Dernière mise à jour le 03/08/2026
RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique
Indications thérapeutiques
Classe pharmacothérapeutique : psychoanaleptiques, anticholinestérasiques - code ATC : N06DA03.
La substance active de RIVASTIGMINE BIOGARAN est la rivastigmine.
La rivastigmine appartient à une classe de substances appelées inhibiteurs de la cholinestérase. Chez les patients atteints de la maladie d’Alzheimer, la disparition de certaines cellules nerveuses au niveau du cerveau entraîne des taux faibles du neurotransmetteur appelé acétylcholine (une substance qui permet aux cellules nerveuses de communiquer entre elles). La rivastigmine agit en bloquant les enzymes responsables de la destruction de l’acétylcholine : l’acétylcholinestérase et la butylcholinestérase. En bloquant ces enzymes, RIVASTIGMINE BIOGARAN permet d’augmenter les taux d’acétylcholine dans le cerveau, contribuant ainsi à diminuer les symptômes de la maladie d’Alzheimer.
RIVASTIGMINE BIOGARAN est utilisé dans le traitement des patients adultes atteints de formes légères à modérément sévères de la maladie d'Alzheimer, une maladie du cerveau qui affecte progressivement la mémoire, la capacité intellectuelle et le comportement.
Présentations
> 30 sachet(s) papier polytéréphtalate (PET) aluminium polyacrylonitrile avec fermeture de sécurité enfant de 1 dispositif(s)
Code CIP : 280 021-3 ou 34009 280 021 3 6
Déclaration de commercialisation : 07/08/2015
Cette présentation n'est pas agréée aux collectivités
Documents de bon usage du médicament
- Maladie d’Alzheimer et maladies apparentées : diagnostic et prise en charge
Auteur : Haute autorité de santé
Type : Recommandation de bonne pratique
Date de mise à jour :Décembre 2011
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : BIOGARAN
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription initiale annuelle réservée à certains spécialistes
- prescription réservée aux médecins spécialistes ou qualifiés en médecine générale titulaires de la capacité de gérontologie
- prescription réservée aux spécialistes et services GERIATRIE
- prescription réservée aux spécialistes et services NEUROLOGIE
- prescription réservée aux spécialistes et services PSYCHIATRIE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 132 233 6
ANSM - Mis à jour le : 03/07/2026
RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Rivastigmine............................................................................................................................ 9 mg
Pour un dispositif transdermique de 5 cm2.
Chaque dispositif transdermique libère 4,6 mg de rivastigmine par 24 heures.
Pour la liste complète des excipients, voir rubrique 6.1.
Dispositif transdermique.
Le médicament se présente sous forme d’un dispositif transdermique de forme ronde, mince, de type matrice composé de trois couches. Il est constitué d’un film de support, d’une matrice (acrylique) contenant la substance active, d’une matrice adhésive (silicone) et d’une membrane libératrice rectangulaire.
La face externe de la couche de support est translucide et de couleur blanche portant les mentions suivantes imprimées en noir : « Rivastigmine 4,6 mg/24 h ».
4.1. Indications thérapeutiques
Traitement symptomatique des formes légères à modérément sévères de la maladie d'Alzheimer.
RIVASTIGMINE BIOGARAN est indiqué chez l’adulte.
4.2. Posologie et mode d'administration
Le traitement doit être instauré et supervisé par un médecin ayant l’expérience du diagnostic et du traitement des patients atteints de la maladie d’Alzheimer. Le diagnostic sera établi selon les critères en vigueur. Comme pour tout traitement instauré chez des patients atteints de démence, le traitement par la rivastigmine ne doit être entrepris que si une personne aidante peut administrer et surveiller régulièrement le traitement.
Posologie
|
Dispositifs transdermiques |
Dose libérée de rivastigmine in vivo par 24 heures |
|
RIVASTIGMINE BIOGARAN 4,6 mg/24 h |
4,6 mg |
|
RIVASTIGMINE BIOGARAN 9,5 mg/24 h |
9,5 mg |
Dose initiale
Le traitement doit être instauré avec 4,6 mg/24 h.
Dose d’entretien
Après un minimum de quatre semaines de traitement et si la posologie est bien tolérée selon le médecin traitant, la dose de 4,6 mg/24 h peut être augmentée à 9,5 mg/24 h, dose quotidienne efficace recommandée, laquelle doit être poursuivie aussi longtemps que le patient continue à présenter un bénéfice thérapeutique.
Augmentation de dose
9,5 mg/24 h est la dose quotidienne efficace recommandée, qui doit être poursuivie aussi longtemps que le patient continue à présenter un bénéfice thérapeutique. Si le traitement à 9,5 mg/24 h est bien toléré, et seulement après un minimum de six mois de traitement, une augmentation de la dose à 13,3 mg/24 h peut être envisagée par le médecin traitant pour obtenir un bénéfice thérapeutique supplémentaire chez les patients qui ont présenté un déclin cognitif significatif (par exemple diminution du MMSE) et/ou un déclin fonctionnel (basé sur le jugement du médecin) tandis qu’ils étaient à la dose quotidienne efficace recommandée de 9,5 mg/24 h (voir rubrique 5.1).
Le bénéfice clinique de la rivastigmine doit être réévalué régulièrement. L’arrêt du traitement doit aussi être envisagé lorsqu’il est évident qu’il n’y a plus de bénéfice thérapeutique à la dose optimale.
En cas de survenue d’effets indésirables gastro-intestinaux, le traitement doit être interrompu temporairement jusqu’à la résolution de ces effets indésirables. Le traitement par dispositif transdermique pourra être repris à la même dose si le traitement n’est pas interrompu pendant plus de trois jours. Dans le cas contraire, le traitement devra être repris avec 4,6 mg/24 h.
Passage des gélules ou de la solution buvable aux dispositifs transdermiques
Sur la base d’une exposition comparable entre la rivastigmine orale et transdermique (voir rubrique 5.2), les patients traités par rivastigmine en gélules ou solution buvable peuvent passer aux dispositifs transdermiques de RIVASTIGMINE BIOGARAN comme suit :
· un patient prenant une dose de 3 mg/jour de rivastigmine orale peut passer aux dispositifs transdermiques 4,6 mg/24 h ;
· un patient prenant une dose de 6 mg/jour de rivastigmine orale peut passer aux dispositifs transdermiques 4,6 mg/24 h ;
· un patient à une dose stable et bien tolérée de 9 mg/jour de rivastigmine orale peut passer aux dispositifs transdermiques 9,5 mg/24 h. Si la dose orale de 9 mg/jour n’est pas stable et bien tolérée, un passage aux dispositifs transdermiques 4,6 mg/24 h est recommandé ;
· un patient prenant une dose de 12 mg/jour de rivastigmine orale peut passer aux dispositifs transdermiques 9,5 mg/24 h.
Après un passage aux dispositifs transdermiques 4,6 mg/24 h et si ceux-ci sont bien tolérés après un minimum de 4 semaines de traitement, la dose de 4,6 mg/24 h doit être augmentée à 9,5 mg/24 h qui est la dose efficace recommandée.
Il est recommandé d’appliquer le premier dispositif transdermique le lendemain de la dernière dose orale.
Populations à risque
· Population pédiatrique : il n’existe pas d’utilisation justifiée de la rivastigmine dans la population pédiatrique dans le traitement de la maladie d’Alzheimer.
· Patients pesant moins de 50 kg : une attention particulière doit être faite pour l’ajustement posologique au-dessus de la dose efficace recommandée de 9,5 mg/24 h chez des patients pesant moins de 50 kg (voir rubrique 4.4). Ils peuvent présenter davantage d’effets indésirables et peuvent être plus susceptibles d’arrêter le traitement à cause de ces effets indésirables.
· Insuffisance hépatique : en raison d’une augmentation de l’exposition au produit chez les insuffisants hépatiques légers à modérés, comme observé avec les formulations orales, les recommandations d’ajustement posologique en fonction de la tolérance individuelle doivent être étroitement suivies. Les patients présentant une insuffisance hépatique cliniquement significative peuvent présenter davantage d’effets indésirables doses-dépendants. Les patients présentant une insuffisance hépatique sévère n’ont pas été étudiés. L’ajustement de la dose chez ces patients doit être réalisée avec prudence (voir rubriques 4.4 et 5.2).
· Insuffisance rénale : aucun ajustement posologique n’est nécessaire chez les patients présentant une insuffisance rénale (voir rubrique 5.2).
Mode d’administration
Les dispositifs transdermiques doivent être appliqués une fois par jour sur une peau saine, propre et sèche, sans pilosité, sur le haut ou le bas du dos, le haut du bras ou la poitrine, à un endroit où il ne risque pas d’être décollé par des vêtements serrés. Du fait de la diminution de la biodisponibilité de la rivastigmine observée lorsque le dispositif transdermique est utilisé sur la cuisse ou l’abdomen, il n’est pas recommandé de l’appliquer sur ces régions du corps.
Le dispositif transdermique ne doit pas être appliqué sur une zone cutanée présentant une rougeur, une irritation ou une coupure. Il faut éviter d’appliquer le dispositif sur la même zone cutanée pendant 14 jours afin de minimiser le risque potentiel d’irritation cutanée.
Les patients et les personnes aidantes doivent être informés des instructions d’administration importantes :
· Le dispositif transdermique du jour précédent doit être enlevé avant d’en appliquer un nouveau chaque jour (voir rubrique 4.9).
· Le dispositif transdermique doit toujours être remplacé par un nouveau après 24 heures. Un seul dispositif transdermique doit être porté à la fois (voir rubrique 4.9).
· Le dispositif transdermique doit être pressé fermement avec la paume de la main pendant au moins 30 secondes jusqu’à ce que les bords adhèrent bien.
· Si le dispositif transdermique se détache, un nouveau dispositif doit être appliqué pour le reste de la journée. Il doit ensuite être remplacé comme d’habitude au même moment le lendemain.
· Le dispositif transdermique peut être utilisé dans toutes les situations de la vie quotidienne, y compris les bains et par temps chaud.
· Le dispositif transdermique ne doit pas être exposé à des sources de chaleur externes (par exemple soleil excessif, sauna, solarium) pendant de longues périodes.
· Le dispositif transdermique ne doit pas être découpé.
Hypersensibilité à la rivastigmine, aux autres dérivés des carbamates ou à l’un des excipients mentionnés à la rubrique 6.1.
Antécédents de réactions au site d’application suggérant une dermatite allergique de contact avec le dispositif transdermique de rivastigmine (voir rubrique 4.4).
4.4. Mises en garde spéciales et précautions d'emploi
L’incidence et la sévérité des effets indésirables augmentent généralement avec l’augmentation des posologies, notamment lors des modifications de dose. Si le traitement est interrompu pendant plus de trois jours, il devra être repris avec 4,6 mg/24 h.
Mésusage du médicament et erreurs de doses entraînant un surdosage
Un mésusage du médicament et des erreurs de doses avec rivastigmine dispositif transdermique ont entraîné des effets indésirables graves dont certains nécessitant une hospitalisation et plus rarement entraînant le décès (voir rubrique 4.9). La plupart des cas de mésusage du médicament et des erreurs de doses étaient liés au fait de ne pas enlever l’ancien dispositif transdermique au moment d’en mettre un nouveau et à l’utilisation simultanée de plusieurs dispositifs transdermiques. Les patients et leurs personnes aidantes doivent être informés des instructions d’administration importantes (voir rubrique 4.2).
Troubles gastro-intestinaux
Des troubles gastro-intestinaux tels que nausées, vomissements et diarrhées sont dose-dépendants et peuvent survenir lors de l’instauration du traitement et/ou de l’augmentation posologique (voir rubrique 4.8). Ces effets indésirables surviennent plus particulièrement chez les femmes. Les patients montrant des signes ou des symptômes de déshydratation résultant de vomissements ou de diarrhées prolongés, si reconnus et pris en charge rapidement, peuvent être traités par des solutions de réhydratation par voie intraveineuse et une diminution de la dose ou un arrêt du traitement. La déshydratation peut avoir de graves conséquences.
Perte de poids
Les patients souffrant de la maladie d’Alzheimer et prenant des inhibiteurs de la cholinestérase, y compris la rivastigmine, peuvent perdre du poids. Durant le traitement par les dispositifs transdermiques de rivastigmine, le poids des patients doit être surveillé.
Bradycardie
Une prolongation de l’intervalle QT à l’électrocardiogramme (ECG) peut se produire chez des patients traités avec certains médicaments inhibiteurs de la cholinestérase y compris la rivastigmine. La rivastigmine peut causer une bradycardie qui constitue un facteur de risque d’apparition de torsades de pointes, principalement chez les patients ayant des facteurs de risque.
La prudence est recommandée chez les patients ayant une prolongation du QTc préexistante ou des antécédents familiaux de prolongation du QTc ou un risque élevé de développer des torsades de pointes ; par exemple, ceux souffrant d’une insuffisance cardiaque décompensée, d’un infarctus du myocarde récent, d’une bradyarythmie, d’une prédisposition à l’hypokaliémie ou à l’hypomagnésémie ou en cas d’utilisation concomitante avec des médicaments connus pour induire une prolongation de l’intervalle QT et/ou des torsades de pointes. Un suivi clinique (ECG) peut aussi être requis (voir rubriques 4.5 et 4.8).
Autres effets indésirables
Les dispositifs transdermiques de RIVASTIGMINE BIOGARAN seront prescrits avec prudence :
· chez les patients présentant une maladie du nœud sinusal ou des troubles de la conduction cardiaque (bloc sino-auriculaire, bloc atrio-ventriculaire) (voir rubrique 4.8) ;
· chez les patients présentant un ulcère gastrique ou duodénal en poussée, ou chez les patients qui y sont prédisposés, la rivastigmine étant susceptible d'augmenter la sécrétion gastrique (voir rubrique 4.8) ;
· chez les patients prédisposés à une rétention urinaire et des convulsions car les cholinomimétiques peuvent induire ou aggraver de telles maladies ;
· chez les patients présentant des antécédents d’asthme ou de bronchopneumopathie obstructive.
Réactions cutanées au site d’application
Des réactions cutanées au site d’application peuvent survenir avec le dispositif transdermique de rivastigmine et sont généralement d’intensité légère à modérée. Les patients et les personnes aidantes doivent être informés en conséquence.
Ces réactions ne sont pas à elles seules un signe de sensibilisation. Cependant, l’utilisation du dispositif transdermique de rivastigmine peut conduire à une dermatite allergique de contact.
Une dermatite allergique de contact doit être suspectée si les réactions au site d’application se propagent au-delà de la taille du dispositif transdermique, s’il y a un signe de réaction locale plus intense (par exemple aggravation de l’érythème, œdème, papules, vésicules) et si les symptômes ne s’améliorent pas significativement dans les 48 heures suivant le retrait du dispositif transdermique. Dans ces cas, le traitement doit être interrompu (voir rubrique 4.3).
Les patients développant des réactions au site d’application suggérant une dermatite allergique de contact au dispositif transdermique de rivastigmine et qui nécessitent encore un traitement par rivastigmine doivent changer pour la rivastigmine orale uniquement après un test allergique négatif et sous surveillance médicale étroite. Il est possible que certains patients sensibles à la rivastigmine suite à une exposition au dispositif transdermique de rivastigmine ne puissent prendre de rivastigmine sous aucune forme.
Après commercialisation de la rivastigmine, il y a eu de rares cas de patients ayant présenté des dermatites allergiques (disséminées) lors de l’administration de rivastigmine quelle que soit la voie d’administration (orale, transdermique). Dans ces cas, le traitement doit être interrompu (voir rubrique 4.3).
Autres mises en garde et précautions
La rivastigmine peut exacerber ou induire les symptômes extrapyramidaux.
Tout contact avec les yeux doit être évité après manipulation des dispositifs transdermiques de RIVASTIGMINE BIOGARAN (voir rubrique 5.3). Les mains doivent être lavées avec de l’eau et du savon après avoir retiré le dispositif transdermique. En cas de contact avec les yeux ou si les yeux deviennent rouges après manipulation du dispositif transdermique, rincez immédiatement avec beaucoup d’eau et consultez votre médecin si les symptômes persistent.
Populations à risque
· Les patients pesant moins de 50 kg peuvent présenter davantage d’effets indésirables et peuvent être plus susceptibles d’arrêter le traitement à cause de ces effets indésirables (voir rubrique 4.2). Ajuster la dose avec précaution et surveiller étroitement ces patients quant à la survenue d’effets indésirables (par exemple nausées importantes ou vomissements) et envisager la réduction de la dose d’entretien à 4,6 mg/24 h en cas de survenue de ce type d’effets indésirables.
· Atteinte hépatique : les patients présentant une insuffisance hépatique cliniquement significative peuvent présenter davantage d’effets indésirables. Les recommandations d’ajustement posologique en fonction de la tolérance individuelle doivent être étroitement suivies. Les patients présentant une atteinte hépatique sévère n’ont pas été étudiés. L’ajustement de la dose chez ces patients doit être réalisé avec prudence (voir rubriques 4.2 et 5.2).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction particulière n’a été réalisée avec les dispositifs transdermiques de rivastigmine.
En tant qu’inhibiteur de la cholinestérase, la rivastigmine peut potentialiser les effets des myorelaxants analogues de la succinylcholine au cours d’une anesthésie. La prudence est recommandée lors du choix des anesthésiques. Des ajustements posologiques ou un arrêt temporaire du traitement peuvent être considérés, si nécessaire.
En raison de ses propriétés pharmacodynamiques et de ses possibles effets additifs, la rivastigmine ne doit pas être administrée simultanément à d'autres cholinomimétiques. La rivastigmine pourrait interférer avec l'activité des anticholinergiques (par exemple oxybutynine, toltérodine).
Les effets additifs conduisant à une bradycardie (pouvant entraîner une syncope) ont été signalés avec l’utilisation concomitante de plusieurs bêtabloquants (y compris de l’aténolol) et de rivastigmine. Les bêtabloquants cardiovasculaires devraient être associés au risque le plus élevé, toutefois des notifications ont aussi été reçues chez des patients utilisant d’autres bêtabloquants. Par conséquent, une attention particulière doit être portée lorsque la rivastigmine est associée à des bêtabloquants ainsi qu’avec d’autres agents bradycardisants (par exemple les produits antiarythmiques de classe III, les antagonistes des canaux calciques, les glucosides digitaliques, la pilocarpine).
Puisque la bradycardie constitue un facteur de risque d’apparition de torsades de pointes, une attention particulière doit être portée et une surveillance clinique (ECG) peut être nécessaire lorsque la rivastigmine est associée à des médicaments favorisant l’apparition de prolongation de l’intervalle QT ou de torsades de pointes tels que les antipsychotiques, à savoir certaines phénothiazines (chlorpromazine, lévomépromazine), les benzamides (sulpiride, sultopride, amisulpride, tiapride, véralipride), pimozide, halopéridol, dropéridol, cisapride, citalopram, diphémanil, érythromycine intraveineuse, halofantrine, mizolastine, méthadone, pentamidine et moxifloxacine.
Des études menées chez des volontaires sains n'ont pas mis en évidence d'interaction pharmacocinétique entre la rivastigmine orale et la digoxine, la warfarine, le diazépam ou la fluoxétine. La rivastigmine orale n'a pas d’incidence sur l'allongement du temps de prothrombine observé sous warfarine. L'administration simultanée de rivastigmine orale et de digoxine n'a pas entraîné d'effet indésirable sur la conduction cardiaque.
Il n’a pas été observé de modification de la cinétique de la rivastigmine ou de risque accru d’effets indésirables cliniquement significatifs en cas d’administration concomitante de rivastigmine avec des médicaments prescrits couramment tels que les antiacides, les antiémétiques, les antidiabétiques, les antihypertenseurs d’action centrale, les inhibiteurs calciques, les agents inotropes, les antiangineux, les anti-inflammatoires non stéroïdiens, les œstrogènes, les analgésiques, les benzodiazépines et les antihistaminiques.
Compte tenu du métabolisme de la rivastigmine et bien que celle-ci soit susceptible d’inhiber le métabolisme d’autres médicaments métabolisés par la butyrylcholinestérase, des interactions médicamenteuses métaboliques paraissent improbables.
4.6. Fertilité, grossesse et allaitement
Grossesse
Chez les femelles gravides, la rivastigmine et/ou ses métabolites traversent le placenta. Il n’est pas déterminé si cela se produit chez l’Homme. Il n’existe pas de données sur l’utilisation de ce médicament chez la femme enceinte. Au cours d’études péri/postnatales menées chez le rat, une augmentation de la durée de gestation a été observée. La rivastigmine ne doit pas être utilisée à moins d’une nécessité absolue.
Allaitement
Chez l’animal, la rivastigmine est excrétée dans le lait. Dans l’espèce humaine il n'existe pas de données concernant le passage de la rivastigmine dans le lait maternel. En conséquence, les femmes traitées par la rivastigmine ne doivent pas allaiter.
Fertilité
Aucun effet indésirable de la rivastigmine n’a été observé sur la fertilité ou la capacité de reproduction chez le rat (voir section 5.3). Les effets de la rivastigmine sur la fertilité chez l’Homme sont inconnus.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
La maladie d’Alzheimer est susceptible de provoquer une dégradation progressive des aptitudes nécessaires à la conduite ou à l’utilisation de machines. De plus, la rivastigmine peut induire une syncope ou un état confusionnel. De ce fait, la rivastigmine a une influence mineure à modérée sur l’aptitude à conduire des véhicules et à utiliser des machines. Par conséquent, chez les patients atteints d’une démence et traités par la rivastigmine, la capacité à continuer de conduire des véhicules ou d’utiliser des machines de maniement complexe, devrait être évaluée régulièrement par le médecin traitant.
Résumé du profil de sécurité
Les réactions cutanées au site d’application (généralement un érythème d’intensité légère à modérée au site d’application) sont les effets indésirables les plus fréquents observés lors de l’utilisation du dispositif transdermique de rivastigmine. Les autres effets indésirables fréquents sont de type gastro‑intestinaux, notamment des nausées et des vomissements.
Selon le système de classification par organe MedDRA, les effets indésirables sont listés dans le Tableau 1 par ordre de fréquence observée. Les catégories de fréquence sont définies selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1000, < 1/100) ; rare (≥ 1/10 000, < 1/1000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Liste tabulée des effets indésirables
Le Tableau 1 présente les effets indésirables décrits chez 1670 patients atteints de la maladie d’Alzheimer traités par les dispositifs transdermiques de rivastigmine dans le cadre d’études cliniques randomisées, en double aveugle, contrôlées versus placebo et versus comparateur actif d’une durée de 24-48 semaines et provenant des données post-commercialisation.
Tableau 1
|
Infections et infestations |
|
|
Fréquent |
Infection urinaire |
|
Troubles du métabolisme et de la nutrition |
|
|
Fréquent |
Anorexie, appétit diminué |
|
Peu fréquent |
Déshydratation |
|
Affections psychiatriques |
|
|
Fréquent |
Anxiété, dépression, état confusionnel, agitation |
|
Peu fréquent |
Agressivité |
|
Fréquence indéterminée |
Hallucinations, impatience, cauchemars |
|
Affections du système nerveux |
|
|
Fréquent |
Céphalée, syncope, sensation vertigineuse |
|
Peu fréquent |
Hyperactivité psychomotrice |
|
Très rare |
Symptômes extrapyramidaux |
|
Fréquence indéterminée |
Aggravation de la maladie de Parkinson, convulsions, tremblements, somnolence, pleurothotonus (Syndrome de la tour de Pise) |
|
Affections cardiaques |
|
|
Peu fréquent |
Bradycardie |
|
Fréquence indéterminée |
Bloc auriculo-ventriculaire, fibrillation auriculaire, tachycardie, maladie du sinus |
|
Affections vasculaires |
|
|
Fréquence indéterminée |
Hypertension |
|
Affections gastro-intestinales |
|
|
Fréquent |
Nausée, vomissement, diarrhée, dyspepsie, douleur abdominale |
|
Peu fréquent |
Ulcère gastrique |
|
Fréquence indéterminée |
Pancréatite |
|
Affections hépato-biliaires |
|
|
Fréquence indéterminée |
Hépatite, élévation des enzymes hépatiques |
|
Affections de la peau et du tissu sous-cutané |
|
|
Fréquent |
Rash |
|
Fréquence indéterminée |
Prurit, érythème, urticaire, vésicules, dermatite allergique (disséminée) |
|
Affections du rein et des voies urinaires |
|
|
Fréquent |
Incontinence urinaire |
|
Troubles généraux et anomalies au site d’administration |
|
|
Fréquent |
Réactions cutanées au site d’application (par exemple érythème*, prurit*, œdème*, dermatite, irritation cutanée), état asthénique (par exemple fatigue, asthénie), pyrexie, perte de poids |
|
Rare |
Chute |
*Lors d’une étude contrôlée de 24 semaines menée chez des patients japonais, érythème, œdème, et prurit au site d’application ont été signalés comme « très fréquents ».
Description des effets indésirables sélectionnés
Lorsque des doses supérieures à 13,3 mg/24 h ont été utilisées dans l’étude contrôlée versus placebo ci-dessus, la fréquence des insomnies et insuffisance cardiaque a été plus élevée qu’avec 13,3 mg/24 h ou le placebo, ce qui semble indiquer une relation dose-effet. Mais ces effets n’ont pas été plus fréquents avec les dispositifs transdermiques de rivastigmine 13,3 mg/24 h qu’avec le placebo.
Les effets indésirables suivants n’ont été observés qu’avec les gélules et la solution buvable de rivastigmine et n’ont pas été décrits dans les études cliniques avec les dispositifs transdermiques de rivastigmine : malaise, confusion, augmentation de la sudation (fréquent) ; ulcères duodénaux, angine de poitrine (rare) ; hémorragie gastro-intestinale (très rare) ; quelques cas de vomissements sévères ont été associés à une rupture de l’œsophage (fréquence indéterminée).
Irritation cutanée
Dans des études cliniques contrôlées en double aveugle, les réactions au site d’application ont été principalement d’intensité légère à modérée. L’incidence des réactions cutanées au site d’application entraînant un arrêt de traitement a été ≤ 2,3 % chez les patients traités avec rivastigmine dispositif transdermique. L’incidence des réactions cutanées au site d’application entraînant un arrêt de traitement a été supérieure au sein de la population asiatique avec respectivement 4,9 % et 8,4 % chez la population chinoise et japonaise.
Dans deux études cliniques de 24 semaines en double aveugle, contrôlées versus placebo, les réactions cutanées ont été évaluées lors de chaque visite à l’aide d’une échelle de cotation de l’irritation cutanée. Quand elle était observée chez des patients traités avec rivastigmine dispositif transdermique, l’irritation de la peau était principalement de sévérité faible à légère. Elle a été évaluée comme sévère chez ≤ 2,2 % des patients de ces études et chez ≤ 3,7 % des patients traités avec rivastigmine dispositif transdermique dans une étude japonaise.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Symptomatologie
La plupart des cas de surdosage accidentel avec la rivastigmine orale n’ont entraîné aucune symptomatologie clinique et presque tous les patients ont poursuivi le traitement par rivastigmine 24 heures après le surdosage.
Une toxicité cholinergique a été rapportée associée à des symptômes muscariniques qui ont été observés lors d’intoxications modérées tels que des myosis, bouffées vasomotrices, troubles gastro‑intestinaux incluant des douleurs abdominales, nausées, vomissements et diarrhée, bradycardie, bronchospasmes et augmentation des sécrétions bronchiques, hyperhidrose, émissions d’urine et/ou défécations involontaires, larmoiements, hypotension et hypersécrétion salivaire.
Dans les cas plus sévères des effets nicotiniques pourraient se développer tels que faiblesse musculaire, fasciculations, convulsions et arrêts respiratoires avec une possible issue fatale.
En outre après la commercialisation, des cas de vertiges, tremblements, maux de tête, somnolence, état confusionnel, hypertension, hallucinations et malaises ont été rapportés. Des cas de surdosage survenus avec le dispositif transdermique de rivastigmine résultant de mésusages/d’erreurs de dosage (application de plusieurs dispositifs transdermiques à la fois) ont été rapportés depuis sa mise sur le marché et rarement lors des essais cliniques.
Prise en charge
La demi-vie plasmatique de la rivastigmine est de 3,4 heures environ et la durée de l’inhibition de l’acétylcholinestérase est d’environ 9 heures : en cas de surdosage asymptomatique, tous les dispositifs transdermiques de rivastigmine doivent être retirés immédiatement ; un délai de 24 heures doit être respecté avant d’appliquer un nouveau dispositif transdermique. En cas de surdosage s’accompagnant de nausées et de vomissements importants, des antiémétiques pourront être utilisés. Les autres effets indésirables feront l’objet d’un traitement symptomatique si nécessaire.
En cas de surdosage massif, l’atropine peut être utilisée. Il est recommandé d’administrer initialement 0,03 mg/kg de sulfate d’atropine par voie intraveineuse, puis d’ajuster les doses ultérieures en fonction de la réponse clinique. L’administration de scopolamine à titre d’antidote n’est pas recommandée.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : psychoanaleptiques, anticholinestérasiques, code ATC : N06DA03.
La rivastigmine est un inhibiteur de l’acétyl et de la butyrylcholinestérase, de type carbamate : on estime qu’elle facilite la neurotransmission cholinergique en ralentissant la dégradation de l’acétylcholine libérée par les neurones cholinergiques intacts sur le plan fonctionnel. La rivastigmine est donc susceptible d’avoir un effet favorable sur les déficits cognitifs dépendants de ces voies cholinergiques au cours de la maladie d’Alzheimer et d’une démence associée à la maladie d’Alzheimer.
La rivastigmine agit sur les enzymes cibles en formant un complexe lié par une liaison covalente qui entraîne une inactivation transitoire des enzymes. Chez le sujet sain jeune, une dose de 3 mg par voie orale entraîne une diminution d’environ 40 % de l’activité de l’acétylcholinestérase (AChE) dans le LCR dans les 1,5 h après administration. L’activité enzymatique revient à son niveau initial 9 heures environ après le pic d’activité inhibitrice. Chez les patients atteints d’une maladie d’Alzheimer, l’inhibition de l’acétylcholinestérase dans le LCR par la rivastigmine orale est dose-dépendante jusqu'à une posologie de 6 mg deux fois par jour, qui a été la dose maximale étudiée. L’inhibition de l’activité de la butyrylcholinestérase dans le LCR chez 14 patients atteints d’une maladie d’Alzheimer, traités par rivastigmine orale, était similaire à l’inhibition de l’activité de l’AChE.
Etudes cliniques dans la maladie d’Alzheimer
L’efficacité des dispositifs transdermiques de rivastigmine chez les patients atteints de la maladie d’Alzheimer a été démontrée dans une étude pivot de 24 semaines en double aveugle contrôlée versus placebo ainsi que dans sa phase d’extension en ouvert et dans une étude de 48 semaines en double aveugle versus comparateur actif.
Etude de 24 semaines contrôlée versus placebo
Les patients inclus dans cette étude contrôlée versus placebo avaient un score MMSE (Mini-Mental State Examination) compris entre 10 et 20. L’efficacité a été établie à l’aide d’échelles d’évaluation indépendantes et spécifiques par domaine qui ont été utilisées à des intervalles réguliers pendant la période thérapeutique de 24 semaines. Ces outils sont l’ADAS-Cog (Alzheimer’s Disease Assessment Scale - Cognitive subscale, évaluation de la performance cognitive), l’ADCS-CGIC (Alzheimer’s Disease Cooperative Study - Clinician’s Global Impression of Change, évaluation compréhensive et globale du patient par le médecin incluant des données recueillies auprès de la personne aidante) et l’ADCS-ADL (Alzheimer’s Disease Cooperative Study - Activities of Daily Living, évaluation réalisée par la personne aidante des activités de la vie quotidienne telles que l’hygiène personnelle, les capacités à se nourrir, s’habiller, les occupations domestiques telles que les courses, le maintien de la capacité à s’orienter dans différents environnements ainsi que l’implication dans des activités en rapport avec l’argent). Le Tableau 2 présente les résultats à 24 semaines pour les trois échelles d’évaluation.
Tableau 2
|
Population ITT – LOCF |
Dispositifs transdermiques de rivastigmine 9,5 mg/24 h |
Gélules de rivastigmine 12 mg/jour |
Placebo |
|
N = 251 |
N = 256 |
N = 282 |
|
|
ADAS-Cog |
|||
|
(n=248) |
(n=253) |
(n=281) |
|
|
Moyenne à l’état initial ± ET |
27,0 ± 10,3 |
27,9 ± 9,4 |
28,6 ± 9,9 |
|
Moyenne de l’écart à 24 semaines ± ET |
- 0,6 ± 6,4 |
- 0,6 ± 6,2 |
1,0 ± 6,8 |
|
Valeur p versus placebo |
0,005*1 |
0,003*1 |
|
|
ADCS-CGIC |
|||
|
(n=248) |
(n=253) |
(n=278) |
|
|
Score moyen ± ET |
3,9 ± 1,20 |
3,9 ± 1,25 |
4,2 ± 1,26 |
|
Valeur p versus placebo |
0,010*2 |
0,009*2 |
|
|
ADCS-ADL |
|||
|
(n=247) |
(n=254) |
(n=281) |
|
|
Moyenne à l’état initial ± ET |
50,1 ± 16,3 |
49,3 ± 15,8 |
49,2 ±16,0 |
|
Moyenne de l’écart à 24 semaines ± ET |
- 0,1 ± 9,1 |
- 0,5 ± 9,5 |
- 2,3 ± 9,4 |
|
Valeur p versus placebo |
0,013*1 |
0,039*1 |
* p ≤ 0,05 versus placebo
ITT : Intent-To-Treat (Intention de traiter) ; LOCF = Last Observation Carried Forward (Dernières observations rapportées)
1 Analyse de covariance avec traitement et pays comme facteurs et valeur initiale comme covariable. Une modification négative de l’ADAS-Cog indique une amélioration. Une modification positive de l’ADCS-ADL indique une amélioration.
2 Sur la base du test CMH (test de van Elteren) stratifié par pays. Un score ADCS-CGIC < 4 indique une amélioration.
Le Tableau 3 présente les résultats pour les patients de l’étude de 24 semaines contrôlée versus placebo ayant obtenu une réponse clinique significative. Une amélioration cliniquement significative était définie à priori comme une amélioration d’au moins 4 points sur l’échelle ADAS-Cog et pas d’aggravation des scores ADCS-CGIC et ADCS-ADL.
Tableau 3
|
Patients présentant une réponse cliniquement significative (%) |
|||
|
Population ITT-LOCF |
Dispositifs transdermiques de rivastigmine 9,5 mg/24 h N = 251 |
Gélules de rivastigmine 12 mg/jour N = 256 |
Placebo
N = 282 |
|
Amélioration d’au moins 4 points sur l’ADAS-Cog sans aggravation des scores ADCS-CGIC et ADCS-ADL |
17,4 |
19,0 |
10,5 |
|
Valeur p versus placebo |
0,037* |
0,004* |
|
*p < 0,05 versus placebo
Comme l’a indiqué la modélisation compartimentale, l’exposition induite par les dispositifs transdermiques 9,5 mg/24 h a été similaire à celle obtenue avec une dose orale de 12 mg/jour.
Etude de 48 semaines contrôlée versus comparateur actif
Les patients inclus dans l’étude contrôlée versus comparateur actif avaient à l’inclusion un score MMSE (Mini-Mental State Examination) entre 10 et 24. L’étude avait pour objectif de comparer l’efficacité du dispositif transdermique à 13,3 mg/24 h versus le dispositif transdermique à 9,5 mg/24 h pendant 48 semaines de traitement en double aveugle chez des patients atteints de la maladie d’Alzheimer et qui ont présenté un déclin cognitif et fonctionnel significatif après une phase de traitement en ouvert pendant 24-48 semaines tandis qu’ils étaient à la dose d’entretien quotidienne recommandée de 9,5 mg/24 h. Le déclin fonctionnel a été évalué par l’investigateur et le déclin cognitif a été défini comme une diminution du score MMSE > 2 points par rapport à la dernière visite ou comme une diminution > 3 points par rapport au score de base. L’efficacité a été établie en utilisant les échelles d’évaluation d’ADAS-Cog (Alzheimer’s Disease Assessment Scale – Cognitive subscale, évaluation de la performance cognitive) et d’ADCS-IADL (Alzheimer’s Disease Cooperative Study – Instrumental Activities of Daily Living) évaluant les activités instrumentales comme le maintien d’un budget, la préparation d’un repas, faire les courses, la capacité à s’orienter dans différents environnements, la capacité à être laissé sans surveillance. Les résultats de l’étude 48 semaines pour ces deux échelles d’évaluation sont résumés dans le Tableau 4.
Tableau 4
|
Population/Visite |
|
Rivastigmine 15 cm2 N=265 |
Rivastigmine 10 cm2 N=271 |
Rivastigmine 15 cm2 |
Rivastigmine 10 cm2 |
|||
|
|
|
n |
Moyenne |
n |
Moyenne |
DLSM |
IC (95 %) |
p (valeur) |
|
ADAS-Cog |
|
|
|
|
|
|
|
|
|
LOCF |
Valeur initiale |
264 |
34,4 |
268 |
34,9 |
|
|
|
|
Semaine 48 Double aveugle |
Résultat |
264 |
38,5 |
268 |
39,7 |
|
|
|
|
|
Variation |
264 |
4,1 |
268 |
4,9 |
-0,8 |
(-2,1, 0,5) |
0,227 |
|
ADCS-IADL |
|
|
|
|
|
|
|
|
|
LOCF |
Valeur initiale |
265 |
27,5 |
271 |
25,8 |
|
|
|
|
Semaine 48 |
Résultat |
265 |
23,1 |
271 |
19,6 |
|
|
|
|
|
Variation |
265 |
-4,4 |
271 |
-6,2 |
2,2 |
(0,8, 3,6) |
0,002* |
IC – intervalle de confiance.
DLSM – difference in least square means (différence entre les moindres carrés).
LOCF – Last Observation Carried Forward (dernière observation reportée)
Résultats d’ADAS-cog : Une différence négative de la DLSM indique une plus grande amélioration avec rivastigmine 15 cm2 qu’avec rivastigmine 10 cm2.
Résultats d’ADCS-IADL : Une différence positive de la DLSM indique une plus grande amélioration avec rivastigmine 15 cm2 qu’avec rivastigmine 10 cm2.
N est le nombre de patients avec une évaluation à la base (dernière évaluation dans la phase initiale en ouvert) et avec au moins 1 évaluation ultérieure (pour la dernière observation reportée).
La DLSM, l’IC (95 %) et la valeur de p sont basés sur un modèle ANCOVA (analyse de la covariance) ajusté en fonction du pays et de la valeur initiale d’ADAS-cog.
* p < 0,05
Source : Etude D2340-Tableau 11-6 et Tableau 11-7
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec la rivastigmine dans tous les sous-groupes de la population pédiatrique dans le traitement des démences liées à la maladie d’Alzheimer (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Absorption
L’absorption de la rivastigmine libérée par les dispositifs transdermiques de rivastigmine est lente. Après la première dose, des concentrations plasmatiques détectables sont observées après 0,5 à 1 heure. La Cmax est atteinte au bout de 10 à 16 heures. Après le pic, les concentrations plasmatiques diminuent lentement pendant la période d’application de 24 heures restante. En cas de doses répétées (comme à l’état d’équilibre), après qu’un dispositif transdermique neuf ait été appliqué, les concentrations plasmatiques commencent par diminuer lentement pendant 40 minutes en moyenne, jusqu'à ce que l’absorption à partir du nouveau dispositif transdermique soit plus rapide que l’élimination, puis les concentrations plasmatiques s’élèvent à nouveau pour atteindre un nouveau pic après 8 heures environ. A l’état d’équilibre, les concentrations résiduelles représentent environ 50 % des concentrations maximales, contrairement à l’administration orale, avec laquelle les concentrations sont pratiquement nulles entre les prises.
Bien que cela soit moins prononcé qu’avec la formulation orale, l’exposition à la rivastigmine (Cmax et ASC) est augmentée de façon surproportionnelle (multiplication par 2,6 et 4,9) en passant de 4,6 mg/24 h à 9,5 mg/24 h et 13,3 mg/24 h respectivement. L’indice de fluctuation (IF), qui mesure la différence relative entre les concentrations maximales et résiduelles ((Cmax-Cmin)/Cmoyen), a été respectivement de 0,58 pour les dispositifs transdermiques de rivastigmine 4,6 mg/24 h, 0,77 pour les dispositifs transdermiques de rivastigmine 9,5 mg/24 h et 0,72 pour les dispositifs transdermiques de rivastigmine 13,3 mg/24 h, ce qui démontre une fluctuation beaucoup moins importante entre les concentrations résiduelles et maximales qu’avec la formulation orale (IF = 3,96 [6 mg/jour] et 4,15 [12 mg/jour]).
La dose de rivastigmine libérée par le dispositif transdermique sur 24 heures (mg/24 h) ne peut directement être égalée à la quantité (mg) de rivastigmine contenue dans la gélule en ce qui concerne la concentration plasmatique sur 24 heures.
Après une dose unique, la variabilité interindividuelle des paramètres pharmacocinétiques de la rivastigmine (normalisés à la dose/kg de poids corporel) a été de 43 % (Cmax) et 49 % (ASC0-24h) avec l’administration transdermique, versus 74 % et 103 % respectivement avec la forme orale. Dans une étude à l’état d’équilibre menée chez des patients atteints de la maladie d’Alzheimer, la variabilité interindividuelle a été de 45 % (Cmax) et 43 % (ASC0-24h) au maximum après l’utilisation du dispositif transdermique et 71 % et 73 % respectivement après l’administration de la forme orale.
Il a été observé une relation entre l’exposition au médicament à l’état d’équilibre (rivastigmine et son métabolite NAP226-90) et le poids corporel chez des patients atteints de la maladie d’Alzheimer. Par rapport à un patient pesant 65 kg, les concentrations de rivastigmine à l’état d’équilibre chez un patient de 35 kg sont multipliées par deux environ, alors que chez un patient pesant 100 kg, elles seront divisées par deux environ. En raison de l’effet du poids sur l’exposition au médicament, une prudence particulière s’impose pendant la période d’augmentation de posologie chez les patients d’un poids très faible (voir rubrique 4.4).
L’exposition (ASC¥) à la rivastigmine (et à son métabolite NAP226-90) a été plus élevée lorsque le dispositif transdermique était appliqué sur le haut du dos, la poitrine ou le haut du bras, et environ 20 à 30 % plus faible lorsqu’il était appliqué sur l’abdomen ou la cuisse.
Il n’a pas été observé d’accumulation plasmatique significative de la rivastigmine ou de son métabolite NAP226-90 chez les patients atteints de la maladie d’Alzheimer, à l’exception des concentrations plasmatiques qui ont été plus élevées le deuxième jour de traitement par le dispositif transdermique que le premier jour.
Distribution
La liaison de la rivastigmine aux protéines est faible (approximativement 40 %). Elle traverse facilement la barrière hémato-encéphalique et son volume de distribution apparent se situe entre 1,8 et 2,7 l/kg.
Biotransformation
La rivastigmine est fortement et rapidement métabolisée ; la demi-vie d’élimination apparente dans le plasma est d’environ 3,4 heures après le retrait du dispositif transdermique. L’élimination est limitée par la vitesse d’absorption (phénomène de « flip-flop »), ce qui explique le t1/2 plus long observé avec le dispositif transdermique (3,4 h) par rapport à une administration orale ou intraveineuse (1,4 à 1,7 h). La rivastigmine est métabolisée essentiellement par hydrolyse en son métabolite NAP226-90 par l’acétylcholinestérase. In vitro, ce métabolite n’exerce qu’une inhibition minime de l’acétylcholinestérase (< 10 %).
Les résultats des études in vitro indiquent qu’aucune interaction pharmacocinétique n’est attendue avec les médicaments métabolisés par les isoenzymes des cytochromes suivants : CYP1A2, CYP2D6, CYP3A4/5, CYP2E1, CYP2C9, CYP2C8, CYP2C19 ou CYP2B6. Les résultats des études effectuées chez l’animal indiquent que les principales isoenzymes du cytochrome P450 ne participent que de façon mineure au métabolisme de la rivastigmine. La clairance plasmatique totale de la rivastigmine est d’environ 130 litres/h après une dose intraveineuse de 0,2 mg et elle n’est plus que de 70 litres/h après une dose intraveineuse de 2,7 mg, ce qui concorde avec sa pharmacocinétique surproportionnelle non linéaire due à la saturation de son élimination.
Le rapport des ASC¥ métabolite/molécule mère est d’environ 0,7 après l’application du dispositif transdermique versus 3,5 après l’administration orale, ce qui indique un métabolisme beaucoup plus faible après l’administration dermique qu’après l’administration orale. La quantité de NAP226-90 formée après l’application du dispositif transdermique est plus faible, probablement du fait de l’absence de métabolisme présystémique (métabolisme de premier passage hépatique), contrairement à l’administration orale.
Elimination
La rivastigmine inchangée est retrouvée sous forme de traces dans les urines ; l’excrétion urinaire est la voie principale d’élimination des métabolites après l’application du dispositif transdermique. Après administration orale de 14C-rivastigmine, l’élimination rénale est rapide et pratiquement complète (> 90 %) en 24 heures. Moins de 1 % de la dose administrée est éliminée dans les selles.
Une analyse pharmacocinétique de population a montré que l’utilisation de nicotine augmente la clairance orale de la rivastigmine de 23 % chez les patients présentant une maladie d’Alzheimer (n = 75 fumeurs et 549 non-fumeurs) suite à une prise orale de gélules de rivastigmine à des doses allant jusqu’à 12 mg/jour.
Populations particulières
Personnes âgées
L’âge n’a pas eu d’effet sur l’exposition à la rivastigmine chez des patients atteints de la maladie d’Alzheimer traités par les dispositifs transdermiques de rivastigmine.
Insuffisance hépatique
Il n’a pas été mené d’étude avec les dispositifs transdermiques de rivastigmine chez des sujets présentant une insuffisance hépatique. Après administration orale chez des sujets atteints d’insuffisance hépatique légère à modérée comparativement à des sujets à fonction hépatique normale, la Cmax de la rivastigmine est augmentée d’environ 60 % et l’ASC est plus que doublée.
Après une dose unique de 3 mg ou 6 mg par voie orale, la clairance orale moyenne de rivastigmine était approximativement de 46 à 63 % plus basse chez les patients présentant une insuffisance hépatique légère à modérée (n = 10, score de Child-Pugh de 5 à 12, prouvé par biopsie) que chez les sujets sains (n = 10).
Insuffisance rénale
Il n’a pas été mené d’étude avec les dispositifs transdermiques de rivastigmine chez des sujets présentant une insuffisance rénale. Sur la base d’une analyse de la population, la clairance de la créatinine n’a montré aucun effet évident sur les concentrations à l’état d’équilibre de la rivastigmine ou de ses métabolites.
Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuffisance rénale (voir section 4.2).
5.3. Données de sécurité préclinique
Les études de toxicité à doses orales et topiques répétées réalisées chez la souris, le rat, le chien et le mini-porc ont uniquement révélé des effets associés à une action pharmacologique exagérée. Il n'a pas été identifié d’organe cible pour la toxicité. Dans les études animales, l’administration orale et topique a été limitée en raison de la sensibilité des modèles animaux utilisés.
La rivastigmine n'est pas mutagène dans une batterie standard de tests in vitro et in vivo, excepté dans un test d’aberrations chromosomiques sur des lymphocytes périphériques humains à des doses représentant 104 fois l’exposition clinique attendue. Le résultat du test in vivo du micronoyau a été négatif. Le métabolite majeur NAP226-90 n’a pas non plus montré de potentiel génotoxique.
Aucun signe de carcinogénicité n’a été mis en évidence dans les études à doses orales et topiques chez la souris et dans une étude à doses orales chez le rat à la dose maximale tolérée. L’exposition à la rivastigmine et à ses métabolites a été à peu près équivalente à celle observée chez l’homme aux doses maximales de rivastigmine sous forme de gélules et de dispositifs transdermiques.
Chez l’animal, la rivastigmine traverse la barrière placentaire et est excrétée dans le lait. Les études menées par voie orale chez les rattes et les lapines gravides n’ont pas mis en évidence de potentiel tératogène de la rivastigmine. Dans les études par administration orale chez les rats mâles et femelles, aucun effet indésirable de la rivastigmine n’a été observé sur la fertilité ou la capacité de reproduction chez la génération parent ou chez la progéniture des parents. Il n’a pas été mené d’études dermatologiques spécifiques chez les animaux gravides.
Les dispositifs transdermiques de rivastigmine n’ont pas induit de phototoxicité et sont considérés comme non allergènes. Dans d’autres études de toxicité dermique, il a été observé un léger effet irritant sur la peau des animaux de laboratoire, y compris des témoins. Cela pourrait indiquer que les dispositifs transdermiques de rivastigmine peuvent induire un érythème léger chez les patients.
Un léger potentiel d’irritation de l’œil/de la muqueuse de la rivastigmine a été identifié dans une étude menée chez le lapin. Les patients et/ou les aidants doivent donc éviter de se toucher les yeux après avoir manipulé le dispositif transdermique (voir rubrique 4.4).
Film
Film de polyester.
Film de polyester recouvert de fluoropolymère.
Matrice délivrant la substance active
Adhésif acrylique, copolymères d’acrylates poly(butylméthacrylate-méthyl-méthacrylate).
Matrice adhésive
Matrice silicone adhésive.
Encre d’impression noire
Résines, cires, noir de carbone, additifs.
Afin d’éviter toute interférence avec les propriétés adhésives du dispositif transdermique, aucune crème, lotion ou poudre ne doit être appliquée sur la zone cutanée où le médicament est collé.
2 ans.
6.4. Précautions particulières de conservation
A conserver dans l’emballage d’origine à l’abri de la lumière.
Conserver le dispositif transdermique dans son sachet jusqu’à utilisation.
Ce médicament ne nécessite pas de conditions particulières de conservation concernant la température.
6.5. Nature et contenu de l'emballage extérieur
Conditionnement primaire
Le sachet de sécurité enfant est composé d’un matériau multilaminé papier/polyéthylène téréphtalate/aluminium/polyacrylonitrile ou papier/polyéthylène téréphtalate/aluminium/polyamide.
Chaque sachet contient un dispositif transdermique.
Emballage extérieur
Les sachets sont conditionnés dans une boîte.
Les dispositifs transdermiques sont disponibles en boîtes de 7, 30, 60 et 90 sachets.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les dispositifs transdermiques usagés doivent être pliés en deux, face adhésive à l’intérieur, replacés dans le sachet d’origine et éliminés en toute sécurité et hors de la portée et de la vue des enfants. Tous les dispositifs transdermiques usagés ou inutilisés doivent être éliminés conformément à la réglementation en vigueur ou rapportés à la pharmacie.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
BIOGARAN
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 280 020 7 5 : 7 sachets (papier/polyéthylène téréphtalate/aluminium/polyacrylonitrile) de 1 dispositif transdermique.
· 34009 280 021 3 6 : 30 sachets (papier/polyéthylène téréphtalate/aluminium/polyacrylonitrile) de 1 dispositif transdermique.
· 34009 587 179 7 0 : 60 sachets (papier/polyéthylène téréphtalate/aluminium/polyacrylonitrile) de 1 dispositif transdermique.
· 34009 587 180 5 2 : 90 sachets (papier/polyéthylène téréphtalate/aluminium/polyacrylonitrile) de 1 dispositif transdermique.
· 34009 303 437 3 9 : 7 sachets (papier polytéréphtalate (PET) aluminium polyamide de 1 dispositif transdermique.
· 34009 303 437 4 6 : 30 sachets (papier polytéréphtalate (PET) aluminium polyamide) de 1 dispositif transdermique.
· 34009 551 160 0 4 : 60 sachets (papier polytéréphtalate (PET) aluminium polyamide) de 1 dispositif transdermique.
· 34009 551 160 1 1 : 90 sachets (papier polytéréphtalate (PET) aluminium polyamide) de 1 dispositif transdermique.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I.
Surveillance particulière nécessaire pendant le traitement.
Prescription initiale annuelle réservée aux médecins spécialistes en neurologie, en psychiatrie, aux médecins spécialistes titulaires du diplôme d’études spécialisées complémentaires de gériatrie et aux médecins spécialistes ou qualifiés en médecine générale titulaires de la capacité de gérontologie.
ANSM - Mis à jour le : 03/07/2026
RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique
Rivastigmine
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d’utiliser RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique ?
3. Comment utiliser RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : psychoanaleptiques, anticholinestérasiques - code ATC : N06DA03.
La substance active de RIVASTIGMINE BIOGARAN est la rivastigmine.
La rivastigmine appartient à une classe de substances appelées inhibiteurs de la cholinestérase. Chez les patients atteints de la maladie d’Alzheimer, la disparition de certaines cellules nerveuses au niveau du cerveau entraîne des taux faibles du neurotransmetteur appelé acétylcholine (une substance qui permet aux cellules nerveuses de communiquer entre elles). La rivastigmine agit en bloquant les enzymes responsables de la destruction de l’acétylcholine : l’acétylcholinestérase et la butylcholinestérase. En bloquant ces enzymes, RIVASTIGMINE BIOGARAN permet d’augmenter les taux d’acétylcholine dans le cerveau, contribuant ainsi à diminuer les symptômes de la maladie d’Alzheimer.
RIVASTIGMINE BIOGARAN est utilisé dans le traitement des patients adultes atteints de formes légères à modérément sévères de la maladie d'Alzheimer, une maladie du cerveau qui affecte progressivement la mémoire, la capacité intellectuelle et le comportement.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique ?
N’utilisez jamais RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique
· Si vous êtes allergique à la rivastigmine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si vous avez eu une réaction allergique à un médicament du même type (les dérivés du carbamate) ;
· si vous avez une réaction cutanée qui se propage au-delà de la taille du dispositif transdermique, s’il y a une réaction locale plus intense (telle que des ampoules, une aggravation de l’inflammation de la peau, un gonflement) et si cela ne s’améliore pas dans les 48 heures suivant le retrait du dispositif transdermique.
Dans ces cas, parlez-en à votre médecin et n’utilisez pas les dispositifs transdermiques de RIVASTIGMINE BIOGARAN.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser RIVASTIGMINE BIOGARAN :
· si vous avez, ou avez eu, une maladie du cœur telle que des battements du cœur irréguliers ou lents, une prolongation de l’intervalle QTc, des antécédents familiaux de prolongation de l’intervalle QTc, des torsades de pointes, ou si vous avez des taux sanguins de potassium ou magnésium faibles ;
· si vous avez, ou avez eu, un ulcère de l’estomac actif ;
· si vous avez, ou avez eu, des difficultés à uriner ;
· si vous avez, ou avez eu, des crises convulsives ;
· si vous avez, ou avez eu, de l’asthme ou une maladie respiratoire sévère ;
· si vous souffrez de tremblements ;
· si vous avez un poids corporel bas ;
· si vous avez des manifestations gastro-intestinales telles que nausées (mal au cœur), vomissements et diarrhées. Vous pourriez vous déshydrater (perte importante de liquide) si les vomissements ou les diarrhées sont prolongés ;
· si vous souffrez d’une maladie du foie.
Dans ces cas, votre médecin vous surveillera plus étroitement pendant que vous utilisez ce médicament.
Si vous n’avez pas utilisé de dispositif transdermique pendant plus de trois jours, n’appliquez pas le prochain avant d’avoir consulté votre médecin.
Enfants et adolescents
Il n’y a pas d’utilisation justifiée de RIVASTIGMINE BIOGARAN dans la population pédiatrique dans le traitement de la maladie d’Alzheimer.
Autres médicaments et RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
RIVASTIGMINE BIOGARAN peut interférer avec des médicaments anticholinergiques dont certains sont des médicaments utilisés pour soulager des crampes d’estomac ou des spasmes (par exemple la dicyclomine), traiter une maladie de Parkinson (par exemple l’amantadine) ou prévenir le mal des transports (par exemple la diphénhydramine, la scopolamine ou la méclizine).
RIVASTIGMINE BIOGARAN ne doit pas être utilisé en même temps que le métoclopramide (un médicament utilisé pour soulager ou prévenir les nausées et vomissements). Prendre ces deux médicaments ensemble pourrait entraîner des problèmes tels que raideur des membres et tremblements des mains.
Si vous devez subir une intervention chirurgicale alors que vous utilisez RIVASTIGMINE BIOGARAN, vous devez en informer votre médecin car il peut augmenter les effets de certains relaxants musculaires durant l’anesthésie.
Prudence lorsque RIVASTIGMINE BIOGARAN est pris en association avec des bêtabloquants (médicaments tels que l’aténolol utilisés pour traiter l’hypertension, l’angine de poitrine, ainsi que d’autres maladies du cœur). Prendre ces deux médicaments ensemble pourrait entraîner des problèmes tels qu’un ralentissement du rythme cardiaque (bradycardie) pouvant amener à un évanouissement ou une perte de conscience.
Prudence lorsque RIVASTIGMINE BIOGARAN est pris en association avec d’autres médicaments pouvant avoir un effet sur votre rythme cardiaque ou sur le système électrique de votre cœur (prolongation de l’intervalle QT).
RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique avec des aliments, boissons et de l’alcool
RIVASTIGMINE BIOGARAN peut être utilisé avec la nourriture, les boissons et l’alcool.
Grossesse et allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Si vous êtes enceinte, les bénéfices de l’utilisation de RIVASTIGMINE BIOGARAN doivent être évalués par rapport aux risques possibles pour votre enfant à naître. RIVASTIGMINE BIOGARAN ne doit pas être utilisé au cours de la grossesse sauf en cas de nécessité clairement définie.
Vous ne devez pas allaiter durant un traitement par les dispositifs transdermiques de RIVASTIGMINE BIOGARAN.
Conduite de véhicules et utilisation de machines
Votre médecin vous dira si votre maladie vous permet de conduire des véhicules et d’utiliser des machines en toute sécurité. Les dispositifs transdermiques de RIVASTIGMINE BIOGARAN sont susceptibles de provoquer des étourdissements et une confusion sévère. Si vous remarquez de tels effets, vous ne devez pas conduire ni utiliser des machines ou effectuer d’autres tâches qui nécessitent de la vigilance.
RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique contient
Sans objet.
3. COMMENT UTILISER RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique ?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
IMPORTANT :
· Retirez le dispositif transdermique précédent avant d’appliquer UN nouveau dispositif.
· Un seul dispositif transdermique par jour.
· Ne découpez pas le dispositif transdermique en morceaux.
· Appuyez fermement sur le dispositif transdermique avec la paume de la main pendant au moins 30 secondes.
Posologie
Début du traitement
Votre médecin vous dira quel est le dosage de RIVASTIGMINE BIOGARAN qui vous convient.
· En général, le traitement débute avec RIVASTIGMINE BIOGARAN 4,6 mg/24 h.
· La dose quotidienne habituelle recommandée est RIVASTIGMINE BIOGARAN 9,5 mg/24 h. Si elle est bien tolérée, le médecin peut envisager d’augmenter la dose à 13,3 mg/24 h.
· Utilisez un seul dispositif transdermique à la fois et remplacez le dispositif transdermique par un nouveau après 24 heures.
Pendant le traitement, votre médecin pourra ajuster la dose pour qu’elle soit adaptée à vos besoins individuels.
Si vous n’avez pas utilisé de dispositif depuis plus de trois jours, n’appliquez pas le prochain dispositif transdermique avant d’avoir consulté votre médecin. Le traitement avec le dispositif transdermique peut être repris à la même dose s’il n’est pas arrêté plus de trois jours. Dans le cas contraire, votre médecin recommencera votre traitement avec RIVASTIGMINE BIOGARAN 4,6 mg/24 h.
RIVASTIGMINE BIOGARAN peut être utilisé avec la nourriture, les boissons et l’alcool.
Où appliquer votre dispositif transdermique RIVASTIGMINE BIOGARAN ?
· Avant d’appliquer le dispositif transdermique, assurez-vous que votre peau est propre et sèche, et sans poils, qu’aucune poudre, huile, crème hydratante ou lotion qui pourrait empêcher le dispositif transdermique d’adhérer correctement sur la peau n’a été appliquée, que votre peau ne présente pas de coupure, d’éruption et/ou d’irritation.
· Enlevez minutieusement le dispositif transdermique précédent avant d’en appliquer un nouveau. Le fait d’avoir plusieurs dispositifs transdermiques collés sur votre corps peut vous exposer à une quantité trop importante de ce médicament, ce qui peut potentiellement être dangereux.
· Appliquez UN SEUL dispositif transdermique par jour sur UNE SEULE des zones possibles montrées sur les diagrammes suivants :
o le haut du bras gauche ou le haut du bras droit ;
o le haut gauche de la poitrine ou le haut droit de la poitrine (évitez de l’appliquer sur les seins) ;
o le haut gauche du dos ou le haut droit du dos ;
o le bas gauche du dos ou le bas droit du dos.
Toutes les 24 heures retirez le dispositif transdermique précédent avant d’appliquer UN nouveau dispositif sur UNE SEULE des zones possibles suivantes
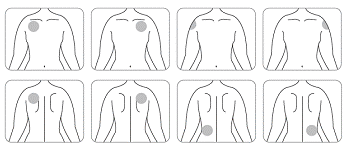
Lors du remplacement du dispositif transdermique, vous devez retirer le dispositif de la veille avant d’appliquer le nouveau dispositif à un endroit différent à chaque fois (par exemple sur le côté droit du corps un jour, puis sur le côté gauche le lendemain, et sur le haut du corps un jour, puis sur le bas du corps le lendemain). N’appliquez pas un nouveau dispositif transdermique 2 fois sur la même zone de la peau pendant 14 jours.
Comment appliquer votre dispositif transdermique RIVASTIGMINE BIOGARAN ?
Les dispositifs RIVASTIGMINE BIOGARAN sont des dispositifs en plastique minces, translucides qui se collent sur la peau. Chaque dispositif transdermique est présenté dans un sachet scellé qui le protège jusqu’à ce que vous soyez prêt à l’appliquer. Vous ne devez ouvrir le sachet ou retirer le dispositif transdermique que juste avant l’application.
Retirez avec précaution le dispositif transdermique existant avant d’appliquer un nouveau dispositif.
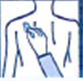
Pour les patients commençant le traitement pour la première fois et pour les patients recommençant RIVASTIGMINE BIOGARAN après un arrêt de traitement, allez directement à la deuxième image.
· Chaque dispositif transdermique est scellé dans son propre sachet protecteur. Le sachet ne doit être ouvert que juste avant d’appliquer le dispositif transdermique.
Découpez le sachet le long de la ligne pointillée avec des ciseaux et sortez le dispositif du sachet.
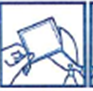
· Une pellicule protectrice recouvre la face adhésive du dispositif transdermique.
Décollez un côté de la pellicule protectrice ; ne touchez pas la partie adhésive du dispositif transdermique avec les doigts.
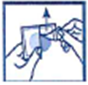
· Posez la face adhésive du dispositif transdermique sur le haut ou le bas du dos, le haut du bras ou la poitrine, puis décollez le second côté de la pellicule protectrice.
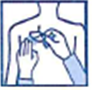
· Appuyez ensuite fermement sur le dispositif transdermique avec la paume de la main pendant au moins 30 secondes pour que les bords adhèrent bien.
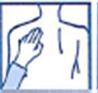
Si cela vous aide, vous pouvez par exemple écrire le jour de la semaine sur le dispositif transdermique avec un stylo à bille fin.
Le dispositif transdermique doit être porté en permanence jusqu’au moment de le remplacer par un dispositif neuf. Vous pouvez essayer différents endroits lorsque vous appliquez un nouveau dispositif transdermique, pour trouver ceux qui sont le plus confortables pour vous et où les vêtements ne frottent pas sur le dispositif.
Comment retirer votre dispositif transdermique de RIVASTIGMINE BIOGARAN ?
Tirez doucement sur un bord du dispositif transdermique pour le décoller lentement de la peau. Dans le cas où un résidu d’adhésif reste sur votre peau, mouillez doucement la zone avec de l’eau chaude et du savon doux ou utilisez une huile pour bébé pour l’enlever. L’alcool ou un autre dissolvant liquide (dissolvant à ongles ou autres solvants) ne doivent pas être utilisés.
Vous devez vous laver les mains avec de l’eau et du savon après avoir retiré le dispositif transdermique. En cas de contact avec les yeux ou si les yeux deviennent rouges après manipulation du dispositif transdermique, rincez immédiatement avec beaucoup d’eau et consultez votre médecin si les symptômes persistent.
Pouvez-vous prendre un bain, nager ou vous mettre au soleil lorsque vous portez votre dispositif transdermique de RIVASTIGMINE BIOGARAN ?
· Les bains, la natation ou les douches n’altèrent pas le dispositif transdermique. Vérifiez que le dispositif transdermique ne s’est pas décollé pendant ces activités.
· Le dispositif transdermique ne doit pas être exposé pendant une longue période à une source de chaleur externe (exemple : soleil excessif, sauna, solarium).
Que faut-il faire si votre dispositif transdermique se détache ?
Si le dispositif transdermique s’est détaché, appliquez-en un nouveau pendant le reste de la journée, puis remplacez-le au moment habituel le lendemain.
Durée du traitement
Quand faut-il appliquer votre dispositif transdermique de RIVASTIGMINE BIOGARAN et pendant combien de temps ?
· Pour que le traitement soit bénéfique, vous devez appliquer un nouveau dispositif transdermique chaque jour, de préférence au même moment de la journée.
· Utilisez un seul dispositif transdermique RIVASTIGMINE BIOGARAN à la fois et remplacez le dispositif transdermique par un nouveau après 24 heures.
Si vous avez utilisé plus de RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique que vous n’auriez dû
Si vous appliquez par erreur plus d’un seul dispositif transdermique, retirez tous les dispositifs collés sur votre peau et prévenez votre médecin que vous avez appliqué accidentellement plus d’un seul dispositif transdermique. Vous pouvez nécessiter une surveillance médicale. Certaines personnes qui ont pris accidentellement trop de rivastigmine ont eu mal au cœur (nausées), des vomissements, des diarrhées, une augmentation de la tension artérielle et des hallucinations. Un ralentissement du rythme cardiaque et un évanouissement peuvent également se produire.
Si vous oubliez d’utiliser RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique
Si vous vous rendez compte que vous avez oublié d’appliquer un dispositif transdermique, appliquez‑en un immédiatement. Vous pourrez appliquer le prochain dispositif au moment habituel le lendemain. N’appliquez pas deux dispositifs transdermiques pour compenser le dispositif transdermique que vous avez oublié d’utiliser.
Si vous arrêtez d’utiliser RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique
Informez votre médecin ou votre pharmacien si vous arrêtez d’utiliser le dispositif transdermique.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Il se peut que vous ayez des effets indésirables plus souvent au début du traitement ou quand la dose est augmentée. Le plus souvent, ces effets indésirables vont disparaître progressivement au fur et à mesure que votre organisme s’habituera au médicament.
Retirez immédiatement le dispositif transdermique et prévenez immédiatement votre médecin, si vous constatez l’un des effets indésirables suivants pouvant devenir grave :
Fréquent (peut affecter jusqu’à 1 patient sur 10) :
· perte d’appétit ;
· sensation de vertige ;
· sensation d’agitation ;
· incontinence urinaire (incapacité à retenir l’urine) ;
· infection urinaire ;
· anxiété ;
· dépression ;
· confusion ;
· maux de tête ;
· évanouissement ;
· troubles de l’estomac comme mal au cœur (nausées) ou vomissements, diarrhées ;
· brûlures d’estomac ;
· douleurs d’estomac ;
· éruptions cutanées ;
· réaction allergique au niveau du site d’application, par exemple vésicules ou inflammation cutanée ;
· sensation de fatigue ou d’affaiblissement ;
· perte de poids ;
· fièvre.
Peu fréquent (peut affecter jusqu’à 1 patient sur 100) :
· troubles du rythme cardiaque comme un ralentissement des battements du cœur ;
· ulcère d’estomac ;
· déshydratation (perte importante de liquide) ;
· hyperactivité (haut niveau d’activité, impatience) ;
· agressivité.
Rare (peut affecter jusqu’à 1 patient sur 1000) :
· chute.
Très rare (peut affecter jusqu’à 1 patient sur 10 000) :
· rigidité des bras ou des jambes ;
· tremblements des mains.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
· signes d’une aggravation d’une maladie de Parkinson - tels que tremblements, raideur, troubles de la marche ;
· inflammation du pancréas - les signes incluent une douleur importante du haut de l’estomac, souvent accompagnée d’un mal au cœur (nausées) ou de vomissements ;
· accélération ou irrégularité des battements cardiaques ;
· augmentation de la pression artérielle ;
· crises convulsives ;
· troubles du foie (jaunissement de la peau, jaunissement du blanc des yeux, coloration anormalement foncée des urines ou nausées inexpliquées, vomissements, fatigue et perte d’appétit) ;
· modifications des tests de votre fonction hépatique ;
· sensation d’impatience ;
· cauchemars ;
· Syndrome de Pise ou Syndrome de la tour de Pise (un trouble caractérisé par une contraction musculaire involontaire accompagnée d’une inclinaison anormale du corps et de la tête d’un côté) ;
· vision de choses qui n’existent pas (hallucinations) ;
· tremblements ;
· somnolence ;
· éruptions cutanées, démangeaisons ;
· rougeur de la peau ;
· ampoules.
Retirez immédiatement le dispositif transdermique et prévenez immédiatement votre médecin, si vous constatez l’un de ces effets indésirables.
Des effets indésirables supplémentaires ont été rapportés avec les gélules ou la solution buvable de rivastigmine et peuvent se produire avec le dispositif transdermique :
Fréquent (peut affecter jusqu’à 1 patient sur 10) :
· hypersécrétion de salive ;
· sensation d’impatience ;
· sensation de malaise général/se sentir malade ;
· tremblements ;
· augmentation de la sudation.
Peu fréquent (peut affecter jusqu’à 1 patient sur 100) :
· battements du cœur irréguliers (par exemple battements cardiaques rapides) ;
· difficulté à s’endormir ;
· chutes accidentelles.
Rare (peut affecter jusqu’à 1 patient sur 1000) :
· crises convulsives ;
· ulcère de l’intestin ;
· douleurs dans la poitrine - probablement causées par un spasme coronaire.
Très rare (peut affecter jusqu’à 1 patient sur 10 000) :
· augmentation de la pression artérielle ;
· inflammation du pancréas - les signes incluent une douleur importante du haut de l’estomac, souvent accompagnée d’un mal au cœur (nausées) ou de vomissements ;
· saignements gastro-intestinaux - présence de sang dans les selles ou lors de vomissements ;
· vision de choses qui n’existent pas (hallucinations) ;
· certains patients ont eu des vomissements violents qui ont pu conduire à une rupture du tube reliant la bouche à l’estomac (œsophage).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique ?
Tenir ce médicament hors de la vue et de la portée des enfants.
A conserver dans l’emballage d’origine à l’abri de la lumière.
Conserver le dispositif transdermique dans son sachet jusqu’à utilisation.
Ce médicament ne nécessite pas de conditions particulières de conservation concernant la température.
Ne pas utiliser un dispositif transdermique endommagé ou dont le sachet était ouvert.
Après avoir retiré le dispositif transdermique, pliez-le en deux, faces adhésives à l’intérieur, et appuyez pour les faire adhérer l’une à l’autre. Remettez le dispositif transdermique usagé dans son sachet et éliminez-le en veillant à ce que les enfants ne puissent pas le manipuler. Ne touchez pas vos yeux avec vos doigts et lavez-vous les mains avec de l’eau et du savon après avoir retiré le dispositif transdermique.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient RIVASTIGMINE BIOGARAN 4,6 mg/24 h, dispositif transdermique
· La substance active est :
Rivastigmine...................................................................................................................... 9 mg
Pour un dispositif transdermique de 5 cm2.
Chaque dispositif transdermique libère 4,6 mg de rivastigmine par 24 heures.
· Les autres composants sont :
Film
Film de polyester.
Film de polyester recouvert de fluoropolymère.
Matrice délivrant la substance active
Adhésif acrylique, copolymères d’acrylates poly(butylméthacrylate-méthyl-méthacrylate).
Matrice adhésive
Matrice silicone adhésive.
Encre d’impression noire
Résines, cires, noir de carbone, additifs.
Chaque dispositif transdermique se présente sous forme d’un dispositif mince composé de trois couches.
La couche extérieure est translucide, de couleur blanche et porte les mentions suivantes imprimées en noir : « Rivastigmine 4,6 mg/24h ».
Chaque dispositif transdermique est contenu dans un sachet de sécurité enfant scellé. Les dispositifs transdermiques sont présentés en boîtes contenant 7, 30, 60 ou 90 sachets.
Titulaire de l’autorisation de mise sur le marché
BIOGARAN
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
Exploitant de l’autorisation de mise sur le marché
BIOGARAN
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
EUROFINS PHAST GMBH
KARDINAL-WENDEL-STRASSE 16
66424 HOMBURG
ALLEMAGNE
OU
ACC GMBH ANALYTICAL CLINICAL CONCEPTS
SCHÖNTALWEG 7 + 9
63849 LEIDERSBACH BAVIERE
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).