Dernière mise à jour le 03/08/2026
RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale
Indications thérapeutiques
Classe pharmacothérapeutique : GLUCOCORTICOIDE LOCAL PAR VOIE NASALE – code ATC : R01AD05.
Ce médicament contient la substance active : budésonide.
Le budésonide est un corticoïde qui exerce une action anti-inflammatoire.
Il est indiqué :
· en traitement de la rhinite allergique saisonnière ou perannuelle chez l’adulte et l’enfant de plus de 6 ans,
· pour soulager les symptômes liés à la présence de polypes dans les fosses nasales chez l’adulte.
Présentations
> 1 flacon(s) en verre brun de 120 dose(s) muni d'une pompe à valve doseuse et d'un embout nasal polypropylène
Code CIP : 351 582-2 ou 34009 351 582 2 2
Déclaration de commercialisation : 16/06/2000
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 5,47 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 6,49 €
- Taux de remboursement :30%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Modéré | Avis du 16/09/2015 | Renouvellement d'inscription (CT) | le service médical rendu par RHINOCORT reste modéré dans les indications de l'AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l\'aide)
ANSM - Mis à jour le : 06/10/2025
RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Budésonide......................................................................................................... 64 microgrammes
Pour une dose
Excipient à effet notoire : sorbate de potassium
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension pour pulvérisation nasale.
4.1. Indications thérapeutiques
Traitement symptomatique de la polypose nasale de l’adulte.
(Remarque : l’instillation nasale de budésonide en cas de polypose nasale améliore les symptômes et entraîne une diminution du volume des polypes mais n’a pas fait la preuve de son efficacité dans la diminution du recours à la polypectomie par voie nasale ni dans la prévention de la récidive de la polypose).
4.2. Posologie et mode d'administration
Posologie
Traitement de la rhinite allergique saisonnière ou perannuelle chez l’adulte et l’enfant de plus de 6 ans :
La dose initiale préconisée est de 256 µg en une à deux prises par jour :
- soit 2 pulvérisations de 64 µg dans chaque narine une fois par jour (le matin)
- soit 1 pulvérisation de 64 µg dans chaque narine deux fois par jour (matin et soir).
Le traitement sera poursuivi en s'efforçant d'abaisser progressivement les doses dès l’amélioration des symptômes (en général 1 à 2 semaines). Une dose d’entretien de 64 µg (soit une pulvérisation) dans chaque narine le matin est suffisante dans la plupart des cas.
Il convient d’informer le patient que l’efficacité du médicament ne se manifeste qu’au bout de quelques jours de traitement. La mise en route et la durée du traitement sont fonction de l’exposition allergénique.
L’avis d’un spécialiste pédiatre est requis pour tout traitement chez l’enfant pendant plus de 2 mois par an.
Traitement symptomatique de la polypose nasale chez l’adulte :
La dose préconisée est de 256 µg en 2 prises par jour, soit 1 pulvérisation de 64 µg dans chaque narine 2 fois par jour (matin et soir).
La dose quotidienne peut être administrée en une prise par jour (soit 2 pulvérisations de 64 µg dans chaque narine) dans les formes peu sévères.
Mode d’administration
Voie nasale.
Agiter le flacon avant emploi.
Lors du premier usage, agiter le flacon et amorcer la pompe par 5 ou 10 mouvements de pulvérisation dans l’air. Si l’appareil n’est pas utilisé quotidiennement, il doit être ré-amorcé de nouveau en pompant simplement une fois dans l’air.
Nettoyage de l'embout nasal :
Il convient de nettoyer régulièrement la partie supérieure en plastique du flacon. Pour cela, le bouchon et l’embout nasal seront ôtés et les parties en plastique seront rincées à l’eau chaude puis séchées complètement avant de les remettre en place.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Troubles de l'hémostase, ou épistaxis.
· Infection oro-bucco-nasale et ophtalmique par herpès virus.
4.4. Mises en garde spéciales et précautions d'emploi
Des effets systémiques peuvent apparaître lors de traitement au long cours avec des doses élevées de corticoïdes par voie nasale. Le risque de retentissement systémique reste néanmoins moins important qu’avec les corticoïdes oraux et peut varier en fonction de la susceptibilité individuelle et de la composition de la spécialité corticoïde utilisée. Les effets systémiques possibles sont : syndrome de Cushing ou tableau cushingoïde, amincissement cutané, hématomes sous cutanés, insuffisance surrénalienne, retard de croissance chez les enfants et les adolescents, diminution de la densité osseuse, cataracte et glaucome et plus rarement, troubles psychologiques et du comportement comprenant hyperactivité psychomotrice, troubles du sommeil, anxiété, dépression ou agressivité (en particulier chez l’enfant). Le risque d’insuffisance surrénale latente après administration prolongée devra être considéré dans les situations susceptibles de déclencher un stress (infection intercurrente, accident, etc..) ou en cas de chirurgie programmée.
Les glucocorticoïdes peuvent augmenter la glycémie. Ceci doit être pris en compte lors de la prescription chez des patients diabétiques.
Il est important de toujours rechercher la posologie minimale efficace de corticoïdes par voie nasale.
L’administration conjointe de corticoïdes par voie nasale chez les patients sous corticothérapie orale au long cours ne dispense pas des précautions nécessaires lors d’une réduction des doses de corticoïdes par voie orale. Celles-ci seront diminuées très progressivement et le sevrage devra être effectué sous surveillance médicale attentive (à la recherche de l’apparition de signes d’insuffisance surrénale aiguë ou subaiguë) se prolongeant au-delà de l’arrêt de la corticothérapie générale. Une attention particulière sera portée lors du passage d’une corticothérapie systémique à un traitement par Rhinocort en cas de suspicion d’inhibition des fonctions surrénaliennes.
L’administration locale par voie nasale de corticoïdes n’est pas recommandée chez les patients présentant des épisodes d’épistaxis graves ou fréquents, une ulcération récente de la cloison nasale, ou ayant subi une intervention ou un traumatisme au niveau du nez, tant que la guérison n’est pas complète.
La perméabilité des fosses nasales doit être assurée pour la diffusion optimale du budésonide dans les fosses nasales. En avertir le patient en lui conseillant de se moucher avant chaque instillation.
En cas de traitement prolongé, des examens détaillés des fosses nasales s'imposent du fait du risque de retentissement sur la muqueuse nasale. La constatation d’une atrophie de la muqueuse doit conduire à la diminution des doses de corticoïdes locaux.
En cas de tuberculose pulmonaire, d’infection mycosique pulmonaire, l’instauration d’une surveillance étroite et d’un traitement adapté s’impose.
Tout contact avec une personne ayant contracté la tuberculose, la rougeole ou la varicelle est à prendre en compte lors de l’instauration du traitement.
En cas d'insuffisance hépatique, l'élimination des corticoïdes est réduite et en conséquence expose les patients à des concentrations systémiques plus élevées et une augmentation du risque d'effets systémiques. La prudence est requise en cas d'insuffisance hépatique.
Les glucocorticoïdes peuvent augmenter la pression intraoculaire. Les patients atteints de glaucome ou ceux ayant des antécédents familiaux de glaucome doivent donc être étroitement surveillés pendant la prise de ce médicament.
Des troubles visuels peuvent apparaitre lors d'une corticothérapie par voie systémique ou locale. En cas de vision floue ou d'apparition de tout autre symptôme visuel apparaissant au cours d'une corticothérapie, le traitement devra être interrompu et un examen ophtalmologique devra être réalisé à la recherche notamment d'une cataracte, d'un glaucome, ou d'une lésion plus rare telle qu'une choriorétinopathie séreuse centrale, décrits avec l'administration de corticostéroïdes par voie systémique ou locale.
Un examen ophtalmologique est également requis en cas d’infection oculaire.
En cas de traitement prolongé, un examen ORL détaillé de la muqueuse nasale doit être réalisé.
Ce médicament contient du sorbate de potassium et peut provoquer des réactions cutanées locales (par exemple dermatite de contact).
Population pédiatrique
Il a été observé un ralentissement de la croissance chez des enfants recevant des corticoïdes par voie nasale aux posologies thérapeutiques. Il est recommandé de surveiller régulièrement la croissance des enfants recevant une corticothérapie au long cours. En cas de ralentissement de la croissance, le traitement devra être réévalué en vue de réduire les doses de corticoïde nasal. Il conviendra de soigneusement peser les bénéfices attendus d’une corticothérapie par voie nasale face aux risques éventuels de ralentissement de la croissance. L’avis d’un spécialiste pédiatre peut être requis.
Sportif
L’attention des sportifs sera attirée sur le fait que cette spécialité contient un principe actif pouvant induire une réaction positive des tests pratiqués lors de contrôle anti-dopage.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Associations déconseillées (voir rubrique 4.4)
+ inhibiteurs puissants du CYP3A4 (bocéprevir, clarithromycine, cobicistat, érythromycine, itraconazole, kétoconazole, posaconazole, ritonavir, télithromycine, voriconazole).
En cas d’utilisation prolongée par voie orale ou inhalée : augmentation des concentrations plasmatiques du budésonide par diminution de son métabolisme hépatique par l’inhibiteur, avec risque d’apparition d’un syndrome cushingoïde voire d’une insuffisance surrénalienne. Préférer un corticoïde non métabolisé.
Le budésonide est principalement métabolisé par le cytochrome P450 3A4. Une augmentation significative des taux sanguins de budésonide peut être observée avec les inhibiteurs puissants du CYP3A4 (ex : produits contenant du cobicistat, kétoconazole, itraconazole, voriconazole, posaconazole, clarithromycine, télithromycine, néfazodone et inhibiteurs des protéases du VIH tels que saquinavir, nelfinavir, indinavir, atazanavir, ritonavir).
L’administration concomitante d’inhibiteurs du CYP3A, y compris de produits contenant du cobicistat, peut augmenter le risque d’effets indésirables systémiques. L’association doit être évitée, sauf si les bénéfices sont supérieurs au risque accru d’effets indésirables systémiques des corticostéroïdes ; dans ce cas, les patients doivent être surveillés en vue de détecter les éventuels effets indésirables systémiques des corticostéroïdes.
Si cette association ne peut être évitée, un intervalle de temps suffisamment long devra être respecté entre l’administration de l’inhibiteur du CYP3A4 et celle du budésonide.
Il n’y a pas d’interaction médicamenteuse significative suite à la prise concomitante de kétoconazole sur courte durée (1 à 2 semaines) avec du budésonide.
+ Acide acétylsalicylique
Majoration du risque hémorragique. Association déconseillée avec des doses anti-inflammatoires d’acide acétylsalicylique (>=1g par prise et/ou >=3g par jour).
Associations à prendre en compte
+ Acide acétylsalicylique
Majoration du risque hémorragique. Association à prendre en compte avec des doses antalgiques ou antipyrétiques (>=500 mg par prise et/ou <3g par jour).
+ Anti-inflammatoires non stéroïdiens
Augmentation du risque d’ulcération et d’hémorragie gastro-intestinale.
+ Fluoroquinolones
Possible majoration du risque de tendinopathie, voire de rupture tendineuse (exceptionnelle), particulièrement chez les patients recevant une corticothérapie prolongée.
+ Héparines
Augmentation du risque hémorragique.
Précaution d’emploi
+ Anticoagulants oraux
Glucocorticoïdes (voies générale et rectale) : impact éventuel de la corticothérapie sur le métabolisme de l'antivitamine K et sur celui des facteurs de la coagulation. Risque hémorragique propre à la corticothérapie (muqueuse digestive, fragilité vasculaire) à fortes doses ou en traitement prolongé supérieur à 10 jours.
Lorsque l'association est justifiée, renforcer la surveillance : le cas échéant, avec les antivitamines K, contrôle biologique au 8e jour, puis tous les 15 jours pendant la corticothérapie et après son arrêt.
+ Anticonvulsivants inducteurs enzymatiques
Diminution des concentrations plasmatiques et de l'efficacité des corticoïdes par augmentation de leur métabolisme hépatique par l'inducteur : les conséquences sont particulièrement importantes chez les addisoniens traités par l'hydrocortisone et en cas de transplantation.
Surveillance clinique et biologique ; adaptation de la posologie des corticoïdes pendant le traitement par l'inducteur et après son arrêt.
+ Cobimétinib
Augmentation du risque hémorragique. Surveillance clinique.
+ Inducteurs enzymatiques
Diminution des concentrations plasmatiques et de l'efficacité des corticoïdes par augmentation de leur métabolisme hépatique par l'inducteur ; les conséquences sont particulièrement importantes chez les addisoniens traités par l'hydrocortisone et en cas de transplantation.
Surveillance clinique et biologique ; adaptation de la posologie des corticoïdes pendant le traitement par l'inducteur et après son arrêt.
+ Topiques gastro-intestinaux, antiacides et adsorbants
Diminution de l’absorption du budésonide.
Par mesure de précaution, il convient de prendre ces topiques ou antiacides à distance de tout autre médicament (plus de 2 heures, si possible)
4.6. Fertilité, grossesse et allaitement
Grossesse
Chez l’animal, l’expérimentation met en évidence un effet tératogène, variable selon les espèces.
Chez l’homme, les études épidémiologiques n’ont pas mis en évidence d’augmentation de la fréquence globale de malformations suite à la prise lors du premier trimestre de grossesse de corticoïdes per os ou de budésonide par voie inhalée.
Aux doses thérapeutiques, l’exposition systémique après une administration nasale de budésonide est plus faible que celle observée par voie inhalée.
Lors de maladies chroniques nécessitant un traitement tout au long de la grossesse, un léger retard de croissance intra-utérin est possible. Une insuffisance surrénalienne néonatale a été exceptionnellement observée après corticothérapie à doses élevées. Il peut être justifié d’observer une période de surveillance clinique (poids, diurèse) et biologique (glycémie) du nouveau-né.
En conséquence, ce médicament peut être prescrit pendant la grossesse si besoin.
Allaitement
Le budésonide est excrété dans le lait maternel. Le nouveau-né allaité reçoit environ 1% de la dose maternelle de budésonide administrée par voie inhalée ajustée au poids corporel.
Sur la base des données relatives au budésonide inhalé et étant donné que l’exposition systémique du budésonide par voie nasale est plus faible que par inhalation, il est attendu que l’exposition chez l’enfant allaité soit faible lors de l’utilisation de budésonide aux doses thérapeutiques.
Cependant, le retentissement biologique ou clinique d’un traitement maternel de longue durée n’est pas évalué à ce jour.
En conséquence, l’allaitement est possible en cas de traitement bref. En cas de traitement chronique, par mesure de précaution, l’allaitement est à éviter.
Fertilité
Il n'y a aucune preuve que le budésonide administré par voie intranasale ait un effet sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables identifiés durant les essais cliniques et depuis la commercialisation du budénoside sont listés ci-dessous par classe de système d’organe. Les fréquences sont définies selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1000, < 1/100) ; rare (≥ 1/10000, < 1/1000) ; très rare (< 1/10000) ; fréquence indéterminée (ne pouvant être estimée sur la base des données disponibles).
Les effets indésirables sont présentés par catégorie de fréquence basée sur 1) Effets indésirables rapportés lors d’études cliniques ou d’études épidémiologiques, si disponibles, ou 2) quand la fréquence ne peut être estimée, la catégorie de fréquence mentionnée est « fréquence indéterminée ».
|
Classes de système d’organe |
Fréquence |
Effets indésirables |
|
Affections du système immunitaire |
Peu fréquent |
Réactions d'hypersensibilité à type d’érythème, urticaire, rash cutané, dermatite, prurit ou angioedème |
|
Rare |
Réaction anaphylactique |
|
|
Affections du système nerveux |
Indéterminée |
Maux de tête |
|
Affections oculaires |
Rare |
Vision floue (voir rubrique 4.4) Pression oculaire augmentée |
|
Indéterminée |
Cataracte |
|
|
Glaucome |
||
|
Affections respiratoires thoraciques et médiastinales |
Fréquent |
Epistaxis Sécrétion hémorragique Inconfort nasal (Irritation nasale) Sécheresse de la muqueuse nasale Douleurs oropharyngées |
|
Rare |
Dysphonie Perforation du septum nasal Ulcération nasale |
|
|
Affections de la peau et du tissu sous-cutané |
Rare |
Ecchymoses |
|
Affections musculo-squelettiques et systémiques |
Peu fréquent |
Contracture musculaire |
|
Infections et infestations |
Indéterminée |
Candidoses nasales et oropharyngées. |
Infections à Candida albicans
Ont été décrits des cas d’infections nasales et pharyngées à Candida albicans lors de traitement par corticoïdes locaux. Il est préférable dans ce cas d’interrompre la corticothérapie par voie nasale et d’envisager la mise en route d’un traitement adapté.
Effets systémiques
Occasionnellement, des signes et symptômes d’effets secondaires systémiques liés aux glucocorticoïdes peuvent survenir lors de l’utilisation de glucocorticoïdes par voie nasale (voir rubrique 4.4).
Le risque d’insuffisance surrénale latente après administration prolongée devra être considéré (voir rubrique 4.4).
Population pédiatrique
Lors de l’administration au long cours de budésonide, un retentissement systémique et sur la croissance en particulier chez l’enfant, n'est pas exclu. Ce risque est majoré en cas d’administration concomitante d’une corticothérapie par voie inhalée ou a fortiori par voie systémique.
La croissance des enfants et des adolescents doit être surveillée régulièrement (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Un surdosage au long cours pourrait entraîner une freination hypophyso-surrénalienne et, s'il se prolongeait, des signes cliniques d'hypercorticisme. Ces symptômes disparaîtront après l'arrêt du traitement qui doit être progressif.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : GLUCOCORTICOIDE LOCAL PAR VOIE NASALE, code ATC : R01AD05.
Le budésonide administré par voie nasale, exerce une activité anti-inflammatoire marquée sur la muqueuse nasale.
Population pédiatrique
Une étude randomisée de 6 semaines, en double aveugle, contrôlée contre placebo en groupes parallèles, a évalué l’efficacité et la tolérance de Rhinocort à la dose de 128 µg une fois par jour chez 202 enfants (âgés de 6 à 16 ans) ayant une rhinite allergique perannuelle.
Les critères primaires d’efficacité étaient le score de symptômes nasaux combinés (somme des scores des symptômes : congestion, écoulement nasal et éternuement mesurés sur une échelle de 0 à 3) et le débit inspiratoire de pointe nasal. Rhinocort a amélioré ces deux critères de façon statistiquement significative comparativement au placebo.
Une étude randomisée de 2 semaines, en double aveugle, contrôlée contre placebo avec groupes parallèles, a évalué l’efficacité et la tolérance de Rhinocort à la dose de 16, 32 et 64 µg une fois par jour chez 400 enfants (âgés de 2 à 5 ans) ayant une rhinite allergique (saisonnière ou perannuelle). Une diminution du score combiné de symptômes nasaux (somme des scores de symptômes nasaux congestion, écoulement nasal et éternuement mesurés sur une échelle de 0 à 3) a été observée par rapport à la valeur de base dans tous les groupes de traitement y compris le groupe placebo. La différence entre les groupes Rhinocort et le groupe placebo n’était pas statistiquement significative.
L'effet de l'administration de Rhinocort sur la croissance a été évalué dans une étude randomisée, en double aveugle contrôlée contre placebo incluant 229 enfants pré-pubères âgés de 4 à 8 ans traités par Rhinocort à la dose de 64 µg par jour ou par placebo, pendant 12 mois après une période sans traitement corticoïde préalable pendant 6 mois. La différence moyenne de vitesse de croissance entre le groupe traité par placebo et le groupe traité par Rhinocort était de 0,27 cm/an (intervalle de confiance à 95% : -0,07 à 0,62).
Influence sur la concentration de cortisol plasmatique
Après une administration de courte durée de Rhinocort, chez des volontaires sains, une réduction dose-dépendante des concentrations de cortisol urinaire et plasmatiques a été observée.
5.2. Propriétés pharmacocinétiques
La biodisponibilité systémique du budésonide administré par voie nasale avec ce dispositif, rapporté à la dose théorique, est de 33 %. Avec cette spécialité, la concentration plasmatique maximale observée chez l’adulte après administration de 256 µg de budésonide a été de 0.64 nmol/l. Elle a été atteinte en moyenne en 0.7 heures. L’aire sous la courbe des concentrations plasmatiques après administration de 256 µg de budésonide avec Rhinocort est de 2,7 nmol.h/litre chez l’adulte.
Distribution
Le budésonide a un volume de distribution de 3 litres/kg. La fixation aux protéines plasmatiques est d’environ 85 à 90%.
Biotransformation
Le budésonide subit un important effet de premier passage hépatique (90%) avec transformation en métabolites pratiquement dénués d’activité glucocorticoïde. L’activité glucocorticoïde des métabolites principaux, 6β-hydroxybudésonide et 16α-hydroxyprednisolone, est inférieure à 1%.
Le budésonide est principalement métabolisé par l’enzyme CYP3A, une sous-famille du cytochrome P450.
Le budésonide ne subit pas d’inactivation métabolique dans les voies respiratoires nasales.
Elimination
Les métabolites sont excrétés principalement dans les urines, inchangés ou après conjugaison. Il n’a pas été détecté de budésonide sous forme inchangée dans les urines.
La clairance plasmatique est élevée (environ 1,2 L/min) et la demi-vie plasmatique après administration intraveineuse est de 2 à 3 heures.
Linéarité/Non linéarité
La cinétique du budésonide est linéaire aux doses thérapeutiques préconisées.
Population pédiatrique
La clairance plasmatique du budésonide est d’environ 0,5 L/min chez les enfants de 4 à 6 ans asthmatiques anciens. La clairance chez l’enfant exprimée par kg de poids est environ 50% plus élevée que chez l’adulte. La demi-vie terminale du budésonide après inhalation est environ de 2,3 heures chez l’enfant asthmatique. Elle est approximativement la même chez l’adulte sain. L’aire sous la courbe des concentrations plasmatiques après administration de 256 µg de budésonide avec Rhinocort est de 5,5 nmol.h/L chez l’enfant. Ceci indique qu’il y a une exposition systémique aux corticoïdes plus grande chez l’enfant que chez l’adulte.
Aux doses cliniquement recommandées les cinétiques du budésonide sont proportionnelles à la dose et l’exposition plasmatique est corrélée au poids du patient. Par conséquent ceci doit être pris en compte lors de l’établissement des posologies chez l’enfant.
5.3. Données de sécurité préclinique
Il a été montré que le budésonide administré par voie orale augmentait l’incidence de tumeurs hépatiques chez le rat mâle à des doses de 25 microgrammes/kg/jour. Ces effets ont également été observés lors d’une étude de suivi au long cours réalisée avec d’autres corticoïdes (prednisolone et triamcinolone acétonide) ; ils sont donc considérés comme étant des effets de classe liés à l’administration de corticoïdes.
Le budésonide n’a eu aucun effet sur la fertilité chez le rat. Un effet tératogène a été mis en évidence chez le rat et le lapin dans des études conduites par voie sous-cutanée.
30 mois
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
120 doses ou 240 doses en flacon pulvérisateur de 10 ml ou de 20 ml en verre brun muni d’une pompe à valve doseuse et d’un embout nasal (polypropylène).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
41 rue Camille Desmoulins
92130 ISSY-LES-MOULINEAUX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 351 582 2 2 : 120 doses en flacon pulvérisateur (verre brun)
· 34009 351 583 9 0 : 240 doses en flacon pulvérisateur (verre brun).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
Date de première autorisation : 28 juillet 1999
Date du dernier renouvellement : 28 juillet 2009
10. DATE DE MISE A JOUR DU TEXTE
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 06/10/2025
RHINOCORT 64 microgrammes par dose, suspension pour pulvérisation nasale
Budésonide
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale ?
3. Comment utiliser RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : GLUCOCORTICOIDE LOCAL PAR VOIE NASALE – code ATC : R01AD05.
Ce médicament contient la substance active : budésonide.
Le budésonide est un corticoïde qui exerce une action anti-inflammatoire.
Il est indiqué :
· en traitement de la rhinite allergique saisonnière ou perannuelle chez l’adulte et l’enfant de plus de 6 ans,
· pour soulager les symptômes liés à la présence de polypes dans les fosses nasales chez l’adulte.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale ?
N’utilisez jamais RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale :
· si vous êtes allergique à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6,
· si vous avez des troubles de la coagulation du sang ou que vous saignez du nez,
· en cas de boutons de fièvre autour de la bouche, du nez ou des yeux.
EN CAS DE DOUTE, IL EST INDISPENSABLE DE DEMANDER L’AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale :
· Si vous prenez déjà un corticostéroïde par voie orale, par exemple en comprimés. Votre médecin vous fournira les instructions nécessaires pour réduire progressivement la dose de corticostéroïdes par voie orale. N'arrêtez pas soudainement de prendre ces comprimés.
· Si vous prenez déjà des corticostéroïdes pour traiter des affections comme l’asthme, les allergies, les éruptions cutanées.
· Si vous avez des ulcères du nez, avez récemment subi une intervention chirurgicale, avez des saignements graves ou fréquents du nez ou avez subi un traumatisme au niveau du nez qui n'est pas complètement guéri. RHINOCORT 64 microgrammes / dose, suspension pour pulvérisation nasale n'est pas recommandé dans ces situations.
· Si vous avez ou avez eu une tuberculose pulmonaire ou une infection pulmonaire causée par un champignon.
· Si vous êtes ou avez été en contact avec une personne ayant contracté la tuberculose, la rougeole ou la varicelle.
· Si vous avez une maladie hépatique chronique ou si vous avez un ictère. Votre médecin vous conseillera sur une dose adaptée.
· Si vous avez ou avez eu un glaucome, si vous avez des antécédents de glaucome dans votre famille, ou si vous avez ou avez eu une cataracte.
· Si vous avez une infection oculaire.
· Si des signes d’infection comme une fièvre persistante apparaissent.
· Si vous êtes diabétique. Les glucocorticoïdes sont susceptibles d’augmenter la glycémie.
Vous devez arrêter votre traitement et contacter votre médecin en cas de vision floue ou d’autres troubles visuels, d’apparition, de persistance, d’aggravation de symptômes, pendant l’utilisation de ce médicament.
L’efficacité sur les symptômes peut n'apparaître que plusieurs jours après le début du traitement.
Pour que ce médicament soit actif, vos narines doivent être libres. Il convient par conséquent de se moucher avant de prendre votre dose.
Si votre obstruction nasale (sensation de nez bouché) persiste malgré la mise en route du traitement, consultez votre médecin afin qu'il réévalue le traitement.
En cas de traitement prolongé, un examen ORL détaillé de la muqueuse nasale doit être réalisé.
Sportifs, attention ce médicament contient un principe actif pouvant induire une réaction positive des tests pratiqués lors de contrôles antidopage.
EN CAS DE DOUTE NE PAS HESITER A DEMANDER L'AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN.
Enfants et adolescents
Ce médicament est indiqué chez l’enfant de plus de 6 ans dans le traitement de la rhinite allergique saisonnière ou perannuelle. Pendant le traitement au long cours, la croissance des enfants et des adolescents devrait être régulièrement contrôlée.
L'administration de ce médicament chez l'enfant doit être réalisée sous le contrôle d'un adulte.
Autres médicaments et RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris des médicaments obtenus sans prescription.
En particulier, avant de commencer à utiliser ce médicament, informez votre médecin si vous prenez :
· des médicaments qui traitent les infections dues à des champignons microscopiques (comme l’itraconazole, le kétoconazole, le posaconazole ou le voriconazole).
· des médicaments antibiotiques (comme l’érythromycine, la clarithromycine, la télithromycine, ou encore la ciprofloxacine, la lévofloxacine ).
· des médicaments anti- VIH (tels que les médicaments contenant le saquinavir, l’atazanavir, l’indinavir, le nelfinavir, le ritonavir ou le cobicistat).
· le boceprevir (un médicament qui traite l’hépatite, une maladie du foie due à une infection par le virus de l’hépatite C).
· des médicaments anti-inflammatoires non stéroïdiens (AINS) (tels que les médicaments contenant de l’acide acétylsalicylique ou aspirine), utilisés pour réduire la douleur, la fièvre et l'inflammation.
· les héparines et les anticoagulants oraux (des médicaments qui empêchent la formation de caillots dans le sang).
· des médicaments anticonvulsivants utilisés dans le traitement de l’épilepsie (comme la carbamazépine, le phénobarbital, la phénytoïne, le valproate de sodium).
· le cobimétinib (un médicament utilisé dans le traitement de certains types de tumeurs de la peau et des muqueuses, appelées mélanomes).
· des médicaments qui agissent sur le métabolisme au niveau du foie, appelés inducteurs enzymatiques (comme le millepertuis).
· des médicaments contenant du charbon activé qui sont utilisés pour soulager les douleurs gastriques, les reflux gastriques ou pour réduire les gaz intestinaux.
Certains médicaments peuvent augmenter les effets de RHINOCORT à 64 microgrammes/dose, suspension pour pulvérisation nasale et votre médecin peut envisager de vous surveiller attentivement si vous prenez ces médicaments.
RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale avec des aliments et boissons
Sans objet.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Ce médicament ne peut être utilisé pendant la grossesse que sur les conseils de votre médecin.
Si vous découvrez que vous êtes enceinte pendant le traitement, consultez votre médecin car lui seul peut juger du traitement le mieux adapté à votre cas.
Ce médicament peut être administré en cure courte pendant l’allaitement.
En cas de traitement prolongé, il est préférable d’éviter l’allaitement pendant ce traitement.
Conduite de véhicules et utilisation de machines
Ce médicament n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale contient du sorbate de potassium (E 202) et peut provoquer des réactions cutanées locales (par exemple dermatite de contact).
3. COMMENT UTILISER RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale ?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Posologie
Traitement de la rhinite allergique saisonnière ou perannuelle chez l'adulte et l'enfant de plus de 6 ans :
La dose initiale préconisée est de 256 µg en une à deux prises par jour :
· soit 2 pulvérisations de 64 µg dans chaque narine 1 fois par jour (le matin de préférence),
· soit 1 pulvérisation de 64 µg dans chaque narine 2 fois par jour (matin et soir).
Dès l’amélioration des symptômes (en général après 1 à 2 semaines), vous devez réduire le nombre de pulvérisations prises au plus petit nombre de manière à toujours contrôler vos symptômes.
Une dose d'entretien de 64 µg (soit une pulvérisation) dans chaque narine le matin est suffisante dans la plupart des cas.
La mise en route et la durée du traitement sont fonction de l'exposition à l'allergène.
L'administration chez l'enfant devra être supervisée par un adulte.
L’avis d’un pédiatre est requis en cas d’utilisation chez l’enfant pendant plus de 2 mois par an.
Traitement symptomatique de la polypose nasale chez l’adulte :
La dose préconisée est de 256 µg en 2 prises par jour :
· soit 1 pulvérisation de 64 µg dans chaque narine 2 fois par jour (matin et soir).
La dose quotidienne peut également être administrée en une prise par jour (ex : 2 pulvérisations de 64 µg dans chaque narine le matin).
Mode et voie d'administration (accompagné des schémas correspondants)
Voie nasale uniquement. Ne pas avaler.
Suivre attentivement les conseils d'utilisation.
Avant d'utiliser ce médicament pour la première fois, agiter le flacon et après avoir enlevé le capuchon de protection, tenir le flacon en position verticale et pomper 5 à 10 fois dans l'air jusqu’à obtention d’une brume uniforme. Si le flacon n'est pas utilisé quotidiennement, pomper une fois dans l'air.
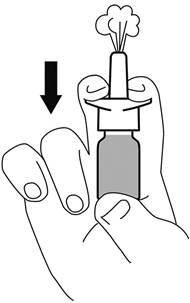
Lors de chaque utilisation :
1. Moucher le nez
Agiter le flacon
Enlever le bouchon de protection
2. Tenir le flacon vertical (voir schéma)
3. Insérer l'extrémité de l’embout dans une narine et pomper le nombre de doses prescrites.
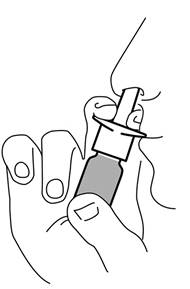
4. Répéter la même opération dans l'autre narine.
5. Essuyer l’embout avec un tissu propre et replacer le bouchon de protection
Nettoyage :
Nettoyer régulièrement la partie supérieure en plastique. Pour cela, enlever le bouchon de protection et essuyer l'embout. Laver les parties en plastique à l'eau chaude et sécher-les complètement avant de les remettre en place. N'essayezpas de nettoyer l'embout nasal en utilisant une épingle ou un objet pointu.
Durée du traitement
Se conformer à l'ordonnance de votre médecin.
Si vous avez utilisé plus de RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale que vous n’auriez dû
Informez immédiatement votre médecin ou votre pharmacien.
Dans tous les cas, se conformer à l’ordonnance de votre médecin et ne pas augmenter ou diminuer la dose sans l’avis de votre médecin ou de votre pharmacien.
Si vous oubliez d’utiliser RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale
Si vous arrêtez d’utiliser RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Si un de ces effets indésirables se produit, arrêter le traitement et consulter le médecin :
· Des réactions de type allergique ont été décrites à la suite de ce traitement : urticaire, démangeaisons, éruptions cutanées, angio-œdème se manifestant par un gonflement du visage, des lèvres, de la langue et/ou de la gorge avec difficulté à respirer et à avaler, sensation de malaise général. Si vous ressentez ces symptômes après l'administration de Rhinocort, vous devez cesser d’utiliser Rhinocort et consulter votre médecin immédiatement.
Autres effets indésirables pouvant survenir :
Fréquent (survient chez 1 patient sur 100 à 1 patient sur 10) :
· Saignements de nez ou présence de sang dans les sécrétions, sécheresse de la muqueuse du nez, irritation ou sensation de brûlure nasale pouvant entraîner des éternuements
· Douleurs dans la bouche et/ou dans la gorge.
Peu fréquent (survient chez 1 patient sur 100 à 1 patient sur 1000) :
· Crampes musculaires.
Rare (survient chez 1 patient sur 1000 à 1 patient sur 10 000) :
· Ulcération de la muqueuse nasale ;
· Perforation de la cloison nasale (cloison séparant les narines) ;
· Réactions anaphylactiques ;
· Ecchymoses ("bleus" sur la peau) amincissement cutané ;
· Hypertonie oculaire (augmentation de la pression à l’intérieur de l’œil);
· Vision floue,
· Dysphonie (raucité de la voix, difficulté à parler et à émettre des sons).
Fréquence indéterminée (la fréquence de survenue n’est pas connue) :
· Glaucome,
· Cataracte (opacification du cristallin de l’œil).
· Maux de tête.
Infections à Candida albicans :
En cas de traitement prolongé, une candidose nasale (infection due à des champignons) peut parfois survenir. Consultez votre médecin afin qu'il envisage avec vous un traitement adapté. Dans ce cas, il est préférable d'interrompre ce traitement jusqu'à guérison de l’infection.
Effets systémiques :
Les corticoïdes nasaux à forte dose et administrés durant une longue durée peuvent avoir un retentissement sur l'organisme. Peuvent apparaitre :
- un hypercorticisme (se manifestant par des symptômes tels qu'augmentation du poids, un aspect lunaire du visage, une fatigue et/ou une augmentation du périmètre abdominal)
- une inhibition de l'activité des glandes surrénales entrainant une insuffisance surrénale pouvant se manifester par des symptômes tels que : fatigue, perte de poids, nausées et diarrhées persistantes, causés par un mauvais fonctionnement de ces glandes.
Si vous ressentez ces symptômes, informez votre médecin.
Effets indésirables supplémentaires chez les enfants et les adolescents
Un ralentissement de croissance peut être observé chez l’enfant et l’adolescent lors d’un traitement au long cours et à forte dose par RHINOCORT.
Votre médecin surveillera régulièrement la croissance des enfants ou des adolescents qui reçoivent un traitement corticoïde au long cours.
Des troubles psychologiques et du comportement tels qu’hyperactivité, troubles du sommeil, anxiété, dépression ou agressivité ont été rarement reportés (spécialement chez les enfants).
En cas de persistance ou d'apparition de nouveaux symptômes, consulter votre médecin.
EN CAS DE DOUTE NE PAS HESITER A DEMANDER L'AVIS DE VOTRE MEDECIN.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ne pas congeler.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient RHINOCORT 64 microgrammes/dose, suspension pour pulvérisation nasale
· La substance active est :
Budésonide......................................................................................................... 64 microgrammes
Pour une dose
· Les autres composants sont : cellulose microcristalline et carboxyméthylcellulose sodique, glucose anhydre, polysorbate 80, édétate disodique, sorbate de potassium (E 202), acide chlorhydrique, eau purifiée.
Ce médicament se présente sous forme de suspension pour pulvérisation nasale.
Flacon de 10 ou 20 ml.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
41 rue Camille Desmoulins
92130 ISSY-LES-MOULINEAUX
Exploitant de l’autorisation de mise sur le marché
41 RUE CAMILLE DESMOULINS
91230 ISSY-LES-MOULINEAUX
18 RUE DE MONTBAZON
37260 MONTS
FRANCE
ou
ASTRAZENECA AB
FORSKARGATAN 18
151 85 - SODERTALJE
SUEDE
ou
MCNEIL AB
NORRBROPLATSEN 2
251 09 - HELSINGBORG
SUEDE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[XXX]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).