mg/5 ml, suspension buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre TALOXA 600 mg/5 ml, suspension buvable ?
3. Comment prendre TALOXA 600 mg/5 ml, suspension buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TALOXA 600 mg/5 ml, suspension buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TALOXA 600 mg/5 ml, suspension buvable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : N03AX10
Qu’est-ce que TALOXA 600 mg/5 ml, suspension buvable ?
TALOXA est utilisé comme un médicament antiépileptique. Les crises d'épilepsie sont également connues sous le nom d'épilepsie ou de crises de convulsions.
Dans quels cas TALOXA 600 mg/5 ml, suspension buvable est-il utilisé ?
TALOXA n’est pas destiné à être utilisé chez l’enfant de moins de 4 ans.
TALOXA est utilisé en association avec d’autres médicaments pour contrôler les convulsions chez l'adulte et l'enfant à partir de 4 ans ayant un syndrome de Lennox-Gastaut non contrôlé par les autres antiépileptiques.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE TALOXA 600 mg/5 ml, suspension buvable ?
Ne prenez jamais TALOXA 600 mg/5 ml, suspension buvable :
· si vous êtes allergique au felbamate ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
· si vous avez des antécédents de maladie du sang (tels qu’anémie, taux de cellules sanguines bas, saignements, hématomes, infections fréquentes), ou de maladies du foie (tels que jaunisse [blanc des yeux jaunes ou peau jaune] ou hépatite). Si vous avez eu des antécédents de maladies du sang ou du foie, prévenez votre médecin avant de prendre TALOXA.
Avertissements et précautions
Adressez-vous à votre médecin ou votre pharmacien avant de prendre TALOXA :
· Si vous avez eu des réactions inhabituelles (telles que urticaire, respiration sifflante ou difficultés à respirer) au médicament ou à l’un de ses autres composants.
· Si vous développez une éruption de la peau, de la fièvre, un gonflement des paupières, du nez ou de la gorge, une urticaire, consultez immédiatement votre médecin.
· Assurez-vous de bien vous hydrater en buvant beaucoup de liquide lors de la prise de ce médicament afin de bien l'éliminer de votre organisme et de prévenir les calculs rénaux dans vos urines.
· Des pensées autodestructrices ou suicidaires ont également été observées chez un petit nombre de personnes traitées par des antiépileptiques tels que le felbamate. Si vous avez ce type de pensées, contactez immédiatement votre médecin.
· Si vous prenez régulièrement TALOXA ou tout autre médicament pour contrôler vos convulsions, ne les arrêtez pas brutalement sans l'avis de votre médecin.
· Si vous êtes une femme en âge de procréer, vous devez utiliser une méthode de contraception efficace durant le traitement et jusqu’à 1 mois après la fin du traitement. Le traitement à base de felbamate peut diminuer l’efficacité des contraceptifs oraux, par conséquent une autre méthode de contraception efficace doit être utilisée.
Tests sanguins
Pendant toute la durée du traitement, votre médecin vous prescrira des analyses de sang fréquentes (toutes les 2 semaines) pour surveiller votre état de santé.
Enfants et adolescents
Pour les enfants et adolescents empruntant des voies de circulation (à bicyclette par exemple), des vertiges et une faiblesse peuvent survenir et doivent être prises en compte (voir rubrique « Conduite de véhicules et utilisation de machines »).
Autres médicaments et TALOXA 600 mg/5 ml, suspension buvable
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Assurez-vous que votre médecin connaît les autres médicaments que vous prenez pour contrôler vos convulsions, car il peut être nécessaire d’ajuster la dose de ces médicaments à vos besoins.
L’efficacité des contraceptifs oraux peut être modifiée par TALOXA. Prévenez votre médecin si vous prenez un contraceptif oral.
Si vous prenez TALOXA en même temps qu'un autre médicament antiépileptique, il est possible que vous remarquiez une augmentation des effets indésirables. Si de telles manifestations deviennent gênantes, consultez votre médecin.
TALOXA 600 mg/5 ml, suspension buvable avec des aliments et des boissons
TALOXA n’est pas altéré par les aliments et peut être pris au moment des repas.
TALOXA ne doit pas être utilisé pendant la grossesse, sauf en cas de nécessité absolue. Si vous êtes une femme en âge de procréer vous devez utiliser une méthode de contraception efficace durant le traitement et jusqu’à 1 mois après la fin du traitement. Le traitement à base de felbamate peut diminuer l’efficacité des contraceptifs oraux, par conséquent une autre méthode de contraception efficace doit être utilisée.
Si vous êtes enceinte ou avez l’intention de l’être, consultez rapidement votre médecin, vous ne devez pas arrêter de prendre votre médicament avant d’en avoir discuté avec votre médecin. Votre médecin peut décider de modifier votre traitement.
TALOXA n’est pas recommandé pour les femmes qui allaitent. Le felbamate passe dans le lait maternel. Cela peut nuire à votre bébé, particulièrement par des troubles sanguins ou hépatiques.
Si vous allaitez votre enfant, votre médecin jugera de l’utilité de continuer à prendre TALOXA.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Il est déconseillé d’utiliser certains outils ou machines. Ne conduisez pas ou n’utilisez pas de machines si vous avez des vertiges ou si vous vous sentez faible lors de la prise de ce médicament. Lorsque TALOXA est donné aux enfants et adolescents, il faut tenir compte des étourdissements et de la sensation de faiblesse pouvant survenir en cas d’emprunt des voies de circulation (à bicyclette, par exemple).
TALOXA 600 mg/5 ml, suspension buvable contient
· 1,05 g de sorbitol dans chaque seringue doseuse de 5,0 ml ce qui équivaut à 210 mg/ml. Le sorbitol est une source de fructose. Si votre médecin vous a informé(e) que vous (ou votre enfant) présentiez une intolérance à certains sucres ou si vous avez été diagnostiqué(e) avec une intolérance héréditaire au fructose (IHF), un trouble génétique rare caractérisé par l’incapacité à décomposer le fructose, parlez-en à votre médecin avant que vous (ou votre enfant) ne preniez ou ne receviez ce médicament. Le sorbitol peut causer une gêne gastro-intestinale et un effet laxatif léger.
· Du méthyl- et des parahydroxybenzoates, qui peuvent provoquer des réactions allergiques (pouvant être retardées).
· Moins de 1 mmol (23 mg) de sodium par 5,0 ml, c’est-à-dire qu’il est essentiellement « sans sodium ».
· 10 mg de benzoate de sodium dans 5 ml, ce qui équivaut à 2 mg/ml.
3. COMMENT PRENDRE TALOXA 600 mg/5 ml, suspension buvable ?
Quelle dose de TALOXA 600 mg/5 ml, suspension buvable prendre ?
· Votre médecin déterminera la dose et l’horaire adaptés à votre cas ou celui de votre enfant pour la prise de ce médicament.
· TALOXA est pris en deux (toutes les 12 heures) ou trois prises (toutes les 8 heures) par jour avec de l’eau. Chez certaines personnes, 4 prises quotidiennes (environ toutes les 6 heures) peuvent être nécessaires.
· Votre médecin vous dira ou dira à votre enfant, ainsi qu'à votre pharmacien, quelle dose en millilitres (ml) et combien de doses vous ou votre enfant devrez prendre chaque jour.
Au cours des premières semaines du traitement :
Il est possible que la dose prescrite soit ajustée par le médecin.
Lorsque la dose définitive est fixée :
Veillez à prendre votre traitement aux mêmes heures chaque jour.
Comment prendre TALOXA 600 mg/5 ml, suspension buvable ?
· Assurez-vous de bien agiter le flacon avant de mesurer chaque dose.
· Votre dose ou celle de votre enfant devra être donnée en utilisant la seringue doseuse qui se trouve dans la boîte.
|
|
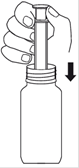
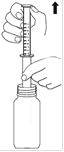
1. Mesure : Votre dose doit être mesurée en utilisant la seringue doseuse de 5,0 ml pour administration orale fournie. La seringue doseuse en plastique pour administration orale est composée de deux parties, un tube opaque et un piston blanc adapté au tube. Le piston comporte des graduations tous les 0,1 ml, démarrant à 0,5 ml (extrémité du piston) et se terminant à 5,0 ml. A. Insérez la seringue doseuse assemblée dans le flacon de TALOXA. B. Tout en maintenant l’embout de la seringue dans la suspension, tirez le piston. Au fur et à mesure que la solution remplit la seringue, vous ou votre enfant verrez apparaître les graduations croissantes du piston. |
|
|
C. Tirez le piston jusqu’à ce que vous ou votre enfant puissiez lire la graduation correspondant à votre dose. D. Retirez la seringue pour administration orale du flacon et vérifiez que la quantité correcte apparaît à l’extrémité de la seringue. Si vous ou votre enfant en avez trop ou trop peu, essayez une nouvelle fois jusqu’à ce que vous ayez la quantité correcte. |
|
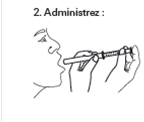
|
2. Administration : Placez la seringue dans votre bouche ou celle de votre enfant et administrez la dose dans la bouche en poussant le piston de la seringue. Avalez la dose. |
|
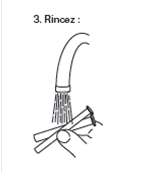
|
3. Rinçage : Démontez la seringue et rincez-la avec de l'eau après chaque utilisation. |
Si vous ou votre enfant avez l’impression que l’effet de TALOXA est trop fort ou trop faible, consultez votre médecin ou votre pharmacien.
Si vous ou votre enfant avez pris plus de TALOXA 600 mg/5 ml, suspension buvable que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous ou votre enfant oubliez de prendre TALOXA 600 mg/5 ml, suspension buvable :
Prenez la dose que vous avez oublié de prendre dès que possible sauf si vous vous en apercevez au moment de la prise de votre prochaine dose.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Il est important de retourner à votre rythme d’administration habituel aussi vite que possible.
Si vous ou votre enfant arrêtez de prendre TALOXA 600 mg/5 ml, suspension buvable :
Sauf avis contraire de votre médecin, il est nécessaire que vous poursuiviez le traitement. Tout arrêt brutal d'un traitement par antiépileptiques, y compris par TALOXA, peut entraîner un risque d’augmentation des crises.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
TALOXA a été associé dans de rares cas à des troubles graves du foie ou du sang, pouvant être mortels.
Contactez immédiatement votre médecin si vous présentez :
· des symptômes inhabituels lors de la prise de TALOXA tels que saignements, bleus, infections fréquentes, fatigue, jaunisse (blanc des yeux jaunes ou peau jaune), perte de poids, vomissements ou douleur abdominale.
· des vésicules dans la bouche, le nez, les yeux ou un gonflement et décollement de la peau, des douleurs aux muscles et aux articulations, de la fièvre, un rash, des vomissements incontrôlés, des ballonnements ou de la constipation.
Effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10)
Perte de poids, perte d’appétit, insomnie, somnolence, instabilité de la marche, vertiges, maux de tête, changements de la vision tels que vision double ou vision anormale, nausées, vomissements, troubles gastriques, douleur abdominale, diarrhée, fatigue.
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100)
Taux anormalement bas de phosphate dans le sang (hypophosphatémie), troubles de la parole, dépression, stupeur, sensation d’anxiété, éruption cutanée (rash), démarche anormale.
Effets indésirables rares (pouvant affecter jusqu’à 1 personne sur 1 000)
Anomalies sanguines incluant thrombocytopénie (diminution des plaquettes), leucopénie (diminution des globules blancs), neutropénie (diminution des globules blancs), et anémie (diminution des globules rouges). Ces anomalies peuvent être combinées, pouvant associer une diminution de ces trois types de cellule et une insuffisance de la moelle osseuse qui produit ces trois types de cellule.
Augmentation de la fréquence des convulsions, choc anaphylactique (réaction allergique sévère de tout le corps), autres réactions allergiques sévères incluant : vésicules dans la bouche, le nez, les yeux et les autres membranes muqueuses (syndrome de Stevens-Johnson, éruption bulleuse), nécrolyse épidermique toxique (gonflement et décollement de la couche supérieure de la peau), douleur dans les muscles ou les articulations, fièvre.
Effets indésirables très rares (pouvant affecter jusqu’à 1 personne sur 10 000)
Constipation, troubles du foie qui peuvent être graves et peuvent inclure une insuffisance hépatique fatale, des cristaux dans les urines.
Effets indésirables supplémentaires chez les enfants et les adolescents
Le profil d’effets indésirables chez les enfants était identique. En plus, des infections des voies respiratoires hautes ont été fréquemment observées chez les enfants ; cependant, le lien de causalité avec le traitement n’est pas probable.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TALOXA 600 mg/5 ml, suspension buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
Avant la première ouverture du flacon
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte/le flacon.
La date de péremption fait référence au dernier jour de ce mois.
Après la première ouverture du flacon
La durée de conservation est d’un mois.
Conserver le flacon soigneusement fermé à l’abri de l’humidité. Jetez le flacon de TALOXA un mois après son ouverture.
A conserver à une température ne dépassant pas 25°C.
Ne pas utiliser TALOXA sans l’avis de votre médecin ou de votre pharmacien si vous remarquez un changement dans l’apparence de la suspension.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient TALOXA 600 mg/5 ml, suspension buvable
· La substance active est :
Felbamate...................................................................................................................... 600 mg
Pour 5 ml de suspension buvable.
· Les autres composants sont :
Sorbitol (E420), glycérol (E422), cellulose dispersible (cellulose microcristalline + carmellose sodique), émulsion de siméthicone, saccharine sodique, parahydroxybenzoate de méthyle (E218), polysorbate 80, parahydroxybenzoate de propyle (E216), benzoate de sodium (E211), prosweet G#859*, eau purifiée.
*Composition du prosweet G#859 : glycérol (E422), vanilline, éthylmaltol.
Qu’est-ce que TALOXA 600 mg/5 ml, suspension buvable et contenu de l’emballage extérieur
Suspension buvable blanche à blanchâtre, opaque et visqueuse.
TALOXA 600 mg/5 ml, suspension buvable se présente sous forme de boîtes de :
flacon de 230 ml (verre)
flacon de 250 ml (verre)
flacon de 450 ml (verre)
flacon de 230 ml (PEHD)
flacon de 250 ml (PEHD)
flacon de 450 ml (PEHD)
Une seringue doseuse de 5,0 ml avec marquage CE est fournie pour administration orale.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
176 RUE MONTMARTRE
75002 PARIS
Exploitant de l’autorisation de mise sur le marché
ORGANON FRANCE
176 RUE MONTMARTRE
75002 PARIS
INDUSTRIEPARK 30
2220 HEIST-OP-DEN-BERG
BELGIQUE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).