Dernière mise à jour le 29/06/2026
BICNU, poudre et solvant pour solution pour perfusion
Indications thérapeutiques
BICNU est utilisé en monothérapie ou en association thérapeutique établie avec d'autres substances anticancéreuses autorisées dans certains types de cancers, comme:
· Tumeurs cérébrales primitives ou secondaires
· Myélomes multiples
· Lymphomes hodgkiniens
· Lymphomes non hodgkiniens
· Mélanomes
· conditionnement préalable à une greffe autologue de cellules souches hématopoïétiques (GCSH) pour le traitement des maladies hématologiques malignes (Maladie de Hodgkin’s / lymphome Non-Hodgkinien).
Présentations
> 1 flacon(s) poudre en verre de 100 mg - 1 flacon(s) solvant en verre de 3 mL
Code CIP : 561 991-6 ou 34009 561 991 6 7
Déclaration de commercialisation : 16/12/2000
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l\'aide)
Autres informations
- Titulaire de l'autorisation : TILLOMED PHARMA GMBH
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux médecins compétents en maladie du sang
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- réservé à l'usage HOSPITALIER
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 191 879 5
ANSM - Mis à jour le : 18/02/2025
BICNU, poudre et solvant pour solution pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour un flacon
Excipient à effet notoire :
Chaque flacon de solvant contient 3 mL de propylène glycol (équivalant à 3,1125 g).
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution pour perfusion.
Poudre : poudre jaunâtre pour reconstitution.
Solvant : liquide visqueux, limpide et incolore.
Apparence de la solution : solution jaune pâle, limpide, exempte de particules visibles.
Le pH et l’osmolarité de la solution pour perfusion prête à l’emploi sont :
pH : 4,0 à 6,8 diluée ou non avec une solution physiologique ou une solution de dextrose à 5 %.
Osmolarité : 320 à 390 mOsmol/l (si diluée dans une solution pour injection de dextrose 50 mg/mL [5 %]) ou dans une solution pour injection de chlorure de sodium 9 mg/mL [0,9 %])
4.1. Indications thérapeutiques
BICNU est utilisé seul ou en association dans le traitement des :
· tumeurs cérébrales primitives ou secondaires,
· myélomes multiples,
· lymphomes hodgkiniens,
· lymphomes non hodgkiniens,
· mélanomes,
· conditionnement préalable à une greffe autologue de cellules souches hématopoïétiques (GCSH) pour le traitement des maladies hématologiques malignes (Maladie de Hodgkin’s / lymphome Non-Hodgkinien).
4.2. Posologie et mode d'administration
Dans toutes les modalités d'administration en monothérapie ou en polychimiothérapie, le délai entre les cures contenant du BICNU ne devra pas être inférieur à 6 semaines.
Dans la majorité des cas, BICNU est prescrit en polychimiothérapie à la posologie moyenne de 150 mg/m² toutes les 6 semaines.
Le produit peut être administré en monothérapie (tumeurs cérébrales primitives).
La posologie chez des sujets non antérieurement traités est de 200 mg/m2 par voie IV, toutes les 6 semaines. Cette dose est habituellement prescrite en une seule injection, mais elle peut être divisée en 2 injections de 100 mg/m2 administrées pendant 2 jours consécutifs.
En traitement de conditionnement préalable à une greffe autologue de cellules souches hématopoïétiques (Forte dose) :
Avant une greffe autologue de cellules souches hématopoïétiques chez des patients souffrant des maladies hématologiques malignes, la carmustine est administrée en association avec d’autres agents chimiothérapeutiques.
Population pédiatrique
BICNU ne doit pas être utilisé chez l’enfant âgé de moins de 5 ans.
Dans le cadre d’un conditionnement préalable à une greffe autologue de cellules souches hématopoïétiques (Forte dose), BICNU est contre-indiquée chez les enfants et les adolescents de moins de 18 ans (voir rubrique 4.3)
Mode d’administration
Pour une utilisation par voie intraveineuse après reconstitution et dilution supplémentaire.
Pour les instructions de reconstitution et de dilution du médicament avant administration, voir rubrique 6.6.
La solution pour perfusion prête à l’emploi résultante doit ensuite être administrée immédiatement par goutte-à-goutte intraveineux sur une période d’une à deux heures à l’abri de la lumière. La durée de la perfusion ne doit pas être inférieure à une heure, sinon cela entraîne des brûlures et des douleurs au site d’injection. Le site d’injection doit être surveillé pendant l’administration.
Modalités de manipulation
La préparation des solutions injectables de cytotoxiques doit être obligatoirement réalisée par un personnel spécialisé et entraîné ayant une connaissance des médicaments utilisés, dans des conditions assurant la protection de l'environnement et surtout la protection du personnel qui manipule. Elle nécessite un local de préparation réservé à cet usage. Il est interdit de fumer, de manger, de boire dans ce local. Les manipulateurs doivent disposer d'un ensemble de matériel approprié à la manipulation notamment blouses à manches longues, masques de protection, calot, lunettes de protection, gants à usage unique stériles, champs de protection du plan de travail, conteneurs et sacs de collecte des déchets. Les excréta et les vomissures doivent être manipulés avec précaution. Les femmes enceintes doivent être averties et éviter la manipulation des cytotoxiques. Tout contenant cassé doit être traité avec les mêmes précautions et considéré comme un déchet contaminé. L'élimination des déchets contaminés se fait par incinération dans des conteneurs rigides étiquetés à cet effet.
Ces dispositions peuvent être envisagées dans le cadre du réseau de cancérologie (circulaire DGS/DH/98 N° 98/188 du 24 mars 1998) en collaboration avec toute structure adaptée et remplissant les conditions requises.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Ne pas administrer aux personnes ayant présenté une diminution du nombre de plaquettes, leucocytes ou érythrocytes lors d'une précédente chimiothérapie ou pour d'autres causes (voir rubrique 4.4).
· Enfant de moins de 5 ans.
· Dans le cadre d’un conditionnement préalable à une greffe autologue de cellules souches hématopoïétiques (Forte dose), BICNU est contre-indiquée chez les enfants et les adolescents de moins de 18 ans
· Grossesse et allaitement (voir rubrique 4.6).
· En association avec les vaccins vivants atténués (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Toxicité pulmonaire : voir rubrique 4.8.
Les injections ne seront répétées que lorsque le nombre de plaquettes et de granulocytes sera redevenu acceptable, respectivement 100 000/mm3 et 4 000/mm3 habituellement après 6 semaines. Les numérations sanguines seront effectuées fréquemment et la cure suivante ne sera pas administrée avant 6 semaines du fait de la toxicité retardée. Les doses seront ajustées en fonction de la réponse hématologique du malade aux doses précédentes.
Le schéma suivant peut servir de guide pour ajuster les doses.
|
Après la 1ère dose |
% 1ère dose à administrer pour renouvellement |
|
|
Leucocytes |
Plaquettes |
|
|
> 4 000 |
> 100 000 |
100 % |
|
3 000 - 3 999 |
75 000 - 99 999 |
100 % |
|
2 000 - 2 999 |
25 000 - 74 999 |
70 % |
|
< 2 000 |
< 25 000 |
50 % |
Une myélosuppression sévère, pouvant survenir en raison de l'utilisation de carmustine à forte dose ou en association avec d'autres agents cytotoxiques, peut entraîner des infections opportunistes, y compris une pneumonie. De telles infections systémiques peuvent conduire à un sepsis et avoir, par la suite, des conséquences fatales ou mortelles. En cas de myélosuppression sévère, le patient doit être surveillé pour détecter tout signe ou symptôme d'infection.
Chez les patients traités par nitroso-urées, des cas de leucémies aiguës et de dysplasie de la moelle osseuse ont été rapportés.
Les fonctions rénale et hépatique doivent être surveillées régulièrement.
La carmustine peut entraîner des effets génotoxiques et provoquer des dégénérescences testiculaires chez plusieurs modèles animaux. Par conséquent, les hommes devant être traités par BICNU doivent être avertis du risque encouru en cas de conception d’un enfant pendant le traitement et jusqu’à 6 mois après le traitement. Une cryoconservation de leur sperme peut être envisagée avant le traitement en raison de la possibilité d’infertilité irréversible dû au traitement par BICNU.
Suite à un contact cutané accidentel avec la solution reconstituée, une hyperpigmentation transitoire des zones atteintes a été rapportée. En cas de contact de la poudre lyophilisée ou de la solution avec la peau ou les muqueuses, rincer immédiatement et abondamment à l’eau.
Ce médicament est déconseillé avec les vaccins vivants atténués, la phénytoïne ou la fosphénytoïne, avec la cimétidine utilisée à des doses supérieures ou égales à 800 mg/j (voir rubrique 4.5).
Ce médicament contient 3112,5 mg de propylène glycol dans chaque unité posologique de 100 mg de carmustine, ce qui est équivalent à 11205 mg de propylène glycol ou 10,8 mL de propylène glycol par dose.
Une surveillance médicale est nécessaire chez les patients souffrant d’insuffisance rénale ou d’une altération de la fonction hépatique, car différents effets indésirables attribués au propylène glycol ont été signalés, notamment un dysfonctionnement rénal (nécrose tubulaire aiguë), une insuffisance rénale aiguë et un dysfonctionnement hépatique.
BICNU doit être administré en injection intraveineuse lente d’une durée comprise entre 1 à 2 heures.
Des réactions au site d'injection peuvent survenir durant l'administration du BICNU (voir rubrique 4.8). Compte-tenu du risque d'extravasation, il est recommandé de surveiller étroitement le site de perfusion pour rechercher des signes d'infiltration durant l'administration du produit. Il n'existe pas de traitement spécifique des réactions d'extravasation à ce jour.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les études d’interaction n’ont été réalisées que chez l’adulte.
INTERACTIONS COMMUNES A TOUS LES CYTOTOXIQUES
Associations contre-indiquées
+ Vaccins vivants atténués
Risque de maladie vaccinale généralisée mortelle. Contre-indication pendant le traitement et pendant les 6 mois suivant l'arrêt de la chimiothérapie
Associations déconseillées (voir rubrique 4.4)
+ Phénytoïne (et, par extrapolation, fosphénytoïne)
Risque de survenue de convulsions par diminution de l'absorption digestive de la seule phénytoïne par le cytotoxique, ou bien risque de majoration de la toxicité ou de perte d'efficacité du cytotoxique par augmentation de son métabolisme hépatique par la phénytoïne ou la fosphénytoïne.
+ Olaparib
Risque de majoration de l’effet myélosuppresseur du cytotoxique
Associations faisant l'objet de précautions d’emploi
+ Antivitamine K
Augmentation du risque thrombotique et hémorragique au cours des affections tumorales. De surcroit, possible interaction entre les AVK et la chimiothérapie. Un contrôle plus fréquent de l'INR est nécessaire
Associations à prendre en compte
+ Flucytosine
Risque de majoration de la toxicité hématologique
+ Immunosuppresseurs
Immunodépression excessive avec risque de syndrome lympho-prolifératif.
INTERACTION SPECIFIQUE A LA CARMUSTINE
Associations déconseillées
+ Cimétidine
Avec la cimétidine utilisée à des doses supérieures ou égales à 800 mg/j : toxicité médullaire accrue (inhibition du métabolisme de la carmustine).
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer/contraception chez les hommes et les femmes
Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace afin d’éviter une grossesse au cours du traitement et au moins 6 mois après le traitement.
Il convient de conseiller aux patients de sexe masculin d’utiliser une méthode de contraception appropriée pendant le traitement par carmustine et pendant au moins 6 mois après le traitement.
Grossesse
Compte tenu des données disponibles, l'utilisation de carmustine est contre-indiquée au cours de la grossesse et chez la femme en âge de procréer n'utilisant pas de contraception efficace. La carmustine est embryotoxique chez les rats et les lapins et tératogène chez les rats à des doses équivalentes à celles utilisées dans l'espèce humaine.
On ignore si la carmustine est excrétée dans le lait. En raison des potentiels effets indésirables graves pouvant survenir chez l'enfant nourri au sein, l'allaitement doit être interrompu pendant la prise de BICNU.
Fertilité
La carmustine peut entraîner des effets génotoxiques et provoquer des dégénérescences testiculaires chez plusieurs modèles animaux. La carmustine peut altérer la fertilité masculine. Il convient d’informer les patients de sexe masculin du risque potentiel d’infertilité et de recourir à des conseils en matière de fertilité/planification familiale avant un traitement par carmustine.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Chez les patients ayant reçu de la carmustine durant l’enfance ou l’adolescence, des cas de fibrose pulmonaire apparue extrêmement tardivement (jusqu’à 17 ans après le traitement) ont été décrits. Une étude de suivi à long terme portant sur 17 patients ayant survécu à des tumeurs cérébrales durant l’enfance a montré que huit d’entre eux ont succombé à une fibrose pulmonaire. Deux de ces huit décès sont survenus au cours des trois premières années de traitement et six d’entre eux 8 à 13 ans après le traitement. L’âge médian des patients décédés au cours du traitement était de 2,5 ans (1-12 ans), l’âge médian des survivants à long terme sous traitement était de 10 ans (5- 16 ans). Tous les patients âgés de moins de 5 ans au moment du traitement sont décédés d’une fibrose pulmonaire; ni la dose de carmustine, ni une dose supplémentaire de vincristine ni une irradiation spinale n’ont eu d’influence sur l’issue fatale. Une fibrose pulmonaire a été diagnostiquée chez tous les survivants disponibles pour un suivi.
Troubles hématologiques principalement :
Toxicité hématologique retardée caractérisée par une thrombocytopénie et par une leucopénie survenant respectivement 4 à 5 semaines et 5 à 6 semaines après l'injection. La thrombopénie est en général plus sévère que la leucopénie. Une anémie peut également se produire mais elle est en général moins sévère. Cette toxicité est dose-dépendante et peut devenir cumulative lorsque les traitements se répètent.
La survenue de leucémies aiguës ou de dysplasies médullaires a été signalée chez des malades recevant un traitement au long cours.
Infections et infestations
· Des cas d’infections opportunistes, incluant des pneumonies, ont été rapportés. Certains ont eu une évolution fatale.
Troubles gastro-intestinaux :
Fréquemment observés dans les 2 heures suivant l'injection : nausées et vomissements pouvant durer 4 à 6 heures, étant dose-dépendants et nécessitant l'utilisation d'antiémétiques.
L’apparition de mucites est également très fréquemment observée
Troubles respiratoires :
La toxicité pulmonaire induite par la carmustine a été rapportée avec une fréquence allant jusqu’à
30 %. Les manifestations précoces surviennent généralement dans les 3 ans de traitement et sont caractérisées par des infiltrats et/ou une fibrose pulmonaire ; des cas d’évolution fatale ont été rapportés. Des cas de fibrose pulmonaire retardée survenant jusque 17 ans après le traitement ont également été rapportés.
Ces troubles surviennent quel que soit l’âge des patients.
La toxicité pulmonaire est dose-dépendante, des doses cumulatives totales comprises entre 1200 et 1500 mg/m2 étant associées à un risque accru de fibrose pulmonaire.
Les facteurs de risque incluent le tabagisme, l’existence d’une pathologie respiratoire, des anomalies radiologiques préexistantes, une irradiation thoracique séquentielle ou concomitante et l’association avec d’autres facteurs entraînant des troubles pulmonaires.
Les patients présentant une capacité vitale ou une capacité de diffusion du monoxyde de carbone inférieure de 70 % aux valeurs théoriques sont particulièrement à risque.
Une surveillance pulmonaire particulière pourra être envisagée.
La toxicité pulmonaire se manifeste également par une pneumopathie inflammatoire et une pneumopathie interstitielle au cours de l’expérience post-commercialisation.
Troubles hépatiques :
Lors de l'administration de fortes doses de BICNU, rares élévations transitoires des transaminases, des phosphatases alcalines et de la bilirubine.
Troubles rénaux :
Altérations rénales (diminution du volume rénal, azotémie, insuffisance rénale) après des doses élevées et prolongées. Ces altérations ont aussi été notées chez les malades recevant des doses plus faibles.
Troubles cardiovasculaires :
· Hypotension, tachycardie.
Egalement notés :
· Brûlures sur le trajet veineux.
· Des perfusions rapides peuvent entraîner des rougeurs de la peau intenses et une suffusion de la
· conjonctive dans les 2 heures et durant environ 4 heures.
· Aménorrhée, azoospermie.
· Quelques cas de neurorétinite, douleur thoracique, céphalées, réactions allergiques ont été rapportés.
Réaction au point d'injection :
Une toxicité locale des tissus mous consécutive à l'extravasation de BICNU a été rapportée. L'infiltration de BICNU peut entraîner gonflement, douleur, érythème, sensation de brûlure et exceptionnellement nécrose de la peau.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
BICNU est un antinéoplasique cytostatique alkylant appartenant au groupe des nitroso-urées. Il agit essentiellement par alkylation de l'ADN et de l'ARN ainsi que par carbamylation des protéines.
L'intérêt particulier de BICNU dans le domaine des cytostatiques est représenté notamment par sa grande solubilité dans les graisses qui favorise alors son passage à travers la barrière hémato-encéphalique.
Mécanisme d’action
Sans objet.
Effets pharmacodynamiques
Sans objet.
Efficacité et sécurité clinique
Sans objet.
Population pédiatrique
Sans objet.
5.2. Propriétés pharmacocinétiques
Administré par voie IV, BICNU est rapidement métabolisé et, après 15 minutes, il n'est pas retrouvé dans le sang de produit non dégradé.
Cependant après administration du produit marqué au C14, des taux prolongés de l'isotope sont retrouvés dans le plasma et les tissus.
L'activité et la toxicité du produit sont vraisemblablement dues à ses métabolites.
Distribution
Du fait de sa grande solubilité dans les lipides et de son absence d'ionisation au pH physiologique, BICNU traverse la barrière méningée.
Biotransformation
Sans objet.
Élimination
Environ 60 à 70 % de la dose totale est excrétée dans les urines après 96 heures, et environ 10 % est éliminée par voie respiratoire sous forme de CO2.
Les taux de radioactivité retrouvés dans le liquide céphalo-rachidien représentent 50 % ou plus des taux plasmatiques.
Linéarité/non-linéarité
Sans objet.
Relations pharmacocinétique/pharmacodynamique
Sans objet.
5.3. Données de sécurité préclinique
Compatibilité / Incompatibilité avec les conteneurs
La solution intraveineuse est instable dans un récipient en chlorure de polyvinyle. La solution de carmustine ne peut être administrée qu'à partir de bouteilles en verre ou de récipients en polypropylène.
Le médicament doit être utilisé conformément aux instructions de la rubrique 6.6, et non mélangé avec d'autres médicaments pharmaceutiques.
3 ans.
Après reconstitution selon les recommandations, la carmustine est stable pendant 480 heures au réfrigérateur (entre 2 °C et 8 °C) et 24 heures à température ambiante (25 °C ± 2 °C) dans un récipient en verre. Avant utilisation, les flacons reconstitués doivent être examinés pour vérifier qu’il n’y a pas de formation de cristaux. Si des cristaux sont observés, ils peuvent être redissous en réchauffant le flacon à température ambiante sous agitation.
La solution reconstituée, de nouveau diluée jusqu’à 500 mL avec du chlorure de sodium à 0,9 % pour injection ou du dextrose à 5 % pour injection dans des récipients en verre ou en polypropylène, est physiquement et chimiquement stable pendant 8 heures à 25 °C ± 2 °C à l’abri de la lumière. Ces solutions sont également stables jusqu’à 48 heures au réfrigérateur (entre 2 °C et 8 °C) et pendant 6 heures supplémentaires à 25 °C ± 2 °C à l’abri de la lumière.
La solution doit être conservée à l’abri de la lumière jusqu’à la fin de l’administration.
Du point de vue microbiologique, sauf si la méthode de reconstitution exclut le risque de contamination microbiologique, le produit doit être utilisé immédiatement. S’il n’est pas utilisé immédiatement, l’utilisateur est responsable des conditions et durées de conservation.
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
100 mg de poudre en flacon (verre) + 3,0 mL de solvant en flacon (verre) ; boîte de 1.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Comparé à d'autres médicaments contenant de la carmustine, le propylène glycol est utilisé comme diluant dans le BICNU à la place de l’éthanol. Le propylène glycol contribue à une meilleure stabilité de la solution reconstituée. Les étapes suivantes décrivent les règles de manipulation à suivre pour garantir une dissolution et une reconstitution optimale du médicament. Ces étapes diffèrent de celles à suivre pour des médicaments à base de carmustine dont le solvant est l’éthanol. Pour connaître les étapes à suivre pour ces médicaments veuillez-vous référer à leurs résumés des caractéristiques de produits en vigueur.
La reconstitution et la dilution de chaque flacon de poudre de solution pour perfusion de BICNU doivent être préparées comme décrit ci-après. Dissoudre la carmustine (100 mg de poudre) dans 3 ml du diluant stérile fourni (propylène glycol pour injection) jusqu'à obtention d'une solution claire. Utiliser le flacon de propylène glycol pour la reconstitution uniquement après avoir atteint la température ambiante et utiliser une aiguille à pores plus larges (aiguille de calibre inférieur à 22) pour prélever le diluant du flacon. Un guide étape par étape pour la reconstitution est fourni ci-dessous.
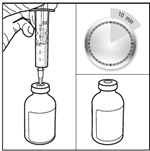
1. Sortir le BICNU du réfrigérateur (2°C-8°C) et le laisser atteindre la température ambiante. Retirer les flacons de produit et de diluant de l’emballage et les laisser à température ambiante pendant au moins 10 minutes. Cela permet que la solution se mélange correctement lors de la reconstitution. Le diluant ne doit être utilisé qu’après que le flacon ait atteint la température ambiante.
|
|
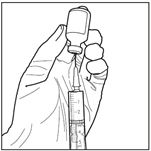
2. Prélever 3 mL du diluant stérile (solution de propylène glycol) avec une seringue stérile. Utiliser une seringue stérile pour prélever aseptiquement 3 mL de diluant du flacon. Assurez-vous de prélever la totalité du volume (3 mL) en utilisant une aiguille de calibre inférieur à 22 pour permettre une injection facile et précise.
|
|
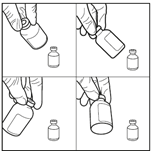
3. Injecter le diluant stérile dans le flacon de carmustine. Injecter les 3 mL de diluant stérile dans le flacon contenant 100 mg de carmustine. Laisser la solution s’hydrater pendant au moins 10 minutes, sans secouer immédiatement. Cette période de repos est nécessaire pour permettre au produit de se dissoudre correctement.
|
|
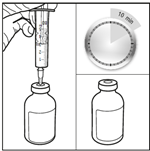
4. Agiter doucement le flacon pour obtenir une solution limpide. Après la période d’hydratation de 10 minutes, ne pas secouer le flacon de bas en haut mais agiter vigoureusement et régulièrement le flacon en mouvement circulaire pendant au moins 60 secondes, jusqu’à obtention d’une solution claire et homogène. Cette agitation est essentielle pour garantir la dissolution complète de la carmustine.
|
|
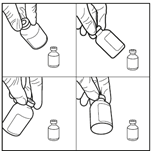
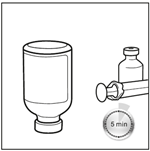
5. Laisser le flacon en position inversée pendant 5 minutes. Après avoir obtenu une solution limpide, placer le flacon en position inversée pendant 5 minutes pour permettre une préparation optimale avant le prélèvement.
Chaque millilitre de la solution reconstituée contient 33,3 mg de carmustine.
|
|
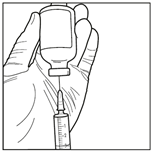
6. Prélever la solution reconstituée de manière aseptique. En maintenant le flacon en position inversée (au moment de l’insertion de l’aiguille et compte tenu de la viscosité de la solution reconstituée, le flacon doit rester le moins longtemps possible en position tête en haut et le flacon doit rester à nouveau en position stationnaire inversée une fois l’aiguille insérée avant le prélèvement), utiliser une seringue stérile pour prélever la solution reconstituée.
|
|
7. Dilution de la solution reconstituée.
La solution reconstituée doit en outre être diluée jusqu’à 500 mL avec une solution injectable de chlorure de sodium à 0,9 % ou une solution injectable de dextrose à 5 %. La solution résultante contient une concentration finale de 0,2 mg/mL de carmustine et doit être conservée à l’abri de la lumière. La solution ainsi préparée ne devra être injectée que par voie IV, sous la forme d'une perfusion lente, d'une durée comprise entre 1 et 2 heures. L’administration doit être effectuée avec un set de perfusion PE sans PVC.
La manipulation de ce cytotoxique par le personnel infirmier ou médical nécessite un ensemble de précautions permettant d'assurer la protection du manipulateur et de son environnement (Voir rubrique 4.2.).
BICNU se présente sous forme de poudre lyophilisée et ne contient pas de conservateur : il faudra donc préparer la solution extemporanément. Pour plus d’informations sur les durées et conditions de conservation des solutions reconstituées et diluées veuillez-vous référer à aux instructions de la rubrique 6.3.
La conservation de la carmustine à 27 °C ou à une température supérieure peut conduire à la liquéfaction de la substance, en raison du point de fusion bas de la carmustine (environ 30,5 °C à 32,0 °C). Un signe de décomposition est l’apparition d’une pellicule huileuse au fond du flacon qui est visible lorsque le flacon est exposé à une lumière vive. Ce médicament ne doit plus être utilisé. Des manifestations physiques se traduisant par des flocons pointus allant jusqu’à une masse solidifiée peuvent survenir dans le flacon non ouvert sans qu’une décomposition de la carmustine n’ait lieu.
Précautions importantes à respecter lors de la reconstitution
Ne pas utiliser les flacons avant qu’ils aient atteint la température ambiante. Il est essentiel de respecter le volume de diluant (3 mL) et le temps d’hydratation (10 minutes) avant de commencer l’agitation. Une fois la solution reconstituée, le flacon doit être manipulé avec précaution, et l’utilisation d’une aiguille de calibre inférieur à 22 est recommandée pour éviter tout dommage au flacon ou contamination du produit.
Ne pas utiliser les flacons conservés à des températures inadéquates (2°C-8°C) pour la reconstitution. Ne pas utiliser moins de 3 mL de diluant pour la reconstitution. Ne pas commencer l'agitation avant un temps de repos de 10 minutes. Ne pas arrêter l'agitation avant 60 secondes ou avant l'obtention d'une solution limpide. Ne pas prélever la solution reconstituée sans avoir maintenu le flacon inversé pendant 5 minutes. Ne pas utiliser une aiguille de calibre supérieur à 22 pour le prélèvement.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
MITTELSTRAßE 5 / 5A
12529 SCHÖNEFELD
ALLEMAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 561 991 6 7 : 100 mg de poudre en flacon (verre) + 3 mL de solvant en flacon (verre) ; boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament réservé à l’usage hospitalier.
Prescription réservée aux spécialistes en oncologie ou en hématologie ou aux médecins compétents en cancérologie ou en maladies du sang.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 18/02/2025
BiCNU, poudre et solvant pour solution pour perfusion
Carmustine
Gardez cette notice. Vous pourriez avoir besoin de la relire.
Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
2. Quelles sont les informations à connaître avant d'utiliser BICNU, poudre et solvant pour solution pour perfusion ?
3. Comment utiliser BICNU, poudre et solvant pour solution pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver BICNU, poudre et solvant pour solution pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE BICNU, poudre et solvant pour solution pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
BICNU est utilisé en monothérapie ou en association thérapeutique établie avec d'autres substances anticancéreuses autorisées dans certains types de cancers, comme:
· Tumeurs cérébrales primitives ou secondaires
· Myélomes multiples
· Lymphomes hodgkiniens
· Lymphomes non hodgkiniens
· Mélanomes
· conditionnement préalable à une greffe autologue de cellules souches hématopoïétiques (GCSH) pour le traitement des maladies hématologiques malignes (Maladie de Hodgkin’s / lymphome Non-Hodgkinien).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER BICNU, poudre et solvant pour solution pour perfusion ?
N’utilisez jamais BICNU, poudre et solvant pour solution pour perfusion :
· si vous êtes allergique (hypersensible) à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous avez connu des épisodes de diminution des cellules sanguines : globules rouges, globules blancs, plaquettes, lors d’un traitement antérieur ou pour d’autres raisons.
· si vous êtes enceinte, ou pensez l’être, ou si vous allaitez.
· si vous avez reçu et allez recevoir un vaccin vivant atténué (voir rubrique « Autres médicaments et BICNU, poudre et solvant pour solution pour perfusion »).
· chez les enfants de moins de 5 ans
· chez les enfants et les adolescents de moins de 18 ans dans le cadre d’un conditionnement préalable à une greffe autologue de cellules souches hématopoïétiques (Forte dose), BICNU est contre-indiquée
En cas de doute, il est indispensable de demander l’avis de votre médecin ou de votre pharmacien.
Avertissements et précautions
Le traitement doit être réalisé d’après les résultats des examens sanguins.
Ce médicament est déconseillé en association avec les vaccins vivants atténués (sauf le vaccin contre la fièvre jaune (voir rubrique « Autres médicaments et BICNU, poudre et solvant pour solution pour perfusion ») (rougeole, rubéole, oreillons, poliomyélite, tuberculose, varicelle), la phénytoïne ou la fosphénytoïne (médicaments utilisés dans le traitement de l’épilepsie), la cimétidine utilisée à des doses supérieures ou égales à 800 mg/j (voir rubrique «Prise ou utilisation d’autres médicaments »).
L'effet indésirable majeur de ce médicament étant la suppression retardée de la moelle osseuse, votre médecin surveillera la numération de la formule sanguine de façon hebdomadaire pendant au moins 6 semaines après l'administration d'une dose. À la dose recommandée, les cures de Carmustine ne doivent pas être données plus fréquemment que toutes les 6 semaines. Le dosage sera confirmé avec la numération de la formule sanguine. La baisse au niveau de la numération de la formule sanguine augmente le risque de certaines infections. Si vous remarquez des symptômes semblables à ceux de l'infection, contactez immédiatement votre médecin.
Ce médicament ne doit pas être utilisé en injection intraveineuse rapide.
Des réactions au site d'injection liées au passage de BICNU hors des vaisseaux peuvent survenir durant l'administration de BICNU (voir rubrique 4).
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser BICNU.
Enfants
N’utilisez jamais BICNU, poudre et solvant pour solution pour perfusion chez l’enfant de moins de 5 ans.
Autres médicaments et BICNU, poudre et solvant pour solution pour perfusion
Ce médicament NE DOIT PAS ETRE UTILISE en association avec le vaccin contre la fièvre jaune (voir rubrique "Ce médicament NE DOIT PAS ETRE UTILISE dans les cas suivants").
Ce médicament est DECONSEILLE en association avec les vaccins vivants atténués (sauf le vaccin contre la fièvre jaune, voir rubrique "Ce médicament NE DOIT PAS ETRE UTILISE dans les cas suivants") (rougeole, rubéole, oreillons, poliomyélite, tuberculose, varicelle), la phénytoïne ou la fosphénytoïne (médicaments utilisés dans le traitement de l'épilepsie), la cimétidine (médicament anti-ulcéreux et anti-reflux gastro-œsophagien) (voir rubrique "Mises en garde spéciales").
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
BICNU, poudre et solvant pour solution pour perfusion avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Ce médicament est contre-indiqué dans ces cas.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Grossesse
Ce médicament est contre-indiqué pendant la grossesse parce qu’il peut être nocif pour votre enfant à naître.
Les femmes en âge d’avoir des enfants doivent utiliser une méthode de contraception efficace pour éviter toute grossesse au cours du traitement par ce médicament et pendant six mois au moins après la fin du traitement. Si vous découvrez que vous êtes enceinte pendant le traitement par ce médicament, informez immédiatement votre médecin.
Les patients de sexe masculin doivent utiliser une contraception appropriée pendant le traitement par ce médicament pendant au moins 6 mois après la fin du traitement afin d’éviter que leur partenaire ne contracte une grossesse.
Allaitement
Vous ne devez pas allaiter pendant le traitement par ce médicament. On ne sait pas si la substance active de BICNU passe dans le lait maternel.
Fertilité
La carmustine peut provoquer une infertilité chez les hommes (nombre faible ou absence de spermatozoïdes). Cela peut affecter votre capacité à concevoir un enfant. Demandez conseil à votre médecin au sujet de la conservation du sperme avant de commencer votre traitement par BICNU.
Sportifs
Sans objet.
Conduite de véhicules et utilisation de machines
Les effets de ce médicament sur votre aptitude à conduire des véhicules et à utiliser des machines ne sont pas connus. Vous devez demander conseil à votre médecin avant de conduire un véhicule ou de manipuler des outils ou des machines car les vertiges sont un effet indésirable rapporté avec ce médicament, pouvant diminuer votre aptitude à conduire des véhicules ou à utiliser des machines.
BiCNU, poudre et solvent pour une solution pour perfusion, contient 3112,5 mg de propylène glycol dans chaque unité posologique de 100 mg de carmustine, ce qui est équivalent à 11205 mg de propylène glycol ou 10,8 mL de propylène glycol par dose.
Le propylène glycol contenu dans ce médicament peut avoir les mêmes effets que la consommation d’alcool et augmenter la probabilité d’effets secondaires.
Ce médicament ne peut être utilisé que sur l’avis de votre médecin. Pendant la prise de ce médicament, votre médecin peut prendre des dispositions pour vous faire subir des examens complémentaires.
3. COMMENT UTILISER BICNU, poudre et solvant pour solution pour perfusion ?
Posologie
BICNU poudre et solvant pour solution pour perfusion vous sera toujours administré par un professionnel de la santé expérimenté dans l'utilisation des médicaments anticancéreux.
Adultes
La posologie est basée sur votre état de santé, votre surface corporelle et votre réponse au traitement. Il est généralement administré au moins toutes les 6 semaines. La dose recommandée de BICNU poudre et solvant pour solution pour perfusion en monothérapie chez les patients non préalablement traités est de 150 à 200 mg / m2 par voie intraveineuse toutes les 6 semaines. Celui-ci peut être administré en dose unique ou divisé en perfusions quotidiennes telles que 75 à 100 mg / m2 sur deux jours successifs. La posologie dépendra également de l'administration de BICNU poudre et solvant pour solution pour perfusion avec d'autres médicaments anticancéreux.
Les doses seront ajustées en fonction de votre réponse au traitement.
La dose recommandée de BICNU administrée en association avec d’autres agents chimiothérapeutiques avant une greffe de cellules souches hématopoïétiques est de 300 à 600 mg/m2 par voie intraveineuse.
Votre hémogramme sera surveillé fréquemment pour éviter la toxicité dans votre moelle osseuse et la dose ajustée si nécessaire.
Mode et voie d’administration
Perfusion intraveineuse dans du sérum salé isotonique.
BICNU doit être administré en perfusion intraveineuse lente.
A l’attention du personnel soignant :
Comme pour tout cytotoxique, la préparation et la manipulation de ce produit nécessitent un ensemble de précautions permettant d’assurer la protection du manipulateur et de son environnement, dans les conditions de sécurité requises pour le patient.
En plus des précautions usuelles pour préserver la stérilité des préparations injectables, il faut :
· mettre une blouse à manches longues et poignets serrés, afin d’éviter toute projection de solution sur la peau,
· porter également un masque chirurgical à usage unique et des lunettes enveloppantes,
· mettre des gants à usage unique en PVC, et non en latex, après lavage aseptique des mains,
· préparer la solution sur un champ de travail,
· arrêter la perfusion, en cas d’injection hors de la veine,
· éliminer tout matériel ayant servi à la préparation de la solution (seringues, compresses, champs, flacon) dans un conteneur réservé à cet effet,
· détruire les déchets toxiques,
· manipuler les excréta et vomissures avec précaution.
Les femmes enceintes doivent éviter la manipulation des cytotoxiques.
En cas de contact de la poudre ou de la solution avec la peau ou les muqueuses, des sensations de brûlure et des taches marron peuvent apparaître ; rincer à l’eau immédiatement et abondamment.
Fréquence d’administration
Le délai entre les cures contenant du BICNU ne devra pas être inférieur à 6 semaines.
Durée du traitement
Une à deux perfusions toutes les 6 semaines.
Dans le cadre d’un conditionnement préalable à une greffe autologue de cellules souches hématopoïétiques (Forte dose) et selon le protocole de conditionnement choisit par votre médecin, BICNU est administré en une dose unique ou sur 3 jours consécutifs.
Utilisation chez les enfants
Ce médicament est contre-indiqué chez les enfants de moins de 5 ans.
Si vous avez utilisé plus de BICNU, poudre et solvant pour solution pour perfusion que vous n’auriez dû :
Sans objet.
Si vous oubliez d’utiliser BICNU, poudre et solvant pour solution pour perfusion :
Si vous arrêtez d’utiliser BICNU, poudre et solvant pour solution pour perfusion :
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Avertissez immédiatement votre médecin ou votre infirmier/ère:
Si vous présentez subitement une respiration sifflante, des difficultés à respirer, un gonflement des paupières, du visage ou des lèvres, une éruption cutanée ou des démangeaisons (en particulier si cela affecte tout votre corps), et si vous sentez que vous allez vous évanouir. Ces symptômes peuvent être des signes de réaction allergique grave.
Autres effets indésirables :
· Myélosuppression tardive (diminution du nombre de cellules sanguines dans la moelle osseuse), ce qui peut accroître la probabilité d’infections en cas de diminution du nombre de globules blancs;
· infections opportunistes, y compris des pneumonies (des cas d’évolution fatale ont été rapportés) ;
· nausées, vomissements améliorés par un traitement antiémétique ;
· apparition de mucites fréquemment observée
· Troubles respiratoires (troubles pulmonaires) avec des difficultés à respirer.
· Ce médicament peut provoquer des lésions pulmonaires (avec une issue fatale possible). Des lésions pulmonaires peuvent survenir plusieurs années après le traitement. Informez immédiatement votre médecin si vous ressentez l’un des symptômes suivants: difficulté respiratoire, toux persistante, douleurs à la poitrine, faiblesse/fatigue persistante ;
· modifications rénales et hépatiques lors de l’utilisation de fortes doses ;
· troubles cardio-vasculaires : hypotension, augmentation de la fréquence cardiaque ;
· rougeur de la peau lors de perfusion rapide ;
· troubles des règles, diminution du sperme ;
· troubles visuels ;
· douleurs dans la poitrine ;
· maux de tête ;
· réactions allergiques ;
· Réaction au point d'injection : passage du produit hors des vaisseaux pouvant entraîner gonflement, douleur, érythème, sensation de brûlure et exceptionnellement, nécrose de la peau.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER BICNU, poudre et solvant pour solution pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage. La date de péremption fait référence au dernier jour de ce mois.
Après reconstitution selon les recommandations, la carmustine pour injection est stable pendant 480 heures au réfrigérateur (entre 2 °C et 8 °C) et 24 heures à température ambiante (25 °C ± 2 °C) dans un récipient en verre. Avant utilisation, les flacons reconstitués doivent être examinés pour vérifier qu’il n’y a pas de formation de cristaux. Si des cristaux sont observés, ils peuvent être redissous en réchauffant le flacon à température ambiante sous agitation.
Conservez les flacons dans leur emballage extérieur afin de les protéger de la lumière. La solution reconstituée, de nouveau diluée jusqu’à 500 mL avec du chlorure de sodium à 0,9 % pour injection ou du dextrose à 5 % pour injection dans des récipients en verre ou en polypropylène, est physiquement et chimiquement stable pendant 8 heures à 25 °C ± 2 °C à l’abri de la lumière. Ces solutions sont également stables jusqu’à 48 heures au réfrigérateur (entre 2 °C et 8 °C) et pendant 6 heures supplémentaires à 25 °C ± 2 °C à l’abri de la lumière.
La solution doit être conservée à l’abri de la lumière jusqu’à la fin de l’administration.
N’utilisez pas ce médicament si vous remarquez des signes visibles de détérioration. Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient BICNU, poudre et solvant pour solution pour perfusion
· La substance active est :
Carmustine...................................................................................................................... 100,00 mg
Pour un flacon
· Flacon de solvant : propylène glycol
Poudre et solvant pour solution pour perfusion. Boîte de 1 flacon de poudre et 1 flacon de solvant.
Titulaire de l’autorisation de mise sur le marché
MITTELSTRAßE 5 / 5A
12529 SCHÖNEFELD
ALLEMAGNE
Exploitant de l’autorisation de mise sur le marché
LES FJORDS - IMMEUBLE OSLO,
19 AVENUE DE NORVÈGE,
91140 VILLEBON-SUR-YVETTE
Suite 2, Stafford House, Strand Road
Portmarnock, Co. Dublin
IrlandE
ou
Tillomed Malta Limited
Malta Life Sciences Park,
LS2.01.06 Industrial Estate,
San Gwann, SGN 3000, Malte
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Informations destinées aux professionnels de santé :
Comparé à d'autres médicaments contenant de la carmustine, le propylène glycol est utilisé comme diluant dans le BICNU à la place de l’éthanol. Le propylène glycol contribue à une meilleure stabilité de la solution reconstituée. Les étapes suivantes décrivent les règles de manipulation à suivre pour garantir une dissolution et une reconstitution optimale du médicament. Ces étapes diffèrent de celles à suivre pour des médicaments à base de carmustine dont le solvant est l’éthanol. Pour connaître les étapes à suivre pour ces médicaments veuillez-vous référer à leurs résumés des caractéristiques de produits en vigueur.
La reconstitution et la dilution de chaque flacon de poudre de solution pour perfusion doivent être préparées comme décrit ci-après. Dissoudre la carmustine (100 mg de poudre) dans 3 ml du diluant stérile fourni (propylène glycol pour injection) jusqu'à obtention d'une solution claire. Utiliser le flacon de propylène glycol pour la reconstitution uniquement après avoir atteint la température ambiante et utiliser une aiguille à pores plus larges (aiguille de calibre inférieur à 22) pour prélever le diluant du flacon. Un guide étape par étape pour la reconstitution est fourni ci-dessous.
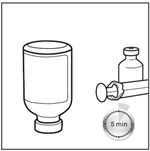
1. Sortir le BICNU du réfrigérateur (2°C-8°C) et le laisser atteindre la température ambiante. Retirer les flacons de produit et de diluant de l’emballage et les laisser à température ambiante pendant au moins 10 minutes. Cela permet que la solution se mélange correctement lors de la reconstitution. Le diluant ne doit être utilisé qu’après que le flacon ait atteint la température ambiante.
|
|
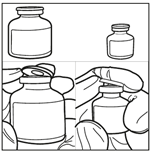
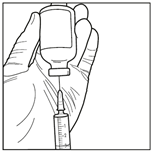
2. Prélever 3 mL du diluant stérile (solution de propylène glycol) avec une seringue stérile. Utiliser une seringue stérile pour prélever aseptiquement 3 mL de diluant du flacon. Assurez-vous de prélever la totalité du volume (3 mL) en utilisant une aiguille de calibre inférieur à 22 pour permettre une injection facile et précise.
|
|
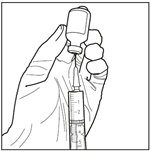
3. Injecter le diluant stérile dans le flacon de carmustine. Injecter les 3 mL de diluant stérile dans le flacon contenant 100 mg de carmustine. Laisser la solution s’hydrater pendant au moins 10 minutes, sans secouer immédiatement. Cette période de repos est nécessaire pour permettre au produit de se dissoudre correctement.
|
|
4. Agiter doucement le flacon pour obtenir une solution limpide. Après la période d’hydratation de 10 minutes, ne pas secouer le flacon de bas en haut mais agiter vigoureusement et régulièrement le flacon en mouvement circulaire pendant au moins 60 secondes, jusqu’à obtention d’une solution claire et homogène. Cette agitation est essentielle pour garantir la dissolution complète de la carmustine.
|
|
5. Laisser le flacon en position inversée pendant 5 minutes. Après avoir obtenu une solution limpide, placer le flacon en position inversée pendant 5 minutes pour permettre une préparation optimale avant le prélèvement.
Chaque millilitre de la solution reconstituée contient 33,3 mg de carmustine.
|
|
6. Prélever la solution reconstituée de manière aseptique. En maintenant le flacon en position inversée (au moment de l’insertion de l’aiguille et compte tenu de la viscosité de la solution reconstituée, le flacon doit rester le moins longtemps possible en position tête en haut et le flacon doit rester à nouveau en position stationnaire inversée une fois l’aiguille insérée avant le prélèvement), utiliser une seringue stérile pour prélever la solution reconstituée.
|
|
7. Dilution de la solution reconstituée.
La solution reconstituée doit en outre être diluée jusqu’à 500 mL avec une solution injectable de chlorure de sodium à 0,9 % ou une solution injectable de dextrose à 5 %. La solution résultante contient une concentration finale de 0,2 mg/mL de carmustine et doit être conservée à l’abri de la lumière. La solution ainsi préparée ne devra être injectée que par voie IV, sous la forme d'une perfusion lente, d'une durée comprise entre 1 et 2 heures. L’administration doit être effectuée avec un set de perfusion PE sans PVC.
La manipulation de ce cytotoxique par le personnel infirmier ou médical nécessite un ensemble de précautions permettant d'assurer la protection du manipulateur et de son environnement (Voir rubrique 4.2.).
BICNU se présente sous forme de poudre lyophilisée et ne contient pas de conservateur : il faudra donc préparer la solution extemporanément. Pour plus d’informations sur les durées et conditions de conservation des solutions reconstituées et diluées veuillez-vous référer à aux instructions de la rubrique 6.3.
La conservation de la carmustine à 27 °C ou à une température supérieure peut conduire à la liquéfaction de la substance, en raison du point de fusion bas de la carmustine (environ 30,5 °C à 32,0 °C). Un signe de décomposition est l’apparition d’une pellicule huileuse au fond du flacon qui est visible lorsque le flacon est exposé à une lumière vive. Ce médicament ne doit plus être utilisé. Des manifestations physiques se traduisant par des flocons pointus allant jusqu’à une masse solidifiée peuvent survenir dans le flacon non ouvert sans qu’une décomposition de la carmustine n’ait lieu.
Précautions importantes à respecter lors de la reconstitution
Ne pas utiliser les flacons avant qu’ils aient atteint la température ambiante. Il est essentiel de respecter le volume de diluant (3 mL) et le temps d’hydratation (10 minutes) avant de commencer l’agitation. Une fois la solution reconstituée, le flacon doit être manipulé avec précaution, et l’utilisation d’une aiguille de calibre inférieur à 22 est recommandée pour éviter tout dommage au flacon ou contamination du produit.
Ne pas utiliser les flacons conservés à des températures inadéquates (2°C-8°C) pour la reconstitution. Ne pas utiliser moins de 3 mL de diluant pour la reconstitution. Ne pas commencer l'agitation avant un temps de repos de 10 minutes. Ne pas arrêter l'agitation avant 60 secondes ou avant l'obtention d'une solution limpide. Ne pas prélever la solution reconstituée sans avoir maintenu le flacon inversé pendant 5 minutes. Ne pas utiliser une aiguille de calibre supérieur à 22 pour le prélèvement.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.