Dernière mise à jour le 01/06/2026
EBASTINE TEVA 10 mg, comprimé orodispersible
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : R06AX22
L’ébastine est un antihistaminique qui soulage des symptômes d’allergie tels que les éternuements, l’écoulement nasal, le larmoiement au cours des rhinites (ou rhino-conjonctivites) allergiques et les éruptions sur la peau avec démangeaisons.
Chez les adultes et les enfants à partir de 12 ans, EBASTINE TEVA est utilisé pour soulager les symptômes de rhinite saisonnière (rhume des foins) et de rhinite allergique perannuelle, y compris dans les cas s’accompagnant d’une conjonctivite allergique.
Chez les adultes de plus de 18 ans, EBASTINE TEVA 10 mg est également utilisé pour réduire les démangeaisons et les papules d’urticaire (plaques cutanées surélevées).
Présentations
> plaquette(s) polyamide aluminium PVC-Aluminium de 30 comprimé(s)
Code CIP : 216 899-1 ou 34009 216 899 1 4
Déclaration de commercialisation : 16/01/2012
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 4,01 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 5,03 €
- Taux de remboursement :30%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 03/04/2024
EBASTINE TEVA 10 mg, comprimé orodispersible
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Ebastine................................................................................................................................ 10 mg
Pour un comprimé orodispersible.
Excipients à effet notoire :
Chaque comprimé orodispersible contient 2,5 mg d’aspartam (E951) et environ 29 mg de lactose monohydraté (correspondant à 28 mg de lactose).
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé blanc, biconvexe et rond d’environ 6,7 mm de diamètre, gravé « E10 » sur une face et lisse sur l’autre.
4.1. Indications thérapeutiques
Traitement symptomatique de l’urticaire.
4.2. Posologie et mode d'administration
Rhinite et rhino-conjonctivite allergique
Réservé à l’adulte et l’enfant à partir de 12 ans : 10 mg d’ébastine une fois par jour.
La dose peut être augmentée à 20 mg d’ébastine une fois par jour en cas de symptômes sévères.
Urticaire
Réservé à l’adulte à partir de 18 ans : 10 mg d’ébastine une fois par jour.
Population pédiatrique
La sécurité et l’efficacité d’EBASTINE TEVA chez les enfants âgés de moins de 12 ans n’ont pas été établies.
Populations particulières
Chez les patients présentant une insuffisance rénale légère, modérée ou sévère, et les patients présentant une insuffisance hépatique légère à modérée, il n’y a pas lieu de prévoir une adaptation posologique.
Il n’y a pas d’expérience avec des doses supérieures à 10 mg chez les patients atteints d’insuffisance hépatique sévère. Par conséquent, la dose ne doit pas dépasser 10 mg chez ces patients.
Le traitement peut être prolongé jusqu’à disparition des symptômes.
Mode d’administration
Voie orale.
Le comprimé orodispersible sera placé sur la langue, où il se dispersera quasi instantanément. Il n’est pas nécessaire de prendre de l’eau ou un autre liquide.
Le comprimé peut être pris pendant ou en dehors des repas.
Durée du traitement
La durée de traitement est établie par le médecin prescripteur.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Un traitement par ébastine à long terme peut entraîner un risque accru de caries chez certains patients en raison d’une bouche sèche. Les patients doivent donc être informés de l'importance d'une hygiène bucco-dentaire rigoureuse.
La prescription d'ébastine doit être prudente chez les patients présentant un syndrome du QTc long, ayant une hypokaliémie, ou recevant un traitement connu comme pouvant induire un allongement de l'intervalle QTc ou comme inhibiteur du CYP450 2J2, 4F12 ou 3A4, tel que les antifongiques azolés et les macrolides (voir rubrique 4.5).
Puisqu’il y a une interaction pharmacocinétique entre les antimycotiques du groupe des imidazoles comme le kétoconazole et l’itraconazole, ou entre les antibiotiques du groupe des macrolides comme l’érythromycine ou ceux du groupe des antituberculeux comme la rifampicine (voir rubrique 4.5) l’ébastine doit être prescrite avec précaution en association avec les médicaments qui contiennent ces substances.
L’ébastine doit être utilisée avec précaution chez les patients atteints d’insuffisance hépatique sévère (voir rubrique 4.2).
Excipient(s)
Aspartam
L’aspartam est hydrolysé dans le tube gastro-digestif lorsqu’il est ingéré par voie orale. L’un des principaux produits de l’hydrolyse est la phénylalanine.
Ceci doit être pris en compte pour les personnes atteintes de phénylcétonurie (PCU).
Lactose
Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Ces interactions résultent d’une augmentation des concentrations plasmatiques de l’ébastine et, dans une moindre mesure, de la carébastine, sans effet pharmacodynamique cliniquement significatif.
Des interactions pharmacocinétiques ont été observées lors de la prise d’ébastine et de rifampicine. Ces interactions peuvent provoquer une baisse des concentrations plasmatiques et réduire les effets antihistaminiques.
Il n’a pas été rapporté d’interaction entre l’ébastine et la théophylline, la warfarine, la cimétidine, le diazépam ou l’alcool.
La prise d’ébastine avec de la nourriture ne modifie pas son effet clinique.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il y a peu de données concernant l’utilisation d’ébastine chez les femmes enceintes. Les études effectuées chez l'animal n’ont pas mis en évidence d'effets délétères directs ou indirects sur la toxicité pour la reproduction (voir rubrique 5.3). Par mesure de précaution, il est préférable d’éviter de prendre de l’ébastine pendant la grossesse.
Le passage de l’ébastine et de ses métabolites dans le lait maternel n’a pas été étudié. La forte liaison aux protéines plasmatiques (> 97 %) de l’ébastine et de son principal métabolite, la carébastine, suggère que le médicament n’est pas excrété dans le lait maternel. L'excrétion d'ébastine dans le lait a été mise en évidence chez la rate. Par mesure de précaution, il est préférable d’éviter de prendre de l’ébastine pendant l’allaitement.
Fertilité
Il n’y a pas de données concernant l’effet d’ébastine sur la fertilité humaine.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Dans une analyse poolée d’essais cliniques contrôlés contre placebo avec 5 708 patients, les effets indésirables les plus fréquemment rapportés étaient la bouche sèche et la somnolence.
Les effets indésirables rapportés lors des essais cliniques chez les enfants (n = 460) ont été similaires à ceux observés chez les adultes.
Le tableau ci-dessous présente les effets indésirables provenant des essais cliniques et de l'expérience post-commercialisation.
|
Classe de systèmes d’organes |
Très fréquent (≥ 1/10) |
Fréquent (≥ 1/100, < 1/10) |
Peu fréquent (≥ 1/1 000, < 1/100) |
Rare (≥ 1/10 000, < 1/1 000) |
Très rare (< 1/10 000) |
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) |
|
Affections du système immunitaire |
|
|
|
Réactions d’hypersensibilité (telles qu’anaphylaxie et angio-œdème) |
|
|
|
Troubles du métabolisme et de la nutrition |
|
|
|
|
|
Augmentation de l’appétit |
|
Affections psychiatriques |
|
|
|
Nervosité, insomnie |
|
|
|
Affections du système nerveux |
Céphalées |
Somnolence |
|
Sensations vertigineuses, hypoesthésie, dysgueusie |
Dysesthésie |
|
|
Affections cardiaques |
|
|
|
Palpitations, tachycardie |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Epistaxis, pharyngite, rhinite |
|
|
|
|
Affections gastro-intestinales |
|
Bouche sèche |
|
Douleurs abdominales, vomissements, nausées, dyspepsie |
|
|
|
Affections hépatobiliaires |
|
|
|
Hépatite, cholestase, anomalie du bilan hépatique (transaminases, gamma-GT, phosphatase alcaline et augmentation de la bilirubine) |
|
|
|
Affections de la peau et du tissu sous-cutané |
|
|
|
Urticaire, éruption cutanée, dermatite |
Exanthème, eczéma |
|
|
Affections des organes de reproduction |
|
|
|
Troubles menstruels |
Dysménorrhée |
|
|
Troubles généraux |
|
|
|
Œdème, asthénie |
|
|
|
Investigations |
|
|
|
|
|
Prise de poids |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
Symptômes
Il n’a pas été observé de signes ou symptômes cliniquement significatifs lors des études utilisant des doses élevées allant jusqu’à 100 mg une fois par jour. Un surdosage peut accroître le risque de sédation et d’effets antimuscariniques. Une fatigue, une bouche sèche et des troubles de l'accommodation ont été observés à des doses comprises entre 300 et 500 mg. A doses élevées, d’éventuels effets cardiovasculaires peuvent survenir.
Traitement
Il n’y a pas d’antidote spécifique pour l’ébastine. Un lavage gastrique, une surveillance des fonctions vitales incluant un ECG et un traitement symptomatique doivent être effectués jusqu'à la guérison complète et pendant au moins 48 heures.
Une surveillance en service de soins intensifs peut être requise en cas d’apparition de troubles du système nerveux central.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
L’ébastine est un antagoniste puissant et hautement sélectif du récepteur H1 de l’histamine. L’ébastine exerce un effet prolongé sur le récepteur et est dénué d’effet anticholinergique.
Propriétés cliniques
Des tests par voie intradermique ont révélé un effet antihistaminique statistiquement et cliniquement significatif débutant 1 heure après l’administration et persistant plus de 24 heures.
Il n’a pas été observé d’allongement de l’intervalle QT ni d’effet indésirable cardiaque après administration du traitement à la dose recommandée lors d’études spécifiques visant à évaluer le retentissement cardiaque de l’ébastine chez des volontaires sains.
Avec des doses supérieures, il n’a pas été rapporté de retentissement sur l’intervalle QTc lors d’administration de doses allant jusqu'à 60 mg/jour, alors qu’une augmentation statistiquement, mais non cliniquement significative de 10 ms (2,7 %) a été observée en cas de surdosage à 100 mg/jour.
5.2. Propriétés pharmacocinétiques
L’ébastine est rapidement absorbée et subit un effet de premier passage hépatique important après une administration orale. Elle est presque totalement métabolisée en carébastine, qui est le métabolite actif. Après une dose orale de 10 mg d’ébastine, des concentrations plasmatiques maximales de 80 à 100 ng/ml de carébastine ont été observées au bout de 2,6 à 4 heures. Après une dose orale unique de 20 mg d’ébastine, la concentration plasmatique du métabolite carébastine a atteint la valeur maximale moyenne de 195 ng/ml au bout de 3 à 6 heures. La demi-vie du métabolite est de 15 à 19 heures ; 66 % sont excrétés dans l’urine sous forme de métabolites conjugués. Après des administrations réitérées d’une dose quotidienne de 10 mg, l’état d’équilibre a été atteint au bout de 3 à 5 jours avec des concentrations plasmatiques de 130 à 160 ng/ml.
Plus de 95 % de l'ébastine et de la carébastine sont liés aux protéines plasmatiques.
Des études in vitro sur microsomes hépatiques humains ont montré que les CYP450 (2J2, 4F12 et 3A4) exerçaient une action prédominante dans la métabolisation de l’ébastine en carébastine. Des augmentations significatives des concentrations plasmatiques de l’ébastine et de la carébastine ont été observées après l’administration concomitante de kétoconazole ou d’érythromycine (tous deux inhibiteurs de CYP450 3A4) (voir rubrique 4.5).
Il n’a pas été observé de modification des paramètres pharmacocinétiques chez des patients âgés comparativement à des adultes jeunes.
Chez des patients présentant une insuffisance rénale légère, modérée ou sévère ou une insuffisance hépatique légère à modérée traités par des doses quotidiennes de 20 mg d’ébastine, les concentrations plasmatiques de l’ébastine et de la carébastine mesurées au 1er et au 5ème jour du traitement étaient similaires à celles déterminées chez des volontaires sains.
La demi-vie d’élimination du métabolite carébastine était prolongée à 23-26 heures chez des patients insuffisants rénaux. La demi-vie mesurée était de 27 heures chez des patients insuffisants hépatiques.
Après administration des comprimés pelliculés d’ébastine avec des aliments, la concentration plasmatique de la carébastine, le métabolite actif principal de l’ébastine, était augmentée d’un facteur 1,5 à 2 et l’aire sous la courbe des concentrations plasmatique (ASC) était augmentée de 50 %, sans modification de Tmax. Il n’a pas été mis en évidence de modification en termes d’efficacité clinique.
5.3. Données de sécurité préclinique
36 mois.
6.4. Précautions particulières de conservation
A conserver dans l’emballage extérieur d’origine à l’abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
Plaquettes pelables OPA/Alu/PVC – Aluminium.
Boîte de 10, 20, 30, 40, 50, 90, 98 ou 100 comprimés orodispersibles.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
100-110 ESPLANADE DU GENERAL DE GAULLE
92931 PARIS LA DEFENSE CEDEX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 216 897 9 2 : 10 comprimés sous plaquettes (OPA/Aluminium/PVC-Aluminium).
· 34009 216 898 5 3 : 20 comprimés sous plaquettes (OPA/Aluminium/PVC-Aluminium).
· 34009 216 899 1 4 : 30 comprimés sous plaquettes {OPA/Aluminium/PVC-Aluminium).
· 34009 216 901 6 3 : 40 comprimés sous plaquettes {OPA/Aluminium/PVC-Aluminium).
· 34009 216 902 2 4 : 50 comprimés sous plaquettes (OPA/Aluminium/PVC-Aluminium).
· 34009 580 831 0 5 : 90 comprimés sous plaquettes (OPA/Aluminium/PVC-Aluminium).
· 34009 580 832 7 3 : 98 comprimés sous plaquettes (OPA/Aluminium/PVC-Aluminium).
· 34009 580 833 3 4 : 100 comprimés sous plaquettes (OPA/Aluminium/PVC-Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II.
ANSM - Mis à jour le : 03/04/2024
EBASTINE TEVA 10 mg, comprimé orodispersible
Ebastine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que EBASTINE TEVA 10 mg, comprimé orodispersible et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre EBASTINE TEVA 10 mg, comprimé orodispersible ?
3. Comment prendre EBASTINE TEVA 10 mg, comprimé orodispersible ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver EBASTINE TEVA 10 mg, comprimé orodispersible ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE EBASTINE TEVA 10 mg, comprimé orodispersible ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : R06AX22
L’ébastine est un antihistaminique qui soulage des symptômes d’allergie tels que les éternuements, l’écoulement nasal, le larmoiement au cours des rhinites (ou rhino-conjonctivites) allergiques et les éruptions sur la peau avec démangeaisons.
Chez les adultes et les enfants à partir de 12 ans, EBASTINE TEVA est utilisé pour soulager les symptômes de rhinite saisonnière (rhume des foins) et de rhinite allergique perannuelle, y compris dans les cas s’accompagnant d’une conjonctivite allergique.
Chez les adultes de plus de 18 ans, EBASTINE TEVA 10 mg est également utilisé pour réduire les démangeaisons et les papules d’urticaire (plaques cutanées surélevées).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE EBASTINE TEVA 10 mg, comprimé orodispersible ?
Ne prenez jamais EBASTINE TEVA 10 mg, comprimé orodispersible :
· si vous êtes allergique à l’ébastine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin avant de prendre EBASTINE TEVA :
· si le taux de potassium dans votre sang est bas ;
· si vous présentez une anomalie cardiaque désignée sous le nom de « syndrome du QTc long » qui est une anomalie identifiée par la réalisation d’un électrocardiogramme (ECG) par l’allongement de l’intervalle QTC ;
· si vous prenez déjà certains antibiotiques ou médicaments utilisés pour le traitement de mycoses : voir « Autres médicaments et EBASTINE TEVA 10 mg, comprimé orodispersible » ci-dessous.
· si la fonction de votre foie est sévèrement altérée (insuffisance hépatique).
EBASTINE TEVA peut provoquer une bouche sèche. Lors d'un traitement à long terme, il est donc primordial d'avoir une bonne hygiène bucco-dentaire (les dents doivent être brossées deux fois par jour) afin de réduire le risque de caries.
Enfants et adolescents
Ce médicament ne doit être utilisé que chez des enfants âgés d’au moins 12 ans. Ne donnez pas ce médicament aux enfants de moins de 12 ans car la sécurité et l’efficacité n’ont pas été établies pour ce groupe d’âge.
Autres médicaments et EBASTINE TEVA 10 mg, comprimé orodispersible
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
EBASTINE TEVA peut affecter ou être affectée par la prise de certains autres médicaments qui contiennent les substances actives suivantes :
· le kétoconazole, l’itraconazole (médicaments utilisés pour le traitement des mycoses) ;
· l’érythromycine (antibiotique) ;
· la rifampicine (médicament utilisé pour le traitement de la tuberculose).
EBASTINE TEVA 10 mg, comprimé orodispersible avec des aliments et boissons
Vous pouvez prendre EBASTINE TEVA pendant ou en dehors des repas.
Grossesse, allaitement et fertilité
L’expérience chez la femme enceinte n’est pas suffisante à ce jour pour établir l’existence ou non d’un risque pour la grossesse lors de la prise d’ébastine. Par conséquent, si vous êtes enceinte, vous ne devez prendre EBASTINE TEVA que si votre médecin considère que le bénéfice attendu l’emporte sur les risques éventuels.
Ne prenez pas EBASTINE TEVA si vous allaitez, car le passage de ce médicament dans le lait maternel n’est pas connu.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
La plupart des patients traités par EBASTINE TEVA peuvent conduire ou mener d’autres activités qui nécessitent une bonne réactivité. Comme avec tout autre médicament, vous devez vérifier votre réaction individuelle après avoir pris EBASTINE TEVA avant de conduire ou de mener des activités complexes, car certains patients peuvent ressentir une somnolence ou des étourdissements.
EBASTINE TEVA 10 mg, comprimé orodispersible contient de l’aspartam
EBASTINE TEVA 10 mg contient 2,5 mg d’aspartam par comprimé. EBASTINE TEVA 20 mg contient 5 mg d’aspartam par comprimé. L’aspartam contient une source de phénylalanine. Peut être dangereux pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de phénylalanine ne pouvant être éliminée correctement.
EBASTINE TEVA 10 mg, comprimé orodispersible contient du lactose
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
EBASTINE TEVA 10 mg, comprimé orodispersible contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE EBASTINE TEVA 10 mg, comprimé orodispersible ?
La dose recommandée est :
|
Indication |
Age |
Dose |
|
Rhinite allergique
En cas de symptômes sévères |
Enfants à partir de 12 ans et adultes |
1 comprimé d’EBASTINE TEVA 10 mg (10 mg d’ébastine) une fois par jour
2 comprimés d’EBASTINE TEVA 10 mg ou un comprimé d’EBASTINE TEVA 20 mg (20 mg d’ébastine) une fois par jour |
|
Urticaire |
Adultes à partir de 18 ans |
1 comprimé d’EBASTINE TEVA 10 mg (10 mg d’ébastine) une fois par jour |
Aucune adaptation de la dose n’est nécessaire chez les patients présentant une insuffisance rénale.
Aucune adaptation de la dose n’est nécessaire chez les patients présentant une insuffisance hépatique légère à modérée.
Il n’y a pas d’expérience avec des doses supérieures à 10 mg chez des patients présentant une insuffisance hépatique sévère. Par conséquent, la dose ne doit pas dépasser 10 mg chez ces patients.
N’appuyez pas sur le comprimé pour le sortir de l’alvéole, car cela risque de l’écraser.
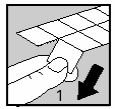
Chaque bande contient des comprimés dans des alvéoles séparées par des perforations. Détachez une alvéole en déchirant le long des lignes prédécoupées (figure 1).
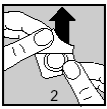
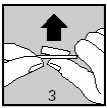
Décollez délicatement la feuille de couverture à partir de l'angle indiqué par la flèche (figures 2 et 3).
Avec les mains sèches, sortez le comprimé de l’alvéole.
Placez le comprimé sur votre langue, où il se dispersera : de l’eau ou un autre liquide n’est pas nécessaire.
Vous pouvez prendre EBASTINE TEVA pendant ou en dehors des repas.
Votre médecin décidera de la durée du traitement.
Si vous avez pris plus d’EBASTINE TEVA 10 mg, comprimé orodispersible que vous n’auriez dû :
Il n’existe aucun antidote spécifique de la substance active, l’ébastine.
Si vous pensez que vous avez pris plus d’EBASTINE TEVA que vous n’auriez dû, dites-le à votre médecin. Selon la sévérité de l’intoxication, il prendra les mesures appropriées (surveillance des fonctions vitales, dont surveillance par ECG pendant au moins 48 heures, traitement symptomatique et lavage gastrique), si nécessaire.
Si vous oubliez de prendre EBASTINE TEVA 10 mg, comprimé orodispersible :
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre EBASTINE TEVA 10 mg, comprimé orodispersible :
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Arrêtez de prendre EBASTINE TEVA et contactez immédiatement votre médecin ou rendez-vous à l’hôpital le plus proche si ce qui suit vous arrive :
· des démangeaisons, de l’urticaire et un gonflement du visage, langue ou gorge pouvant entraîner des difficultés à avaler ou pour respirer. Ceci peut être des signes de réactions d’hypersensibilité telles qu’anaphylaxie et angio-œdème à Ebastine Teva qui sont des effets indésirables rares (pouvant affecter jusqu’à 1 personne sur 10 000).
Les autres effets indésirables incluent :
Effets indésirables très fréquents pouvant affecter plus de 1 personne sur 10 :
· maux de tête.
Effets indésirables fréquents pouvant affecter jusqu’à 1 personne sur 10 :
· envie de dormir ;
· bouche sèche.
Effets indésirables peu fréquents pouvant affecter jusqu’à 1 personne sur 100 :
· saignements de nez ;
· maux de gorge (pharyngite) ;
· écoulement nasal (rhinite).
Effets indésirables rares pouvant affecter jusqu’à 1 personne sur 1 000) :
· nervosité ;
· insomnie ;
· somnolence ;
· étourdissements ;
· perception tactile modifiée ;
· diminution de la sensation du toucher ;
· perception du goût modifiée ;
· palpitations (accélération cardiaque, pouls irrégulier) ;
· accélération du pouls ;
· douleurs abdominales ;
· vomissements ;
· nausées ;
· indigestion ;
· hépatite (inflammation du foie) ;
· cholestase (diminution ou disparition de l’écoulement de la bile) ;
· anomalie du bilan hépatique ;
· éruption cutanée, urticaire, éruption cutanée étendue ;
· troubles menstruels ;
· œdème (accumulation d’eau dans les tissus) ;
· faiblesse (asthénie).
Effets indésirables très rares pouvant affecter jusqu’à 1 personne sur 10 000 :
· dysesthésie (sensation anormale) ;
· eczéma, inflammation de la peau ;
· douleurs menstruels.
Fréquence indéterminée : ne peut être estimée sur la base des données disponibles
· prise de poids ;
· augmentation de l’appétit.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER EBASTINE TEVA 10 mg, comprimé orodispersible ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et la plaquette après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver dans l’emballage extérieur d’origine à l’abri de la lumière.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient EBASTINE TEVA 10 mg, comprimé orodispersible
· La substance active est :
Ebastine.......................................................................................................................... 10 mg
Pour un comprimé orodispersible.
· Les autres composants sont :
Cellulose microcristalline, lactose monohydraté, amidon de maïs, croscarmellose sodique, aspartam (E951), arôme menthe poivrée, silice colloïdale anhydre et stéarate de magnésium.
Qu’est-ce que EBASTINE TEVA 10 mg, comprimé orodispersible et contenu de l’emballage extérieur
Comprimé orodispersible.
Comprimé blanc, biconvexe et rond, gravé « E10 » sur une face et lisse sur l’autre.
Boîtes de 10, 20, 30, 40, 50, 90, 98 ou 100 comprimés orodispersibles.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
100-110 ESPLANADE DU GENERAL DE GAULLE
92931 PARIS LA DEFENSE CEDEX
Exploitant de l’autorisation de mise sur le marché
100-110 ESPLANADE DU GENERAL DE GAULLE
92931 PARIS LA DEFENSE CEDEX
SWENSWEG 5, 2031 GA HAARLEM
PAYS-BAS
OU
TEVA PHARMACEUTICAL WORKS PRIVATE LIMITED COMPANY
PALLAGI ÚT 13, 4042 DEBRECEN
HONGRIE
OU
TEVA CZECH INDUSTRIES S.R.O.
OSTRAVSKÁ 29, Č.P. 305
747 70 OPAVA -KOMÁROV
REPUBLIQUE TCHEQUE
OU
TEVA PHARMA, S.L.U.
C/ C, N° 4, POLIGONO INDUSTRIAL MALPICA
50016 ZARAGOZA
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).