Dernière mise à jour le 01/06/2026
SPOTOF 1 g/10 ml, solution buvable
Indications thérapeutiques
Classe pharmacothérapeutique : ANTIFIBRINOLYTIQUE, code ATC : B02AA02.
Qu’est-ce que SPOTOF 1 g/10 ml, solution buvable ?
SPOTOF contient de l’acide tranexamique. Cette substance appartient à une famille de médicaments appelés les antifibrinolytiques.
Le rôle de ce médicament est de traiter certains types de saignements.
Dans quel cas est-il utilisé ?
Ce médicament est utilisé pour traiter :
· Les saignements provoqués par la dégradation trop rapide des caillots sanguins, due à une libération importante d’enzymes appelées activateurs du plasminogène,
· Les saignements au cours d’un traitement par un médicament qui dégrade les caillots sanguins (fibrinolytique),
· Les saignements entretenus par la dégradation locale de caillots sanguins, comme par exemple :
o Les règles abondantes,
o Les saignements en dehors des règles,
o Les saignements digestifs,
o Certains types de saignements, provoquant la présence de sang dans les urines, dus à des maladies de la prostate, de la vessie, à des calculs, à des opérations chirurgicales au niveau de la prostate et de l’appareil urinaire,
o Les saignements pouvant survenir pendant certaines opérations chirurgicales de la gorge et du nez (opération des végétations ou des amygdales).
Présentations
> 5 ampoule(s) en verre brun de 10 ml
Code CIP : 338 996-1 ou 34009 338 996 1 5
Déclaration de commercialisation : 15/05/1997
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 2,13 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 3,15 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 03/05/2022
SPOTOF 1 g/10 ml, solution buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour une ampoule de 10 ml.
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
· Accidents hémorragiques dus à un état fibrinolytique primitif généralisé.
· Accidents hémorragiques au cours d'un traitement à effet fibrinolytique.
· Accidents hémorragiques entretenus par une fibrinolyse locale, comme c'est le cas dans :
o ménorragies et métrorragies :
§ par dysfonctionnement hormonal,
§ secondaires à des lésions traumatiques ou infectieuses, ou dégénératives de l'utérus.
o hémorragies digestives,
o hématuries d'origine basse :
§ des adénomes prostatiques,
§ des néoplasies malignes prostatiques et vésicales,
§ des lithiases,
§ et plus généralement des affections urinaires hémorragiques, au décours des interventions chirurgicales prostatiques et des actes chirurgicaux intéressant le tractus urinaire.
o hémorragies opératoires oto-rhino-laryngologiques (adénoïdectomies et amygdalectomies).
4.2. Posologie et mode d'administration
Chez l'adulte, la posologie se situe, suivant le cas à traiter, de 2 à 4 g par 24 heures à répartir en 2 ou 3 prises (soit 2 à 4 ampoules par jour).
Population pédiatrique
Chez l'enfant, pour les indications actuellement approuvées et décrites à la rubrique 4.1, la posologie est de l'ordre de 20 mg/kg/jour. Cependant, les données d’efficacité, de posologie et de sécurité d’emploi pour ces indications sont limitées.
Mode d’administration
Voie orale.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Thrombose veineuse ou artérielle aiguë (voir rubrique 4.4).
· Etats fibrinolytiques réactionnels à une coagulopathie de consommation, à l'exception d'états associés à une activation prédominante du système fibrinolytique avec une hémorragie grave aigüe (voir rubrique 4.4).
· Insuffisance rénale grave (risque d'accumulation).
· Antécédent de convulsions.
4.4. Mises en garde spéciales et précautions d'emploi
Evénements thrombo-emboliques
Les facteurs de risque de maladie thrombo-embolique doivent être pris en compte avant l’utilisation d’acide tranexamique. Chez les patients présentant des antécédents de maladies thrombo-emboliques ou chez les patients ayant une incidence accrue d’événements thrombo-emboliques dans leurs antécédents familiaux (patients à haut risque de thrombophilie), l’acide tranexamique ne doit être administré qu’en présence d’une forte indication médicale, après consultation d’un spécialiste de l’hémostase et sous une stricte surveillance médicale (voir rubrique 4.3).
Oestro-progestatifs
L’acide tranexamique doit être administré avec prudence chez les patientes sous oestro-progestatifs (contraception orale et hormonothérapie substitutive) du fait du risque accru de thrombose (voir rubrique 4.5).
Le traitement doit être arrêté immédiatement en cas de survenue de troubles thromboemboliques artériels ou veineux.
Troubles visuels
Il faut prêter attention aux troubles visuels possibles, notamment une déficience visuelle, une vision trouble, une perturbation de la vision des couleurs. Si nécessaire, le traitement doit être interrompu. Des examens ophtalmologiques réguliers (examens de l’œil et notamment acuité visuelle, vision des couleurs, fond de l’oeil, champ visuel, etc.) sont indiqués lors d’une utilisation continue et prolongée d’acide tranexamique. Si des changements ophtalmiques pathologiques sont observés, particulièrement en présence de maladies de la rétine, le médecin doit consulter un spécialiste afin de décider de la nécessité d’utiliser à long terme l’acide tranexamique dans chaque cas individuel.
Précautions d'emploi
Convulsions
Des cas de convulsions ont été rapportés en association avec un traitement par l’acide tranexamique. Lors d’une chirurgie de pontage aortocoronarien (PAC), la plupart de ces cas ont été signalés après une injection intraveineuse (IV) d’acide tranexamique à des doses élevées. Lorsque les doses plus faibles recommandées d’acide tranexamique étaient utilisées, l’incidence des crises post-opératoires était similaire à celle constatée chez des patients non traités.
Hématurie
En cas d’hématurie provenant des voies urinaires supérieures, il y a un risque d’obstruction urétrale.
Insuffisance rénale
En cas d'insuffisance rénale entraînant un risque d'accumulation, la posologie d'acide tranexamique sera réduite en fonction de la créatininémie.
Lorsque la créatinine sérique est comprise :
· entre 120 et 250 micromol/l, la posologie sera de 10 mg/kg, deux fois par jour,
· entre 250 et 500 micromol/l, la posologie sera de 10 mg/kg, une fois par 24 heures,
· de 500 micromol/l et plus, la posologie sera de 5 mg/kg, toutes les 24 heures.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée. Un traitement simultané par des anticoagulants doit être administré sous la stricte surveillance d’un médecin spécialisé dans ce domaine.
Les médicaments qui agissent sur l’hémostase doivent être administrés avec précaution aux patients traités par l’acide tranexamique.
Par ailleurs, l’effet antifibrinolytique du médicament peut être antagonisé avec des thrombolytiques.
Il existe un risque théorique d’augmentation de la formation de thrombus, tel qu’avec les œstrogènes.
4.6. Fertilité, grossesse et allaitement
Grossesse
Par mesure de précaution, il est préférable de ne pas utiliser l'acide tranéxamique au cours du premier trimestre de la grossesse. En effet, les données cliniques sont insuffisantes bien que les données animales n'aient pas mis en évidence d'effet tératogène.
Des données cliniques limitées lors de l'utilisation d'acide tranéxamique dans certaines complications hémorragiques des second et troisième trimestres de la grossesse n'ont pas mis en évidence d'effet foetotoxique majeur.
L'acide tranéxamique est excrété dans le lait maternel. En conséquence, l'allaitement est déconseillé pendant la durée du traitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Tableau reprenant la liste des effets indésirables
Les effets indésirables signalés sont présentés dans le tableau ci-dessous. Les effets indésirables sont repris selon les classes principales de systèmes d’organes MedDRA. Au sein de chaque classe de systèmes d’organes, les effets indésirables sont présentés par fréquence. Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité. Les fréquences ont été définies comme suit : très fréquent (≥1/10) ; fréquent (≥1/100 à <1/10) ; peu fréquent (≥1/1000 à <1/100) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classe de systèmes d’organes MedDRA |
Fréquence |
Effets indésirables |
|
Affections gastro-intestinales |
Fréquent |
Troubles digestifs tels que : |
|
|
|
Diarrhée |
|
|
|
Vomissements |
|
|
|
Nausées |
|
Affections du système nerveux |
Fréquence indéterminée |
Convulsions, particulièrement en cas de facteurs de risque ou d’antécédents de convulsion ainsi qu’en cas de mésusage (voir les rubriques 4.3 et 4.4) |
|
Affections oculaires |
Fréquence indéterminée |
Troubles visuels, dont des troubles de la perception des couleurs |
|
Affections vasculaires |
Fréquence indéterminée |
Malaise avec hypotension, avec ou sans perte de conscience (généralement après une injection intraveineuse trop rapide, exceptionnellement après une administration par voie orale) |
|
|
|
Thrombose artérielle ou veineuse susceptible de survenir dans n’importe quel site. |
|
Troubles généraux |
Peu fréquent |
Dermite allergique |
|
|
Fréquence indéterminée |
Réactions d’hypersensibilité incluant des réactions anaphylactiques (urticaire, angio- œdème, choc anaphylactique) et des éruptions cutanées diverses. |
|
Affections de la peau et du tissu sous-cutané |
Fréquence indéterminée |
Erythème pigmenté fixe |
|
Affection du rein |
Fréquence indéterminée |
Insuffisance rénale aiguë une à une nécrose corticale rénale |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
La prise en charge d’un surdosage consiste en un traitement symptomatique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : ANTIFIBRINOLYTIQUE, code ATC : B02AA02.
L'acide tranéxamique développe une action anti-hémorragique par inhibition des activités fibrinolytiques de la plasmine.
Il se forme ainsi un complexe entre l'acide tranéxamique et le plasminogène, l'acide tranéxamique restant lié au plasminogène lors de sa transformation en plasmine.
La plasmine, liée à l'acide tranéxamique, aurait, vis-à-vis de la fibrine, une activité considérablement diminuée par rapport à celle de la plasmine libre.
Enfin, il ressort de diverses études que, in vivo, l'acide tranéxamique à fortes doses exerce une activité freinatrice sur l'activation du système complément.
5.2. Propriétés pharmacocinétiques
Par voie orale (20 mg/kg), absorption rapide avec concentration sanguine maximale entre la 2ème et la 3ème heure, le produit n'étant plus retrouvé à la 6ème heure.
Distribution
· dans le compartiment cellulaire,
· dans le liquide céphalorachidien de façon retardée.
Le volume de distribution est de 33 p. 100 de la masse corporelle.
Elimination
La demi-vie d'élimination est d'environ 1 heure.
Quatre-vingt dix pour cent de la dose administrée sont éliminés sous forme inchangée par voie urinaire dans les 12 premières heures (excrétion glomérulaire sans réabsorption tubulaire).
Insuffisance rénale
Les concentrations plasmatiques sont augmentées en cas d’insuffisance rénale.
Grossesse
L'acide tranéxamique traverse le placenta.
5.3. Données de sécurité préclinique
Une action épileptogène a été observée chez l'animal lors d'utilisations intracérébrales.
*Composition de l’arôme naturel cerise : alcoolat obtenu par distillation d’une macération de cerises.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
10 ampoules (verre jaune) de 10 ml.
6.6. Précautions particulières d’élimination et de manipulation
Pas d'exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
36 RUE BRUNEL
75017 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 338 996-15 : 10 ml en ampoule (verre brun). Boîte de 5.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 03/05/2022
SPOTOF 1 g/10 ml, solution buvable
acide tranexamique
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que SPOTOF 1 g/10 ml, solution buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre SPOTOF 1 g/10 ml, solution buvable ?
3. Comment prendre SPOTOF 1 g/10 ml, solution buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver SPOTOF 1 g/10 ml, solution buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE SPOTOF 1 g/10 ml, solution buvable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : ANTIFIBRINOLYTIQUE, code ATC : B02AA02.
Qu’est-ce que SPOTOF 1 g/10 ml, solution buvable ?
SPOTOF contient de l’acide tranexamique. Cette substance appartient à une famille de médicaments appelés les antifibrinolytiques.
Le rôle de ce médicament est de traiter certains types de saignements.
Dans quel cas est-il utilisé ?
Ce médicament est utilisé pour traiter :
· Les saignements provoqués par la dégradation trop rapide des caillots sanguins, due à une libération importante d’enzymes appelées activateurs du plasminogène,
· Les saignements au cours d’un traitement par un médicament qui dégrade les caillots sanguins (fibrinolytique),
· Les saignements entretenus par la dégradation locale de caillots sanguins, comme par exemple :
o Les règles abondantes,
o Les saignements en dehors des règles,
o Les saignements digestifs,
o Certains types de saignements, provoquant la présence de sang dans les urines, dus à des maladies de la prostate, de la vessie, à des calculs, à des opérations chirurgicales au niveau de la prostate et de l’appareil urinaire,
o Les saignements pouvant survenir pendant certaines opérations chirurgicales de la gorge et du nez (opération des végétations ou des amygdales).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE SPOTOF 1 g/10 ml, solution buvable ?
Ne prenez jamais SPOTOF 1 g/10 ml, solution buvable:
· si vous êtes allergique à l’acide tranéxamique ou à l’un des autres composants contenus dans ce médicament mentionnés à la rubrique 6.
· si vous souffrez actuellement d’une thrombose artérielle ou veineuse (caillots dans les artères tels que infarctus du myocarde ou accident vasculaire cérébral) ou dans les veines tels que thrombose veineuse profonde ou embolie pulmonaire.
· Si vous souffrez d’une affection, dénommée « coagulopathie de consommation » dans laquelle des caillots sanguins se forment dans l’ensemble des vaisseaux de l’organisme.
· si vous avez des problèmes rénaux,
· si vous avez des antécédents de convulsions,
Si vous pensez être dans l’un de ces cas, ou en cas de doute informez votre medecin avant de prendre SPOTOF 1g/10 ml, solution buvable..
Avertissements et précautions
Prévenez immédiatement votre médecin en cas d’apparition des signes suivants : douleur inhabituelle dans les jambes, faiblesses dans les membres (bras et jambes), douleur de la poitrine, pouls irrégulier ou essoufflement soudain, perte de connaissance, confusion, maux de tête importants et inhabituels, vertiges, troubles de la vue, difficultés à parler (élocution ralentie) ou perte de la parole.
Si vous vous trouvez dans l’un des cas suivants, informez votre médecin afin qu’il puisse décider si SPOTOF 1g/10 ml, solution buvable vous convient :
· si vous avez présenté du sang dans les urines car SPOTOF 1 g/10 ml, solution buvable peut entraîner une obstruction des voies urinaires,
· si vous présentez un risque de survenue de caillot sanguin,
· vous prenez un contraceptif oral ou un traitement substitutif de la ménopause,
· si vous présentez (ou avez déjà présenté) des convulsions ou si vous prenez un traitement contre l’épilepsie et/ou les convulsions (antiépileptique), SPOTOF 1 g/10 ml, solution buvable ne doit pas vous être prescrit,
· Si vous êtes sous traitement de longue durée par SPOTOF 1 g/10 ml, solution buvable, il faut prêter attention à de possibles perturbations de la vision des couleurs et, si nécessaire, le traitement doit être interrompu. Des examens ophtalmologiques réguliers (examens de l’œil et notamment acuité visuelle, vision des couleurs, fond de l’œil, champ visuel, (etc.) sont indiqués lors de l’utilisation continue et prolongée de SPOTOF 1 g/10 ml, solution buvable. Si des changements ophtalmiques pathologiques sont observés, particulièrement en présence de maladies de la rétine, votre médecin doit consulter un spécialiste afin de décider de la nécessité d’utiliser à long terme SPOTOF 1 g/10 ml, solution buvable dans votre cas personnel.
· si vous avez une maladie des reins (insuffisance rénale chronique). Dans ce cas, votre médecin adaptera la dose à votre état. ;
Adressez-vous à votre médecin ou pharmacien avant de prendre SPOTOF 1 g/10 ml, solution buvable.
Enfants
Sans objet.
Autres médicaments et SPOTOF 1 g/10 ml, solution buvable
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament. Il peut s’agir de médicaments obtenus sans ordonnance, de vitamines, de minéraux, de produits phytothérapeutiques ou de compléments alimentaires.
Vous devez tout spécialement informer votre médecin si vous prenez :
· d’autres médicaments susceptibles de favoriser la coalgulation du sang, dénommés antifibronolytiques,
· des médicament qui préviennent la formation de caillots sanguins, dénommés thrombolytiques,
· des contraceptifs oraux.
SPOTOF 1 g/10 ml, solution buvable avec les aliments, les boissons et l’alcool
Sans objet.
Grossesse
Il est préférable de ne pas utiliser ce médicament pendant les 3 premiers mois de grossesse. Si vous découvrez que vous êtes enceinte pendant le traitement, consultez rapidement votre médecin, lui seul pourra adapter le traitement à votre état.
Allaitement
Il est déconseillé d’allaiter pendant la durée du traitement.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Faites attention, ce médicament provoque parfois des vertiges. Si vous ressentez cet effet, ne conduisez pas et n’utilisez pas de machine.
SPOTOF 1 g/10 ml, solution buvable contient :
Sans objet.
3. COMMENT PRENDRE SPOTOF 1 g/10 ml, solution buvable ?
Posologie
Votre médecin déterminera la dose que vous devrez prendre.
Respectez toujours la dose prescrite par votre médecin. En cas de doute, consultez votre médecin ou votre pharmacien.
Chez l’adulte, vous ne pas devez pas dépasser 4 g par 24 heures, correspondant à 4 ampoules de solution buvable par 24 heures.
Mode d’administration
Ce médicament est à prendre par voie orale.
|
|
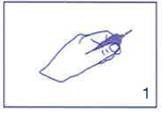
Tenir l’ampoule, dans la main, entre le pouce et l’index, comme sur le schéma. |
|
|
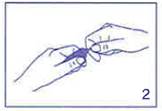
Saisir la pointe entre le pouce et l’index de l’autre main. |
|
|
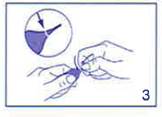
Appliquer une légère pression avec le pouce de cette main au niveau de l’anneau de précassure, en résistant avec l’index de l’autre main. Ne pas exercer de torsion |
|
|
Renouveler l’ensemble de l’opération pour l’ouverture de l’autre pointe, en prenant soin de bien positionner la partie ouverte, au-dessus du verre |

Fréquence d’administration
Vous devez prendre ce médicament en 2 à 3 prises par 24 heures.
Durée du traitement
Votre médecin vous précisera pendant combien de temps vous devez prendre ce médicament.
Si vous avez pris plus de SPOTOF 1 g/10 ml, solution buvable que vous n’auriez dû :
Si vous avez reçu plus de SPOTOF 1 g/10 ml, solution buvable que la dose recommandée, il se peut que vous soyez sujet à une diminution transitoire de la tension artérielle, des vertiges, des maux de têtes, des convulsions.
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de prendre SPOTOF 1 g/10 ml, solution buvable :
Si vous arrêtez de prendre SPOTOF 1 g/10 ml, solution buvable :
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables suivants ont été observés avec SPOTOF 1 g/10 ml, solution buvable :
Fréquent (peuvent affecter jusqu’à 1 personne sur 10)
· Effets sur l’estomac et les intestins : nausées, vomissements, diarrhées
Peu fréquent (peuvent affecter entre 1 et 10 personnes sur 1000)
· Effets sur la peau : éruption cutanée
Fréquence indéterminée (ne peut être estimée au vu des données disponibles)
· Malaise avec hypotension (hypotension artérielle), parfois accompagné de perte de connaissance
· Caillots sanguins, susceptibles de survenir dans n’importe quelle partie du corps. Vous pourrez reconnaître cet effet notamment par :
o une douleur inhabituelle dans les jambes, une faiblesse dans les membres (bras, jambes),
o une douleur de la poitrine, un pouls irrégulier, un essoufflement soudain,
o une perte de connaissance, une confusion, des maux de tête importants et inhabituels, des vertiges, des troubles de la vue, une difficulté à parler (élocution ralentie) ou une perte de la parole.
· Effets sur le système nerveux : convulsions
· Effets sur les yeux : perturbations de la vision, notamment un trouble de la vision des couleurs.
· Effets sur le système immunitaire : réactions allergiques. Vous pourrez les reconnaître notamment par des boutons et/ou des rougeurs sur la peau, une urticaire, un brusque gonflement du visage et du cou pouvant entraîner une difficulté à respirer (œdème de Quincke), un malaise brutal avec baisse importante de la pression artérielle (choc anaphylactique).
· Effets sur la peau : éruption de plaques brunes ou violacées pouvant laisser une coloration sur la peau (érythème pigmenté fixe).
· Problèmes rénaux d’apparition soudaine.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmière. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER SPOTOF 1 g/10 ml, solution buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage. La date de préemption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient SPOTOF 1 g/10 ml, solution buvable
· La substance active est :
Acide tranéxamique....................................................................................................... 500 mg
Pour une ampoule de 10 ml
· Les autres composants excipients sont :
Arôme naturel cerise, acide chlorhydrique concentré ou solution aqueuse concentrée à 30 pour cent d’hydroxyde de sodium, eau purifiée.
Qu’est-ce que SPOTOF 1 g/10 ml, solution buvable et contenu de l’emballage extérieur
Titulaire de l’autorisation de mise sur le marché
36 RUE BRUNEL
75017 PARIS
Exploitant de l’autorisation de mise sur le marché
36 RUE BRUNEL
75017 PARIS
1-3 ALLEE DE LA NESTE
31770 COLOMIERS
ou
COOPERATION PHARMACEUTIQUE FRANCAISE
Place Lucien Auvert
77020 MELUN
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).