Dernière mise à jour le 03/08/2026
ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie
Indications thérapeutiques
ICATIBANT ZENTIVA contient la substance active icatibant.
ICATIBANT ZENTIVA est utilisé pour le traitement des symptômes de l’angio-œdème héréditaire (AOH) chez les adultes, les adolescents et les enfants âgés de 2 ans et plus.
Dans l’AOH, les taux d’une substance présente dans la circulation sanguine, appelée bradykinine, sont plus élevés, ce qui provoque des symptômes tels que gonflement, douleurs, nausées et diarrhée.
ICATIBANT ZENTIVA bloque l’activité de la bradykinine et, par conséquent, stoppe la progression des symptômes d’une crise d’AOH.
Présentations
> 1 seringue préremplie en verre de 3 mL avec 1 aiguille
Code CIP : 34009 302 375 5 7
Déclaration de commercialisation : 01/04/2022
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : ZENTIVA France
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 204 450 9
ANSM - Mis à jour le : 18/11/2021
ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Icatibant................................................................................................................................. 30 mg
sous forme d’acétate d'icatibant
Pour une seringue préremplie de 3 mL.
Chaque mL de cette solution contient 10 mg d’icatibant.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable en seringue préremplie.
La solution est un liquide limpide et incolore, dont le pH est compris entre 5,2 et 5,8 et dont l’osmolalité est comprise entre 270 et 330 mOsm/kg.
4.1. Indications thérapeutiques
ICATIBANT ZENTIVA est indiqué dans le traitement symptomatique des crises aiguës d’angio-œdème héréditaire (AOH) chez les adultes, les adolescents et les enfants âgés de 2 ans et plus présentant une carence en inhibiteur de la C1 estérase.
4.2. Posologie et mode d'administration
ICATIBANT ZENTIVA doit être administré sous la supervision d’un professionnel de santé.
Posologie
Adultes
La dose recommandée chez les adultes est une injection unique d’ICATIBANT ZENTIVA 30 mg par voie sous-cutanée.
Dans la majorité des cas, une seule injection d’ICATIBANT ZENTIVA suffit à traiter une crise. En cas de soulagement insuffisant ou de récurrence des symptômes, une deuxième injection d’ICATIBANT ZENTIVA peut être administrée 6 heures plus tard. Si la deuxième injection produit un soulagement insuffisant ou en cas de récurrence des symptômes, une troisième injection d’ICATIBANT ZENTIVA peut être administrée de nouveau 6 heures plus tard. Il convient de ne pas dépasser 3 injections d’ICATIBANT ZENTIVA sur une période de 24 heures.
Lors des essais cliniques, 8 injections d’icatibant par mois ont été administrées au maximum.
Population pédiatrique
La dose recommandée d’ICATIBANT ZENTIVA déterminée en fonction du poids corporel chez les enfants et adolescents (âgés de 2 à 17 ans) est présentée dans le tableau 1 ci-dessous.
Tableau 1 : Schéma posologique chez les patients pédiatriques
|
Poids corporel |
Dose (volume à injecter) |
|
12 kg à 25 kg |
10 mg (1,0 mL) |
|
26 kg à 40 kg |
15 mg (1,5 mL) |
|
41 kg à 50 kg |
20 mg (2,0 mL) |
|
51 kg à 65 kg
|
25 mg (2,5 mL) |
|
> 65 kg |
30 mg (3,0 mL) |
Dans l’essai clinique, il n’a pas été administré plus de 1 injection d’icatibant par crise d’AOH.
Aucun schéma posologique ne peut être recommandé chez les enfants âgés de moins de 2 ans ou pesant moins de 12 kg car la sécurité et l’efficacité dans ce groupe pédiatrique n’ont pas été établies.
Personnes âgées
Des données limitées sont disponibles chez les patients âgés de plus de 65 ans.
Il a été démontré que les personnes âgées présentent une exposition systémique accrue à l’icatibant. L’importance de ceci en termes de sécurité d’emploi de l’icatibant n’est pas connue (voir rubrique 5.2).
Insuffisance hépatique
Aucun ajustement de la dose n’est nécessaire chez les patients présentant une insuffisance hépatique.
Insuffisance rénale
Aucun ajustement de la dose n’est nécessaire chez les patients présentant une insuffisance rénale.
Mode d’administration
La voie d’administration d’ICATIBANT ZENTIVA est la voie sous-cutanée, de préférence dans la région abdominale.
ICATIBANT ZENTIVA solution injectable doit être injecté lentement en raison du volume à administrer.
Chaque seringue d’ICATIBANT ZENTIVA est à usage unique.
Se reporter à la notice pour les instructions d’utilisation.
Administration par un soignant/auto-administration
La décision de recourir à l’administration d’ICATIBANT ZENTIVA par un soignant ou à l’auto-administration ne doit être prise que par un médecin expérimenté dans le diagnostic et le traitement de l’angio-œdème héréditaire (voir rubrique 4.4).
Adultes
ICATIBANT ZENTIVA peut être auto-administré ou administré par un soignant uniquement après une formation à la technique de l’injection sous-cutanée dispensée par un professionnel de santé.
Enfants et adolescents âgés de 2 à 17 ans
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Œdème laryngé
Les patients souffrant d’œdèmes laryngés doivent être traités dans un établissement médical approprié après injection jusqu’à ce que le médecin estime qu’ils peuvent quitter l’établissement.
Cardiopathie ischémique
Dans des conditions ischémiques, une détérioration de la fonction cardiaque et une diminution du débit sanguin coronaire seraient théoriquement provoquées par l’antagonisme du récepteur de la bradykinine de type 2. Il convient donc d’être prudent lors de l’administration d’icatibant aux patients présentant une cardiopathie ischémique aiguë ou une angine de poitrine instable (voir rubrique 5.3).
Accident vasculaire cérébral
Bien que certaines données prouvent un effet bénéfique du blocage du récepteur B2 immédiatement après un accident vasculaire cérébral, il existe une possibilité théorique que l’icatibant puisse atténuer les effets neuroprotecteurs positifs de phase tardive de la bradykinine. Ainsi, il conviendrait d’être prudent dans l’administration de l’icatibant aux patients dans les semaines suivant un accident vasculaire cérébral.
Administration par un soignant/auto-administration
Chez les patients n’ayant jamais reçu d’icatibant, il convient d’instaurer le premier traitement au sein d’un établissement médical ou sous la supervision d’un médecin.
En cas de soulagement insuffisant ou de récurrence des symptômes après une auto-administration ou l’administration par un soignant, il est recommandé que le patient ou le soignant consulte un médecin. Chez les adultes, les doses suivantes qui peuvent être nécessaires pour la même crise doivent être administrées au sein d’un établissement médical (voir rubrique 4.2). Il n’existe pas de données sur l’administration de doses supplémentaires pour la même crise chez les adolescents ou les enfants.
Les patients souffrant d’œdèmes laryngés doivent toujours consulter un médecin et rester sous observation au sein d’un établissement médical, même si l’injection a été administrée à domicile.
Population pédiatrique
L’expérience du traitement de plus d’une crise d’AOH par icatibant dans la population pédiatrique est limitée.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction pharmacocinétique des médicaments impliquant le CYP450 n’est attendue (voir rubrique 5.2).
La co-administration d’icatibant avec des inhibiteurs de l’enzyme de conversion de l’angiotensine (ECA) n’a pas été étudiée. Les inhibiteurs de l’ECA sont contre-indiqués chez les patients souffrant d’AOH en raison de l’augmentation possible des taux de bradykinine.
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
Aucune donnée clinique n’est disponible concernant l’exposition des femmes enceintes à l’icatibant. Des études menées chez l’animal ont mis en évidence des effets sur l’implantation utérine et la mise bas (voir rubrique 5.3) mais le risque potentiel pour l’Homme n’est pas connu.
L’icatibant ne doit être utilisé pendant la grossesse que si le bénéfice escompté justifie le risque potentiel pour le fœtus (par exemple, pour traiter des œdèmes laryngés susceptibles de mettre en jeu le pronostic vital).
L’icatibant est excrété dans le lait des rates allaitantes à des concentrations similaires à celles retrouvées dans le sang maternel. Aucun effet n’a été constaté dans le développement post-natal des rats nouveau-nés.
On ne sait pas si l’icatibant est excrété dans le lait maternel humain mais il est recommandé aux femmes allaitantes souhaitant prendre ICATIBANT ZENTIVA de ne pas allaiter pendant les 12 heures qui suivent l’administration du traitement.
Fertilité
Chez le rat et le chien, l’utilisation répétée d’icatibant a eu des effets sur les organes reproducteurs. L’icatibant n’a eu aucun effet sur la fertilité des souris mâles et des rats mâles (voir rubrique 5.3). Dans une étude menée chez 39 hommes et femmes adultes sains ayant reçu 3 doses de 30 mg à intervalle de 6 heures tous les 3 jours pour un total de 9 doses, aucune modification cliniquement significative des taux d’hormones sexuelles à l’inclusion et après stimulation par la GnRH n’a été observée chez les femmes ou les hommes. L’icatibant n’a pas eu d’effets significatifs sur le taux de progestérone de la phase lutéale et la fonction lutéale ou sur la durée du cycle menstruel chez les femmes, ni sur le nombre, la motilité et la morphologie des spermatozoïdes chez les hommes. Il est peu probable que le schéma posologique utilisé dans cette étude soit maintenu en pratique clinique.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Dans les études cliniques d’enregistrement, 999 crises d’AOH au total ont été traitées par 30 mg d’icatibant administré par voie sous-cutanée par un professionnel de santé. L’icatibant 30 mg SC a été administré par un professionnel de santé à 129 sujets sains et 236 patients atteints d’AOH.
La quasi-totalité des sujets ayant reçu de l’icatibant en injection sous-cutanée lors des essais cliniques ont présenté des réactions au site d’injection (caractérisées par des irritations cutanées, un œdème, une douleur, des démangeaisons, un érythème, une sensation de brûlure). Ces réactions ont généralement été de sévérité légère à modérée, transitoires et se sont résolues sans intervention.
Liste des effets indésirables sous forme de tableau
La fréquence des effets indésirables répertoriés dans le tableau 2 est définie à l’aide de la convention suivante :
Très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000).
Tous les effets indésirables rapportés après la commercialisation sont présentés en italique.
Tableau 2 : Effets indésirables rapportés avec l’icatibant
|
Classe de systèmes d’organes |
Fréquence |
Terme préférentiel |
|
Affections du système nerveux |
Fréquent |
Sensations vertigineuses Céphalées |
|
Affections gastro-intestinales |
Fréquent |
Nausées |
|
Affections de la peau et du tissu sous-cutané |
Fréquent |
Rash Erythème Prurit |
|
Fréquence indéterminée |
Urticaire |
|
|
Troubles généraux et anomalies au site d’administration |
Très fréquent |
Réactions au site d’injection* |
|
Fréquent |
Fièvre |
|
|
Investigations |
Fréquent |
Transaminases augmentées |
|
* Ecchymose au point d'injection, hématome au site d’injection, brûlure au point d’injection, érythème au point d’injection, hypoesthésie au site d’injection, irritation au point d’injection, engourdissement au site d’injection, œdème au point d’injection, douleur au point d’injection, sensation de pression au site d’injection, prurit au point d’injection, gonflement au point d’injection, urticaire au point d’injection et chaleur au niveau du site d’injection. |
||
Population pédiatrique
Au total, 32 patients pédiatriques (8 enfants âgés de 2 à 11 ans et 24 adolescents âgés de 12 à 17 ans) atteints d’AOH ont été exposés au traitement par icatibant au cours des études cliniques. Trente-et-un patients ont reçu une dose unique d’icatibant et 1 patient (un adolescent) a reçu l’icatibant pour deux crises d’AOH (deux doses au total). L’icatibant était administré en injection sous-cutanée à la dose de 0,4 mg/kg de poids corporel, jusqu’à une dose maximale de 30 mg.
La majorité des patients pédiatriques ayant été traités par icatibant en injection sous-cutanée ont présenté des réactions au site d’injection telles qu’érythème, gonflement, sensation de brûlure, douleur cutanée et démangeaisons/prurit ; elles étaient de sévérité légère à modérée et concordaient avec les réactions rapportées chez les adultes. Deux patients pédiatriques ont présenté des réactions au site d’injection qui ont été évaluées comme sévères et qui se sont complètement résolues dans les 6 heures. Ces réactions ont inclus érythème, gonflement, sensation de brûlure et sensation de chaleur.
Il n’a pas été observé de modifications cliniquement significatives des taux d’hormones sexuelles lors des études cliniques.
Description d’effets indésirables sélectionnés
Immunogénicité
Pendant le traitement en administrations répétées chez les adultes dans les études de phase III contrôlées, une positivité transitoire pour les anticorps anti-icatibant a été observée dans de rares cas. L’efficacité a été maintenue chez tous les patients. Un patient traité par icatibant était positif pour les anticorps anti-icatibant avant et après le traitement. Ce patient a été suivi pendant 5 mois et la recherche d’anticorps anti-icatibant a été négative lors des prélèvements ultérieurs. Aucune réaction d’hypersensibilité ou anaphylactique n’a été rapportée avec l’icatibant.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Aucune donnée clinique concernant le surdosage n’est disponible.
Une dose de 3,2 mg/kg administrée par voie intraveineuse (environ 8 fois la dose thérapeutique) a provoqué un érythème, des démangeaisons, des bouffées congestives ou une hypotension transitoires chez des sujets sains. Aucune intervention thérapeutique n’a été nécessaire.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L’AOH (une maladie autosomique dominante) est provoqué par une absence ou un dysfonctionnement de l’inhibiteur de C1 estérase. Les crises d’AOH s’accompagnent d’une libération accrue de bradykinine, qui constitue le principal médiateur dans le développement des symptômes cliniques.
L’AOH se manifeste par des crises intermittentes d’œdème sous-cutané et/ou sous-muqueux touchant les voies respiratoires supérieures, la peau et l’appareil gastro-intestinal. Une crise dure généralement de 2 à 5 jours.
L’icatibant est un antagoniste compétitif sélectif des récepteurs de la bradykinine de type 2 (B2). C’est un décapeptide de synthèse ayant une structure similaire à celle de la bradykinine, mais comportant 5 acides aminés non protéinogènes. Dans l’AOH, les concentrations accrues de bradykinine constituent le principal médiateur dans le développement des symptômes cliniques.
Effets pharmacodynamiques
Chez des volontaires sains jeunes recevant l’icatibant aux doses de 0,8 mg/kg sur 4 heures, de 1,5 mg/kg/jour ou 0,15 mg/kg/jour pendant 3 jours, le développement de l’hypotension, de la vasodilatation et de la tachycardie réflexe induites par la bradykinine a pu être prévenu. L’icatibant a été un antagoniste compétitif lorsque la dose de provocation de bradykinine a été multipliée par quatre.
Efficacité et sécurité clinique
Les données d’efficacité sont issues d’une étude de phase II en ouvert initiale et de trois études de phase III contrôlées.
Les études cliniques de phase III (FAST-1 et FAST-2) étaient des études randomisées en double aveugle contrôlées, dont le plan expérimental était identique à l’exception du comparateur (une étude contrôlée versus acide tranexamique oral et une étude versus placebo). Au total, 130 patients ont été randomisés pour recevoir une dose de 30 mg d’icatibant (63 patients) ou le comparateur (acide tranexamique, 38 patients ou placebo, 29 patients). Les épisodes ultérieurs d’AOH ont été traités dans le cadre d’une extension en ouvert. Les patients présentant des symptômes d’angio-œdème laryngé ont reçu un traitement en ouvert par icatibant. Le critère d’évaluation de l’efficacité principal était le délai jusqu’au début du soulagement des symptômes, évalué à l’aide d’une échelle visuelle analogique (EVA). Le tableau 3 présente les résultats d’efficacité de ces études.
L’étude FAST-3 était une étude randomisée, contrôlée versus placebo, en groupes parallèles, menée chez 98 patients adultes (âge médian : 36 ans). Les patients ont été randomisés pour recevoir l’icatibant 30 mg ou le placebo en injection sous-cutanée. Un sous-groupe de patients de cette étude présentait des crises aiguës d’AOH malgré l’administration d’androgènes, d’antifibrinolytiques ou d’inhibiteurs de C1. Le critère d’évaluation principal était le délai jusqu’au début du soulagement des symptômes, évalué par le score composite en 3 items d’une échelle visuelle analogique (EVA-3), consistant en évaluations de l’œdème cutané, de la douleur cutanée et de la douleur abdominale. Le tableau 4 présente les résultats d’efficacité de l’étude FAST-3.
Dans ces études, le délai médian jusqu’au début du soulagement des symptômes a été plus court chez les patients traités par icatibant (2,0 2,5 et 2,0 heures, respectivement) par rapport à l’acide tranexamique (12,0 heures) et au placebo (4,6 et 19,8 heures). L’effet du traitement par icatibant a été confirmé par les critères d’évaluation de l’efficacité secondaires.
Dans une analyse intégrée de ces études de phase III contrôlées, le délai jusqu’au début du soulagement des symptômes et le délai jusqu’au début du soulagement du symptôme primaire ont été similaires quels que soient la tranche d’âge, le sexe, l’origine ethnique, le poids et l’utilisation ou non d’androgènes ou d’antifibrinolytiques.
La réponse a également été uniforme lors des crises répétées dans les études de phase III contrôlées. Au total, 237 patients ont été traités par 1 386 doses d’icatibant 30 mg pour 1 278 crises aiguës d’AOH. Pour les 15 premières crises traitées par icatibant (1 114 doses pour 1 030 crises), on a décrit un délai médian jusqu’au début du soulagement des symptômes similaire entre les crises (2,0 à 2,5 heures). 92,4 % de ces crises d’AOH ont été traitées par une dose unique d’icatibant.
Tableau 3. Résultats d’efficacité des études FAST-1 et FAST-2
|
Etude clinique contrôlée d’ICATIBANT versus acide tranexamique ou placebo : résultats d’efficacité |
|||||
|
FAST-2 |
FAST-1 |
||||
|
|
Icatibant |
Acide tranexamique |
|
Icatibant |
Placebo |
|
Nombre de sujets de la population ITT |
36 |
38 |
Nombre de sujets de la population ITT |
27 |
29 |
|
Score EVA initial (mm) |
63,7 |
61,5 |
Score EVA initial (mm) |
69,3 |
67,7 |
|
Modification après 4 heures par rapport au score initial |
-41,6 |
-14,6 |
Modification après 4 heures par rapport au score initial |
-44,8 |
-23,5 |
|
Différence entre les traitements (IC à 95 %, valeur de p) |
-27,8 (-39,4 ; -16,2) p < 0,001 |
Différence entre les traitements (IC à 95 %, valeur de p) |
-23,3 (-37,1 ; -9,4) p = 0,002 |
||
|
Modification après 12 heures par rapport au score initial |
-54,0 |
-30,3 |
Modification après 12 heures par rapport au score initial |
-54,2 |
-42,4 |
|
Différence entre les traitements (IC à 95 %, valeur de p) |
-24,1 (-33,6 ; -14,6) p < 0,001 |
Différence entre les traitements (IC à 95 %, valeur de p) |
-15,2 (-28,6 ; -1,7) p = 0,028 |
||
|
Délai médian jusqu’au début du soulagement des symptômes (heures) |
|
|
Délai médian jusqu’au début du soulagement des symptômes (heures) |
|
|
|
Tous épisodes (N = 74) |
2,0 |
12,0 |
Tous épisodes (N = 56) |
2,5 |
4,6 |
|
Taux de réponse (%, IC) 4 heures après le début du traitement |
|
|
Taux de réponse (%, IC) 4 heures après le début du traitement |
|
|
|
Tous épisodes (N = 74) |
80,0 (63,1 ; 91,6) |
30,6 (16,3 ; 48,1) |
Tous épisodes (N = 56) |
66,7 (46,0 ; 83,5) |
46,4 (27,5 ; 66,1) |
|
Délai médian jusqu’au début du soulagement des symptômes : tous symptômes (heures) : Douleur abdominale Œdème cutané Douleur cutanée |
1,6 2,6 1,5 |
3,5 18,1 12,0 |
Délai médian jusqu’au début du soulagement des symptômes : tous symptômes (heures) : Douleur abdominale Œdème cutané Douleur cutanée |
2,0 3,1 1,6 |
3,3 10,2 9,0 |
|
Délai médian jusqu’au soulagement quasi complet des symptômes (heures) |
|
|
Délai médian jusqu’au soulagement quasi complet des symptômes (heures) |
|
|
|
Tous épisodes (N = 74) |
10,0 |
51,0 |
Tous épisodes (N = 56) |
8,5 |
19,4 |
|
Délai médian jusqu’à la régression des symptômes, évaluation par le patient (heures) |
|
|
Délai médian jusqu’à la régression des symptômes, évaluation par le patient (heures) |
|
|
|
Tous épisodes (N = 74) |
0,8 |
7,9 |
Tous épisodes (N = 56) |
0,8 |
16,9 |
|
Délai médian jusqu’à l’amélioration globale du patient, évaluation par le médecin (heures) |
|
|
Délai médian jusqu’à l’amélioration globale du patient, évaluation par le médecin (heures) |
|
|
|
Tous épisodes (N = 74) |
1,5 |
6,9 |
Tous épisodes (N = 56) |
1,0 |
5,7 |
Tableau 4. Résultats d’efficacité de l’étude FAST-3
|
Résultats d’efficacité : FAST-3 ; phase contrôlée population ITT |
||||
|
Critère d’évaluation |
Statistique |
Icatibant |
Placebo |
Valeur de p |
|
|
|
(n = 43) |
(n=45) |
|
|
Critère d’évaluation principal |
|
|
|
|
|
Délai jusqu’au début du soulagement des symptômes Score EVA composite (heures) |
Médiane |
2,0 |
19,8 |
< 0,001 |
|
Autres critères d’évaluation |
|
|
|
|
|
Délai jusqu’au début du soulagement du symptôme primaire (heures) |
Médiane |
1,5 |
18,5 |
< 0,001 |
|
Variation du score EVA composite 2 heures après le traitement |
Moyenne |
-19,74 |
-7,49 |
< 0,001 |
|
Variation du score composite de symptômes évalués par le patient 2 heures après le traitement |
Moyenne |
-0,53 |
-0,22 |
< 0,001 |
|
Variation du score composite de symptômes évalués par l’investigateur 2 heures après le traitement |
Moyenne |
-0,44 |
-0,19 |
< 0,001 |
|
Délai jusqu’au soulagement quasi-complet des symptômes (heures) |
Médiane |
8,0 |
36,0 |
0,012 |
|
Délai jusqu’à la régression initiale des symptômes, évaluation par le patient (heures) |
Médiane |
0,8 |
3,5 |
< 0,001 |
|
Délai jusqu’à la régression initiale des symptômes, évaluation visuelle par l’investigateur (heures) |
Médiane |
0,8 |
3,4 |
< 0,001 |
Au total, 66 patients présentant des crises d’AOH touchant le larynx ont été traités dans ces études cliniques de phase III contrôlées. Les résultats ont été similaires à ceux observés chez les patients ayant présenté des crises d’AOH non laryngé en termes de délai jusqu’au début du soulagement des symptômes.
Population pédiatrique
Une étude en ouvert non randomisée, en un seul bras (HGT-FIR-086), a été menée chez 32 patients au total. Tous les patients ont reçu au moins une dose d’icatibant (0,4 mg/kg de poids corporel jusqu’à une dose maximale de 30 mg) et la majorité des patients ont été suivis pendant au moins 6 mois. Onze patients étaient au stade prépubertaire et 21 patients étaient au stade pubertaire ou postpubertaire.
La population d’analyse de l’efficacité était composée de 22 patients (11 patients au stade prépubertaire et 11 patients au stade pubertaire/postpubertaire) qui avaient été traités par icatibant pour une crise d’AOH.
Le critère d’évaluation principal était le délai jusqu’au début du soulagement des symptômes, mesuré à l’aide d’un score composite d’évaluation des symptômes par l’investigateur. Le délai jusqu’au soulagement des symptômes était défini comme la durée (en heures) nécessaire pour observer une amélioration de 20 % des symptômes.
Globalement, le délai médian jusqu’au début du soulagement des symptômes a été de 1 heure (intervalle de confiance à 95 % : 1,0 ; 1,1 heure). Une heure et deux heures après le traitement, un début de soulagement des symptômes a été observé chez 50 % et 90 % des patients, respectivement.
Globalement, le délai médian jusqu’aux symptômes minimaux (moment le plus proche après le traitement où tous les symptômes étaient légers ou absents) était de 1,1 heure (intervalle de confiance à 95 % : 1,0 ; 2,0 heures).
5.2. Propriétés pharmacocinétiques
La pharmacocinétique de l’icatibant a été caractérisée par des études utilisant à la fois l’administration intraveineuse et sous-cutanée à des volontaires sains et à des patients. Le profil pharmacocinétique de l’icatibant chez les patients souffrant d’AOH était similaire à celui des volontaires sains.
Absorption
Après administration sous-cutanée, la biodisponibilité absolue de l’icatibant est de 97 %. Le temps nécessaire pour atteindre la concentration maximale est d’environ 30 minutes.
Distribution
Le volume de distribution de l’icatibant (Vss) est d’environ 20– 25 L. La liaison aux protéines plasmatiques est de 44 %.
Biotransformation
L’icatibant est métabolisé de façon importante par les enzymes protéolytiques en métabolites inactifs qui sont principalement excrétés dans les urines.
Des études in vitro ont confirmé que l’icatibant n’était pas dégradé par les voies métaboliques oxydatives, qu’il n’était pas un inhibiteur des principales isoenzymes (CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 et 3A4) du cytochrome P450 (CYP) et qu’il n’était pas un inducteur des CYP 1A2 et 3A4.
Elimination
L’icatibant est principalement éliminé par le métabolisme avec moins de 10 % de la dose excrétée dans les urines sous forme non modifiée. La clairance est d’environ 15– 20 L/h et est indépendante de la dose. La demi-vie terminale plasmatique est d’environ 1– 2 heures.
Populations particulières
Personnes âgées
Les données suggèrent une diminution, liée à l’âge, de la clairance, qui entraîne une exposition supérieure d’environ 50– 60 % chez les personnes âgées (75– 80 ans) par rapport aux patients de 40 ans.
Sexe
Les données semblent indiquer qu’il n’y a pas de différence de la clairance entre les sujets de sexe masculin et féminin après correction pour le poids corporel.
Insuffisance hépatique et rénale
Des données limitées suggèrent que l’exposition à l’icatibant n’est pas influencée par une insuffisance hépatique ou rénale.
Origine ethnique
Les informations sur l’effet des origines ethniques sont limitées. Les données d’exposition disponibles semblent indiquer qu’il n’existe pas de différence de la clairance entre les sujets non blancs (n = 40) et blancs (n = 132).
Population pédiatrique
La pharmacocinétique de l’icatibant a été caractérisée chez des patients pédiatriques atteints d’AOH dans l’étude HGT-FIR-086 (voir rubrique 5.1). Après administration d’une dose unique par voie sous-cutanée (0,4 mg/kg jusqu’à un maximum de 30 mg), le temps jusqu’à la concentration maximale est d’environ 30 minutes et la demi-vie terminale est d’environ 2 heures. Il n’a pas été observé de différences de l’exposition à l’icatibant chez les patients présentant ou non une crise d’AOH. Le modèle pharmacocinétique de population utilisant à la fois les données chez les adultes et chez les patients pédiatriques a démontré que la clairance de l’icatibant était corrélée au poids corporel, des valeurs de clairance plus faibles étant observées pour les poids corporels plus faibles dans la population pédiatrique atteinte d’AOH. Sur la base de la modélisation de la posologie en fonction du poids, l’exposition prédite à l’icatibant dans la population pédiatrique atteinte d’AOH (voir rubrique 4.2) est plus faible que celle observée dans les études menées chez des patients adultes atteints d’AOH.
5.3. Données de sécurité préclinique
Des études à doses répétées menées pendant une période maximale de 6 mois chez le rat et de 9 mois chez le chien ont montré une diminution dose-dépendante du taux d’hormones sexuelles circulantes, ainsi qu’un retard réversible de la maturation sexuelle dû à l’administration répétée d’icatibant chez les deux espèces.
L’exposition journalière maximale définie par l’aire sous la courbe (ASC) à la dose sans effet nocif observé (NOAEL) lors de l’étude de 9 mois menée chez le chien était 2,3 fois l’ASC chez l’adulte après une dose sous-cutanée de 30 mg. Une NOAEL n’était pas mesurable dans l’étude chez le rat ; toutefois, tous les résultats issus de cette étude ont montré un effet soit totalement, soit partiellement réversible chez les rats traités. Une hypertrophie surrénalienne a été observée chez le rat à toutes les doses testées. Une réversibilité de l’hypertrophie surrénalienne a été constatée une fois le traitement par icatibant interrompu. La pertinence clinique de l’exploration des glandes surrénales n’est pas connue.
L’icatibant n’a eu aucun effet sur la fertilité des souris mâles (dose maximale : 80,8 mg/kg/jour) et des rats mâles (dose maximale : 10 mg/kg/jour).
Au cours d’une étude de 2 ans visant à évaluer le potentiel carcinogène de l’icatibant chez le rat, des doses quotidiennes produisant des niveaux d’exposition atteignant environ 2 fois le niveau obtenu après administration d’une dose thérapeutique chez l’Homme n’ont eu aucun effet sur l’incidence ni sur la morphologie des tumeurs. Ces résultats n’indiquent aucun potentiel carcinogène pour l’icatibant.
Lors d’une batterie classique de tests in vitro et in vivo, l’icatibant n’a montré aucun signe de génotoxicité.
L’icatibant n’est pas tératogène lorsqu’il est administré par injection SC pendant le développement embryonnaire et fœtal précoce chez le rat (dose maximale de 25 mg/kg/jour) et chez le lapin (dose maximale de 10 mg/kg/jour). L’icatibant est un antagoniste puissant de la bradykinine et, par conséquent, à des doses élevées, le traitement peut avoir des effets sur le processus d’implantation utérine et sur la stabilité utérine ultérieure en début de gestation. Ces effets sur l’utérus se manifestent également plus tard au cours de la gestation où l’icatibant présente un effet tocolytique entraînant le retard de la mise bas chez le rat, avec une souffrance fœtale accrue et une mort périnatale lors de l’administration de doses élevées (10 mg/kg/jour).
Dans une étude de recherche de dose en administration sous-cutanée d’une durée de 2 semaines chez le rat juvénile, la dose maximale tolérée a été établie à 25 mg/kg/jour. Une atrophie des testicules et des épididymes a été observée dans l’étude pivot de toxicité juvénile au cours de laquelle des rats sexuellement immatures ont été traités à la dose de 3 mg/kg/jour pendant 7 semaines ; les anomalies microscopiques observées étaient partiellement réversibles. Des effets similaires de l’icatibant sur les tissus reproducteurs ont été observés chez des rats et des chiens sexuellement matures. Ces anomalies tissulaires étaient compatibles avec les effets observés sur les gonadotrophines et semblent être réversibles pendant la période sans traitement ultérieure.
L’icatibant n’a provoqué aucune modification de la conduction cardiaque in vitro (canal hERG) ou in vivo chez les chiens sains ou différents modèles de chien (régulation du rythme ventriculaire, effort physique et ligature coronaire) chez lesquels aucune modification hémodynamique associée n’a été observée. Il a été démontré que l’icatibant aggrave l’ischémie cardiaque induite chez plusieurs modèles non cliniques, mais il n’a pas été prouvé qu’il ait un effet délétère systématique dans les cas d’ischémie aiguë.
Chlorure de sodium, acide acétique glacial, hydroxyde de sodium (pour ajustement du pH), eau.
2 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25 °C. Ne pas congeler.
6.5. Nature et contenu de l'emballage extérieur
3 mL de solution dans une seringue préremplie de 3 mL (verre transparent de type I) avec bouchon piston en chlorobutyle gris et embout Luer avec butée en polypropylène blanc.
Une aiguille séparée 25G, 16 mm, sera incluse pour réaliser une injection.
Conditionnement unitaire contenant une seringue préremplie et une aiguille ou conditionnement multiple contenant trois seringues préremplies et trois aiguilles.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
La solution doit être limpide et incolore, et exempte de particules visibles.
Utilisation dans la population pédiatrique
La dose appropriée à administrer est déterminée en fonction du poids corporel (voir rubrique 4.2).
Si la dose requise est inférieure à 30 mg (3 mL), les accessoires ci-dessous sont nécessaires pour prélever et administrer la dose appropriée :
· adaptateur (raccord Luer Lock femelle proximal et/ou distal) ;
· seringue graduée de 3 mL (recommandée).
La seringue préremplie d’icatibant et tous les autres composants sont à usage unique.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Toutes les aiguilles et seringues doivent être éliminées dans un collecteur d’aiguilles.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
35 RUE DU VAL DE MARNE
75013 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 375 5 7 : 3 mL en seringue préremplie (verre) + 1 aiguille. Boîte de 1.
· 34009 302 375 6 4 : 3 mL en seringue préremplie (verre) + 1 aiguille. Boîte de 3.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 18/11/2021
ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie
Icatibant
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie ?
3. Comment utiliser ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie ET DANS QUELS CAS EST-IL UTILISE ?
ICATIBANT ZENTIVA contient la substance active icatibant.
ICATIBANT ZENTIVA est utilisé pour le traitement des symptômes de l’angio-œdème héréditaire (AOH) chez les adultes, les adolescents et les enfants âgés de 2 ans et plus.
Dans l’AOH, les taux d’une substance présente dans la circulation sanguine, appelée bradykinine, sont plus élevés, ce qui provoque des symptômes tels que gonflement, douleurs, nausées et diarrhée.
ICATIBANT ZENTIVA bloque l’activité de la bradykinine et, par conséquent, stoppe la progression des symptômes d’une crise d’AOH.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie ?
N’utilisez jamais ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie
· si vous êtes allergique à l’icatibant ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin avant de prendre ICATIBANT ZENTIVA :
· si vous souffrez d’angine de poitrine (réduction du débit sanguin vers le muscle cardiaque) ;
· si vous avez récemment eu un accident vasculaire cérébral.
Certains des effets indésirables liés à ICATIBANT ZENTIVA sont similaires aux symptômes de votre maladie. Si vous remarquez que les symptômes de la crise empirent après l’administration de ce médicament, informez-en immédiatement votre médecin.
De plus :
· Il est impératif que vous ou un soignant ayez suivi une formation sur la technique de l’injection sous-cutanée (injection sous la peau) avant que vous ou un soignant puissiez injecter ce médicament.
· Immédiatement après une auto-injection d’ICATIBANT ZENTIVA ou une administration par un soignant formé en cas d’œdème laryngé (obstruction des voies respiratoires supérieures), veuillez vous rendre dans un établissement médical.
· Si vos symptômes ne disparaissent pas après une injection d’ICATIBANT ZENTIVA en auto-administration ou par un soignant, consultez un médecin concernant des injections supplémentaires de ce médicament. Chez les patients adultes, jusqu’à 2 injections supplémentaires pourront être administrées en 24 heures.
Enfants et adolescents
L’utilisation de ce médicament n’est pas recommandée chez les enfants âgés de moins de 2 ans ou pesant moins de 12 kg car il n’a pas été étudié chez ces patients.
Autres médicaments et ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie
Informez votre médecin si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
ICATIBANT ZENTIVA n’est pas connu pour interagir avec d’autres médicaments. Si vous prenez un médicament appelé « inhibiteur de l’enzyme de conversion de l’angiotensine » (ECA) (par exemple, captopril, énalapril, ramipril, quinapril, lisinopril) qui est utilisé pour réduire votre pression artérielle ou pour toute autre raison, vous devez en informer votre médecin avant de recevoir ce médicament.
ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie avec des aliments et boissons
Sans objet.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de commencer à utiliser ce médicament.
Si vous allaitez, vous ne devez pas le faire pendant les 12 heures qui suivent la dernière prise de ce médicament.
Conduite de véhicules et utilisation de machines
Ne conduisez pas de véhicules et n’utilisez pas de machines si vous vous sentez fatigué ou si vous avez des sensations vertigineuses suite à une crise d’AOH ou après avoir utilisé ce médicament.
ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie contient du sodium
La solution injectable contient moins de 1 mmol (23 mg) de sodium, c.-à-d. qu’elle est essentiellement « sans sodium ».
3. COMMENT UTILISER ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie ?
Si vous n’avez jamais reçu ICATIBANT ZENTIVA, votre première dose de ce médicament vous sera toujours injectée par votre médecin ou votre infirmier/ère. Votre médecin vous indiquera quand vous pourrez rentrer chez vous en toute sécurité.
Il est possible, après discussion avec votre médecin ou votre infirmier/ère et suite à une formation adéquate sur la technique de l’injection sous-cutanée (injection sous la peau), que l’on vous autorise à vous auto-injecter ICATIBANT ZENTIVA ou qu’un soignant vous injecte ce médicament en cas de crise d’AOH.
Il est important que l’injection sous-cutanée (sous la peau) d’ICATIBANT ZENTIVA soit pratiquée dès les tout premiers symptômes d’angio-œdème. Le professionnel de santé qui s’occupe de vous vous montrera (à vous et/ou à votre soignant) comment procéder à une injection de ce médicament en toute sécurité conformément aux instructions fournies dans cette notice.
Quand et à quelle fréquence utiliser ICATIBANT ZENTIVA
Votre médecin a déterminé la dose exacte de ce médicament et vous indiquera à quelle fréquence vous devez l’utiliser.
Adultes
· La dose recommandée d’ICATIBANT ZENTIVA est d’une injection (3 mL, 30 mg) administrée en sous-cutané (sous la peau) dès que vous remarquez l’apparition d’une crise d’angio-œdème (par exemple en cas d’augmentation du gonflement cutané, notamment lorsqu’il affecte le visage et le cou, ou en cas de douleur abdominale plus importante).
· Si vous ne ressentez aucun soulagement des symptômes 6 heures après l’injection, vous devez consulter un médecin concernant des injections supplémentaires d’ICATIBANT ZENTIVA. Chez les adultes, jusqu’à 2 injections supplémentaires pourront être administrées dans les 24 heures.
· Vous ne devez pas recevoir plus de 3 injections en 24 heures et, si vous avez besoin de plus de 8 injections en un mois, vous devez consulter votre médecin.
Enfants et adolescents âgés de 2 à 17 ans
· La dose recommandée d’ICATIBANT ZENTIVA est d’une injection de 1 mL jusqu’à un maximum de 3 mL en fonction du poids, administrée en sous-cutané (sous la peau) dès que vous remarquez l’apparition d’une crise d’angio-œdème (par exemple, en cas d’augmentation du gonflement cutané, notamment lorsqu’il affecte le visage et le cou, ou en cas de douleur abdominale plus importante).
· Voir la rubrique Instructions d’utilisation pour les informations sur la dose à injecter.
· En cas de doute sur la dose à injecter, adressez-vous à votre médecin, pharmacien ou infirmier/ère.
· Si les symptômes s’aggravent ou ne s’améliorent pas, vous devez immédiatement consulter un médecin.
Administration d’ICATIBANT ZENTIVA
ICATIBANT ZENTIVA est conçu pour être administré par injection sous-cutanée (sous la peau). Chaque seringue ne doit être utilisée qu’une fois.
Ce médicament est injecté au moyen d’une aiguille courte dans le tissu adipeux, sous la peau, dans l’abdomen (ventre).
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
Les instructions suivantes, présentées étape par étape, concernent :
· l’auto-administration (adultes),
· l’administration par un soignant ou un professionnel de santé chez les adultes, les adolescents et les enfants âgés de plus de 2 ans (pesant au moins 12 kg).
Ces instructions sont organisées comme suit :
1) Informations générales
2a) Préparation de la seringue pour les enfants et adolescents (2 à 17 ans) pesant 65 kg ou moins
2b) Préparation de la seringue et de l’aiguille pour l’injection (tous les patients)
3) Préparation du site d’injection
4) Injection de la solution
5) Elimination du kit d’injection
Instructions étape par étape pour l’injection
|
1) Informations générales |
||||||||||
|
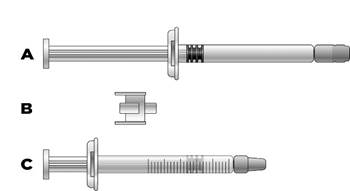
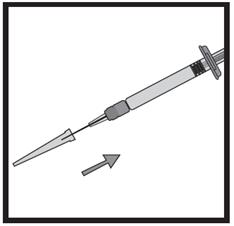
· Nettoyez le plan de travail (la surface) utilisé avant de commencer. · Lavez-vous les mains à l’eau et au savon. · Retirez la seringue préremplie de la boîte. · Retirez le capuchon situé à l’extrémité de la seringue préremplie en dévissant celui-ci. · Une fois le capuchon retiré, posez la seringue préremplie sur une surface plane. |
||||||||||
|
2a) Préparation de la seringue pour les enfants et adolescents (2 à 17 ans) pesant 65 kg ou moins : |
||||||||||
|
Informations importantes destinées aux professionnels de santé et aux soignants : Si la dose est inférieure à 30 mg (3 mL), le matériel suivant est nécessaire pour extraire la dose appropriée (voir ci-dessous) : a) Seringue préremplie d’ICATIBANT ZENTIVA (contenant la solution d’icatibant) b) Raccord (adaptateur) c) Seringue graduée de 3 mL
Le volume à injecter en mL nécessaire doit être prélevé dans une seringue graduée de 3 mL vide (voir le tableau ci-dessous). Tableau 1 : Schéma posologique chez les enfants et adolescents
Chez les patients pesant plus de 65 kg, utiliser le contenu total de la seringue préremplie (3 mL).
1) Retirez les capuchons à chaque extrémité du raccord.
2) Vissez le raccord sur la seringue préremplie. 3) Fixez la seringue graduée à l’autre extrémité du raccord en veillant à ce que les deux connexions soient bien verrouillées.
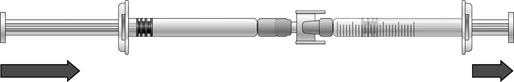
Transfert de la solution d’icatibant dans la seringue graduée : 1) Pour commencer à transférer la solution d’icatibant, appuyez sur le piston de la seringue préremplie (à l’extrême gauche sur l’illustration ci-dessous).
2) Si la solution d’icatibant ne commence pas à passer dans la seringue graduée, tirez légèrement sur le piston de la seringue graduée jusqu’à ce que la solution d’icatibant commence à s’écouler dans la seringue graduée (voir l’illustration ci-dessous).
3) Continuez à appuyer sur le piston de la seringue préremplie jusqu’à ce que le volume d’injection (dose) nécessaire soit transféré dans la seringue graduée. Voir le tableau 1 pour les informations sur la posologie. |
||||||||||
|
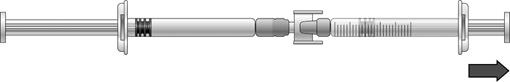
S’il y a de l’air dans la seringue graduée : · Retournez les seringues connectées de façon à ce que la seringue préremplie soit en haut (voir l’illustration ci-dessous).
· Appuyez sur le piston de la seringue graduée afin que tout l’air repasse dans la seringue préremplie (il peut être nécessaire de répéter cette étape plusieurs fois). · Prélevez le volume nécessaire de solution d’icatibant. |
||||||||||
|
4) Retirez la seringue préremplie et le raccord de la seringue graduée. 5) Eliminez la seringue préremplie et le raccord dans le collecteur d’aiguilles. |
||||||||||
|
2b) Préparation de la seringue et de l’aiguille pour l’injection : tous les patients (adultes, adolescents et enfants) |
||||||||||
|
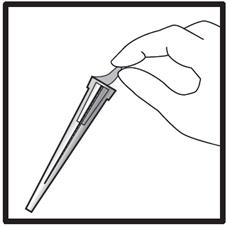
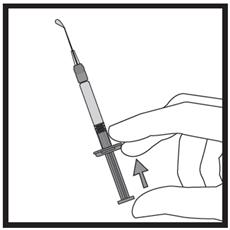
· Retirez de la plaquette l’étui contenant l’aiguille. · Retirez le film protecteur de l’étui (assurez-vous que l’aiguille reste bien dans son étui).
· Munissez-vous de la seringue et maintenez-la fermement. Fixez soigneusement l’aiguille à la seringue préremplie contenant la solution incolore. · Introduisez la seringue dans l’étui contenant l’aiguille et vissez la seringue à l’aiguille. · Retirez l’aiguille de son étui en tirant sur le corps de la seringue. Ne tirez pas sur le piston. · La seringue est à présent prête pour l’injection. |
||||||||||
|
3) Préparation du site d’injection |
||||||||||
|
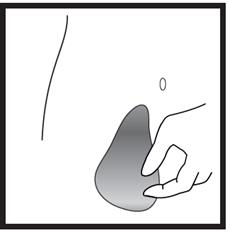
· Choisissez le site d’injection. L’injection doit être pratiquée dans un pli de la peau sur le côté gauche ou droit de votre ventre à environ 5– 10 cm au-dessous de votre nombril. Cette zone doit se trouver à au moins 5 cm de toute cicatrice éventuelle. Ne pas choisir une zone tuméfiée (gonflée), présentant des ecchymoses (bleus) ou douloureuse. · Nettoyez le site d’injection à l’aide d’un coton imbibé d’alcool et laissez sécher. |
||||||||||
|
4) Injection de la solution |
||||||||||
|
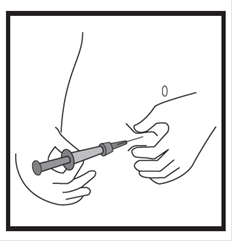
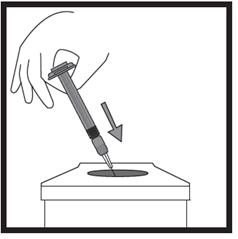
· Maintenez la seringue avec les deux doigts d’une main, le pouce étant positionné sur le piston. · Vérifiez l’absence de bulles d’air dans la seringue en appuyant sur le piston jusqu’à l’apparition d’une première goutte à l’extrémité de l’aiguille.
· Maintenez la seringue selon un angle compris entre 45 et 90 degrés par rapport à la surface de la peau, l’aiguille étant dirigée vers la peau. · Tandis que vous maintenez la seringue d’une main, utilisez l’autre main pour former un pli de peau entre le pouce et l’index au site d’injection que vous avez désinfecté au préalable. · Tout en maintenant le pli de peau, mettez la seringue en contact avec la peau et introduisez l’aiguille rapidement dans le pli de peau. · Appuyez lentement sur le piston tout en gardant la position initiale de votre main jusqu’à ce que l’intégralité du liquide soit injectée dans la peau et que plus aucun liquide ne reste dans la seringue. · Appuyez lentement sur le piston ; une injection doit prendre environ 30 secondes. · Relâchez le pli de peau et retirez doucement l’aiguille. |
||||||||||
|
5) Elimination du kit d’injection |
||||||||||
|
· Jetez la seringue, l’aiguille et l’étui protecteur de l’aiguille dans le conteneur prévu à cet effet (conteneur pour objets piquants/coupants/tranchants destiné à l’élimination des déchets dangereux), afin d’éviter toute blessure à quiconque en cas de mauvaise manipulation. |


Si vous avez utilisé plus d’ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie que vous n’auriez dû
Sans objet.
Si vous oubliez d’utiliser ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie
Sans objet.
Si vous arrêtez d’utiliser ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Informez immédiatement votre médecin si vous remarquez que les symptômes de votre crise s’aggravent après avoir reçu ce médicament.
Très fréquent (pouvant affecter plus de 1 personne sur 10) :
Réactions supplémentaires au site d’injection (sensation de pression, ecchymose (« bleu »), diminution de la sensibilité et/ou engourdissement, éruption cutanée accompagnée de démangeaisons et sensation de chaleur).
Fréquent (pouvant affecter jusqu’à 1 personne sur 10) :
Nausées
Maux de tête
Sensations vertigineuses
Fièvre
Démangeaisons
Eruption cutanée
Rougeur de la peau
Anomalies des tests de la fonction hépatique
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
Urticaire
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et la boîte après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25 °C. Ne pas congeler.
N’utilisez pas ce médicament si vous remarquez que l’emballage de la seringue ou de l’aiguille est endommagé ou s’il existe des signes visibles de détérioration, par exemple si la solution est opaque, si elle contient des particules ou si sa couleur a changé.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ICATIBANT ZENTIVA 30 mg, solution injectable en seringue préremplie
· La substance active est :
Icatibant........................................................................................................................... 30 mg
sous forme d’acétate d'icatibant
Pour une seringue préremplie de 3 mL.
Chaque mL de cette solution contient 10 mg d’icatibant.
· Les autres composants sont : chlorure de sodium, acide acétique glacial, hydroxyde de sodium (pour ajustement du pH) et eau (voir rubrique 2).
ICATIBANT ZENTIVA se présente sous la forme d’une solution injectable limpide et incolore disponible dans une seringue de verre préremplie de 3 mL. Une aiguille hypodermique est incluse dans l’emballage.
ICATIBANT ZENTIVA est disponible en conditionnement unitaire contenant une seringue préremplie et une aiguille ou en conditionnement multiple contenant trois seringues préremplies et trois aiguilles.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
35 RUE DU VAL DE MARNE
75013 PARIS
Exploitant de l’autorisation de mise sur le marché
ZENTIVA FRANCE
35 RUE DU VAL DE MARNE
75013 PARIS
KW20A KORDIN INDUSTRIAL PARK
PAOLA PLA 3000
MALTE
ou
EUROFINS PROXY LABORATORIES B.V.
ARCHIMEDESWEG 25
2333 CM LEIDEN
PAYS-BAS
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).