Dernière mise à jour le 29/06/2026
HALDOL 2 mg/ml, solution buvable
Indications thérapeutiques
Le nom de votre médicament est HALDOL 2 mg/ml, solution buvable.
HALDOL contient une substance active appelée halopéridol. Il appartient à un groupe de médicaments appelés « antipsychotiques ».
HALDOL est utilisé chez les adultes, les adolescents et les enfants pour traiter des maladies affectant les pensées, les sensations ou le comportement. Cela comprend des problèmes de santé mentale (tels que la schizophrénie et le trouble bipolaire) et des problèmes comportementaux.
Ces maladies peuvent provoquer chez vous :
· un état de confusion (délire)
· voir, entendre, ressentir ou sentir des choses qui ne sont pas réelles (hallucinations)
· croire des choses qui ne sont pas réelles (idées délirantes)
· une suspicion anormale (paranoïa)
· un fort sentiment d’excitation, d’agitation, d’enthousiasme, de l’impulsivité ou de l’hyperactivité
· un comportement très agressif, hostile ou violent.
Chez les adolescents et les enfants, HALDOL est utilisé pour traiter la schizophrénie chez les patients âgés de 13 à 17 ans, et pour traiter les problèmes comportementaux chez les patients âgés de 6 à 17 ans.
HALDOL est également utilisé :
· chez les adolescents et les enfants âgés de 10 à 17 ans et chez les adultes pour traiter les mouvements ou émissions de sons incontrôlables (tics), par exemple dans le syndrome de Gilles de la Tourette sévère
· chez les adultes, pour aider à contrôler les mouvements associés à la maladie de Huntington.
HALDOL est parfois utilisé lorsque les autres médicaments ou traitements n’ont pas fonctionné ou ont provoqué des effets indésirables inacceptables.
Présentations
> 1 flacon(s) compte-gouttes polyéthylène basse densité (PEBD) de 30 ml avec fermeture de sécurité enfant
Code CIP : 269 211-4 ou 34009 269 211 4 9
Déclaration de commercialisation : 18/08/2014
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 1,88 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 2,90 €
- Taux de remboursement :65%
· États psychotiques particuliers (c'est à dire maladies psychiques dans lesquelles le malade peut perdre contact avec la réalité), chez l'adulte.
· Chorées (mouvements anormaux), maladie des tics de Gilles de la Tourette, chez l'enfant de plus de 3 ans et l'adulte.
. Troubles graves du comportement, en particulier chez l'autiste (enfant). ; JOURNAL OFFICIEL ; 07/08/13
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Non précisé | Avis du 25/07/2018 | Extension d'indication non sollicitée | Le laboratoire ne demande pas l’inscription des spécialités HALDOL 1 mg et 5 mg, comprimés et HALDOL 2 mg/ml, solution buvable dans cette indication et rappelle que de ce fait ces spécialités ne sont pas remboursables/agréées aux collectivités dans l’indication : « Traitement de l’agressivité persistante et des symptômes psychotiques chez les patients présentant une démence d’Alzheimer modérée à sévère ou une démence vasculaire en cas d’échec des traitements non pharmacologiques et lorsqu’il existe un risque de préjudice pour le patient lui-même ou autrui. » |
| Important | Avis du 25/01/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par HALDOL 5 mg comprimé et HALDOL 2 mg/ml solution buvable reste important dans le traitement des états psychotiques aigus et chroniques chez l’adulte. Le service médical rendu par HALDOL 2 mg/ml solution buvable et HALDOL 5 mg/ml solution injectable reste important dans le traitement des vomissements lors de traitements antimitotiques post-radiothérapiques chez l’adulte. Le service médical rendu par HALDOL 2 mg/ml solution buvable reste important dans le traitement des chorées (mouvements anormaux), maladie des tics de Gilles de la Tourette chez l’adulte et l’enfant de plus de 3 ans. Le service médical rendu par HALDOL solution injectable reste important dans le traitement de courte durée des états d'agitation et d'agressivité au cours des états psychotiques aigus et chroniques. Le service médical rendu par HALDOL DECANOAS reste important dans le traitement au long cours des états psychotiques chroniques. |
| Modéré | Avis du 25/01/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par HALDOL 1 mg comprimé et HALDOL 2 mg/ml solution buvable reste modéré dans le traitement symptomatique de courte durée de l'anxiété de l'adulte en cas d'échec des thérapeutiques habituelles. Le service médical rendu par HALDOL 2 mg/ml solution buvable reste modéré dans la prise en charge des troubles graves du comportement de l’enfant. La Commission de la transparence a tenu compte du caractère très limité des données cliniques disponibles dans cette indication et d’une place mal définie des antipsychotiques dans la prise en charge des troubles graves du comportement avec agitation et agressivité de l’enfant. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 20/07/2016 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à la présentation déjà inscrite. |
| V (Inexistant) | Avis du 15/05/2013 | Inscription (CT) | Il s’agit d’un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres présentations déjà inscrites. |
| V (Inexistant) | Avis du 17/11/2010 | Extension d'indication | La spécialité HALDOL 2 mg/ml, solution buvable remplace la spécialité HALDOL 0,5 mg/ml solution buvable dont l'AMM a été abrogée le 18 décembre 2009 et n'apporte pas d'amélioration du service médical rendu (ASMR V) dans ces extensions d'indications. |
ANSM - Mis à jour le : 08/04/2026
HALDOL 2 mg/ml, solution buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Halopéridol.............................................................................................................................. 2 mg
Pour 1 ml.
Flacon avec bouchon compte-gouttes : chaque goutte de la solution buvable contient 0,1 mg d’halopéridol.
Excipients à effet notoire :
Chaque ml de la solution buvable contient 1,9 mg de parahydroxybenzoate de méthyle.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution limpide et incolore.
4.1. Indications thérapeutiques
Patients adultes âgés de 18 ans et plus
· Traitement de la schizophrénie et du trouble schizo-affectif.
· Traitement aigu du délire en cas d’échec des traitements non pharmacologiques.
· Traitement des épisodes maniaques modérés à sévères associés au trouble bipolaire de type I.
· Traitement de l’agitation psychomotrice aiguë associée aux troubles psychotiques ou aux épisodes maniaques du trouble bipolaire de type I.
· Traitement de l’agressivité persistante et des symptômes psychotiques chez les patients présentant une démence d’Alzheimer modérée à sévère ou une démence vasculaire en cas d’échec des traitements non pharmacologiques et lorsqu’il existe un risque de préjudice pour le patient lui-même ou autrui.
· Traitement des tics, notamment du syndrome de Gilles de la Tourette, chez les patients sévèrement atteints, après échec des prises en charge éducatives, psychologiques et des autres traitements pharmacologiques.
· Traitement des mouvements choréiques légers à modérés de la maladie de Huntington en cas d’inefficacité ou d’intolérance aux autres traitements.
Patients pédiatriques
Traitement :
· De la schizophrénie chez les adolescents âgés de 13 à 17 ans en cas d’échec ou d’intolérance aux autres traitements pharmacologiques.
· De l’agressivité sévère persistante chez les enfants et les adolescents âgés de 6 à 17 ans atteints d’autisme ou de troubles envahissants du développement, en cas d’échec ou d’intolérance aux autres traitements.
· Des tics, notamment du syndrome de Gilles de la Tourette, chez les enfants et les adolescents âgés de 10 à 17 ans sévèrement atteints, après échec des prises en charge éducatives, psychologiques et des autres traitements pharmacologiques.
4.2. Posologie et mode d'administration
Adultes
Il est recommandé d’initier le traitement à faible dose, celle-ci pouvant ensuite être ajustée en fonction de la réponse du patient. Les patients doivent toujours recevoir la dose minimale efficace (voir rubrique 5.2).
Les doses recommandées pour HALDOL solution buvable sont présentées dans le tableau 1.
Tableau 1 : doses d’halopéridol recommandées chez les adultes âgés de 18 ans et plus
|
Traitement de la schizophrénie et du trouble schizo-affectif · 2 à 10 mg/jour par voie orale, en prise unique ou en 2 prises distinctes. Les patients connaissant un premier épisode schizophrénique répondent généralement à une dose de 2 à 4 mg/jour, tandis que chez les patients ayant présentés des épisodes schizophréniques multiples, des doses allant jusqu’à 10 mg/jour peuvent être nécessaires. · La dose peut être ajustée tous les 1 à 7 jour(s). · Chez la majorité des patients, les doses supérieures à 10 mg/jour n’ont pas montré une plus grande efficacité que les doses inférieures et peuvent être associées à une incidence accrue de symptômes extrapyramidaux. Le rapport bénéfice/risque doit être évalué au cas par cas lorsque des doses supérieures à 10 mg/jour sont envisagées. · La dose maximale est de 20 mg/jour car, au-delà, les risques en termes de sécurité sont supérieurs aux bénéfices cliniques apportés par le traitement. |
|
Traitement aigu du délire en cas d’échec des traitements non pharmacologiques · 1 à 10 mg/jour par voie orale, en prise unique ou en 2 à 3 prises distinctes. · Le traitement doit être initié à la plus faible dose possible, et la dose doit être ajustée par paliers toutes les 2 à 4 heures si l’agitation persiste, jusqu’à un maximum de 10 mg/jour. |
|
Traitement des épisodes maniaques modérés à sévères associés au trouble bipolaire de type I · 2 à 10 mg/jour par voie orale, en prise unique ou en 2 prises distinctes. · La dose peut être ajustée tous les 1 à 3 jour(s). · Chez la majorité des patients, les doses supérieures à 10 mg/jour n’ont pas montré une plus grande efficacité que les doses inférieures et peuvent être associées à une incidence accrue de symptômes extrapyramidaux. Le rapport bénéfice/risque doit être évalué au cas par cas lorsque des doses supérieures à 10 mg/jour sont envisagées. · La dose maximale est de 15 mg/jour car, au-delà, les risques en termes de sécurité sont supérieurs aux bénéfices cliniques apportés par le traitement. · La pertinence de la poursuite du traitement par HALDOL doit être évaluée rapidement après l’initiation du traitement (voir rubrique 4.4). |
|
Traitement de l’agitation psychomotrice aiguë associée aux troubles psychotiques ou aux épisodes maniaques du trouble bipolaire de type I · 5 à 10 mg par voie orale, à renouveler au bout de 12 heures si nécessaire, sans dépasser un maximum de 20 mg/jour. · La pertinence de la poursuite du traitement par HALDOL doit être évaluée rapidement après l’initiation du traitement (voir rubrique 4.4). · Si le patient recevait précédemment de l’halopéridol en injection intramusculaire, le traitement oral par HALDOL doit être initié en appliquant un rapport de conversion de dose initiale de 1/1, et suivi d’un ajustement de la dose en fonction de la réponse clinique. |
|
Traitement de l’agressivité persistante et des symptômes psychotiques chez les patients présentant une démence d’Alzheimer modérée à sévère ou une démence vasculaire en cas d’échec des traitements non pharmacologiques et lorsqu’il existe un risque de préjudice pour le patient lui-même ou autrui · 0,5 à 5 mg/jour par voie orale, en prise unique ou en 2 prises distinctes. · La dose peut être ajustée tous les 1 à 3 jour(s). · La nécessité de poursuivre le traitement doit être réévaluée dans un délai maximal de 6 semaines. |
|
Traitement des tics, notamment du syndrome de Gilles de la Tourette, chez les patients sévèrement atteints, après échec des prises en charge éducatives, psychologiques et des autres traitements pharmacologiques · 0,5 à 5 mg/jour par voie orale, en prise unique ou en 2 prises distinctes. · La dose peut être ajustée tous les 1 à 7 jour(s). · La nécessité de poursuivre le traitement doit être réévaluée tous les 6 à 12 mois.
|
|
Traitement des mouvements choréiques légers à modérés de la maladie de Huntington en cas d’inefficacité ou d’intolérance aux autres traitements · 2 à 10 mg/jour par voie orale, en prise unique ou en 2 prises distinctes. · La dose peut être ajustée tous les 1 à 3 jour(s). |
HALDOL, solution buvable doit être utilisé pour l’administration de doses uniques inférieures à 1 mg que les comprimés d’HALDOL ne permettent pas d’obtenir.
HALDOL, solution buvable en flacon compte-gouttes est destinée à être utilisée pour les doses uniques allant jusqu’à 2 mg d’halopéridol (équivalentes à 20 gouttes).
HALDOL, solution buvable en flacon avec seringue doseuse pour administration orale est destinée à être utilisée pour les doses uniques d’halopéridol de 0,5 mg et plus (équivalent à 0,25 ml et plus).
Le nombre de gouttes ou le volume (ml) requis pour obtenir une dose unique donnée avec HALDOL, solution buvable sont indiqués dans le tableau 2.
Tableau 2 : Table de conversion pour HALDOL, solution buvable (2 mg/mL)
|
mg d’halopéridol |
Nombre de gouttes d’HALDOL (flacon compte-gouttes) |
mL d’HALDOL (flacon avec seringue doseuse pour administration orale) |
|
0,1 mg |
1 goutte |
- |
|
0,2 mg |
2 gouttes |
- |
|
0,3 mg |
3 gouttes |
- |
|
0,4 mg |
4 gouttes |
- |
|
0,5 mg |
5 gouttes |
0,25 mL |
|
1 mg |
10 gouttes |
0,5 mL |
|
2 mg |
20 gouttes |
1 mL |
|
5 mg |
- |
2,5 mL |
|
10 mg |
- |
5 mL |
|
15 mg |
- |
7,5 mL |
|
20 mg |
- |
10 mL |
Sevrage thérapeutique
Un arrêt progressif de l’halopéridol est conseillé (voir rubrique 4.4).
Oubli de dose
En cas d’oubli, il est recommandé que les patients prennent la dose suivante à l’heure habituelle et qu’ils ne prennent pas de dose double.
Populations particulières
Personnes âgées
Les études cliniques évaluant l’halopéridol par voie orale dans le traitement des tics, notamment du syndrome de Gilles de la Tourette, n’incluaient pas de patients âgés de 65 ans et plus.
Chez les patients âgés, il est recommandé d’instaurer le traitement en utilisant les doses d’halopéridol suivantes :
· Traitement de l’agressivité persistante et des symptômes psychotiques chez les patients présentant une démence d’Alzheimer modérée à sévère ou une démence vasculaire en cas d’échec des traitements non pharmacologiques et lorsqu’il existe un risque de préjudice pour le patient lui-même ou autrui : 0,5 mg/jour.
· Pour toutes les autres indications : la moitié de la plus faible dose utilisée chez l’adulte.
La dose d’halopéridol peut être ajustée en fonction de la réponse du patient au traitement. Une augmentation prudente et progressive de la dose est recommandée chez les patients âgés.
Chez les patients âgés, la dose maximale est de 5 mg/jour.
Des doses supérieures à 5 mg/jour ne doivent être envisagées que chez les patients qui ont préalablement toléré des doses supérieures et après réévaluation du rapport bénéfice/risque pour chaque patient.
Insuffisance rénale
L’influence de l’insuffisance rénale sur la pharmacocinétique de l’halopéridol n’a pas été évaluée. Aucun ajustement de la dose n’est recommandé, néanmoins il est conseillé de procéder avec prudence lors de l’utilisation du traitement chez des patients atteints d’insuffisance rénale. Cependant, en cas d’insuffisance rénale sévère, il peut être nécessaire d’utiliser une dose initiale plus faible et d’ajuster ensuite la dose par paliers plus petits et plus espacés que chez les patients ne présentant pas d’insuffisance rénale (voir rubrique 5.2).
Insuffisance hépatique
L’influence de l’insuffisance hépatique sur la pharmacocinétique de l’halopéridol n’a pas été évaluée. L’halopéridol étant très largement métabolisé dans le foie, il est recommandé de réduire la dose initiale de moitié et d’ajuster la dose par paliers plus petits et plus espacés que chez les patients ne présentant pas d’insuffisance hépatique (voir rubriques 4.4 et 5.2).
Population pédiatrique
Les doses recommandées pour HALDOL solution buvable sont présentées dans le tableau 3.
Tableau 3 : doses d’halopéridol recommandées chez les patients pédiatriques
|
Traitement de la schizophrénie chez les adolescents âgés de 13 à 17 ans en cas d’échec ou d’intolérance aux autres traitements pharmacologiques · La dose recommandée est de 0,5 à 3 mg/jour par voie orale à répartir de préférence en plusieurs prises (2 à 3 prises par jour). · Il est recommandé d’évaluer le rapport bénéfice/risque au cas par cas lorsqu’une dose supérieure à 3 mg/jour est envisagée. · La dose maximale recommandée est de 5 mg/jour. · La durée du traitement doit être évaluée au cas par cas. |
|
Traitement de l’agressivité sévère persistante chez les enfants et les adolescents âgés de 6 à 17 ans atteints d’autisme ou de troubles envahissants du développement, en cas d’échec ou d’intolérance aux autres traitements · La dose recommandée est de 0,5 à 3 mg/jour chez les enfants âgés de 6 à 11 ans et de 0,5 à 5 mg/jour chez les adolescents âgés de 12 à 17 ans, par voie orale à répartir de préférence en plusieurs prises (2 à 3 prises par jour). · La nécessité de poursuivre le traitement doit être réévaluée au bout de 6 semaines. |
|
Traitement des tics, notamment du syndrome de Gilles de la Tourette, chez les enfants et les adolescents âgés de 10 à 17 ans sévèrement atteints, après échec des prises en charge éducatives, psychologiques et des autres traitements pharmacologiques · La dose recommandée est de 0,5 à 3 mg/jour chez les enfants et les adolescents âgés de 10 à 17 ans, par voie orale à répartir de préférence en plusieurs prises (2 à 3 prises par jour). · La nécessité de poursuivre le traitement doit être réévaluée tous les 6 à 12 mois. |
La sécurité et l’efficacité d’HALDOL solution buvable n’ont pas été établies chez les enfants dont l’âge est inférieur à celui défini dans les indications. Aucune donnée n’est disponible concernant les enfants âgés de moins de 3 ans.
Mode d’administration
HALDOL solution buvable doit être administré par voie orale. La solution peut être mélangée à de l’eau pour en faciliter l’administration, mais elle ne doit pas être mélangée à un autre liquide. Une fois diluée, la solution doit être prise immédiatement.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· État comateux.
· Dépression du système nerveux central (SNC).
· Maladie de Parkinson.
· Démence à corps de Lewy.
· Paralysie supranucléaire progressive.
· Allongement connu de l’intervalle QTc ou syndrome du QT long congénital.
· Infarctus du myocarde aigu récent.
· Insuffisance cardiaque non compensée.
· Antécédents d’arythmies ventriculaires ou de torsades de pointes.
· Hypokaliémie non corrigée.
· Traitement concomitant par des médicaments allongeant l’intervalle QT (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Mortalité accrue chez les personnes âgées atteintes de démence
De rares cas de mort subite ont été signalés chez des patients atteints de troubles psychiatriques traités par des antipsychotiques, notamment par l’halopéridol (voir rubrique 4.8).
Les patients âgés atteints de psychose liée à une démence et traités par des antipsychotiques sont exposés à un risque accru de mortalité. L’analyse de 17 études contrôlées contre placebo (d’une durée modale de 10 semaines), portant essentiellement sur des patients traités par des antipsychotiques atypiques, a révélé un risque de mortalité compris, chez les patients traités, entre 1,6 et 1,7 fois le risque correspondant chez les patients sous placebo. Sur la durée d’une étude contrôlée typique de 10 semaines, le taux de mortalité a été d’environ 4,5 % chez les patients traités par des antipsychotiques, contre 2,6 % environ dans le groupe placebo. Bien que les causes de mortalité aient été diverses, la plupart des décès sont apparus être d’origine cardiovasculaire (insuffisance cardiaque, mort subite, par exemple) ou infectieuse (pneumonie, par exemple). Les études observationnelles semblent indiquer que le traitement par l’halopéridol chez les patients âgés est également associé à une mortalité accrue. Cette association pourrait être plus importante avec l’halopéridol qu’avec les antipsychotiques atypiques ; elle est plus marquée pendant les 30 premiers jours suivant le début du traitement et persiste pendant au moins 6 mois. Il n’a pas été clairement établi dans quelle mesure cette association est imputable au médicament ou plutôt liée aux conditions du patient.
Effets cardiovasculaires
Outre les cas de mort subite, des allongements de l’intervalle QTc et/ou des arythmies ventriculaires ont été signalés avec l’halopéridol (voir rubriques 4.3 et 4.8). Le risque de survenue de ces événements semble être plus élevé à forte dose, à forte concentration plasmatique, chez les patients prédisposés ou en cas d’administration par voie parentérale, en particulier intraveineuse.
La prudence est conseillée chez les patients présentant une bradycardie, une maladie cardiaque, des antécédents familiaux d’allongement du QTc ou des antécédents de consommation importante d’alcool. La prudence est également requise chez les patients susceptibles de présenter des concentrations plasmatiques élevées (voir rubrique 4.4, Métaboliseurs lents du CYP2D6).
Il est recommandé de réaliser un ECG à l’initiation traitement. La nécessité d’effectuer des ECG de contrôle pendant le traitement pour surveiller l’allongement de l’intervalle QTc et les arythmies ventriculaires doit être évaluée chez tous les patients. En cas d’allongement du QTc en cours de traitement, il est recommandé de réduire la dose, mais la prise d’halopéridol doit être interrompue si le QTc dépasse 500 ms.
Les déséquilibres électrolytiques tels que l’hypokaliémie et l’hypomagnésémie augmentent le risque d’arythmies ventriculaires et doivent donc être corrigés avant de commencer le traitement par l’halopéridol. Par conséquent, un bilan initial des électrolytes suivi de contrôles réguliers est recommandé.
Des cas de tachycardie et d’hypotension (notamment d’hypotension orthostatique) ont également été signalés (voir rubrique 4.8). La prudence est recommandée lors de l’administration d’halopéridol chez des patients enclins à l’hypotension ou à l’hypotension orthostatique.
Événements vasculaires cérébraux
Lors des études cliniques randomisées, contrôlées contre placebo, menées chez des patients atteints de démence, une augmentation d’un facteur 3 environ du risque d’événements vasculaires cérébraux indésirables a été observée avec certains antipsychotiques atypiques. Les études observationnelles qui ont comparé le taux d’Accident Vasculaire Cérébral (AVC) chez les patients âgés exposés à des antipsychotiques, tous types confondus, au taux d’AVC chez ceux non exposés à des médicaments de ce type ont constaté que le taux d’AVC était plus élevé parmi les patients exposés. Cette majoration pourrait être plus importante avec l’ensemble des butyrophénones, y compris l’halopéridol. Le mécanisme à l’origine de cette augmentation du risque n’est pas connu. Une augmentation du risque ne peut être exclue chez les autres populations de patients. HALDOL doit être utilisé avec prudence chez les patients présentant des facteurs de risque d’AVC.
Syndrome malin des neuroleptiques
L’halopéridol a été associé à des cas de syndrome malin des neuroleptiques, une réaction idiosyncrasique rare caractérisée par une hyperthermie, une rigidité musculaire généralisée, une instabilité du système nerveux autonome, des troubles de la conscience et une augmentation des taux sériques de créatine phosphokinase. L’hyperthermie est souvent un signe précoce de ce syndrome. Le traitement antipsychotique doit être immédiatement interrompu et un traitement symptomatique approprié doit être mis en place, ainsi qu’une surveillance rapprochée.
Dyskinésie tardive
Une dyskinésie tardive peut apparaître chez certains patients traités au long cours ou après l’arrêt du médicament. Le syndrome est principalement caractérisé par des mouvements répétitifs involontaires de la langue, du visage, de la bouche ou de la mâchoire. Les manifestations peuvent être permanentes chez certains patients. Le syndrome peut être occulté par la réinstauration du traitement, l’augmentation de la dose ou le passage à un antipsychotique différent. Si des signes et symptômes de dyskinésie tardive apparaissent, l’arrêt de tous les antipsychotiques, y compris d’HALDOL, doit être envisagé.
Symptômes extrapyramidaux
Des symptômes extrapyramidaux peuvent survenir (par exemple, tremblement, rigidité, hypersalivation, bradykinésie, akathisie, dystonie aiguë). L’utilisation d’halopéridol a été associée à l’apparition d’une akathisie, caractérisée par une sensation subjective d’agitation désagréable ou éprouvante et un besoin de bouger, souvent accompagnés d’une incapacité à rester assis ou debout sans bouger. Cet effet survient le plus souvent pendant les premières semaines du traitement. Chez les patients développant ces symptômes, une augmentation de la dose peut être délétère.
Une dystonie aiguë peut survenir pendant les premiers jours du traitement par HALDOL, mais son apparition a également été signalée plus tardivement et à la suite d’augmentations de la dose. Les symptômes dystoniques peuvent comprendre le torticolis, les grimaces faciales, le trismus, la protrusion linguale et les mouvements oculaires anormaux, notamment la crise oculogyre (liste non exhaustive). Le risque de survenue de ces réactions est plus important chez les patients de sexe masculin et les plus jeunes. En cas de dystonie aiguë, il peut être nécessaire d’arrêter la prise du médicament.
Si nécessaire, il est possible de prescrire des médicaments antiparkinsoniens de type anticholinergique pour corriger les symptômes extrapyramidaux, mais il est recommandé de ne pas les prescrire de façon systématique à titre préventif. Lorsqu’un traitement concomitant par un antiparkinsonien est requis, il peut être nécessaire de le poursuivre après l’arrêt d’HALDOL, s’il est excrété plus rapidement que l’halopéridol, afin d’éviter l’apparition ou l’aggravation de symptômes extrapyramidaux. Le risque d’augmentation de la pression intra-oculaire doit être pris en compte lorsque des médicaments anticholinergiques, y compris des médicaments antiparkinsoniens, sont administrés en concomitance avec HALDOL.
Crises épileptiques/convulsions
La survenue de crises épileptiques déclenchées par l’halopéridol a été signalée. La prudence est requise chez les patients atteints d’épilepsie ou présentant des prédispositions aux crises épileptiques (sevrage alcoolique et lésions cérébrales, par exemple).
Sécurité hépatobiliaire
L’halopéridol étant métabolisé par le foie, il est conseillé d’ajuster la dose et de procéder avec prudence chez les patients atteints d’insuffisance hépatique (voir rubriques 4.2 et 5.2). Des cas isolés d’anomalies de la fonction hépatique ou d’hépatite, le plus souvent cholestatique, ont été signalés (voir rubrique 4.8).
Sécurité endocrinienne
La thyroxine peut favoriser l’apparition d’effets toxiques de l’halopéridol. Chez les patients atteints d’hyperthyroïdie, le traitement antipsychotique ne devra être utilisé qu’avec précaution et devra toujours être accompagné d’un traitement visant à rétablir l’euthyroïdie.
Les effets hormonaux des antipsychotiques comprennent l’hyperprolactinémie, laquelle peut entraîner une galactorrhée, une gynécomastie et une oligoménorrhée ou une aménorrhée (voir rubrique 4.8). Les études de cultures tissulaires semblent indiquer que le développement des cellules au sein des tumeurs mammaires humaines pourrait être stimulé par la prolactine. Bien qu’aucune association claire n’ait été démontrée entre l’administration d’antipsychotiques et les tumeurs mammaires humaines lors des études cliniques et épidémiologiques, la prudence est recommandée en présence d’antécédents médicaux pertinents. HALDOL doit être utilisé avec précaution chez les patients présentant une hyperprolactinémie préexistante ou de possibles tumeurs prolactino‑dépendantes (voir rubrique 5.3).
Des cas d’hypoglycémie et de syndrome de sécrétion inappropriée d’hormone antidiurétique ont été signalés avec l’halopéridol (voir rubrique 4.8).
Thromboembolie veineuse
Des cas de thromboembolie veineuse (TEV) ont été signalés avec les antipsychotiques. Étant donné que les patients traités par des antipsychotiques présentent souvent des facteurs de risque acquis de TEV, tous les facteurs de risque éventuels de TEV doivent être identifiés avant et pendant le traitement par HALDOL et des mesures préventives doivent être prises.
Réponse au traitement et sevrage
Dans le cadre de la schizophrénie, la réponse au traitement antipsychotique peut être différée.
Si les antipsychotiques sont arrêtés, la réapparition des symptômes liés à la pathologie sous-jacente peut ne pas être perceptible pendant plusieurs semaines ou mois.
Dans de très rares cas, des symptômes de sevrage aigus (incluant des nausées, des vomissements et des insomnies) ont été signalés après l’arrêt brutal d’antipsychotiques à haute dose. Un arrêt progressif est conseillé par mesure de précaution.
Patients atteints de dépression
Il est recommandé de ne pas utiliser HALDOL seul lorsque la dépression est l’affection prédominante chez un patient. Le médicament peut être associé à des antidépresseurs pour traiter les patients chez lesquels coexistent une dépression et une psychose (voir rubrique 4.5).
Passage d’une phase maniaque à une phase dépressive
Lors du traitement des épisodes maniaques chez les patients atteints de troubles bipolaires, il existe un risque de passage de la phase maniaque à une phase dépressive. Il est important de surveiller ce passage à un épisode dépressif et les risques associés, tels qu’un comportement suicidaire, afin de pouvoir intervenir le cas échéant.
Métaboliseurs lents du CYP2D6
HALDOL doit être utilisé avec précaution chez les patients connus pour être des métaboliseurs lents du cytochrome P450 (CYP) 2D6 et recevant en parallèle un inhibiteur du CYP3A4.
Population pédiatrique
Les données de sécurité disponibles concernant la population pédiatrique indiquent un risque de survenue de symptômes extrapyramidaux, notamment une dyskinésie tardive, et d’une sédation. Les données de sécurité disponibles concernant l’usage à long terme sont limitées.
Excipients d’HALDOL
HALDOL, solution buvable contient du parahydroxybenzoate de méthyle et peut provoquer des réactions allergiques (éventuellement retardées).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les études d’interaction n’ont été réalisées que chez l’adulte.
Effets cardiovasculaires
L’utilisation d’HALDOL est contre-indiquée en association avec les médicaments connus pour allonger l’intervalle QTc (voir rubrique 4.3), par exemple :
· les anti-arythmiques de classe IA (p. ex., disopyramide, quinidine).
· les anti-arythmiques de classe III (p. ex., amiodarone, dofétilide, dronédarone, ibutilide, sotalol).
· certains antidépresseurs (p. ex., citalopram, escitalopram).
· certains antibiotiques (p. ex., azithromycine, clarithromycine, érythromycine, lévofloxacine, moxifloxacine, télithromycine).
· d’autres antipsychotiques (p. ex., dérivés de la phénothiazine, sertindole, pimozide, ziprasidone).
· certains antifongiques (p. ex., pentamidine).
· certains antipaludéens (p. ex., halofantrine).
· certains traitements gastro-intestinaux (p. ex., dolasétron).
· certains médicaments utilisés pour le traitement du cancer (p. ex., torémifène, vandétanib).
· certains autres médicaments (p. ex., bépridil, méthadone).
Cette liste n’est pas exhaustive.
La prudence est conseillée lorsque HALDOL est utilisé en association avec des médicaments connus pour provoquer un déséquilibre électrolytique (voir rubrique 4.4).
Médicaments pouvant augmenter les concentrations plasmatiques de l’halopéridol
L’halopéridol est métabolisé par plusieurs voies (voir rubrique 5.2). Les principales voies sont la glucurono-conjugaison et la réduction cétonique. Le système enzymatique du cytochrome P450 est également impliqué, en particulier le CYP3A4 et, dans une moindre mesure, le CYP2D6. L’inhibition de ces voies de métabolisation par un autre médicament ou la réduction de l’activité enzymatique du CYP2D6 peuvent conduire à une augmentation des concentrations de l’halopéridol. Les effets de l’inhibition du CYP3A4 et de la réduction de l’activité enzymatique du CYP2D6 peuvent être cumulatifs (voir rubrique 5.2). D’après les informations limitées et parfois contradictoires qui sont disponibles, l’augmentation potentielle des concentrations plasmatiques de l’halopéridol, lors de l’administration concomitante d’un inhibiteur du CYP3A4 et/ou du CYP2D6, peut être comprise entre 20 % et 40 % bien que, dans certains cas, des augmentations allant jusqu’à 100 % aient été rapportées. Les médicaments susceptibles de provoquer une augmentation des concentrations plasmatiques de l’halopéridol (d’après l’expérience clinique ou le mécanisme d’interaction médicamenteuse) comprennent, par exemple :
· les inhibiteurs du CYP3A4 : alprazolam, fluvoxamine, indinavir, itraconazole, kétoconazole, néfazodone, posaconazole, saquinavir, vérapamil, voriconazole.
· les inhibiteurs du CYP2D6 : bupropion, chlorpromazine, duloxétine, paroxétine, prométhazine, sertraline, venlafaxine.
· les inhibiteurs combinés du CYP3A4 et du CYP2D6 : fluoxétine, ritonavir.
· des médicaments dont le mécanisme est incertain : buspirone.
Cette liste n’est pas exhaustive.
L’augmentation des concentrations plasmatiques de l’halopéridol peut entraîner une majoration du risque d’effets indésirables, notamment d’allongement du QTc (voir rubrique 4.4). Des allongements du QTc ont été observés lorsque l’halopéridol a été administré avec une association des inhibiteurs métaboliques que sont le kétoconazole (400 mg/jour) et la paroxétine (20 mg/jour).
Chez les patients prenant de l’halopéridol en concomitance avec des médicaments de ce type, il est recommandé de surveiller les signes ou symptômes d’une majoration ou d’une prolongation des effets pharmacologiques de l’halopéridol et de réduire la dose d’HALDOL si nécessaire.
Médicaments pouvant réduire les concentrations plasmatiques de l’halopéridol
L’administration concomitante d’halopéridol et d’inducteurs enzymatiques puissants du CYP3A4 peut entraîner une diminution progressive des concentrations plasmatiques de l’halopéridol au point d’en réduire potentiellement l’efficacité, par exemple :
· Carbamazépine, phénobarbital, phénytoïne, rifampicine, millepertuis (Hypericum perforatum).
Cette liste n’est pas exhaustive.
Une induction enzymatique peut être observée au bout de quelques jours de traitement. L’induction enzymatique atteint généralement son niveau maximal en l’espace de 2 semaines environ et peut ensuite persister pendant une durée similaire après l’arrêt du traitement par le médicament. En cas de traitement concomitant par des inducteurs du CYP3A4, il est recommandé de maintenir les patients sous surveillance et d’augmenter la dose d’HALDOL si nécessaire. Après l’arrêt de l’inducteur du CYP3A4, la concentration de l’halopéridol peut augmenter progressivement et il peut donc être nécessaire de réduire la dose d’HALDOL.
Le valproate de sodium est connu pour inhiber la glucurono-conjugaison mais n’altère pas les concentrations plasmatiques de l’halopéridol.
Effets de l’halopéridol sur les autres médicaments
L’halopéridol peut amplifier la dépression du SNC induite par l’alcool ou par les médicaments dépresseurs du SNC, notamment les hypnotiques, les sédatifs ou les analgésiques puissants. Une majoration de l’effet sur le SNC a également été rapportée en association avec la méthyldopa.
L’halopéridol peut antagoniser l’action de l’adrénaline et des autres médicaments sympathomimétiques (stimulants tels que les amphétamines, par exemple) et inverser les effets hypotenseurs des antagonistes adrénergiques tels que la guanéthidine.
L’halopéridol peut antagoniser les effets de la lévodopa et des autres agonistes de la dopamine.
L’halopéridol est un inhibiteur du CYP2D6. L’halopéridol inhibe le métabolisme des antidépresseurs tricycliques (imipramine, désipramine, par exemple), ce qui conduit à une augmentation des concentrations plasmatiques de ces médicaments.
Autres formes d’interactions
Dans de rares cas, les symptômes suivants ont été signalés lors de l’utilisation concomitante de lithium et d’halopéridol : encéphalopathie, symptômes extrapyramidaux, dyskinésie tardive, syndrome malin des neuroleptiques, syndrome cérébelleux aigu et coma. La plupart de ces symptômes ont été réversibles. Il n’a pas été clairement établi s’il s’agit là d’une entité clinique distincte.
En cas de traitement concomitant par le lithium et par HALDOL, il est néanmoins conseillé d’arrêter immédiatement le traitement si des symptômes de ce type apparaissent.
Un antagonisme des effets de la phénindione, un anticoagulant, a été rapporté.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les nouveau-nés exposés aux antipsychotiques (y compris l’halopéridol) pendant le troisième trimestre de la grossesse courent un risque de réactions indésirables, notamment des symptômes extrapyramidaux et/ou des symptômes de sevrage, dont la sévérité et la durée peuvent varier, après l’accouchement. Des cas d’agitation, d’hypertonie, d’hypotonie, de tremblement, de somnolences, de détresse respiratoire et de troubles alimentaires ont été signalés. Par conséquent, il est recommandé de surveiller étroitement les nouveau-nés.
Allaitement
L’halopéridol est excrété dans le lait maternel. De faibles quantités d’halopéridol ont été détectées dans le plasma et l’urine de nouveau-nés allaités par des mères traitées par l’halopéridol. Il n’existe pas de données suffisantes concernant les effets de l’halopéridol chez les nouveau-nés allaités. Une décision doit être prise soit d’interrompre l’allaitement soit d’interrompre le traitement avec HALDOL en prenant en compte le bénéfice de l’allaitement pour l’enfant au regard du bénéfice du traitement pour la femme.
Fertilité
L’halopéridol augmente le taux de prolactine. L’hyperprolactinémie peut inhiber la GnRH hypothalamique, entraînant une réduction de la sécrétion de gonadotrophine par l’hypophyse. Ceci peut inhiber la fonction de reproduction en altérant la stéroïdogenèse gonadique chez les femmes comme chez les hommes (voir rubrique 4.4).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
La sécurité de l’halopéridol a été évaluée chez 284 patients traités par l’halopéridol dans le cadre de 3 études cliniques contrôlées contre placebo et chez 1 295 patients traités par l’halopéridol dans le cadre de 16 études cliniques en double aveugle contrôlées contre comparateur actif.
D’après les données de sécurité compilées issues de ces études cliniques, les effets indésirables les plus fréquemment signalés ont été : syndrome extrapyramidal (34 %), insomnies (19 %), agitation (15 %), hyperkinésie (13 %), céphalées (12 %), trouble psychotique (9 %), dépression (8 %), prise de poids (8 %), tremblement (8 %), hypertonie (7 %), hypotension orthostatique (7 %), dystonie (6 %) et somnolences (5 %).
Par ailleurs, la sécurité du décanoate d’halopéridol a été évaluée chez 410 patients dans le cadre de 3 études comparatives (1 comparant le décanoate d’halopéridol à la fluphénazine et 2 comparant la formulation sous forme de décanoate à l’halopéridol oral), de 9 études en ouvert et d’une étude dose-réponse.
Le tableau 4 présente les effets indésirables qui ont été :
· signalés au cours des études cliniques avec l’halopéridol ;
· signalés au cours des études cliniques avec le décanoate d’halopéridol en lien avec la fraction active ;
· identifiés dans le cadre de la surveillance post-commercialisation avec l’halopéridol et le décanoate d’halopéridol.
La fréquence des effets indésirables repose sur (ou estimée d’après) les essais cliniques ou les études épidémiologiques concernant l’halopéridol et classée suivant la convention ci-dessous :
Très fréquent : ≥ 1/10
Fréquent : ≥ 1/100, < 1/10
Peu fréquent : ≥ 1/1 000, < 1/100
Rare : ≥ 1/10 000, < 1/1 000
Très rare : < 1/10 000
Fréquence indéterminée : ne peut être estimée sur la base des données disponibles.
Les effets indésirables sont présentés par classe de système d’organes et par ordre décroissant de gravité au sein de chaque catégorie de fréquence.
Tableau 4 : effets indésirables
|
Classe de système d’organes |
Effet indésirable |
||||
|
|
Fréquence |
||||
|
|
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Fréquence indéterminée |
|
Affections hématologiques et du système lymphatique |
|
|
Leucopénie |
|
Pancytopénie Agranulocytose Thrombopénie Neutropénie |
|
Affections du système immunitaire |
|
|
Hypersensibilité |
|
Réaction anaphylactique |
|
Affections endocriniennes |
|
|
|
Hyperprolactinémie |
Sécrétion inappropriée d’hormone antidiurétique |
|
Troubles du métabolisme et de la nutrition |
|
|
|
|
Hypoglycémie |
|
Affections psychiatriques |
Agitation Insomnies |
Trouble psychotique
|
État de confusion Perte de la libido Diminution de la libido Nervosité |
|
|
|
Affections du système nerveux |
Syndrome extrapyramidal Hyperkinésie Céphalées |
Dyskinésie tardive Akathisie Bradykinésie Dyskinésie Dystonie Hypokinésie Sensations vertigineuses Somnolences Tremblement |
Convulsion Parkinsonisme Sédation Contractions musculaires involontaires |
Syndrome malin des neuroleptiques Dysfonction motrice Nystagmus |
Akinésie Signe de la roue dentée Faciès figé
|
|
Affections oculaires |
|
Crise oculogyre Troubles de la vision |
Vision floue |
|
|
|
Affections cardiaques |
|
|
Tachycardie |
|
Fibrillation ventriculaire Torsades de pointes Tachycardie ventriculaire Extrasystoles |
|
Affections vasculaires |
|
Hypotension |
|
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Dyspnée |
Bronchospasme |
Œdème laryngé Laryngospasme |
|
Affections gastro-intestinales |
|
Vomissements Nausées Constipation Sécheresse buccale Hypersécrétion salivaire |
|
|
|
|
Affections hépatobiliaires |
|
Anomalies du bilan hépatique |
Hépatite Ictère |
|
Insuffisance hépatique aiguë Cholestase |
|
Affections de la peau et du tissu sous-cutané |
|
Éruption cutanée |
Réaction de photosensibilité Urticaire Prurit Hyperhidrose |
|
Angio-œdème Vascularite leucocytoclasique |
|
Affections musculo-squelettiques et systémiques |
|
|
Torticolis Spasmes musculaires Raideur musculo-squelettique |
Trismus Fasciculations |
Rhabdomyolyse |
|
Affections du rein et des voies urinaires |
|
Rétention urinaire |
|
|
|
|
Affections gravidiques, puerpérales et périnatales |
|
|
|
|
Syndrome de sevrage médicamenteux chez le nouveau-né (voir rubrique 4.6) |
|
Affections des organes de reproduction et du sein |
|
Dysfonction érectile |
Aménorrhée Galactorrhée Dysménorrhée Douleur mammaire Gêne mammaire |
Ménorragie Troubles menstruels Dysfonction sexuelle |
Priapisme Gynécomastie |
|
Troubles généraux et anomalies au site d’administration |
|
|
Hyperthermie Œdème Troubles de la démarche |
|
Mort subite Œdème de la face Hypothermie |
|
Investigations |
|
Prise de poids Perte de poids |
|
Allongement du QT à l’électrocardiogramme |
|
Des allongements de l’intervalle QT à l’électrocardiogramme, des arythmies ventriculaires (fibrillation ventriculaire, tachycardie ventriculaire), des torsades de pointes et des morts subites ont été signalés avec l’halopéridol.
Effets de classe des antipsychotiques
Des arrêts cardiaques ont été signalés avec les antipsychotiques.
Des cas de thromboembolie veineuse, y compris des cas d’embolie pulmonaire et de thrombose veineuse profonde, ont été signalés avec les antipsychotiques. La fréquence de ces événements n’est pas connue.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
Signes et symptômes
Le surdosage de l’halopéridol se manifeste par une exagération des effets pharmacologiques et indésirables connus du médicament. Les symptômes prédominants sont des réactions extrapyramidales sévères, une hypotension et une sédation. Les réactions extrapyramidales se présentent sous la forme d’une rigidité musculaire et d’un tremblement généralisé ou localisé. Il est également possible qu’une hypertension survienne au lieu d’une hypotension.
Dans les cas extrêmes, le patient paraîtra comateux, avec une dépression respiratoire et une hypotension suffisamment sévères pour entraîner un état comparable à l’état de choc. Le risque d’arythmies ventriculaires, potentiellement associées à un allongement du QTc, doit être envisagé.
Traitement
Il n’existe aucun antidote spécifique. Le traitement sera symptomatique. L’efficacité du charbon actif n’a pas été établie. La dialyse ne permettant d’éliminer que de très faibles quantités d’halopéridol, son utilisation n’est pas recommandée pour le traitement du surdosage (voir rubrique 5.2).
Si le patient est comateux, ses voies aériennes doivent être maintenues dégagées au moyen d’une canule oropharyngée ou d’une sonde endotrachéale. Une ventilation artificielle peut être nécessaire en cas de dépression respiratoire.
Il est recommandé de surveiller l’ECG et les signes vitaux et de poursuivre la surveillance jusqu’à ce que l’ECG soit normal. Il est également recommandé de traiter les arythmies sévères en prenant les mesures anti‑arythmiques appropriées.
L’hypotension et le collapsus cardiovasculaire peuvent être contrebalancés au moyen d’un remplissage vasculaire, de plasma ou d’albumine concentrée et d’agents vasopresseurs tels que la dopamine ou la noradrénaline. L’adrénaline ne doit pas être utilisée car elle pourrait provoquer une hypotension profonde en présence d’halopéridol.
En cas de réaction extrapyramidale sévère, il est recommandé d’administrer un médicament antiparkinsonien par voie parentérale.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L’halopéridol est un antipsychotique de la famille des butyrophénones. Il s’agit d’un puissant antagoniste central des récepteurs dopaminergiques de type 2 qui, aux doses recommandées, exerce une faible activité alpha‑1 anti-adrénergique et n’a aucune activité anti-histaminergique ou anticholinergique.
Effets pharmacodynamiques
L’halopéridol inhibe les idées délirantes et les hallucinations en conséquence directe du blocage de la signalisation dopaminergique au sein de la voie mésolimbique. L’effet de blocage central de la dopamine exerce une activité sur les noyaux gris centraux (faisceaux nigro-striés). L’halopéridol provoque une sédation psychomotrice efficace, ce qui explique son effet positif sur la manie et les autres syndromes d’agitation.
L’action sur les noyaux gris centraux est probablement à l’origine des effets indésirables moteurs extrapyramidaux (dystonie, akathisie et parkinsonisme).
Les effets anti‑dopaminergiques de l’halopéridol sur les cellules lactotropes de l’anté-hypophyse expliquent l’hyperprolactinémie due à l’inhibition tonique de la sécrétion de prolactine médiée par la dopamine.
5.2. Propriétés pharmacocinétiques
La biodisponibilité moyenne de l’halopéridol administré sous forme de comprimé ou de solution buvable est de 60 % à 70 %. Les concentrations plasmatiques maximales de l’halopéridol sont généralement atteintes 2 à 6 heures après une prise orale. Une forte variabilité interindividuelle des concentrations plasmatiques a été constatée. L’état d’équilibre est atteint en l’espace d’une semaine après l’instauration du traitement.
Distribution
Le taux de liaison moyen de l’halopéridol avec les protéines plasmatiques est d’environ 88 % à 92 % chez l’adulte. Le taux de liaison avec les protéines plasmatiques est soumis à une forte variabilité interindividuelle. L’halopéridol est rapidement distribué dans les divers tissus et organes, comme indiqué par son large volume de distribution (moyennes de 8 à 21 L/kg après administration intraveineuse). L’halopéridol traverse facilement la barrière hémato-encéphalique. Il traverse également la barrière placentaire et est excrété dans le lait maternel.
Biotransformation
L’halopéridol est très largement métabolisé dans le foie. Les principales voies métaboliques de l’halopéridol chez l’être humain comprennent la glucurono-conjugaison, la réduction cétonique, la N‑désalkylation oxydative et la formation de métabolites pyridinium. Les métabolites de l’halopéridol ne sont pas considérés comme contribuant de façon significative à son activité ; cependant, la voie de réduction représente environ 23 % de la biotransformation et une rétroconversion du métabolite réduit de l’halopéridol en halopéridol ne peut être totalement exclue. Les enzymes CYP3A4 et CYP2D6 du cytochrome P450 sont impliquées dans le métabolisme de l’halopéridol. L’inhibition ou l’induction du CYP3A4, ou l’inhibition du CYP2D6, peuvent altérer le métabolisme de l’halopéridol. La réduction de l’activité enzymatique du CYP2D6 peut entraîner une augmentation des concentrations de l’halopéridol.
Élimination
La demi‑vie d’élimination finale de l’halopéridol est en moyenne de 24 heures (intervalle de valeurs moyennes : 15 à 37 heures) après administration orale. La clairance apparente de l’halopéridol après administration extravasculaire est comprise entre 0,9 et 1,5 L/h/kg et elle est réduite chez les métaboliseurs lents du CYP2D6. La réduction de l’activité enzymatique du CYP2D6 peut entraîner une augmentation des concentrations de l’halopéridol. La variabilité interindividuelle (coefficient de variation, %) de la clairance de l’halopéridol a été estimée à 44 % lors d’une analyse pharmacocinétique de population chez des patients atteints de schizophrénie.
Après administration intraveineuse de l’halopéridol, 21 % de la dose ont été éliminés dans les selles et 33 % dans les urines. Moins de 3 % de la dose sont excrétés sous forme inchangée dans les urines.
Linéarité/non linéarité
Il existe une relation linéaire entre la dose d’halopéridol et les concentrations plasmatiques chez l’adulte.
Populations particulières
Personnes âgées
Les concentrations plasmatiques de l’halopéridol ont été plus élevées chez les patients âgés que chez les adultes plus jeunes après administration de la même dose. Les résultats issus d’études cliniques de taille limitée semblent indiquer une clairance plus faible et une demi‑vie d’élimination plus longue de l’halopéridol chez les patients âgés. Les résultats se situent dans les limites de la variabilité observée au niveau de la pharmacocinétique de l’halopéridol. Un ajustement de la dose est recommandé chez les patients âgés (voir rubrique 4.2).
Insuffisance rénale
L’influence de l’insuffisance rénale sur la pharmacocinétique de l’halopéridol n’a pas été évaluée. Un tiers environ de la dose d’halopéridol est excrété dans les urines, principalement sous forme de métabolites. Moins de 3 % de l’halopéridol administré sont éliminés sous forme inchangée dans les urines. Les métabolites de l’halopéridol ne sont pas considérés comme contribuant de façon significative à son activité, bien qu’une rétroconversion en halopéridol ne puisse être exclue dans le cas du métabolite réduit de l’halopéridol. Même si l’altération de la fonction rénale ne semble pas devoir affecter l’élimination de l’halopéridol de façon cliniquement significative, il est conseillé de procéder avec prudence en présence d’une insuffisance rénale, en particulier en cas d’insuffisance sévère, en raison de la longue demi‑vie de l’halopéridol et de son métabolite réduit, ainsi que de l’éventualité d’une accumulation (voir rubrique 4.2).
Étant donnés le volume de distribution important de l’halopéridol et son fort taux de liaison avec les protéines plasmatiques, la dialyse ne permet de l’éliminer qu’en très faible quantité.
Insuffisance hépatique
L’influence de l’insuffisance hépatique sur la pharmacocinétique de l’halopéridol n’a pas été évaluée. Cependant, l’insuffisance hépatique peut avoir des effets significatifs sur la pharmacocinétique de l’halopéridol dans la mesure où le médicament est très largement métabolisé dans le foie. Par conséquent, il est conseillé d’ajuster la dose et de procéder avec prudence chez les patients atteints d’insuffisance hépatique (voir rubriques 4.2 et 4.4).
Population pédiatrique
Des données limitées concernant les concentrations plasmatiques ont été obtenues lors des études pédiatriques réalisées chez 78 patients présentant diverses pathologies (schizophrénie, trouble psychotique, syndrome de Tourette, autisme) traités par des doses orales d’halopéridol allant jusqu’à un maximum de 30 mg/jour. Ces études ont été menées principalement chez des enfants et des adolescents âgés de 2 à 17 ans. Les concentrations plasmatiques mesurées à divers moments et après diverses durées de traitement ont été soit indétectables soit comprises dans un intervalle allant jusqu’à un maximum de 44,3 ng/mL. Comme chez l’adulte, une forte variabilité interindividuelle des concentrations plasmatiques a été constatée. La demi‑vie a eu tendance à être plus courte chez l’enfant que chez l’adulte.
Lors de 2 études menées chez des enfants traités par l’halopéridol pour des tics et des syndromes de Gilles de la Tourette, une réponse positive a été associée aux concentrations plasmatiques de 1 à 4 ng/mL.
Relations pharmacocinétique/pharmacodynamique
Concentrations thérapeutiques
D’après les données publiées à la suite de multiples études cliniques, une réponse thérapeutique est obtenue chez la plupart des patients atteints de schizophrénie aiguë ou chronique à des concentrations plasmatiques de 1 à 10 ng/mL. Des concentrations plus élevées peuvent être nécessaires chez une partie des patients en raison de la forte variabilité pharmacocinétique interindividuelle de l’halopéridol.
Chez les patients connaissant un premier épisode de schizophrénie, une réponse au traitement peut être obtenue à des concentrations de seulement 0,6 à 3,2 ng/mL, comme estimé d’après la mesure du taux d’occupation des récepteurs D2 et en partant de l’hypothèse qu’un taux d’occupation des récepteurs D2 de 60 % à 80 % est le plus adapté pour obtenir une réponse au traitement et limiter les symptômes extrapyramidaux. En moyenne, ces concentrations devraient pouvoir être atteintes avec des doses de 1 à 4 mg par jour.
En raison de la forte variabilité interindividuelle de la pharmacocinétique de l’halopéridol et de la relation concentration‑effet, il est recommandé d’ajuster la dose d’halopéridol au cas par cas selon la réponse au traitement chez le patient et en tenant compte des données indiquant une latence de 5 jours avant obtention de la moitié de la réponse maximale au traitement. Une mesure des concentrations plasmatiques de l’halopéridol peut être envisagée dans certains cas.
Effets cardiovasculaires
Le risque d’allongement du QTc augmente avec la dose et les concentrations plasmatiques de l’halopéridol.
Symptômes extrapyramidaux
Des symptômes extrapyramidaux peuvent survenir dans l’intervalle thérapeutique, mais leur fréquence est généralement plus élevée aux doses conduisant à des concentrations supérieures aux concentrations thérapeutiques.
5.3. Données de sécurité préclinique
Lors d’une étude sur la cancérogénicité de l’halopéridol, des augmentations doses-dépendantes des adénomes hypophysaires et des carcinomes mammaires ont été observées chez les souris femelles. Ces tumeurs pourraient avoir été causées par l’antagonisation prolongée des récepteurs dopaminergiques D2 et l’hyperprolactinémie. La pertinence clinique de ces tumeurs constatées chez les rongeurs n’est pas connue chez l’homme.
Plusieurs études in vitro publiées ont montré que l’halopéridol bloque le canal hERG des cellules cardiaques. Dans un certain nombre d’études in vivo, l’administration intraveineuse d’halopéridol chez certains modèles animaux a provoqué un allongement significatif du QTc à des doses d’environ 0,3 mg/kg, conduisant à une Cmax plasmatique au moins 7 à 14 fois supérieure aux concentrations plasmatiques thérapeutiques de 1 à 10 ng/mL qui ont été efficaces chez la majorité des patients au cours des études cliniques. Ces doses intraveineuses, qui ont entraîné un allongement du QTc, n’ont pas provoqué d’arythmies. Lors de certaines études effectuées chez l’animal, des doses intraveineuses d’halopéridol plus élevées, de 1 mg/kg ou plus, ont entraîné un allongement du QTc et/ou des arythmies ventriculaires à une Cmax au moins 38 à 137 fois supérieure aux concentrations plasmatiques thérapeutiques qui ont été efficaces chez la majorité des patients au cours des études cliniques.
Parahydroxybenzoate de méthyle (E 218), acide lactique, eau purifiée.
3 ans.
Après la première ouverture : 3 mois.
6.4. Précautions particulières de conservation
Pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Solution buvable – flacon compte-gouttes :
15 ml ou 30 ml de solution dans un flacon compte-gouttes en polyéthylène basse densité avec bouchon scellé et sécurité-enfant en polyéthylène haute densité.
Solution buvable – flacon avec seringue doseuse pour administration orale :
· flacon en verre ambré de 100 ml avec bouchon scellé et sécurité-enfant en polyéthylène haute densité et une seringue doseuse de 2,5 ml pour administration orale en polyéthylène basse densité, graduée en millilitres et/ou milligrammes.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Vision Exchange Building
Triq it-Territorjals, Zone 1
Central Business District
Birkirkara, CBD 1070
MALTE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 304 717 2 2 : 15 ml en flacon (polyéthylène) compte-gouttes.
· 34009 553 301 4 1 : 15 ml en flacon (polyéthylène) compte-gouttes, boîte de 50.
· 34009 553 300 8 0 : 195 ml en flacon (polyéthylène) avec pipette compte-gouttes, boîte de 4.
· 34009 269 211 4 9 : 30 ml en flacon (polyéthylène) compte-gouttes.
· 34009 300 599 20 : 100 ml en flacon (verre ambré) avec seringue doseuse pour administration orale, boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 08/04/2026
HALDOL 2 mg/ml, solution buvable
Halopéridol
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que HALDOL 2 mg/ml, solution buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre HALDOL 2 mg/ml, solution buvable ?
3. Comment prendre HALDOL 2 mg/ml, solution buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver HALDOL 2 mg/ml, solution buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE HALDOL 2 mg/ml, solution buvable ET DANS QUELS CAS EST-IL UTILISE ?
Le nom de votre médicament est HALDOL 2 mg/ml, solution buvable.
HALDOL contient une substance active appelée halopéridol. Il appartient à un groupe de médicaments appelés « antipsychotiques ».
HALDOL est utilisé chez les adultes, les adolescents et les enfants pour traiter des maladies affectant les pensées, les sensations ou le comportement. Cela comprend des problèmes de santé mentale (tels que la schizophrénie et le trouble bipolaire) et des problèmes comportementaux.
Ces maladies peuvent provoquer chez vous :
· un état de confusion (délire)
· voir, entendre, ressentir ou sentir des choses qui ne sont pas réelles (hallucinations)
· croire des choses qui ne sont pas réelles (idées délirantes)
· une suspicion anormale (paranoïa)
· un fort sentiment d’excitation, d’agitation, d’enthousiasme, de l’impulsivité ou de l’hyperactivité
· un comportement très agressif, hostile ou violent.
Chez les adolescents et les enfants, HALDOL est utilisé pour traiter la schizophrénie chez les patients âgés de 13 à 17 ans, et pour traiter les problèmes comportementaux chez les patients âgés de 6 à 17 ans.
HALDOL est également utilisé :
· chez les adolescents et les enfants âgés de 10 à 17 ans et chez les adultes pour traiter les mouvements ou émissions de sons incontrôlables (tics), par exemple dans le syndrome de Gilles de la Tourette sévère
· chez les adultes, pour aider à contrôler les mouvements associés à la maladie de Huntington.
HALDOL est parfois utilisé lorsque les autres médicaments ou traitements n’ont pas fonctionné ou ont provoqué des effets indésirables inacceptables.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE HALDOL 2 mg/ml, solution buvable ?
Ne prenez jamais HALDOL 2 mg/ml, solution buvable si :
· vous êtes allergique à l’halopéridol ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6
· votre niveau de conscience est altéré ou si vos réactions deviennent anormalement lentes ;
· vous avez une maladie de Parkinson
· vous avez un type de démence appelé « démence à corps de Lewy »
· vous avez une paralysie supranucléaire progressive (PSP)
· vous avez une maladie cardiaque appelée « allongement de l’intervalle QT » ou tout autre trouble du rythme cardiaque mis en évidence par un tracé anormal sur l’électrocardiogramme (ECG)
· vous avez une insuffisance cardiaque ou avez récemment eu une crise cardiaque
· le taux de potassium dans votre sang est faible et que vous n’avez pas reçu de traitement pour cela
· vous prenez l’un des médicaments mentionnés dans la rubrique « Autres médicaments et HALDOL - Ne prenez pas HALDOL si vous prenez d’autres médicaments pour traiter ».
Ne prenez pas ce médicament si vous vous trouvez dans l’une des situations décrites ci-dessus. En cas de doute, adressez-vous à votre médecin ou pharmacien avant de prendre HALDOL.
Avertissements et précautions
Effets indésirables graves
HALDOL peut provoquer des problèmes cardiaques, des difficultés à contrôler les mouvements du corps ou des membres et un effet indésirable grave appelé « syndrome malin des neuroleptiques ». Il peut également entraîner des réactions allergiques sévères et des caillots sanguins. Vous devez avoir connaissance des effets indésirables graves possibles pendant la prise d’HALDOL car vous pourriez avoir besoin d’un traitement médical en urgence, le cas échéant. Voir « Soyez attentif aux effets indésirables graves » dans la rubrique 4.
Personnes âgées et personnes atteintes de démence
Une légère augmentation de la mortalité et des accidents vasculaires cérébraux a été signalée chez les personnes âgées atteintes de démence prenant des antipsychotiques. Adressez-vous à votre médecin ou pharmacien avant de prendre HALDOL si vous êtes âgé(e), en particulier si vous souffrez de démence.
Adressez-vous à votre médecin ou pharmacien si vous présentez :
· un rythme cardiaque lent, une maladie cardiaque, ou si quelqu’un de votre famille proche est décédé subitement à cause de problèmes cardiaques
· une tension artérielle basse ou des vertiges lorsque vous vous levez ou vous asseyez
· un faible taux de potassium ou de magnésium (ou d’un autre électrolyte) dans votre sang. Votre médecin déterminera comment traiter ce problème
· des antécédents de saignement dans le cerveau, ou si votre médecin vous a indiqué que vous risquiez plus que les autres d’avoir un AVC
· une épilepsie ou avez déjà fait des crises épileptiques (convulsions)
· des problèmes de reins, de foie ou de thyroïde
· un taux élevé de l’hormone appelée « prolactine » dans votre sang, ou un cancer pouvant être provoqué par des taux élevés de prolactine (comme le cancer du sein)
· des antécédents de caillots sanguins, ou si un membre de votre famille a des antécédents de caillots sanguins
· une dépression ou si vous êtes atteint(e) de trouble bipolaire et commencez à vous sentir déprimé(e).
Vous pourriez avoir besoin d’une surveillance plus étroite et il pourrait être nécessaire de modifier la quantité d’HALDOL que vous prenez.
Si vous n’êtes pas sûr(e) de savoir si vous êtes concerné(e) par l’une des situations ci-dessus, adressez-vous à votre médecin ou pharmacien avant de prendre HALDOL.
Contrôles médicaux
Votre médecin pourra décider de réaliser un électrocardiogramme (ECG) avant ou pendant votre traitement par HALDOL. L’ECG mesure l’activité électrique de votre cœur.
Analyses sanguines
Votre médecin pourra décider de contrôler les taux de potassium ou de magnésium (ou d’un autre électrolyte) dans votre sang avant ou pendant votre traitement par HALDOL.
Enfants âgés de moins de 6 ans
HALDOL ne doit pas être utilisé chez les enfants âgés de moins de 6 ans car il n’a pas été convenablement étudié chez cette classe d’âge.
Autres médicaments et HALDOL 2 mg/ml, solution buvable
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Ne prenez pas HALDOL si vous prenez d’autres médicaments pour traiter :
· des problèmes cardiaques (comme l’amiodarone, le dofétilide, le disopyramide, la dronédarone, l’ibutilide, la quinidine et le sotalol)
· une dépression (comme le citalopram et l’escitalopram)
· une psychose (comme la fluphénazine, la lévomépromazine, la perphénazine, le pimozide, la prochlorpérazine, la promazine, le sertindole, la thiorizadine, la trifluopérazine, la triflupromazine et la ziprasidone)
· une infection bactérienne (comme l’azithromycine, la clarithromycine, l’érythromycine, la lévofloxacine, la moxifloxacine et la télithromycine)
· une infection fongique (comme la pentamidine)
· le paludisme (comme l’halofantrine)
· les nausées et vomissements (comme le dolasétron)
· un cancer (comme le torémifène et le vandétanib).
Prévenez également votre médecin si vous prenez du bépridil (pour des douleurs thoraciques ou pour faire baisser votre tension artérielle) ou de la méthadone (comme antidouleur ou pour traiter une toxicomanie).
Ces médicaments pourraient augmenter le risque de problèmes cardiaques ; par conséquent, si vous prenez l’un de ces médicaments, parlez-en à votre médecin et ne prenez pas HALDOL (voir « Ne prenez jamais HALDOL si »).
Une surveillance particulière pourrait être nécessaire si vous prenez du lithium en même temps qu’HALDOL. Prévenez immédiatement votre médecin et arrêtez de prendre les deux médicaments si vous présentez :
· une fièvre inexplicable ou des mouvements incontrôlables
· une confusion, une désorientation, des maux de tête, des problèmes d’équilibre et des somnolences.
Ces signes indiquent une maladie grave.
Certains médicaments pourraient altérer le fonctionnement d’HALDOL ou augmenter le risque de problèmes cardiaques
Informez votre médecin si vous prenez :
· de l’alprazolam ou de la buspirone (pour l’anxiété)
· de la duloxétine, de la fluoxétine, de la fluvoxamine, de la néfazodone, de la paroxétine, de la sertraline, du millepertuis (Hypericum perforatum) ou de la venlafaxine (pour la dépression)
· du bupropion (pour la dépression ou l’aide à l’arrêt du tabac)
· de la carbamazépine, du phénobarbital ou de la phénytoïne (pour l’épilepsie)
· de la rifampicine (pour une infection bactérienne)
· de l’itraconazole, du posaconazole ou du voriconazole (pour une infection fongique)
· du kétoconazole en comprimés (pour traiter le syndrome de Cushing)
· de l’indinavir, du ritonavir ou du saquinavir (pour une infection par le virus de l’immunodéficience humaine, le VIH)
· de la chlorpromazine ou de la prométhazine (pour des nausées et vomissements)
· du vérapamil (pour la pression artérielle ou les problèmes cardiaques).
Prévenez également votre médecin si vous prenez un autre médicament pour faire baisser votre tension artérielle, tel qu’un diurétique.
Si vous prenez l’un de ces médicaments, votre médecin devra peut-être modifier votre dose d’HALDOL.
HALDOL peut altérer le fonctionnement des types de médicaments suivants
Informez votre médecin si vous prenez des médicaments pour :
· vous calmer ou vous aider à dormir (tranquillisants)
· la douleur (antalgiques puissants)
· la dépression (« antidépresseurs tricycliques »)
· faire baisser votre tension artérielle (comme la guanéthidine et la méthyldopa)
· des réactions allergiques sévères (adrénaline)
· un trouble de déficit de l’attention avec ou sans hyperactivité (TDAH) ou une narcolepsie (médicaments appelés « stimulants »)
· une maladie de Parkinson (comme la lévodopa)
· fluidifier le sang (phénindione).
Adressez-vous à votre médecin avant de prendre HALDOL si vous prenez l’un de ces médicaments.
HALDOL 2 mg/ml, solution buvable avec de l’alcool
La consommation d’alcool pendant le traitement par HALDOL peut entraîner des somnolences et une perte de vigilance. Vous devez donc faire attention à la quantité d’alcool que vous consommez. Parlez avec votre médecin de la consommation d’alcool pendant le traitement par HALDOL et indiquez-lui en quelle quantité vous en consommez.
Grossesse, allaitement et fertilité
Grossesse : si vous êtes enceinte, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin. Votre médecin pourra vous conseiller de ne pas prendre HALDOL pendant votre grossesse.
Chez les nouveau-nés dont les mères ont pris HALDOL pendant les 3 derniers mois de la grossesse (dernier trimestre), les problèmes suivants pourraient survenir :
· tremblements musculaires, raideur ou faiblesse des muscles
· somnolences ou agitation
· difficultés à respirer ou à s’alimenter.
La fréquence exacte de ces problèmes n’est pas connue. Si vous avez pris HALDOL pendant votre grossesse et que l’un de ces effets indésirables apparaît chez votre enfant, contactez votre médecin.
Allaitement : si vous allaitez ou prévoyez d’allaiter, parlez-en à votre médecin. En effet, de petites quantités du médicament pourraient passer dans le lait maternel et être absorbées par l’enfant. Votre médecin vous expliquera quels sont les risques et les bénéfices liés à l’allaitement pendant le traitement par HALDOL.
Fertilité : HALDOL pourrait augmenter le taux d’une hormone appelée « prolactine », ce qui pourrait altérer la fertilité masculine et féminine. Si vous avez des questions à ce sujet, adressez-vous à votre médecin.
Conduite de véhicules et utilisation de machines
HALDOL peut altérer votre aptitude à conduire des véhicules ou à utiliser des machines. Des effets indésirables, tels que la somnolence, peuvent altérer votre vigilance, en particulier lors de la première utilisation ou après administration d’une dose élevée. Ne conduisez pas de véhicules et n’utilisez pas d’outils ou de machines sans en avoir discuté au préalable avec votre médecin.
HALDOL 2 mg/ml, solution buvable contient du parahydroxybenzoate de méthyle
Le parahydroxybenzoate de méthyle peut provoquer des réactions allergiques (éventuellement retardées).
3. COMMENT PRENDRE HALDOL 2 mg/ml, solution buvable ?
Quelle quantité devez-vous prendre ?
Votre médecin vous indiquera la quantité d’HALDOL à prendre et la durée du traitement. Votre médecin vous indiquera également si vous devez prendre HALDOL en une fois ou en plusieurs prises chaque jour. Il peut falloir un certain temps pour que le médicament fasse pleinement effet. En principe, votre médecin vous prescrira d’abord une faible dose, qu’il ajustera ensuite selon vos besoins. Il est très important que vous preniez une dose adaptée.
La dose d’halopéridol que vous prendrez dépendra :
· de votre âge
· de la maladie traitée
· de l’état de vos reins ou votre foie
· des autres médicaments que vous prenez.
Adultes
· La dose sera normalement comprise entre 0,5 mg et 10 mg par jour.
· Votre médecin pourra l’ajuster de façon à trouver la dose qui vous convient le mieux.
· La dose maximale pouvant être utilisée chez l’adulte dépend de la maladie pour laquelle vous êtes traité et varie entre 5 mg et 20 mg par jour.
Personnes âgées
· En principe, les personnes âgées commenceront le traitement à 0,5 mg par jour ou avec la moitié de la plus faible dose utilisée chez l’adulte.
· La quantité d’HALDOL à prendre sera ensuite ajustée jusqu’à ce que le médecin trouve la dose qui vous convient le mieux.
· La dose maximale pouvant être utilisée chez les personnes âgées est de 5 mg par jour sauf si votre médecin décide qu’une dose supérieure est nécessaire.
Enfants et adolescents âgés de 6 à 17 ans
· La dose sera normalement comprise entre 0,5 mg et 3 mg par jour.
· Jusqu’à l’âge de 17 ans, les adolescents traités pour une schizophrénie ou des troubles comportementaux peuvent recevoir une dose plus élevée, pouvant aller jusqu’à 5 mg par jour.
Prise d’HALDOL 2 mg/ml, solution buvable
· HALDOL doit être administré par voie orale.
· Vous pouvez mélanger HALDOL solution buvable avec de l’eau avant de le prendre, mais ne le mélangez pas avec un autre liquide.
|
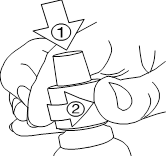
Flacon compte-gouttes : · Retirez le bouchon du flacon en appuyant sur le bouchon tout en tournant dans le sens contraire des aiguilles d'une montre. · Retournez le flacon en tenant une cuillère en dessous. · Appuyez doucement sur les côtés du flacon et comptez le nombre de gouttes que vous devez prendre. · Buvez immédiatement la solution. · Refermez le flacon. |
|
Flacon avec seringue doseuse pour administration orale :
Vous devez prendre la solution en utilisant la seringue doseuse pour administration orale.
|
· Posez le flacon sur une surface plane. · Retirez le bouchon du flacon en appuyant sur le bouchon tout en tournant dans le sens contraire des aiguilles d'une montre (figure 1). · Un piston se trouve à une extrémité de la seringue doseuse pour administration orale. Placez l’autre extrémité dans la solution, à l’intérieur du flacon. · Tout en maintenant la collerette du bas de la seringue doseuse pour administration orale, tirez la collerette située au sommet du piston vers le haut. Tirez jusqu’à ce que le repère correspondant au nombre de millilitres (ml) ou milligrammes (mg) requis soit visible (figure 2). · Sortez l’ensemble de la seringue doseuse pour administration orale du flacon en tenant la collerette du bas (figure 3). · Versez le contenu de la seringue doseuse pour administration orale dans une cuillère ou un verre. Pour cela, poussez la collerette du haut vers le bas tout en continuant de maintenir la collerette du bas. · Buvez immédiatement la solution. · Refermez le flacon, puis rincez la seringue doseuse pour administration orale avec un peu d’eau. |
|
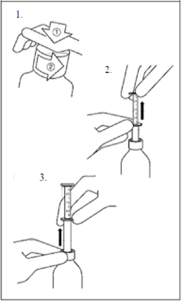
Si vous avez pris plus d’HALDOL 2 mg/ml, solution buvable que vous n’auriez dû
Si vous avez pris plus d’HALDOL que vous n’auriez dû ou si une autre personne a pris HALDOL, contactez un médecin ou rendez-vous immédiatement aux urgences de l’hôpital le plus proche.
Si vous oubliez de prendre HALDOL 2 mg/ml, solution buvable
· Si vous avez oublié de prendre une dose, prenez la dose suivante à l’heure habituelle. Continuez ensuite le traitement comme indiqué par votre médecin.
· Ne prenez pas de dose double.
Si vous arrêtez de prendre HALDOL 2 mg/ml, solution buvable
Sauf indication contraire de votre médecin, l’arrêt du traitement par HALDOL doit être progressif. L’arrêt brutal du traitement pourrait entraîner des effets tels que :
· Nausées et vomissements
· Troubles du sommeil.
Veillez à toujours suivre scrupuleusement les instructions de votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Soyez attentif aux effets indésirables graves
Informez immédiatement votre médecin si vous remarquez ou suspectez l’un des effets indésirables suivants. Vous pourriez avoir besoin d’un traitement médical en urgence.
Problèmes cardiaques :
· Rythme cardiaque anormal, empêchant le cœur de fonctionner normalement et pouvant provoquer une perte de conscience
· Rythme cardiaque anormalement rapide
· Battements de cœur supplémentaires
Les problèmes cardiaques sont peu fréquents chez les personnes prenant HALDOL (peuvent affecter jusqu’à 1 personne sur 100). Des cas de mort subite se sont produits chez des patients prenant ce médicament, mais la fréquence exacte de ces décès n’est pas connue. Des arrêts cardiaques (cœur qui s’arrête de battre) se sont également produits chez des personnes prenant des antipsychotiques.
Problème grave appelé « syndrome malin des neuroleptiques », qui provoque une forte fièvre, une raideur musculaire sévère, une confusion et une perte de conscience. Sa survenue est rare chez les personnes prenant HALDOL (peut affecter jusqu’à 1 personne sur 1 000).
Problèmes de contrôle des mouvements du corps ou des membres (syndrome extrapyramidal), notamment :
· des mouvements de la bouche, de la langue, de la mâchoire et parfois des membres (dyskinésie tardive)
· une agitation ou des difficultés à rester assis sans bouger, une amplification des mouvements du corps
· des mouvements du corps ralentis ou réduits, des mouvements saccadés ou erratiques
· un tremblement ou une raideur musculaire, un pas traînant
· une incapacité à bouger
· une expression figée du visage pouvant faire penser à un masque.
Ces problèmes sont très fréquents chez les personnes prenant HALDOL (peuvent affecter plus d’une personne sur 10). Si vous présentez l’un de ces effets, un médicament supplémentaire pourra vous être prescrit.
Réaction allergique sévère, avec notamment :
· un gonflement du visage, des lèvres, de la bouche, de la langue ou de la gorge
· des difficultés à avaler ou à respirer
· une éruption cutanée avec démangeaisons (urticaire).
Les réactions allergiques sont peu fréquentes chez les personnes prenant HALDOL (peuvent affecter jusqu’à 1 personne sur 100).
Caillots sanguins dans les veines, généralement dans les jambes (thrombose veineuse profonde, TVP). Ce problème a été signalé chez des personnes prenant des antipsychotiques. Les signes d’une TVP dans la jambe comprennent un gonflement, une douleur et une rougeur au niveau de la jambe, mais le caillot peut également se déplacer vers les poumons et provoquer une douleur thoracique et des difficultés à respirer. Les caillots sanguins peuvent être très graves ; vous devez donc prévenir immédiatement votre médecin si vous remarquez l’un de ces problèmes.
Informez immédiatement votre médecin si vous remarquez l’un des effets indésirables graves mentionnés ci-dessus.
Autres effets indésirables
Informez votre médecin si vous remarquez ou suspectez l’un des effets indésirables suivants.
Très fréquent (pouvant affecter plus d’une personne sur 10) :
· Agitation
· Troubles du sommeil
· Maux de tête
Fréquent (pouvant affecter jusqu’à 1 personne sur 10) :
· Graves problèmes de santé mentale, comme croire des choses qui ne sont pas vraies (illusions) ou voir, ressentir, entendre ou sentir des choses qui ne sont pas réelles (hallucinations)
· Dépression
· Tension musculaire anormale
· Sensations vertigineuses, y compris en s’asseyant ou en se levant
· Somnolences
· Mouvement des yeux vers le haut ou mouvements yeux rapides impossibles à contrôler
· Problèmes de vision, tels qu’une vision floue
· Tension artérielle basse
· Nausées, vomissements
· Constipation
· Bouche sèche ou augmentation de la production de salive
· Éruption cutanée
· Incapacité à uriner ou à vider complètement la vessie
· Difficultés à obtenir et maintenir une érection (impuissance)
· Prise ou perte de poids
· Modifications mises en évidence par les analyses sanguines visant à contrôler le fonctionnement du foie
Peu fréquent (pouvant affecter jusqu’à 1 personne sur 100) :
· Effets sur les cellules sanguines – diminution de tous les types de cellules sanguines, y compris des réductions sévères des globules blancs et une diminution des plaquettes (les cellules qui aident le sang à coaguler)
· Confusion
· Perte ou diminution du désir sexuel
· Crises épileptiques (convulsions)
· Raideur des muscles et des articulations
· Spasmes, secousses ou contractions musculaires impossibles à contrôler, y compris un spasme de la nuque faisant pencher la tête d’un côté
· Troubles de la marche
· Essoufflement
· Inflammation du foie ou problème de foie entraînant un jaunissement de la peau ou des yeux (jaunisse)
· Sensibilité accrue de la peau au soleil
· Démangeaisons
· Transpiration excessive
· Modifications du cycle menstruel (règles), telles qu’une absence de règles ou des règles prolongées, abondantes, douloureuses
· Production inattendue de lait maternel
· Douleur ou gêne à la poitrine
· Température corporelle élevée
· Gonflement dû à l’accumulation de liquide dans le corps
Rare (pouvant affecter jusqu’à 1 personne sur 1 000) :
· Taux élevé de l’hormone appelée « prolactine » dans le sang
· Rétrécissement des voies aériennes dans les poumons, entraînant des difficultés à respirer
· Difficultés ou incapacité à ouvrir la bouche
· Problèmes lors des rapports sexuels
Les effets indésirables suivants ont également été signalés, mais leur fréquence exacte n’est pas connue :
· Taux élevé d’hormone antidiurétique dans le sang (syndrome de sécrétion inappropriée d’hormone antidiurétique)
· Faible taux de sucre dans le sang
· Gonflement autour du larynx ou bref spasme des cordes vocales, pouvant entraîner des difficultés à parler ou à respirer
· Défaillance soudaine du foie
· Diminution du flux de bile dans les canaux biliaires
· Peau qui s’écaille ou qui pèle
· Inflammation des petits vaisseaux sanguins, entraînant une éruption cutanée accompagnée de petits boutons rouges ou violets
· Destruction du tissu musculaire (rhabdomyolyse)
· Érection persistante et douloureuse du pénis
· Augmentation du volume des seins chez les hommes
· Température corporelle basse
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER HALDOL 2 mg/ml, solution buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette du flacon ou sur la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois. Une fois le flacon ouvert, à utiliser dans les 3 mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient HALDOL 2 mg/ml, solution buvable
Flacon compte-gouttes :
· La substance active est :
Halopéridol..................................................................................................................... 0,1 mg
Pour 1 goutte (chaque ml de solution buvable contient 2 mg).
· Les autres composants sont : parahydroxybenzoate de méthyle (E218), acide lactique et eau purifiée.
Flacon avec seringue doseuse pour administration orale :
· La substance active est :
Halopéridol........................................................................................................................ 2 mg
Pour 1 ml.
· Les autres composants sont : parahydroxybenzoate de méthyle (E218), acide lactique et eau purifiée.
Qu’est-ce que HALDOL 2 mg/ml, solution buvable et contenu de l’emballage extérieur
Flacon compte-gouttes :
Ce médicament se présente sous forme de solution limpide et incolore. Il est conditionné dans un flacon compte-gouttes avec bouchon scellé et sécurité-enfant et contient 15 ml ou 30 ml de solution.
Toutes les présentations peuvent ne pas être commercialisées.
Flacon avec seringue doseuse pour administration orale :
Ce médicament se présente sous forme de solution limpide et incolore. Il est conditionné dans un flacon ambré avec bouchon scellé et sécurité-enfant et contient 100 ml de solution. Une seringue doseuse pour administration orale est fournie dans la boîte, graduée en millilitres, milligrammes ou les deux.
Titulaire de l’autorisation de mise sur le marché
Essential Pharma Limited
Vision Exchange Building
Triq it-Territorjals, Zone 1
Central Business District
Birkirkara, CBD 1070
MALTE
Exploitant de l’autorisation de mise sur le marché
Batiment A Vaise Business Centre
3 Place Giovanni Da Verrazzano
69009 LYON
TURNHOUTSEWEG 30
2340 BEERSE
BELGIQUE
Ou
Essential Pharma Limited
Vision Exchange Building
Triq it-Territorjals, Zone 1
Central Business District
Birkirkara, CBD 1070
MALTE
Ou
LUSOMEDICAMENTA - SOCIEDADE TÉCNICA FARMACÊUTICA, S.A.
ESTRADA CONSIGLIERI PEDROSO
69-B QUELUZ DE BAIXO
2730-055 BARCARENA
PORTUGAL
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants :
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Flacon compte-gouttes :
HALDOL, solution buvable en flacon compte-gouttes est destiné à être utilisé pour les doses uniques allant jusqu’à 2 mg d’halopéridol (l’équivalent de 20 gouttes).
Le nombre de gouttes requis pour obtenir une dose unique donnée avec HALDOL, solution buvable est indiqué ci-dessous.
Table de conversion pour HALDOL, solution buvable (2 mg/mL)
|
mg d’halopéridol |
Nombre de gouttes d’HALDOL (flacon compte-gouttes) |
|
0,1 mg |
1 goutte |
|
0,2 mg |
2 gouttes |
|
0,3 mg |
3 gouttes |
|
|
|
|
0,4 mg |
4 gouttes |
|
0,5 mg |
5 gouttes |
|
1 mg |
10 gouttes |
|
2 mg |
20 gouttes |
Flacon avec seringue doseuse pour administration orale :
HALDOL, solution buvable fournie en flacon avec seringue doseuse pour administration orale est destinée à être utilisée pour les doses uniques d’halopéridol de 0,5 mg et plus (équivalent à 0,25 ml et plus).
La quantité (mL) requise pour obtenir une dose unique donnée avec HALDOL, solution buvable est indiquée ci-dessous.
Table de conversion pour HALDOL, solution buvable (2 mg/mL)
|
mg d’halopéridol |
mL d’HALDOL (flacon avec seringue doseuse pour administration orale) |
|
0.5 mg |
0.25 mL |
|
1 mg |
0.5 mL |
|
2 mg |
1 mL |
|
5 mg |
2.5 mL |
|
10 mg |
5 mL |
|
15 mg |
7.5 mL |
|
20 mg |
10 mL |