Dernière mise à jour le 01/06/2026
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, contient la substance active clofarabine. La clofarabine fait partie d’une famille de médicaments appelés « médicaments anticancéreux ». La clofarabine permet de stopper la multiplication de globules blancs anormaux et finit par les détruire. Son effet est optimal sur les cellules qui se multiplient très rapidement, comme les cellules cancéreuses.
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, est utilisé pour le traitement de la leucémie aiguë lymphoblastique (LAL) chez les enfants (≥ 1 an), les adolescents et les jeunes adultes jusqu'à 21 ans dont les traitements précédents n’ont pas donné de résultats ou lorsque ces traitements ne sont plus efficaces. La leucémie aiguë lymphoblastique est due à une multiplication anormale de certains types de globules blancs.
Présentations
> 1 flacon en verre de 20 mL
Code CIP : 34009 550 390 7 5
Déclaration d'arrêt de commercialisation : 21/05/2025
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 21/02/2018 | Inscription (CT) | Le service médical rendu par CLOFARABINE MYLAN 1 mg/ml est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 21/02/2018 | Inscription (CT) | Cette spécialité n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à la spécialité princeps EVOLTRA 1 mg/ml, déjà inscrite. |
Autres informations
- Titulaire de l'autorisation : VIATRIS SANTE
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- prescription réservée aux spécialistes et services PEDIATRIE
- réservé à l'usage HOSPITALIER
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 122 995 3
ANSM - Mis à jour le : 04/03/2024
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Clofarabine.............................................................................................................................. 1 mg
Pour 1 ml de solution.
Chaque flacon de 20 ml contient 20 mg de clofarabine.
Excipient à effet notoire : chaque flacon de 20 ml contient 180 mg de chlorure de sodium, ce qui équivaut à 3,08 mmoles (ou 70,77mg) de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution à diluer pour perfusion.
Solution limpide, presque incolore : pH compris entre 4,5 et 7,5 et osmolarité comprise entre 270 et 310 mOsm/l.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Le traitement devra être mis en place et supervisé par un médecin expérimenté dans la prise en charge des patients atteints de leucémies aiguës.
Population adulte (y compris sujet âgé) :
Les données actuelles, ne permettent pas d’établir un profil concernant la sécurité et l’efficacité de la clofarabine chez le patient adulte (voir rubrique 5.2).
Population pédiatrique :
Enfants et adolescents (≥ 1 an)
La dose recommandée en monothérapie est de 52 mg/m² de surface corporelle administrée par perfusion intraveineuse de 2 heures par jour pendant 5 jours consécutifs. La surface corporelle sera calculée à partir de la taille et du poids réels du patient mesurés avant le début de chaque cycle. Les cycles thérapeutiques seront répétés toutes les 2 à 6 semaines (à partir du premier jour du cycle précédent) après reconstitution d’une hématopoïèse normale (à savoir, un nombre absolu de neutrophiles ≥ 0,75 × 109/l) et un retour aux valeurs initiales des fonctions organiques. Une réduction de dose de 25 % peut être légitime chez les patients souffrant de toxicités importantes (voir ci- dessous). L’expérience chez des patients recevant plus de 3 cycles thérapeutiques est actuellement limitée (voir rubrique 4.4).
La majorité des patients répondant à la clofarabine obtiennent une réponse après 1 ou 2 cycles de traitement (voir rubrique 5.1). Par conséquent, le rapport bénéfice/risque potentiel associé à la poursuite du traitement chez des patients ne montrant aucune amélioration hématologique et/ou clinique après 2 cycles de traitement devra être évalué par le médecin traitant (voir rubrique 4.4).
Chez l’enfant dont le poids < 20 kg) :
Un temps de perfusion > 2 heures devra être envisagé afin d’aider à une réduction des symptômes d’anxiété et d’irritabilité et d’éviter toute concentration maximale de clofarabine excessive chez ce type de patient (voir rubrique 5.2).
Chez l’enfant âgé de < 1 an) :
Aucune donnée concernant la pharmacocinétique, la sécurité ou l’efficacité de la clofarabine n’est disponible chez le nourrisson. Par conséquent, il reste encore à établir une recommandation de posologie efficace et sûre pour ce type de patients.
Réduction de dose chez les patients souffrant de toxicités hématologiques :
Si le nombre absolu de neutrophiles n’est pas récupéré dans les 6 semaines suivant le début du cycle de traitement, une ponction/biopsie médullaire devra être effectuée afin de déterminer si la maladie est potentiellement réfractaire. En cas de leucémie persistante non évidente, il est recommandé que la dose du cycle suivant soit réduite de 25 % par rapport à la dose précédente une fois un nombre absolu de neutrophiles ≥ 0,75 × 109/l récupéré. En cas de nombre absolu de neutrophiles < 0,5 × 109/l pendant une période supérieure à 4 semaines depuis le début du dernier cycle, il est recommandé de réduire de 25 % la dose du cycle suivant.
Réduction de dose chez les patients souffrant de toxicités non-hématologiques :
Événements infectieux
En cas d’infection cliniquement significative développée par le patient, le traitement par clofarabine pourra être interrompu jusqu’à ce que l’infection soit cliniquement contrôlée. Une fois l’infection contrôlée, le traitement pourra être repris avec administration de la dose totale. En cas d’apparition d’une seconde infection cliniquement significative, le traitement par clofarabine sera interrompu jusqu’à ce que l’infection soit cliniquement contrôlée puis potentiellement repris avec une réduction de dose de 25 %.
Événements non-infectieux
Si un patient souffre d’une ou plusieurs toxicités sévères (Critères de Toxicité CTC-NCI – Toxicités de Grade 3 excluant les nausées et les vomissements), le traitement sera différé jusqu’à disparition des toxicités et retour aux paramètres initiaux ou jusqu’à l’obtention d’un grade non sévère avec bénéfice potentiel de la poursuite du traitement par clofarabine supérieur au risque d’une telle poursuite thérapeutique. Il sera ensuite recommandé d’administrer la clofarabine selon une réduction de dose de 25 %.
Si un patient souffre de la même toxicité sévère une deuxième fois, le traitement sera différé jusqu’à ce que l’on observe un retour de la toxicité aux paramètres initiaux ou jusqu’à l’obtention d’un grade non sévère avec bénéfice potentiel de la poursuite du traitement par clofarabine supérieur au risque d’une telle poursuite thérapeutique. Il sera ensuite recommandé d’administrer la clofarabine selon une réduction de dose supplémentaire de 25 %.
Tout patient souffrant d’une toxicité sévère une troisième fois, d’une toxicité sévère ne disparaissant pas en 14 jours (voir critères d’exclusions ci-dessus) ou d’une toxicité menaçant le pronostic vital ou invalidante (Toxicité de Grade 4 – Critères CTC-NCI) devra interrompre le traitement par clofarabine (voir rubrique 4.4).
Populations particulières :
Chez le patient atteint d’insuffisance rénale
Les données limitées disponibles indiquent que la clofarabine peut s'accumuler chez les patients dont la clairance de la créatinine est diminuée (voir rubriques 4.4 et 5.2). L’utilisation de la clofarabine est contre-indiquée chez les patients atteints d’insuffisance rénale sévère (voir rubrique 4.3) et une attention particulière sera portée aux patients atteints d’insuffisance rénale légère à modérée (voir rubrique 4.4).
Chez les patients atteints d'insuffisance rénale modérée (clairance de la créatinine 30 – < 60 ml/min), la dose doit être réduite de 50% (voir rubrique 5.2).
Chez le patient atteint d’insuffisance hépatique
Il n’y a pas d’expérience chez l’insuffisant hépatique (bilirubine sérique > 1,5 x LSN avec ASAT et ALAT > 5 x LSN), alors que le foie constitue un organe cible de la toxicité présumé. Par conséquent il est contre-indiqué d’utiliser la clofarabine chez des patients atteints d’insuffisance hépatique sévère (voir rubrique 4.3) et une attention particulière sera portée aux patients atteints d’insuffisance hépatique légère à modérée (voir rubrique 4.4).
Mode d’administration
La posologie recommandée devra être administrée par perfusion intraveineuse, bien que le produit soit administré par cathéter veineux central au cours des études cliniques. CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, ne doit pas être mélangé ou administré de manière concomitante avec d’autres médicaments sur la même ligne de perfusion intraveineuse (voir rubrique 6.2). Pour les instructions concernant la filtration et la dilution du médicament avant administration voir rubrique 6.6.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Utilisation chez le patient atteint d’insuffisance rénale sévère ou d’insuffisance hépatique sévère.
Allaitement (voir rubrique 4.6).
4.4. Mises en garde spéciales et précautions d'emploi
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, est un agent antinéoplasique puissant entraînant des réactions indésirables hématologiques et non-hématologiques potentiellement importantes (voir rubrique 4.8). Les paramètres suivants devront faire l’objet d’une surveillance étroite chez les patients sous traitement par clofarabine :
· Hémogramme et numération plaquettaire obtenus à intervalles réguliers, plus fréquemment chez le patient atteint de cytopénie.
· Surveillance de la fonction rénale et hépatique avant et au cours du traitement actif, ainsi qu’au terme de la thérapie. Une interruption immédiate du traitement par clofarabine aura lieu en cas d’augmentation prononcée de la créatinine, des enzymes hépatiques et/ou de la bilirubine.
· Évaluation de l’état respiratoire, de la pression sanguine, de l’équilibre hydroélectrolytique et du poids tout au long de la période d’administration de clofarabine sur 5 jours et immédiatement après cette période.
Affections hématologiques et du système lymphatique
La myélosuppression doit être anticipée. Elle est généralement réversible et semble dose-dépendante. Une myélosuppression sévère, y compris une neutropénie, une anémie et une thrombopénie, a été observée chez les patients traités par clofarabine. Des hémorragies, y compris des hémorragies cérébrales, gastro-intestinales et pulmonaires ont été rapportées et peuvent être fatales. La majorité de ces cas étaient associés à une thrombopénie (voir rubrique 4.8). En outre, au début du traitement, la plupart des patients inclus dans les études cliniques présentaient une atteinte hématologique en rapport avec la leucémie. En raison de l’immunodéficience préexistante de ces patients et d’une neutropénie prolongée pouvant résulter du traitement par clofarabine, les patients présentent un risque élevé d’infections opportunistes sévères, à type de septicémie sévère d’évolution parfois fatale. Les patients doivent faire l’objet d’une surveillance clinique particulière des signes et symptômes d’une infection et doivent être traités immédiatement.
La survenue d’entérocolites, notamment de colite neutropénique, d’inflammation du cæcum et de colite C. difficile, a été rapportée au cours du traitement par clofarabine. Elles ont été constatées plus fréquemment dans les 30 jours après traitement, et en cas de polychimiothérapie. Ces entérocolites peuvent se compliquer de nécrose, de perforation ou de sepsis et peuvent être d’issue fatale (voir rubrique 4.8). Les patients doivent faire l’objet d’une surveillance quant à l’apparition de signes et symptômes d’entérocolite.
Affections de la peau et du tissu sous-cutané
Des cas de syndrome de Stevens-Johnson (SJS) et de nécrolyse épidermique toxique (Syndrome de Lyell), dont certains d’issue fatale, ont été rapportés (voir rubrique 4.8). Le traitement par clofarabine doit être arrêté en cas de rash avec exfoliation ou de rash bulleux ou en cas de suspicion de SJS ou de syndrome de Lyell.
Tumeurs bénignes et malignes (incl kystes et polypes) et affections du système immunitaire
L’administration de clofarabine s’accompagne d’une réduction rapide des cellules leucémiques périphériques.
Les patients sous clofarabine devront être évalués et leurs paramètres surveillés afin de détecter tout signe et symptôme de syndrome de lyse tumorale et de libération de cytokines (ex. tachypnée, tachycardie, hypotension, œdème pulmonaire) susceptible d’apparaître au cours d’un Syndrome de Réponse Inflammatoire Systémique (SRIS) d’un syndrome de fuite capillaire et/ou d’un dysfonctionnement organique (voir rubrique 4.8).
· L’administration prophylactique d’allopurinol pourra être envisagée si une hyperuricémie est attendue (lyse tumorale).
· Les patients devront être hydratés par voie intraveineuse pendant les 5 jours de traitement par clofarabine, afin de réduire les effets de lyse tumorale et tout autre évènement.
· L’utilisation de stéroïdes prophylactiques (par ex., 100 mg/m2 d’hydrocortisone du Jour 1 au Jour 3) pourra permettre de prévenir les signes ou symptômes de SRIS ou de fuite capillaire.
Le traitement par clofarabine sera immédiatement interrompu en cas d’apparition de signes ou de symptômes précoces du SRIS/du syndrome de fuite capillaire ou de dysfonctionnement organique prononcé et un traitement d’appoint adéquat sera mis en place. En outre, le traitement par clofarabine devra être interrompu si le patient présente une hypotension au cours des 5 jours d’administration, quel qu’en soit la cause. La poursuite du traitement par clofarabine (avec généralement administration d’une dose plus faible) peut être envisagée lorsque l’état des patients est stable et après retour aux valeurs normales des fonctions organiques.
La majorité des patients répondant à la clofarabine obtiennent une réponse après 1 ou 2 cycles de traitement (Cf. rubrique 5.1). Par conséquent, chez les patients ne montrant aucune amélioration hématologique et/ou clinique après 2 cycles de traitement, le rapport bénéfice/risque potentiel associé à la poursuite du traitement devra être évalué par le médecin traitant.
Affections cardiaques
Les patients atteints d’une affection cardiaque ou recevant des médicaments connus pour leurs effets sur la pression sanguine ou la fonction cardiaque devront faire l’objet d’une surveillance étroite au cours du traitement par clofarabine (voir rubriques 4.5 et 4.8).
Affections du rein et des voies urinaires
Il n’y a pas d'étude clinique chez l’enfant atteint d’insuffisance rénale (définie dans les études cliniques par une créatinine sérique ≥ 2 x LSN par rapport à l’âge), alors que la clofarabine est principalement éliminée par le rein. Les données de pharmacocinétique indiquent que la clofarabine peut s’accumuler chez les patients présentant une diminution de la clairance de la créatinine (voir rubrique 5.2). Par conséquent, une attention particulière sera portée lors de l’utilisation de la clofarabine chez des patients atteints d’insuffisance rénale légère à modérée (voir rubriques 4.2 pour les ajustements de dose). Le profil de tolérance de la clofarabine n'a pas été établi chez les patients atteints d'une insuffisance rénale grave ni chez les patients recevant une transplantation rénale (voir rubrique 4.3). L’utilisation concomitante de médicaments néphrotoxiques et de produits éliminés par sécrétion tubulaire, tels que les AINS, l’amphotéricine B, le méthotrexate, les aminosides, les organoplatines, le foscarnet, les pentamidines, les immunosuppresseurs ciclosporine et tacrolimus, l’aciclovir et le valganciclovir, devra être évitée, en particulier au cours de la période de 5 jours d’administration de la clofarabine ; il est préférable de ne pas administrer de médicaments néphrotoxiques (voir rubriques 4.5 et 4.8). Des cas d’insuffisance rénale ou d’insuffisance rénale aigüe ont été observés à la suite d’infections, de sepsis et d’un syndrome de lyse tumorale (voir rubrique 4.8). Les patients doivent faire l’objet d’une surveillance compte-tenu du risque de néphrotoxicité, le traitement par clofarabine devra être interrompu si nécessaire.
Une augmentation de la fréquence et de la gravité des effets indésirables, en particulier des infections, une myélosuppression (neutropénie) et une toxicité hépatique, a été observée lorsque la clofarabine est utilisée en association. A cet égard, les patients recevant un traitement par clofarabine en association devront faire l’objet d’une surveillance étroite.
Les patients traités par clofarabine peuvent présenter des vomissements et diarrhées ; ils doivent donc être informés des mesures appropriées à prendre pour éviter toute déshydratation. Il doit être indiqué aux patients de consulter un médecin en cas de vertiges, de syncopes répétées ou de baisse du débit urinaire. L’administration prophylactique d’antiémétiques doit être envisagée.
Affections hépatobiliaires
Il n’y a pas d’expérience chez l’insuffisant hépatique (bilirubine sérique > 1,5 x LSN avec ASAT et ALAT > 5 x LSN), alors que le foie constitue un organe cible de la toxicité présumée. Par conséquent, une attention particulière sera portée lors de l’utilisation de la clofarabine chez des patients atteints d’insuffisance hépatique légère à modérée (voir rubriques 4.2 et 4.3). L’utilisation concomitante de médicaments associés à une toxicité hépatique devra être évitée autant que possible (voir rubriques 4.5 et 4.8). Si un patient présente une toxicité hématologique de neutropénie de grade 4 (nombre absolu de neutrophiles < 0,5 x 109/l) durant ≥ 4 semaines, la dose devra alors être réduite de 25 % au cours du cycle suivant.
Tout patient souffrant d’une toxicité non hématologique sévère (Toxicité de Grade 3 – Critères CTC- NCI) une troisième fois, d’une toxicité sévère ne disparaissant pas en 14 jours (à l’exception des nausées/des vomissements) ou d’une toxicité non hématologique et non infectieuse menaçant le pronostic vital ou invalidante (Toxicité de Grade 4 - Critères CTC-NCI) devra interrompre le traitement par clofarabine (voir rubrique 4.2).
Chez les patients ayant reçu antérieurement une transplantation de cellules souches hématopoïétiques, un traitement par clofarabine (40 mg/m2) en association avec de l’étoposide (100 mg/m2) et du cyclophosphamide (440 mg/m2) est associé à un risque accru d’hépatotoxicité suggestive d’une maladie veino-occlusive (MVO). Lors du suivi post commercialisation, des cas de maladie veino- occlusive hépatique, parfois d’issue fatale, ont été rapportés chez des patients pédiatriques et adultes traités par clofarabine. Des cas d’hépatite et d’insuffisance hépatique, dont certains d’issue fatale, ont été rapportés avec le traitement par clofarabine (voir rubrique 4.8).
La plupart de ces patients avaient reçu des chimiothérapies de conditionnement incluant du busulfan, du melphalan et/ou une association cyclophosphamide et irradiation corporelle totale. Des événements hépatotoxiques graves ont été observés lors d’une étude de phase 1/2 avec la clofarabine administrée en association chez des patients pédiatriques présentant une leucémie aiguë en rechute ou réfractaire.
Les données actuellement disponibles concernant la sécurité et l’efficacité de la clofarabine administrée pendant plus de 3 cycles de traitement sont limitées.
Excipient à effet notoire :
Ce médicament contient 70,77 mg de sodium par flacon, ce qui équivaut à 3,54 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
On n’a pas détecté que la clofarabine était métabolisée par le système enzymatique du cytochrome P450 (CYP). Par conséquent, il est peu probable que la clofarabine puisse interagir avec des substances actives ayant une activité inhibitrice ou inductrice d’enzymes du cytochrome P450. En outre, il est peu probable que la clofarabine inhibe l’une des 5 isoformes principales du CYP humain (1A2, 2C9, 2C19, 2D6 et 3A4) ou qu’elle soit un inducteur de 2 de ces isoformes (1A2 et 3A4) aux concentrations plasmatiques obtenues suite à une perfusion intraveineuse de 52 mg/m²/jour. De ce fait, il n’est pas envisagé que la clofarabine puisse modifier le métabolisme de substances actives qui sont des substrats connus de ces enzymes.
La clofarabine est principalement éliminée par le rein. De ce fait, l’utilisation concomitante de médicaments néphrotoxiques et de produits éliminés par sécrétion tubulaire, tels que les AINS, l’amphotéricine B, le méthotrexate, les aminosides, les organoplatines, le foscarnet, les pentamidines, les immunosuppresseurs ciclosporine et tacrolimus, l’aciclovir et le valganciclovir, devra être évitée, en particulier au cours de la période de 5 jours d’administration de la clofarabine (voir rubriques 4.4, 4.8 et 5.2).
Le foie constitue un organe cible potentiel de la toxicité. Par conséquent, l’utilisation concomitante de médicaments associés à une toxicité hépatique devra être évitée autant que possible (voir rubriques 4.4 et 4.8).
Les patients recevant des médicaments connus pour leurs effets sur la pression sanguine ou la fonction cardiaque devront faire l’objet d’une surveillance étroite au cours du traitement par clofarabine (voir rubriques 4.4 et 4.8).
4.6. Fertilité, grossesse et allaitement
Contraception chez les hommes et les femmes
En raison du risque génotoxique de la clofarabine (voir rubrique 5.3), les femmes en âge d’avoir des enfants doivent utiliser des méthodes de contraception efficaces tout au long du traitement par clofarabine et pendant 6 mois après la fin du traitement.
Les hommes doivent utiliser des méthodes de contraception efficaces et être informés de ne pas concevoir d’enfant pendant le traitement par clofarabine et pendant 3 mois après la fin du traitement
Grossesse
Il n’existe pas de données concernant l’utilisation de la clofarabine chez la femme enceinte. Des études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction comprenant une tératogénicité (voir rubrique 5.3). La clofarabine est susceptible d’être à l’origine de malformations graves pour l’enfant lorsqu’elle est administrée pendant la grossesse. Par conséquent, CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, ne doit pas être utilisé au cours de la grossesse, en particulier au cours du premier trimestre, à moins d’une nécessité absolue (à savoir, uniquement si le bénéfice potentiel pour la mère est supérieur aux risques encourus par le fœtus). En cas de grossesse survenant au cours du traitement par clofarabine, la patiente devra impérativement être informée des dangers potentiels encourus par le fœtus.
La possibilité que la clofarabine ou ses métabolites puissent passer dans le lait maternel humain reste inconnue. Le passage de la clofarabine dans le lait n’a pas fait l’objet d’études chez l’animal. Néanmoins, étant donné la possibilité de réactions indésirables graves pour le nouveau-né, il est impératif d’interrompre l’allaitement avant, pendant et dans les 2 semaines qui suivent la fin du traitement par CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion (voir rubrique 4.3).
Fertilité
On a observé des toxicités proportionnelles à la dose administrée sur les organes reproducteurs mâles chez la souris, le rat et le chien, ainsi que des toxicités sur les organes reproducteurs femelles chez la souris (voir rubrique 5.3). Les conséquences du traitement par clofarabine sur la fertilité chez l’être humain étant inconnues, il est recommandé de discuter avec le/la patient(e) de ses intentions et de programmer la grossesse le cas échéant.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de tolérance
La quasi-totalité des patients (98%) a présenté au moins un évènement indésirable considéré comme lié à la clofarabine par l’investigateur de l’étude. Les évènements indésirables les plus fréquemment rapportés étaient les suivants: nausée (61% des patients), vomissements (59%), neutropénie fébrile (35%), céphalées (24%), rash (21%), diarrhée (20%), prurit (20%), pyrexie (19%), syndrome d’érythrodysesthésie palmo-plantaire (15%), fatigue (14%), anxiété (12%), inflammation muqueuse (11%) et bouffées congestives (11%). Soixante-huit patients (59%) ont manifesté au moins un effet indésirable grave lié à la clofarabine. Un patient a arrêté le traitement en raison d’une hyperbilirubinémie de grade 4 considérée comme liée à la clofarabine, après avoir reçu une dose de 52 mg/m2/jour de clofarabine. Trois patients sont décédés suite à des évènements indésirables considérés par l’investigateur de l’étude comme liés à la clofarabine : un patient suite à une détresse respiratoire, des lésions hépatocellulaires et un syndrome de fuite capillaire ; un patient suite à un sepsis à ERV et une défaillance multiorganique et un patient suite à un choc septique et une défaillance multiorganique.
Tableau reprenant la liste des effets indésirables
Les informations suivantes sont basées sur des données issues d’études cliniques au cours desquelles 115 patients (> 1 et ≤ 21 ans) souffrant soit de LAL, soit de leucémie aiguë myéloïde (LAM) ont reçu au moins une dose de clofarabine à la posologie recommandée de 52 mg/m2 par jour x 5. Les effets indésirables sont énumérés par classe de systèmes d'organes et par fréquence (très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) et très rare (< 1/10 000)) dans le tableau ci-dessous.
Les effets indésirables rapportés après commercialisation sont également inclus dans le tableau ci-dessous sous la catégorie de fréquence « indéterminée » (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables doivent être présentés suivant un ordre décroissant de gravité.
Chez les patients atteints de LAL ou de LAM à un stade avancé la causalité des effets indésirables peut être difficile à évaluer étant donné la diversité des symptômes liée à l’affection sous-jacente, à sa progression et à la co-administration de nombreux médicaments.
|
Effets indésirables considérées comme liées à la clofarabine et rapportées à des fréquences ≥1/1000 (c’est à dire chez > 1/115 patients) au cours des études cliniques et après commercialisation |
|
|
Infections et infestations |
Fréquent : choc septique*, sepsis, bactériémie, pneumonie, herpes zoster, herpès simplex, candidose buccale Fréquence indéterminée: Colite C. difficile |
|
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes) |
Fréquent : syndrome de lyse tumorale* |
|
Affections hématologiques et du système lymphatique |
Très fréquent : neutropénie fébrile Fréquent : neutropénie |
|
Affections du système immunitaire |
Fréquent : hypersensibilité |
|
Troubles du métabolisme et de la nutrition |
Fréquent : anorexie, diminution de l’appétit, déshydratation Fréquence indéterminée : hyponatrémie |
|
Affections psychiatriques |
Très fréquent : anxiété Fréquent : agitation, instabilité psychomotrice, modification de l’état mental |
|
Affections du système nerveux |
Très fréquent : céphalées Fréquent : somnolence, neuropathie périphérique, paresthésie, vertiges, tremblements |
|
Affections de l’oreille et du labyrinthe |
Fréquent : hypoacousie |
|
Affections cardiaques |
Fréquent : épanchement péricardique*, tachycardie* |
|
Affections vasculaires |
Très fréquent : bouffées congestives* Fréquent : hypotension*, syndrome de fuite capillaire, hématomes |
|
Affections respiratoires, thoraciques et médiastinales |
Fréquent : détresse respiratoire, épistaxis, dyspnée, tachypnée, toux |
|
Affections gastro-intestinales |
Très fréquent : vomissements, nausées, diarrhée Fréquent : hémorragie buccale, saignement des gencives, hématémèse, douleurs abdominales, stomatite, douleur de l’abdomen supérieur, proctalgie, ulcération buccale Fréquence indéterminée : Pancréatite élévation de l’amylase et de la lipase sériques, entérocolite, colite neutropénique, inflammation du cæcum |
|
Affections hépatobiliaires |
Fréquent : hyperbilirubinémie, ictère, maladie veino- occlusive, augmentation des aminotransférases ALT* (alanine) et AST* (aspartate), insuffisance hépatique Peu fréquent : hépatite |
|
Troubles généraux et anomalies au site d’administration |
Très fréquent : fatigue, pyrexie, inflammation muqueuse Fréquent : défaillance multiorganique, syndrome de réponse inflammatoire systémique*, douleurs, frissons, irritabilité, œdème, œdème périphérique, sensations de chaleur, sensation de mal-être, |
|
Affections de la peau et du tissu sous- cutané |
Très fréquent : syndrome d’érythrodysesthésie palmo- plantaire, prurit Fréquent : éruption maculopapulaire, pétéchies, érythème, rash prurigineux, exfoliation cutanée, rash généralisé, alopécie, hyperpigmentation cutanée, érythème généralisé, rash érythémateux, sécheresse cutanée, hyperhidrose Fréquence indéterminée : Syndrome de Stevens Johnson (SJS), syndrome de Lyell (nécrolyse épidermique toxique) |
|
Affections musculo-squelettiques et systémiques |
Fréquent : douleurs aux extrémités, myalgie, douleurs osseuses, douleurs de la paroi thoracique, arthralgie, douleurs au niveau du cou et du dos |
|
Affections du rein et des voies urinaires |
Fréquent : hématurie*, insuffisance rénale, insuffisance rénale aigüe |
|
Investigations |
Fréquent : perte de poids |
|
Lésions, intoxications et complications liées aux procédures |
Fréquent : contusions |
* = voir ci-dessous
**Tous les effets indésirables survenant au moins deux fois (c.-à-d. 2 effets ou plus (1,7 %)) sont mentionnés dans ce tableau
Description des effets indésirables susmentionnés
Atteintes hématologiques et du système lymphatique
Les anomalies hématologiques les plus fréquemment observées chez les patients traités par clofarabine ont été : anémie (83,3 % ; 95/114), leucopénie (87,7 % ; 100/114), lymphopénie (82,3 % ; 93/113), neutropénie (63,7 % ; 72/113), et thrombopénie (80,7 % ; 92/114). La majorité de ces effets indésirables ont été de grade ≥ 3.
Lors du suivi post commercialisation, des cytopénies prolongées (thrombopénie, anémie, neutropénie et leucopénie) et des insuffisances médullaires ont été rapportées. Des événements hémorragiques ont été observés dans le contexte de thrombopénie. Des hémorragies, y compris des hémorragies cérébrales, gastro-intestinales et pulmonaires ont été rapportées et peuvent être fatales (voir rubrique 4.4).
Affections vasculaires
Soixante-quatre patients sur 115 (55,7 %) ont présenté au moins un événement indésirable vasculaire.
23 patients sur 115 ont présenté un trouble vasculaire considéré comme lié à la clofarabine, les plus fréquemment rapportés étant les bouffées congestives (13 événements ; non graves) et l’hypotension (5 événements ; tous considérés comme graves ; voir rubrique 4.4). Néanmoins, la majorité de ces poussées hypotensives ont été observées chez des patients atteints d’infections sévères confondantes.
Affections cardiaques
Au moins un événement cardiaque a été rapporté par 50 % des patients. 11 événements survenus chez 115 patients ont été considérés comme liés à la clofarabine mais aucun d’entre eux n’a été jugé grave et l’affection cardiaque la plus fréquemment rapportée était la tachycardie (35 %) (voir rubrique 4.4) ; chez 6,1 % des patients (7/115), la tachycardie a été considérée comme liée à la clofarabine. La plupart des événements indésirables d’origine cardiaque ont été observés au cours des 2 premiers cycles de traitement.
L’épanchement péricardique et la péricardite ont été rapportés chez 9 % (10/115) des patients. Trois de ces événements indésirables ont été évalués par la suite comme étant liés à la clofarabine : épanchement péricardique (2 événements ; dont 1 grave) et péricardite (1 événement ; non grave). Chez la majorité des patients (8/10), l’épanchement péricardique et la péricardite ont été jugés asymptomatiques et d’importance clinique légère ou nulle d’après l’examen échocardiographique. L’épanchement péricardique était lui cliniquement significatif chez 2 patients présentant un état hémodynamique précaire.
Infections et infestations
Quarante-huit pour cent des patients présentaient une ou plusieurs infections persistantes avant le début du traitement par clofarabine. Au total, après traitement par clofarabine, 83 % des patients ont présenté au moins 1 infection, fongique, virale ou bactérienne (Cf. rubrique 4.4). 21 événements (18,3 %) ont été considérés comme liés à la clofarabine, parmi lesquels une infection liée au cathéter (1 événement), une septicémie (2 événements) et un choc septique (2 événements ; 1 décès (Cf. ci- dessus)) ont été considérés comme graves.
Lors du suivi post commercialisation, des infections bactériennes, fongiques et virales ont été rapportées, dont certaines d’issue fatale. Ces infections peuvent entraîner un choc septique, une insuffisance respiratoire, une insuffisance rénale et/ou une défaillance multiviscérale.
Affections du rein et des voies urinaires
Au moins un événement rénal ou urinaire a été rapporté chez 41 patients sur 115 (35,7 %). La toxicité rénale la plus fréquente chez les patients pédiatriques était l’augmentation du taux de créatinine. Un taux accru de créatinine de grade 3 ou 4 a été observé chez 8 % des patients. Des médicaments néphrotoxiques ou un syndrome de lyse tumorale avec ou sans hyperuricémie peuvent contribuer à une toxicité rénale (Cf. rubriques 4.3 et 4.4). Une hématurie a été observée chez 13 % de l’ensemble des patients. Quatre événements indésirables rénaux survenus chez 115 patients ont été considérées comme liées à la clofarabine ; aucun n’a été jugé grave : hématurie (3 événements) et insuffisance rénale aiguë (1 événement) (Cf. rubriques 4.3 et 4.4).
Affections hépatobiliaires
Le foie constitue un organe cible potentiel de la toxicité de la clofarabine. 25,2 % des patients ont présenté au moins un événement hépatobiliaire (Cf. rubriques 4.3 et 4.4). Six événements ont été considérés comme liés à la clofarabine, parmi lesquels : cholécystite aiguë (1 événement), cholélithiase (1 événement), lésion hépatocellulaire (1 événement ; décès du patient (voir ci-dessus)) et hyperbilirubinémie (1 événement ; avec interruption du traitement (voir ci-dessus)) a été considéré comme grave. Deux cas de maladie veino-occlusive (MVO) chez des patients pédiatriques (1,7 %) ont été considérés comme liés au médicament à l’étude.
Des cas de MVO hépatique d’issue fatale ont été rapportés lors du suivi post commercialisation chez des patients pédiatriques et adultes (voir rubrique 4.4).
De plus, 50 des 113 patients sous clofarabine ont présenté une élévation de grade ≥3 du taux d’ALAT, 36 patients sur 100 ont montré des taux d’ASAT élevés et 15 patients sur 114 des taux de bilirubine élevés. La majorité des augmentations d’ALAT et d’ASAT ont eu lieu dans les 10 jours ayant suivi l’administration de clofarabine et sont revenus à un grade ≤ 2 dans les 15 jours. Lorsque des données de suivi sont disponibles, la majorité des augmentations de la bilirubine revient à un grade ≤ 2 dans les 10 jours.
Syndrome de réponse inflammatoire systémique (SRIS) ou syndrome de fuite capillaire
Le SRIS et le syndrome de fuite capillaire (signes et symptômes de libération de cytokine par ex., tachypnée, tachycardie, hypotension, œdème pulmonaire) ont été rapportés comme événements indésirables chez 5 % (6 sur 115) des patients pédiatriques (5 LAL, 1 LAM) (Cf. rubrique 4.4). Treize cas de syndrome de lyse tumorale, de syndrome de fuite capillaire ou de SRIS ont été rapportés ; SRIS (2 événements, tous deux considérés comme graves), syndrome de fuite capillaire (4 événements, dont 3 considérés comme graves et associés) et syndrome de lyse tumorale
(7 événements, dont 6 considérés comme associés et 3 comme graves).
Des cas de syndrome de fuite capillaire, dont certains d’issue fatale, ont été rapportés lors du suivi post commercialisation (voir rubrique 4.4).
Affections gastro-intestinales
Des cas d’entérocolite, notamment de colite neutropénique, d’inflammation du cæcum et de colite à C. difficile, ont été rapportés au cours du traitement par la clofarabine. L’entérocolite peut entraîner une nécrose, une perforation ou des complications septiques et peut être associée à une issue fatale (voir rubrique 4.4).
Affections de la peau et du tissu sous-cutané
Des cas de syndrome de Stevens-Johnson (SJS) et de nécrolyse épidermique toxique (Syndrome de Lyell), dont certains d’issue fatale, ont été rapportés chez des patients qui recevaient ou avaient récemment été traités avec de la clofarabine. D’autres affections exfoliatives ont également été rapportées.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Symptômes
Aucun cas de surdosage n’a été rapporté. Des symptômes de surdosage sont néanmoins considérés comme possibles, à savoir : les nausées, les vomissements, la diarrhée et la myélosuppression sévère. A ce jour, la dose quotidienne la plus forte administrée à l’être humain est de 70 mg/m² pendant 5 jours consécutifs (2 patients pédiatriques LAL). Les toxicités observées chez ces patients comprenaient : les vomissements, une hyperbilirubinémie, des transaminases élevées et une éruption maculopapulaire.
Conduite à tenir
Aucun antidote spécifique n’est connu. Une interruption immédiate du traitement, une surveillance étroite du patient et un maintien des fonctions vitales approprié sont recommandées.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
La clofarabine est un antimétabolite nucléoside purique. Son activité antitumorale serait due à mécanismes :
· Inhibition de l’ADN polymérase α entraînant la terminaison de l’élongation d’une chaîne d’ADN et/ou la synthèse/réparation de l’ADN.
· Inhibition de la ribonucléotide réductase avec réduction de la concentration cellulaire en désoxynucléotide triphosphate (dNTP).
· Rupture de l’intégrité de la membrane mitochondriale avec libération du cytochrome C et d’autres facteurs pro-apoptotiques entraînant l’apoptose même chez des lymphocytes non en division.
La clofarabine doit tout d’abord se répandre ou être transportée dans les cellules cibles où elle va être séquentiellement phosphorylée en mono- et diphosphate par des kinases intracellulaires pour finalement donner le conjugué actif : le clofarabine 5’-triphosphate. La clofarabine montre une forte affinité pour l’une des enzymes phosphorylantes d’activation, la désoxycytidine kinase, dépassant celle du substrat naturel, la désoxycytidine.
En outre, la clofarabine présente une résistance plus forte à la dégradation cellulaire par l’adénosine désaminase et une sensibilité moindre au clivage phosphorolytique comparé aux autres substances actives de sa classe. Par contre, l’affinité du clofarabine triphosphate pour l’ADN polymérase α et la ribonucléotide réductase est similaire, voire supérieure à celle de la désoxyadénosine triphosphate.
Effets pharmacodynamiques :
Les études in vitro ont montré que la clofarabine inhibe la croissance cellulaire et qu’elle est cytotoxique envers un grand nombre de lignées cellulaires de tumeurs solides et de tumeurs hématologiques prolifératives. On a également observé son activité contre les lymphocytes et les macrophages en phase de quiescence. En outre, on a observé que la clofarabine retardait la croissance tumorale et, dans certains cas, entraînait la régression tumorale chez toute une gamme de xénogreffes tumorales murines et humaines implantées chez la souris.
Efficacité et sécurité clinique :
Efficacité clinique : Afin de permettre une évaluation systématique des réponses observées chez les patients, un comité indépendant chargé de l’analyse des réponses en ouvert (Independent Response Review Panel - IRRP) a déterminé les taux de réponse suivants en se basant sur les définitions du Children’s Oncology Group :
|
RC = Rémission complète |
Patients répondant à chacun des critères suivants : · Pas d’argument en faveur de blastes circulants ou d’affection extramédullaire · Moelle osseuse M1 (≤ 5 % de blastes) · Récupération des taux systémiques (plaquettes ≥ 100 x 109/l et nombre absolu des neutrophiles ≥ 1,0 x 109/l) |
|
RCp = Rémission complète en l’absence de récupération totale des plaquettes |
· Patients répondant à tous les critères de RC à l’exception d’une récupération de la numération plaquettaire à > 100 x 109/l |
|
RP = Rémission partielle |
Patients répondant à chacun des critères suivants : · Disparition totale des blastes circulants · Moelle osseuse M2 (≥ 5 % et ≤ 25 % de blastes) et apparition de cellules progéniteurs normales · Moelle M1 ne pouvant être classifiée RC ou RCp |
|
Taux de Rémission Globale (RG) |
· (Nombre de patients en RC + Nombre de patients en RCp) ÷ Nombre de patients éligibles ayant reçu de la clofarabine |
La sécurité et l’efficacité de la clofarabine ont été évaluées au cours d’une étude non-comparative en ouvert de phase I à doses croissantes portant sur 25 patients pédiatriques atteints de leucémie en rechute ou réfractaire (17 LAL ; 8 LAM) ayant connu un échec au traitement standard ou chez qui aucun autre traitement n’existait. L’administration de doses a débuté à 11,25 avec une progression à 15, 30, 40, 52 et 70 mg/m²/jour par perfusion intraveineuse pendant 5 jours toutes les 2 à 6 semaines selon la toxicité et la réponse. 9 des 17 patients atteints de LAL ont été traités par clofarabine, à raison d’une dose de 52 mg/m2/jour. Sur les 17 patients atteints de LAL, 2 ont réussi à obtenir une rémission complète (12 % ; RC) et 2 une rémission partielle (12 % ; RP) à des doses variables. Les facteurs dose-limitants de cette étude étaient l’hyperbilirubinémie, des transaminases élevées et une éruption maculopapulaire apparus à une dose de 70 mg/m²/jour (2 patients LAL ; voir rubrique 4.9).
Une étude multicentrique non-comparative de phase II en ouvert de la clofarabine a été menée afin de déterminer le taux de rémission globale (RG) chez des patients lourdement prétraités (≤ 21 ans au moment du diagnostic initial) avec LAL en rechute ou réfractaire selon la classification FAB (French- American-British classification). La dose maximum tolérée identifiée au cours de l’étude de phase I décrite ci-dessus de 52 mg/m²/jour de clofarabine a été administrée par perfusion intraveineuse pendant 5 jours consécutifs toutes les 2 à 6 semaines. Le tableau ci-dessous résume les résultats d’efficacité clé de cette étude.
Les patients atteints de LAL ne devaient pas avoir été éligibles pour un traitement au potentiel curatif supérieur et devaient avoir vécu au moins deux récidives et/ou être réfractaire à deux traitements, c’est à dire qu’ils n’ont pas réussi à obtenir de rémission suite à au moins deux régimes thérapeutiques antérieurs. Avant d’être inclus dans cette étude, 58 des 61 patients (95 %) ont reçu entre 2 et 4 traitements d’induction différents et 18/61 (30 %) de ces patients ont reçu au moins 1 greffe antérieure de cellules souches hématopoïétiques. L’âge médian des patients traités (37 de sexe masculin, 24 de sexe féminin) était de 12 ans.
L’administration de la clofarabine a entraîné une réduction spectaculaire et rapide des cellules leucémiques périphériques chez 31 des 33 patients (94 %) dont le nombre absolu de blastes était mesurable initialement. Les 12 patients ayant obtenu une rémission globale (RC + RCp) montraient un temps de survie médian de 69,5 semaines à la date de recueil des données. Des réponses ont été observées chez les différents immunophénotypes de LAL, notamment ceux du type pré-B et T. Bien que le taux de transplantation ne constituait pas un critère d’évaluation pour cette étude, 10/61 patients (16 %) ont pu subir une greffe de cellules souches hématopoïétiques après le traitement par clofarabine (3 ayant obtenu une RC, 2 après une RCp, 3 après une RP, 1 patient considéré comme en échec du traitement par le comité indépendant d’analyse des réponses et 1 considéré comme impossible à évaluer par ce même comité). Les durées de réponse ne sont pas comparables car certains patients ont reçu une greffe de cellules souches hématopoïétiques.
|
Résultats d’efficacité issus de l’étude pivot portant sur des patients (≤ 21 ans au moment du diagnostic initial) atteints de LAL en rechute ou réfractaire après au moins deux régimes thérapeutiques antérieurs |
||||
|
Catégorie de Réponse |
ITT* patients (n = 61) |
Durée médiane de la rémission (semaines) (IC à 95 %) |
Temps de progression médian (semaines)** (IC à 95 %) |
Survie globale médiane (semaines) (IC à 95 %) |
|
Rémission globale (RC + RCp) |
12 (20 %) |
32,0 (9,7 à 47,9) |
38,2 (15,4 à 56,1) |
69,5 (58,6 à -) |
|
RC |
7 (12 %) |
47,9 (6,1 à -) |
56,1 (13,7 à -) |
72,4 (66,6 à -) |
|
RCp |
5 (8 %) |
28,6 (4,6 à 38,3) |
37,0 (9,1 à 42) |
53,7 (9,1 à -) |
|
RP |
6 (10 %) |
11,0 (5,0 à -) |
14,4 (7,0 à -) |
33,0 (18,1 à -) |
|
RC + RCp + RP |
18 (30 %) |
21,5 (7,6 à 47,9) |
28,7 (13,7 à 56,1) |
66,6 (42,0 à -) |
|
Échec au traitement |
33 (54 %) |
N/A |
4,0 (3,4 à 5,1) |
7,6 (6,7 à 12,6) |
|
Impossibles à évaluer |
10 (16 %) |
N/A |
||
|
Totalité des patients |
61 (100 %) |
N/A |
5,4 (4,0 à 6,1) |
12,9 (7,9 à 18,1) |
|
*ITT = en intention de traiter. ** Les patients en vie et en rémission au moment du dernier suivi ont été censurés dans l’analyse à ce même moment. |
||||
|
Durée de rémission individuelle et données de survie des patients ayant obtenu une RC ou RCp |
|||
|
Meilleure réponse |
Temps écoulé jusqu’à rémission globale (semaines) |
Durée de remission (semaines) |
Survie globale (semaines) |
|
Patients n’ayant pas subi de transplantation |
|||
|
RC |
5,7 |
4,3 |
66,6 |
|
RC |
14,3 |
6,1 |
58,6 |
|
RC |
8,3 |
47,9 |
66,6 |
|
RCp |
4,6 |
4,6 |
9,1 |
|
RC |
3,3 |
58,6 |
72,4 |
|
RCp |
3,7 |
11,7 |
53,7 |
|
Patients ayant subi une transplantation alors qu’ils étaient en rémission continue* |
|||
|
RCp |
8,4 |
11,6+ |
145,1+ |
|
RC |
4,1 |
9,0+ |
111,9+ |
|
RCp |
3,7 |
5,6+ |
42,0 |
|
RC |
7,6 |
3,7+ |
96,3+ |
|
Patients ayant subi une transplantation après un traitement alternatif ou une rechute* |
|||
|
RCp |
4,0 |
35,4 |
113,3+** |
|
RC |
4,0 |
9,7 |
89,4*** |
|
* Durée de rémission censurée au moment de la transplantation ** Patient ayant subi une transplantation suite à un traitement alternatif *** Patient ayant subi une transplantation suite à une rechute |
|||
Une autorisation de mise sur le marché « sous circonstances exceptionnelles » a été délivrée pour le médicament de référence contenant de la clofarabine.
Cela signifie qu’en raison de la rareté de cette maladie il n’a pas été possible d’obtenir des informations complètes concernant ce médicament.
L’Agence européenne des médicaments réévaluera chaque année toute nouvelle information qui pourrait être disponible, et, si nécessaire, ce RCP sera mis à jour, en accord avec le RCP du médicament de référence.
5.2. Propriétés pharmacocinétiques
La pharmacocinétique de la clofarabine a été étudiée chez 40 patients âgés de 2 à 19 ans et atteints de LAL ou de LAM en rechute ou réfractaire. Les patients ont été inclus dans une étude unique de phase I (n = 12) et deux études de phase II (n = 14 / n = 14) de sécurité et d’efficacité au cours desquelles ils ont reçu des doses multiples de clofarabine par perfusion intraveineuse (voir rubrique 5.1).
|
Pharmacocinétique des patients âgés de 2 à 19 ans atteints de LAL ou de LAM en rechute ou réfractaire suite à l’administration de doses multiples de clofarabine par perfusion intraveineuse |
||
|
Paramètre |
Estimations fondées sur une analyse non-compartimentale (n = 14/n = 14) |
Estimations fondées sur une autre analyse |
|
Distribution : |
||
|
Volume de distribution (état d’équilibre) |
172 l/m2 |
|
|
Liaison des protéines plasmatiques |
|
47,1 % |
|
Albumine sérique |
|
27,0 % |
|
Élimination : |
||
|
β demi-vie de la clofarabine |
5,2 heures |
|
|
Demi-vie du clofarabine triphosphate |
|
> 24 heures |
|
Clairance systémique |
28,8 l/h/m2 |
|
|
Clairance rénale |
10,8 l/h/m2 |
|
|
Dose éliminée par les urines |
57 % |
|
Une analyse multidimensionnelle a montré que la pharmacocinétique de la clofarabine dépendait du poids et bien que la numération leucocytaire ait été identifiée comme ayant un impact sur la pharmacocinétique de la clofarabine, ce facteur n’a pas semblé suffisant pour permettre de déterminer un régime posologique pour chacun des patients en fonction de leur numération leucocytaire. Une perfusion intraveineuse de 52 mg/m² de clofarabine a produit une exposition équivalente quel qu’ait été le poids des patients. Par contre, la Cmax est inversement proportionnelle au poids du patient et, par conséquent, les enfants de faible poids pourront présenter une Cmax plus élevée au terme de la perfusion par rapport à un enfant « typique » de 40 kg ayant reçu la même dose de clofarabine par m². De ce fait, des temps de perfusion plus longs devront être envisagés chez les enfants pesant < 20 kg (voir rubrique 4.2).
Biotransformation et élimination
La clofarabine est éliminée à la fois par voie rénale et non-rénale. Après 24 heures, environ 60 % de la dose est éliminée dans les urines sous forme inchangée. Les taux de clairance de la clofarabine semblent être bien plus élevés que la filtration glomérulaire suggérant que la filtration et la sécrétion tubulaire font partie des mécanismes de l’élimination rénale. Par contre, étant donné que la clofarabine n’a pas été détectée comme étant métabolisée par le système enzymatique du cytochrome P450 (CYP), les voies d’élimination non-rénales restent actuellement inconnues.
Aucune différence apparente de la pharmacocinétique n’a été observée entre les patients atteints de LAL et les patients atteints de LAM ni entre les hommes et les femmes.
Aucun impact sur l’efficacité ou la toxicité n’a été établi dans cette population après exposition à la clofarabine ou au clofarabine triphosphate.
Populations spécifiques :
Adultes (> 21 et < 65 ans)
Les données actuellement disponibles étant insuffisantes, elles ne nous permettent pas d’établir un profil de sécurité et d’efficacité de la clofarabine chez le patient adulte. Cependant, la pharmacocinétique de la clofarabine chez l’adulte souffrant de LAM en rechute ou réfractaire suite à l’administration d’une dose unique de 40 mg/m² de clofarabine par perfusion intraveineuse sur 1 heure était comparable à celle décrite ci-dessus pour les patients âgés de 2 à 19 ans avec LAL et LAM en rechute ou réfractaire suite à l’administration de 52 mg/m² de clofarabine par perfusion intraveineuse sur 2 heures pendant 5 jours consécutifs.
Sujets âgés (≥ 65 ans)
Les données actuellement disponibles étant insuffisantes, elles ne nous permettent pas d’établir le profil de sécurité et d’efficacité de la clofarabine chez les patients âgés de 65 ans ou plus.
Insuffisance rénale
A ce jour, on ne dispose que de données limitées sur la pharmacocinétique de la clofarabine chez les patients pédiatriques présentant une diminution de la clairance de la créatinine. Ces données indiquent néanmoins que la clofarabine aurait tendance à s’accumuler chez ce type de patients (voir le schéma ci-dessous).
Les données de pharmacocinétiques de population obtenues chez des patients adultes et pédiatriques suggèrent que les patients atteints d'une insuffisance rénale modérée stable (clairance de la créatinine 30 - < 60 ml/min) recevant une dose réduite de 50 % présentent une exposition à la clofarabine similaire à ceux dont la fonction rénale est normale et qui reçoivent une dose standard.
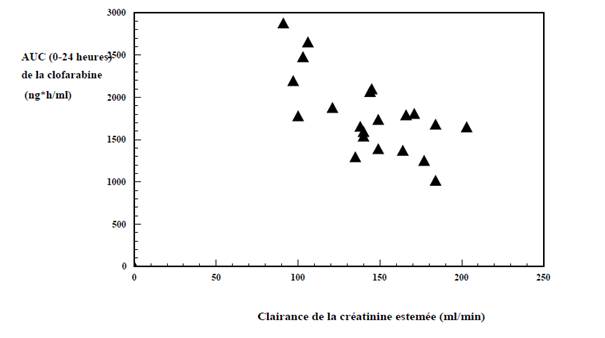
AUC0-24 heures de la clofarabine par rapport à la clairance de la créatinine estimée initialement chez des patients âgés de 2 à 19 ans avec LAL ou LAM en rechute ou réfractaire (n = 11 / n = 12) suite à l’administration de doses multiples de clofarabine par perfusion intraveineuse (estimation de la clairance de la créatinine d’après la formule de Schwartz)
Insuffisance hépatique
Il n’y a pas d’expérience chez l’insuffisant hépatique (bilirubine sérique > 1,5 x LSN avec ASAT et ALAT > 5 x LSN), alors que le foie est un organe cible potentiel de la toxicité (voir rubriques 4.3 et 4.4).
5.3. Données de sécurité préclinique
Des effets cardiaques ont été observés chez le rat, lesquels répondaient à la définition d’une cardiomyopathie et ont entraîné l’apparition de signes d’insuffisance cardiaque suite à des cycles répétés de traitement. L’incidence de ces toxicités était à la fois dépendante de la dose de clofarabine administrée et de la durée du traitement. On a rapporté ces toxicités à des taux d’exposition (Cmax) approximativement 7 à 13 fois supérieurs (après 3 cycles de doses ou plus) ou 16 à 35 fois supérieurs (après un ou plusieurs cycles de doses) aux taux d’expositions cliniques. Les effets minimes observés à des doses inférieures suggèrent qu’il existe un seuil de toxicité pour le cœur et que la pharmacocinétique plasmatique non linéaire du rat peut avoir joué un rôle dans les effets observés. Le risque potentiel pour l’être humain reste inconnu.
On a rapporté une néphropathie glomérulaire chez le rat à des taux d’exposition 3 à 5 fois plus élevés que l’AUC clinique après 6 cycles d’administration de doses de clofarabine. Cette affection se caractérisait par un épaississement mineur de la membrane basale glomérulaire avec lésion tubulaire uniquement légère et non associé à des modifications des paramètres sériques.
On a observé des effets hépatiques chez le rat suite à une administration chronique de clofarabine. Ces effets hépatiques illustrent probablement une superposition de modifications dégénératives et régénératrices résultant des cycles de traitement et n’étaient pas associés à des modifications des paramètres sériques. Un argument histologique en faveur de conséquences hépatiques a été observé chez le chien suite à une administration aiguë de doses élevées, lequel n’était pas non plus accompagné de modifications des paramètres sériques.
Des toxicités dose-dépendantes sur les organes reproducteurs mâles ont été observées chez la souris, le rat et le chien. Ces effets comprenaient une dégénérescence bilatérale de l’épithélium séminifère avec rétention des spermatides et atrophie des cellules interstitielles chez le rat à des taux d’exposition exagérés (150 mg/m²/jour), ainsi qu’une dégénérescence cellulaire de l’épididyme et une dégénérescence de l’épithélium séminifère chez le chien à des taux d’exposition cliniquement pertinents (> 7,5 mg/m2/jour de clofarabine).
Une atrophie retard de l’ovaire ou une dégénérescence et apoptose de la muqueuse utérine ont été observées chez la souris femelle à la dose uniquement utilisée de 225 mg/m2/jour de clofarabine.
La clofarabine était tératogène chez le rat et le lapin. Une augmentation des pertes post- implantatoires, une réduction des poids corporels fœtaux et une réduction des tailles des portées associée à une augmentation du nombre de malformations (externes marquées, tissus souples) et d’altérations du squelette (notamment un retard de l’ossification) ont été rapportées chez le rat recevant des doses produisant approximativement 2 à 3 fois l’exposition clinique (54 mg/m²/jour) et chez le lapin recevant 12 mg/m²/jour de clofarabine (absence de données sur l’exposition chez le lapin). Le seuil de toxicité congénitale a été considéré de 6 mg/m²/jour chez le rat et de 1,2 mg/m²/jour chez le lapin. Le taux d’effets non observables de toxicité maternelle chez le rat était de 18 mg/m²/jour et de plus de 12 mg/m²/jour chez le lapin. Aucune étude sur la fertilité n’a été réalisée.
Des études de la génotoxicité ont montré que la clofarabine n’était pas mutagène au cours du test de mutation reverse bactérienne, mais qu’elle entraînait des effets clastogènes au cours du test d’aberration chromosomique inactivé sur des cellules d’ovaire de hamster chinois (CHO) et du test du micronoyau in vivo chez le rat.
Aucune étude de carcinogénicité n’a été réalisée.
Chlorure de sodium, eau pour préparations injectables
3 ans
Le médicament dilué est chimiquement et physiquement stable pendant 3 jours à une température comprise entre 2°C et 8°C et à température ambiante (jusqu’à 25°C).
Toutefois, d’un point de vue microbiologique, il doit être utilisé immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation après dilution relèvent de la seule responsabilité de l’utilisateur et ne devraient normalement pas dépasser 24 heures à une température comprise entre 2°C et 8°C sauf en cas de dilution réalisée en conditions aseptiques dûment contrôlées et validées.
6.4. Précautions particulières de conservation
Pour les conditions de conservation du médicament après dilution, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon en verre de type I muni d’un bouchon en caoutchouc bromobutyl, d’un opercule de type « flip-off » en polypropylène et d’une bague de sertissage en aluminium. Les flacons contiennent 20 ml de solution à diluer pour perfusion et sont disponibles dans une boîte.
Chaque boîte peut contenir 1, 3, 4, 10 ou 20 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Précautions particulières d’administration
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, doit être reconstitué avant administration. La solution sera filtrée grâce à un filtre seringue stérile de 0,2 micromètre et ensuite diluée dans une solution pour perfusion intraveineuse de chlorure de sodium de 9 mg/ml (0,9 %) afin d’obtenir un volume total correspondant aux exemples présentés dans le tableau ci-dessous. Notons que le volume de dilution final dépendra de l’état clinique du patient et sera déterminé à la discrétion du médecin. (Si l’utilisation d’un filtre seringue de 0,2 micromètre n’est pas possible, la solution à diluer pour perfusion devra être pré-filtrée avec un filtre de 5 micromètres, diluée, puis administrée par l’intermédiaire d’un filtre de 0,22 micromètre pré-implanté sur la ligne de perfusion).
|
Suggestion d’un profil de dilution respectant la posologie recommandée de 52 mg/m²/jour de clofarabine |
||
|
Surface corporelle (m2) |
Solution à diluer (ml)* |
Volume dilué total |
|
≤ 1,44 |
≤ 74,9 |
100 ml |
|
1,45 à 2,40 |
75,4 à 124,8 |
150 ml |
|
2,41 à 2,50 |
125,3 à 130,0 |
200 ml |
|
* Chaque ml de solution à diluer contient 1 mg de clofarabine. Chaque flacon de 20 ml contient 20 mg de clofarabine. Par conséquent, pour les patients ayant une surface corporelle ≤ 0,38 m2, le contenu partiel d’un seul flacon sera suffisant pour obtenir la posologie quotidienne recommandée de clofarabine. A l’inverse, pour les patients dont la surface corporelle est > 0,38 m2, les contenus de 1 à 7 flacons seront nécessaires pour obtenir la posologie quotidienne recommandée de clofarabine. |
||
Vérifiez que le médicament dilué est bien une solution limpide et incolore. Procédez à une inspection visuelle afin d’éliminer la possibilité de matière particulaire et de décoloration avant de pratiquer l’administration.
Les solutions pour perfusion préparées comme indiqué ci-dessus sont compatibles avec les poches de perfusion en polypropylène et en PVC.
Instructions de manipulation
Les procédures concernant la manipulation des agents antinéoplasiques devront être strictement suivies. Les médicaments cytotoxiques devront être manipulés avec précaution.
Il est recommandé d’utiliser des gants jetables et un équipement de protection pour manipuler CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion. En cas de contact avec les yeux, la peau ou les muqueuses, procédez à un rinçage immédiat et abondant à l’eau claire.
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, ne doit en aucun cas être manipulé par la femme enceinte.
Elimination
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, est à usage unique seulement. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1 RUE DE TURIN
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 550 390 7 5 : Boîte de 1 flacon (verre de type I) de 20 mL.
· 34009 550 390 8 2 : Boîte de 3 flacons (verre de type I) de 20 mL.
· 34009 550 390 9 9 : Boîte de 4 flacons (verre de type I) de 20 mL.
· 34009 550 391 0 5 : Boîte de 10 flacons (verre de type I) de 20 mL.
· 34009 550 391 1 2 : Boîte de 20 flacons (verre de type I) de 20 mL.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament réservé à l’usage hospitalier.
Prescription réservée aux spécialistes en oncologie, en hématologie ou en pédiatrie, ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 04/03/2024
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion
Clofarabine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion ?
3. Comment utiliser CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, contient la substance active clofarabine. La clofarabine fait partie d’une famille de médicaments appelés « médicaments anticancéreux ». La clofarabine permet de stopper la multiplication de globules blancs anormaux et finit par les détruire. Son effet est optimal sur les cellules qui se multiplient très rapidement, comme les cellules cancéreuses.
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, est utilisé pour le traitement de la leucémie aiguë lymphoblastique (LAL) chez les enfants (≥ 1 an), les adolescents et les jeunes adultes jusqu'à 21 ans dont les traitements précédents n’ont pas donné de résultats ou lorsque ces traitements ne sont plus efficaces. La leucémie aiguë lymphoblastique est due à une multiplication anormale de certains types de globules blancs.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion ?
N’utilisez jamais CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion:
· si vous êtes allergique à la clofarabine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous allaitez (merci de lire la rubrique ci-dessous intitulée « Grossesse et allaitement » ;
· si vous souffrez de graves problèmes rénaux (du rein) ou hépatiques (du foie).
Si vous correspondez à l’une de ces situations, parlez-en à votre médecin. Si vous êtes parent d’un enfant traité par CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, vous devez indiquer à votre médecin si l’une de ces situations correspond à votre enfant.
Avertissements et précautions
Adressez-vous à votre médecin avant d’utiliser CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion.
Si vous correspondez à l’une de ces situations, parlez-en à votre médecin. Il est possible que CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, ne soit pas indiqué dans votre situation :
· si vous avez souffert d’une grave réaction après avoir utilisé ce médicament dans le passé;
· si vous souffrez d’un problème rénal (du rein) ou si vous avez déjà souffert d’un tel problème dans le passé ;
· si vous souffrez d’un problème hépatique (du foie) ou si vous avez déjà souffert d’un tel problème dans le passé ;
· si vous souffrez d’un problème cardiaque (du cœur) ou si vous avez déjà souffert d’un tel problème dans le passé.
Indiquez immédiatement à votre médecin ou à la personne qui s’occupe de vous si vous souffrez de l’un des symptômes suivants, auquel cas il est possible que vous deviez interrompre le traitement :
· si vous avez de la fièvre (température élevée) : étant donné que la clofarabine réduit le nombre des cellules sanguines fabriquées par la moelle osseuse, il est possible que vous soyez plus sensible aux infections ;
· si vous avez des difficultés à respirer, si votre respiration est rapide ou si vous êtes essoufflé ;
· si vous constatez un changement de votre rythme cardiaque (battements du cœur) ;
· si vous avez des vertiges (ou des étourdissements) ou si vous vous évanouissez, il est possible que vous souffriez d’une faible tension ;
· si vous avez envie de vomir ou si vous avez la diarrhée (selles molles) ;
· si votre urine est plus sombre que d’habitude : il est important de boire de grandes quantités d’eau pour éviter la déshydratation ;
· si vous présentez une éruption avec des cloques ou des ulcères dans la bouche ;
· si vous perdez l’appétit, si vous avez des nausées (envie de vomir), des vomissements, de la diarrhée, des urines de coloration foncée et des selles de coloration claire, une douleur à l’estomac, une jaunisse (coloration jaune de la peau et des yeux) ou si vous ne vous sentez pas bien dans l’ensemble, cela pourrait être les symptômes d’une inflammation du foie (hépatite) ou d’une atteinte hépatique (insuffisance hépatique) ;
· Si vous n’urinez pas ou peu, ou si vous souffre de somnolence, nausées, vomissements, essoufflements, perte d’appétit et/ou faiblesse (cela peut être les signes d’une insuffisance rénale aiguë/insuffisance rénale).
Si vous êtes parent d’un enfant traité par CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, vous devez indiquer à votre médecin si l’une de ces situations correspond à votre enfant.
Tout au long du traitement par CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, votre médecin procédera à des prises de sang régulières, ainsi qu’à d’autres analyses afin de surveiller votre état de santé. Étant donné la manière dont fonctionne ce médicament, il aura nécessairement des conséquences sur votre sang et sur d’autres organes.
Parlez de contraception avec votre médecin. Les jeunes hommes et les jeunes femmes devront obligatoirement utiliser une contraception efficace pendant et après le traitement. Voir la rubrique intitulée « Grossesse et allaitement » ci-dessous. CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, est susceptible de nuire à la fois aux organes reproducteurs masculins et féminins. Demandez à votre médecin de vous expliquer ce qu’il est possible de faire pour vous protéger ou pour vous permettre d’avoir des enfants.
Enfants et adolescents
Sans objet.
Autres médicaments et CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion
Informez votre médecin si vous utilisez ou avez récemment utilisé :
· médicaments pour les maladies cardiaques (maladies du cœur) ;
· tout médicament susceptible de modifier votre pression sanguine ;
· médicaments ayant des conséquences sur votre foie ou sur vos reins ;
· tout autre médicament, y compris ceux obtenus sans ordonnance.
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
La clofarabine ne peut pas être utilisée au cours de la grossesse, sauf si cela est vraiment nécessaire.
Femmes en âge d’avoir des enfants (qui peuvent tomber enceintes): il est impératif que vous utilisiez une contraception efficace pendant le traitement par clofarabine. et pendant 6 mois après la fin du traitement. La clofarabine peut être nuisible au fœtus lorsqu’elle est utilisée par la femme enceinte. Si vous êtes enceinte ou si vous entamez une grossesse au cours du traitement par clofarabine, parlez-en immédiatement à votre médecin.
Les hommes doivent eux aussi utiliser une contraception efficace et être informés de ne pas concevoir d’enfant pendant le traitement par clofarabine et pendant 3 mois après la fin du traitement.
Si vous allaitez, vous devez cesser d’allaiter votre enfant avant le début du traitement. Il est interdit d’allaiter pendant votre traitement et au cours des 2 semaines qui suivent la fin de votre traitement.
Conduite de véhicules et utilisation de machines
Il est déconseillé de conduire ou d’utiliser certains outils ou machines si vous avez des vertiges, des étourdissements ou des évanouissements.
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion contient du sodium.
Ce médicament contient 70,77 mg de sodium (composant principal du sel de cuisine/table) par flacon. Cela équivaut à 3,54 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
3. COMMENT UTILISER CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion ?
Votre médecin identifiera la dose qu’il vous faut en fonction de votre taille, de votre poids et de votre état de santé. Avant de vous administrer CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, le produit sera dilué dans une solution de chlorure de sodium (sel et eau). Informez votre médecin si vous suivez un régime sans sel car il est possible de devoir modifier la manière dont on vous administrera le traitement.
Votre médecin vous administrera CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, une fois par jour pendant 5 jours. Le médicament vous sera administré par perfusion, c’est-à-dire par l’intermédiaire d’un long tube fin qui vous est introduit dans une veine (goutte à goutte) ou par l’intermédiaire d’un petit dispositif médical inséré sous la peau (port-à-cath ou chambre implantable) si l’on vous en a implanté un (ou à votre enfant). La perfusion dure 2 heures. Si vous (ou votre enfant) pesez moins de 20 kg, le temps de perfusion pourra être plus long.
Le médecin sera chargé de surveiller votre état de santé et de modifier la dose selon votre réponse au traitement. Il est important que vous buviez beaucoup d’eau afin d’éviter la déshydratation.
Si vous avez utilisé plus de CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion que vous n’auriez dû
Si vous pensez que vous avez peut-être reçu trop de médicament, parlez-en à votre médecin immédiatement.
Si vous oubliez d’utiliser CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion
Votre médecin vous indiquera quand recevoir ce médicament. Si vous pensez que vous avez manqué une dose, parlez-en à votre médecin immédiatement.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin.
Si vous arrêtez d’utiliser CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Très fréquents (peuvent toucher plus d’1 patient sur 10) :
· anxiété, maux de tête, fièvre, fatigue ;
· nausées (se sentir barbouillé) et vomissements, diarrhée (selles molles) ;
· rougeurs de la peau, démangeaisons et inflammation de la peau, inflammation des parois muqueuses comme la bouche et d’autres parties du corps ;
· il est possible que vous souffriez de plus d’infections que d’habitude étant donné que CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, peut diminuer le nombre de certains types de cellules sanguines de votre corps ;
· éruptions cutanées pouvant entraîner des démangeaisons, des rougeurs, des douleurs ou une desquamation, notamment au niveau de la paume des mains et de la plante des pieds, ainsi que l’apparition de petits boutons rougeâtres ou violacés sous la peau.
Fréquents (peuvent toucher jusqu’à 1 patient sur 10) :
· infections du sang, pneumonie, zona, contamination de l’implant, infections de la bouche comme le muguet et les boutons de fièvre ;
· changements de vos paramètres sanguins, modifications des globules blancs ;
· réactions allergiques ;
· sensation de soif et urines plus sombres ou moins abondantes que d’habitude, diminution ou perte de l’appétit, perte de poids ;
· agitation, irritabilité ou nervosité ;
· se sentir engourdi ou faible dans les bras et les jambes, engourdissement de la peau, somnolence, vertiges, tremblements ;
· problèmes d’audition ;
· amas d’eau autour du cœur, pouls (battements du cœur) rapide ;
· tension faible, bosse due à un mauvais bleu ;
· rupture de petits vaisseaux sanguins, respiration rapide, saignements de nez, difficultés à respirer, essoufflement, toux ;
· vomissements de sang, douleurs à l’estomac, douleur dans le bas du dos ;
· saignements à l’intérieur de la tête, de l’estomac, des intestins ou des poumons, de la bouche ou des gencives, aphtes, inflammation de la muqueuse buccale ;
· jaunissement de la peau et des yeux (également connu sous le nom de jaunisse) ou autres troubles hépatiques (du foie) ;
· bleus, perte des cheveux, changements de la couleur de la peau, augmentation de la quantité de sueur, sécheresse de la peau ou autres problèmes de peau ;
· douleur dans la poitrine ou dans les os, douleur au niveau du cou ou du dos, douleur dans les membres, les muscles ou les articulations ;
· sang dans les urines ;
· défaillance de certains organes, douleur, augmentation de la tension musculaire, rétention d’eau et tuméfaction (gonflement) de certaines parties du corps, notamment les bras et les jambes, changements de l’état mental, sensation de chaleur, de froid ou sensation bizarre ;
· la clofarabine est susceptible d’avoir des conséquences sur les concentrations de certaines substances du sang. Votre médecin procédera donc à des prises de sang régulières afin de vérifier que votre corps fonctionne correctement ;
· atteinte hépatique (insuffisance hépatique) ;
· une quantité d'urine diminuée ou un arrêt total de l'urine, somnolence, nausées, vomissements, essoufflements, perte d’appétit et/ou faiblesse (cela peut être les signes d’une insuffisance rénale aigüe/insuffisance rénale).
Effets indésirables peu fréquents (peuvent toucher plus d’1 patient sur 100) :
· inflammation du foie (hépatite).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette du flacon et sur la boîte après EXP.
Ne pas congeler.
Une fois préparé et dilué, CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, doit être utilisé immédiatement ou dans les 24 heures s’il est conservé au réfrigérateur (entre 2°C et 8°C).
Ne jetez aucun médicament au tout-à-l’égout. Demandez à votre pharmacien comment éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion
· La substance active est la clofarabine
Chaque ml contient 1 mg de clofarabine.
Chaque flacon de 20 ml contient 20 mg de clofarabine.
· Les autres composants sont : le chlorure de sodium et de l’eau pour préparations injectables.
CLOFARABINE VIATRIS est une solution à diluer pour perfusion limpide, presqu’incolore, qui doit être reconstituée et diluée avant utilisation.
Ce médicament est disponible en flacons en verre de 20 mL. Chaque boîte contient 1, 3, 4, 10 ou 20 flacons.
Titulaire de l’autorisation de mise sur le marché
1 RUE DE TURIN
69007 LYON
Exploitant de l’autorisation de mise sur le marché
1 RUE DE TURIN
69007 LYON
MICROWEG 22
6545 CM, NIJMEGEN
PAYS-BAS
OU
SYNTHON HISPANIA S.L.
CASTELLO 1, POLIGONO LAS SALINAS
08830 SANT BOI DE LLOBREGAT
ESPAGNE
OU
SYNTHON S.R.O.
BRNENSKA 32/CP.
597, BLANSKO, 678 01
REPUBLIQUE TCHEQUE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
CLOFARABINE VIATRIS contient la même substance active et agit de la même façon qu’un médicament de référence déjà autorisé dans l’Union européenne. Une autorisation de mise sur le marché sous circonstances exceptionnelles a été délivrée pour le médicament de référence de CLOFARABINE VIATRIS.
Cela signifie qu’en raison de la rareté de cette maladie, il n’a pas été possible d’obtenir des informations complètes concernant le médicament de référence.
L’Agence européenne des médicaments réévaluera chaque année toute nouvelle information qui pourrait être disponible, et toute mise à jour pour le médicament de référence sera aussi incluse dans l’information de CLOFARABINE VIATRIS, dont cette notice.
---------------------------------------------------------------------------------------------------------------------------------------
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Précautions particulières d’administration
CLOFARABINE VIATRIS 1 mg/ml, solution à diluer pour perfusion, doit être reconstitué avant administration. La solution sera filtrée grâce à un filtre seringue stérile de 0,2 micromètre et ensuite diluée dans une solution pour perfusion intraveineuse de chlorure de sodium de 9 mg/ml (0,9 %) afin d’obtenir un volume total correspondant aux exemples présentés dans le tableau ci-dessous. Notons que le volume de dilution final dépendra de l’état clinique du patient et sera déterminé à la discrétion du médecin. (Si l’utilisation d’un filtre seringue de 0,2 micromètre n’est pas possible, la solution à diluer pour perfusion devra être pré-filtrée avec un filtre de 5 micromètres, diluée, puis administrée par l’intermédiaire d’un filtre de 0,22 micromètre pré-implanté sur la ligne de perfusion).
|
Suggestion d’un profil de dilution respectant la posologie recommandée de 52 mg/m²/jour de clofarabine |
||
|
Surface corporelle (m2) |
Solution à diluer (ml)* |
Volume dilué total |
|
≤ 1,44 |
≤ 74,9 |
100 ml |
|
1,45 à 2,40 |
75,4 à 124,8 |
150 ml |
|
2,41 à 2,50 |
125,3 à 130,0 |
200 ml |
|
* Chaque ml de solution à diluer contient 1 mg de clofarabine. Chaque flacon de 20 ml contient 20 mg de clofarabine. Par conséquent, pour les patients ayant une surface corporelle ≤ 0,38 m2, le contenu partiel d’un seul flacon sera suffisant pour obtenir la posologie quotidienne recommandée de clofarabine. A l’inverse, pour les patients dont la surface corporelle est > 0,38 m2, les contenus de 1 à 7 flacons seront nécessaires pour obtenir la posologie quotidienne recommandée de clofarabine. |
||
Vérifiez que le médicament dilué est bien une solution limpide et incolore. Procédez à une inspection visuelle afin d’éliminer la possibilité de matière particulaire et de décoloration avant de pratiquer l’administration.
Le médicament dilué est chimiquement et physiquement stable pendant 3 jours à une température comprise entre 2°C et 8°C et à température ambiante (jusqu’à 25°C). D’un point de vue microbiologique, il doit être utilisé immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation après dilution relèvent de la seule responsabilité de l’utilisateur et ne devraient normalement pas dépasser 24 heures à une température comprise entre 2°C et 8°C sauf en cas de dilution réalisée en conditions aseptiques dûment contrôlées et validées. Ne pas congeler.
Instructions de manipulation
Les procédures concernant la manipulation des agents antinéoplasiques devront être strictement suivies. Les médicaments cytotoxiques devront être manipulés avec précaution.
Il est recommandé d’utiliser des gants jetables et un équipement de protection pour manipuler clofarabine. En cas de contact avec les yeux, la peau ou les muqueuses, procédez à un rinçage immédiat et abondant à l’eau claire.
Clofarabine ne doit en aucun cas être manipulé par la femme enceinte.
Dispositif
Clofarabine est à usage unique seulement. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.