Dernière mise à jour le 03/08/2026
RIASTAP 1 g, poudre pour solution injectable/perfusion
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : B02BB01
Qu’est-ce que RIASTAP ?
RIASTAP contient du fibrinogène humain qui est une protéine importante de la coagulation sanguine. Le déficit en fibrinogène signifie que le sang ne coagule pas aussi rapidement qu'il le devrait, ce qui a pour conséquence une plus grande tendance au saignement. Le remplacement du fibrinogène humain par RIASTAP rétablira les mécanismes de coagulation.
Dans quel cas est utilisé RIASTAP ?
RIASTAP est utilisé pour le traitement de l’hémorragie chez les patients présentant un déficit congénital en fibrinogène (hypo- ou afibrinogénémie) avec une tendance au saignement.
Présentations
> 1 flacon(s) en verre muni d'un bouchon sans latex (caoutchouc bromobutyle), serti d'une capsule en aluminium surmontée d'un disque en plastique avec 1 filtre et 1 dispositif de prélèvement
Code CIP : 494 884-2 ou 34009 494 884 2 1
Déclaration de commercialisation : 19/08/2011
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 09/03/2011 | Inscription (CT) | Le service médical rendu par cette spécialité est important. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| I (Majeur) | Avis du 09/03/2011 | Inscription (CT) | RIASTAP partage l'amélioration du service médical rendu majeure (niveau I) de CLOTTAFACT® dans la prise en charge des patients atteints d'un déficit constitutionnel en fibrinogène. |
Autres informations
- Titulaire de l'autorisation : CSL BEHRING GmbH
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 117 966 0
ANSM - Mis à jour le : 24/09/2024
RIASTAP 1g, poudre pour solution injectable/perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
RIASTAP se présente sous la forme d’une poudre pour solution injectable ou pour perfusion contenant 1 g de fibrinogène humain par flacon.
Le produit reconstitué avec 50 ml d’eau pour préparations injectables contient approximativement 20 mg/ml de fibrinogène humain.
Le contenu de fibrinogène est déterminé conformément à la monographie de la Pharmacopée Européenne pour fibrinogène humain.
Excipient(s) à effet notoire : Sodium jusqu’à 164 mg (7,1 mmol) par flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution injectable/ perfusion.
Poudre blanche.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Posologie
La posologie et la durée du traitement de substitution dépendent de la sévérité des troubles, de l’importance et du lieu de l’hémorragie, et de l’état clinique du patient.
La concentration en fibrinogène (fonctionnel) doit être déterminée afin de calculer la posologie individuelle. La posologie et la fréquence de l'administration doivent être adaptées de façon individuelle grâce à des dosages plasmatiques réguliers en fibrinogène et au contrôle continu de l'état clinique du patient et des autres traitements de substitution utilisés.
Les concentrations plasmatiques normales en fibrinogène sont comprises entre 1,5 à 4,5 g/l. Le seuil critique de fibrinogène plasmatique au-dessous duquel les hémorragies peuvent survenir est approximativement de 0,5 à 1,0 g/l. En cas d'intervention chirurgicale majeure, la surveillance étroite du traitement de substitution par des tests de coagulation est essentielle.
Dose initiale :
Si la concentration en fibrinogène du patient n'est pas connue, la dose recommandée est de 70 mg par kilogramme de poids corporel administrée par voie intraveineuse.
Dose suivante :
La concentration cible (1 g/l) pour des événements mineurs (par exemple épistaxis, saignement intramusculaire ou ménorragie) doit être maintenue pendant au moins trois jours. La concentration cible (1,5 g/l) pour des événements majeurs (par exemple traumatisme crânien ou hémorragie intracrânienne) doit être maintenue pendant sept jours.
|
Dose de fibrinogène = |
[Concentration cible (g/l) – Concentration mesurée (g/l)] |
|
(mg/kg de poids corporel) |
0,017 (g/l par mg/kg de poids corporel)
|
Posologie pour les nouveau-nés, les enfants en bas âge et les enfants :
Les données disponibles obtenues lors des études cliniques concernant les posologies de Riastap chez l’enfant sont limitées. Compte tenu de ces études et de la longue expérience clinique avec les spécialités contenant du fibrinogène, la posologie recommandée dans le traitement chez l’enfant est la même que celle chez l’adulte.
Mode d’administration
Injection ou perfusion intraveineuse.
RIASTAP doit être reconstitué selon les modalités décrites dans la rubrique 6.6. Avant administration, la solution reconstituée doit être amenée à la température ambiante ou corporelle, puis être injectée ou perfusée lentement à un débit confortable pour le patient. Le débit d'injection ou de perfusion ne doit pas dépasser approximativement 5 ml par minute.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Chez les patients présentant des antécédents de maladie coronarienne ou d'infarctus du myocarde, les patients présentant une affection hépatique, les patients en situation péri- ou post opératoires, chez les nouveau-nés, les patients à risque thromboembolique ou de coagulation intravasculaire disséminée (CIVD), le bénéfice potentiel d’un traitement par un concentré de fibrinogène humain devra être évalué par rapport au risque de complications thromboemboliques. Une étroite surveillance devra également être mise en place.
En cas d’allergie ou de réaction anaphylactique, l’administration devra être immédiatement interrompue. En cas de choc anaphylactique, le traitement médical standard du choc devra être instauré.
Dans le cas de traitement de substitution avec des facteurs de coagulation dans d'autres insuffisances congénitales, la formation d'anticorps neutralisant a été observée, mais il n'existe actuellement aucune donnée dans le cas du fibrinogène.
RIASTAP contient jusqu’à 164 mg (7,1 mmol) de sodium par flacon, soit 11,5 mg (0,5 mmol) de sodium par kg de poids corporel chez un patient recevant la posologie initiale recommandée de 70 mg/kg de poids corporel. Ceci est à prendre en considération chez les patients suivant un régime hyposodé.
Sécurité virale
Les mesures habituelles de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection clinique des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de plasma ainsi que la mise en œuvre dans le procédé de fabrication d’étapes efficaces pour l’inactivation/élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents ou d’autres types d’agents infectieux.
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le VIH, le VHB et VHC ainsi que contre les virus non enveloppés comme le VHA et le parvovirus B19.
Une vaccination appropriée (hépatite A et B) doit être envisagée chez les patients recevant régulièrement ou de façon réitérée des produits dérivés du plasma humain.
Il est fortement recommandé que chaque fois que Riastap est administré à un patient, le nom et le numéro de lot du produit soient enregistrés afin de maintenir la traçabilité entre le patient et le lot du médicament.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
4.6. Fertilité, grossesse et allaitement
Grossesse
Aucune étude de reproduction animale n’a été menée avec RIASTAP (voir rubrique 5.3). Compte tenu de l’origine humaine de la substance active, celle-ci est métabolisée de la même manière que les protéines propres au patient. S’agissant de constituants physiologiques du sang humain, aucun effet secondaire sur la reproduction ou le fœtus n’est attendu.
L’innocuité de RIASTAP au cours de la grossesse n’a pas été évaluée par des essais cliniques contrôlés.
L’expérience clinique acquise avec le concentré de fibrinogène dans le traitement des complications obstétricales indique qu’aucun effet nocif sur l’évolution de la grossesse, le développement du fœtus ou du nouveau-né n’est attendu.
On ne sait pas si RIASTAP est excrété dans le lait humain. L’utilisation de RIASTAP chez la femme allaitante n’a pas été évaluée par des essais cliniques.
Le risque de transmission à l’enfant allaité n’est pas exclu. La décision d’arrêter l’allaitement, ou d’arrêter ou de suspendre momentanément le traitement par RIASTAP doit prendre en compte le bénéfice de l’allaitement pour l’enfant et le bénéfice du traitement pour la mère.
Fertilité
Aucune donnée n’est disponible.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Tableau résumant les effets indésirables
Ce tableau combine les effets indésirables observés dans les essais cliniques et l'expérience post-commercialisation. Les fréquences présentées dans le tableau ont été fondées sur des analyses regroupées de deux essais cliniques, promus par la firme et contrôlés par un placebo, réalisés en chirurgie aortique avec ou sans autres interventions chirurgicales [BI3023-2002 (n=61) et BI3023_3002 (n=152)], selon la convention suivante : très fréquent (≥1/10) ; fréquent (≥1/100 à <1/10) ; peu fréquent (≥1/1.000 à <1/100) ; rare (≥1/10.000 à <1/1.000) ; très rare (<1/10.000). Pour les effets indésirables spontanés post-commercialisation, la fréquence des rapports est classée comme indéterminée.
Compte tenu du fait que ces essais ont été réalisés uniquement sur la population étroite de la chirurgie aortique, les taux d’effets indésirables observés dans ces essais peuvent ne pas refléter les taux observés en pratique clinique et sont inconnus pour les paramètres cliniques en dehors de l’indication étudiée.
|
Classe des systèmes d’organe (SOC, MedDRA) |
Effets indésirables |
Fréquence (en chirurgie aortique avec ou sans d'autres interventions chirurgicales) |
|
Troubles généraux et au site d'administration |
Fièvre |
Très fréquent |
|
Troubles du système immunitaire |
Réaction anaphylactique (incluant le choc anaphylactique) |
Peu fréquent |
|
|
Réactions allergiques (y compris urticaire généralisée, éruption cutanée, dyspnée, tachycardie, nausées, vomissements, frissons, fièvre, douleurs à la poitrine, toux, diminution de la pression artérielle) |
Indéterminée |
|
Troubles vasculaires |
Evènements thromboemboliques* (voir la rubrique 4.4) |
Fréquent** |
* Des cas isolés ont été mortels.
** Basé sur les résultats de deux essais cliniques (chirurgie aortique avec ou sans autres procédures chirurgicales), le taux d'incidence globale des évènements thromboemboliques était plus faible chez les sujets traités par fibrinogène (n=8, 7,4%) par rapport au placebo (n=11, 10,4%).
Pour la sécurité relative aux agents transmissibles, voir la rubrique 4.4.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En cas de surdosage, le risque de développer des complications thromboemboliques est augmenté.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antihémorragique, fibrinogène humain, code ATC : B02BB01.
Le fibrinogène humain (facteur I de coagulation), en présence de thrombine, de facteur XIII activé et d’ions calcium, forme un caillot hémostatique de fibrine tridimensionnel, stable et élastique.
L'administration d’un concentré de fibrinogène humain génère une augmentation de la concentration en fibrinogène plasmatique circulant et peut temporairement corriger le défaut de coagulation des patients déficients en fibrinogène.
L’étude pivot de phase II a évalué la pharmacocinétique en dose unique (voir rubrique 5.2 Propriétés Pharmacocinétiques) et a également fourni des données d'efficacité par l’intermédiaire du critère de fermeté maximale du caillot (FMC) et des données de sécurité.
Pour chaque sujet, le FMC a été déterminé avant (valeur de référence) et une heure après l’administration d’une dose unique de 70 mg/kg de poids corporel de RIASTAP. RIASTAP s’est révélé efficace dans l’augmentation de la stabilité du caillot mesurée par thromboélastométrie chez des patients ayant un déficit congénital en fibrinogène (afibrinogénémie). L'efficacité hémostatique dans les épisodes de saignements aigus et sa corrélation avec le FMC sont en cours de vérification dans une étude post-marketing.
5.2. Propriétés pharmacocinétiques
Le produit est administré par voie intraveineuse et est immédiatement disponible dans le plasma à une concentration correspondante à la posologie administrée.
Une étude pharmacocinétique a évalué la pharmacocinétique à dose unique avant et après l'administration de concentré humain de fibrinogène chez les sujets avec afibrinogénémie. Cette étude prospective, en ouvert, non contrôlée et multicentrique a inclus 5 femmes et 10 hommes âgés de 8 à 61 ans (2 enfants, 3 adolescents, 10 adultes). La dose médiane était de 77,0 mg/kg de poids corporel (intervalle compris entre 76,6 – 77,4 mg/kg).
Du sang a été prélevé chez 15 sujets (14 mesurables) pour déterminer l'activité de fibrinogène de base et jusqu'à 14 jours après la fin de la perfusion. En outre, la récupération incrémentale in vivo (RIV), définie comme l’augmentation maximale des concentrations plasmatiques en fibrinogène mesurée par mg/kg de poids corporel a été déterminée par paliers jusqu'à 4 heures après la perfusion. La récupération incrémentale in vivo médiane était de 1,7 (intervalle compris entre 1,30-2,73) mg/dl par mg/kg de poids corporel. Le tableau suivant fournit les résultats pharmacocinétiques.
Résultats pharmacocinétiques pour l’activité du fibrinogène
|
Paramètre (n=14) |
Moyenne ± SD |
Médiane (intervalle) |
|
t1/2 [h] |
78,7 ± 18,13 |
77,1 (55,73-117,26) |
|
Cmax [g/l] |
1,4 ± 0,27 |
1,3 (1,00-2,10) |
|
ASC par dose de 70 mg/kg [hmg/ml] |
124,3 ± 24,16 |
126,8 (81,73-156,40) |
|
Partie extrapolée d’ASC [%] |
8,4 ± 1,72 |
7,8 (6,13-12,14) |
|
Cl [ml/h/kg] |
0,59 ± 0,13 |
0,55 (0,45-0,86) |
|
TMR [h] |
92,8 ± 20,11 |
85,9 (66,14-126,44) |
|
Vss [ml/kg] |
52,7 ± 7,48 |
52,7 (36,22-67,67) |
|
RIV [mg/dl par mg/kg de poids corporel] |
1,8 ± 0,35 |
1,7 (1,30-2,73) |
t1/2 = demi-vie terminale d'élimination
h = heure
Cmax = concentration maximale dans un délai de 4 heures
ASC = aire sous la courbe
Cl = clairance
TMR = temps moyen résiduel
Vss = volume de distribution à l’état d’équilibre
SD = Ecart type
RIV = récupération in vivo
5.3. Données de sécurité préclinique
Des études précliniques par administrations répétées (toxicité chronique, cancérogénicité et mutagénicité) ne peuvent pas être convenablement réalisées dans les modèles animaux conventionnels du fait du développement d’anticorps lors de l'administration des protéines humaines hétérologues.
La stabilité physico-chimique du produit reconstitué a été démontrée pendant 8 heures à température ambiante (max. + 25 °C). D’un point de vue microbiologique, le produit doit être utilisé immédiatement après reconstitution. Cependant, si le produit n’est pas administré immédiatement, la durée de conservation ne doit pas dépasser 8 heures à température ambiante (max. + 25 °C). La solution reconstituée ne doit pas être conservée au réfrigérateur.
6.4. Précautions particulières de conservation
À conserver à une température ne dépassant pas 25°C.
Ne pas congeler.
Conserver le flacon dans l'emballage extérieur à l'abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon (verre type II, incolore) muni d’un bouchon sans latex (caoutchouc bromobutyle), serti d’une capsule en aluminium surmontée d’un disque en plastique.
Boîte de 1 g (Figure 1)
1. Un flacon contenant 1 g de fibrinogène humain
2. Filtre : Filtre seringue Pall®
3. Dispositif de prélèvement : dispositif de prélèvement Mini-Spike®
 Figure 1
Figure 1
6.6. Précautions particulières d’élimination et de manipulation
Instructions générales
· La reconstitution et le prélèvement doivent être effectués en conditions aseptiques.
· Les produits reconstitués doivent être inspectés visuellement avant administration pour mettre en évidence la présence éventuelle de particules ou un changement de coloration.
· La solution est incolore à jaunâtre, claire à légèrement opalescente et de pH neutre. N’utilisez pas de solution trouble ou contenant des dépôts.
Reconstitution
· Amener le flacon de solvant et celui de poudre non ouverts à la température de la pièce ou à la température corporelle (ne pas dépasser +37°C).
· RIASTAP doit être reconstitué avec de l’eau pour préparations injectables (50 ml, non fourni).
· Se laver les mains ou mettre des gants avant de reconstituer le produit.
· Retirer la capsule protectrice du flacon de RIASTAP pour découvrir la partie centrale du bouchon de perfusion.
· Nettoyer la surface du bouchon de perfusion avec une solution antiseptique et laisser sécher.
· Transférer le solvant dans le flacon à l’aide d’un dispositif de transfert approprié. S’assurer de l'imprégnation complète de la poudre.
· Remuer le flacon avec un léger mouvement de rotation jusqu'à dissolution complète de la poudre et obtention de la solution prête à administrer. Eviter de secouer le flacon car cela pourrait générer l’apparition de mousse. Généralement, la poudre se dissout en 5 minutes environ. Cela ne devrait pas prendre plus de 15 minutes pour se dissoudre complétement.
· Ouvrir le blister en plastique contenant le dispositif de prélèvement (dispositif de prélèvement Mini-Spike®) fourni avec RIASTAP (Figure 2).
 Figure 2
Figure 2
· Prendre le dispositif de prélèvement fourni et l’insérer dans le bouchon du flacon contenant le produit reconstitué (Figure 3).
 Figure 3
Figure 3
· Après avoir inséré le dispositif de prélèvement, retirer le capuchon. Après avoir retiré le capuchon, ne pas toucher la surface exposée.
· Ouvrir le blister du filtre (Filtre seringue Pall®) fourni avec RIASTAP (Figure 4).
 Figure 4
Figure 4
· Visser la seringue sur le filtre (Figure 5).
 Figure 5
Figure 5
· Visser la seringue avec le filtre sur le dispositif de prélèvement (Figure 6).
 Figure 6
Figure 6
· Aspirer le produit reconstitué dans la seringue (Figure 7).
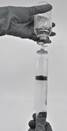 Figure 7
Figure 7
· Une fois terminé, retirer le filtre, le dispositif de prélèvement et le flacon vide de la seringue, les jeter selon la réglementation en vigueur et procéder à l’administration comme d’habitude.
· Le produit reconstitué doit être administré immédiatement par une ligne séparée d’injection / de perfusion.
· Eviter que du sang ne pénètre dans les seringues remplies de produit.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
EMIL-VON-BEHRING-STRASSE 76
35041 MARBURG
ALLEMAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 494 884 2 1 : Flacon (verre type II, incolore) muni d’un bouchon sans latex (caoutchouc bromobutyle), serti d’une capsule en aluminium surmontée d’un disque en plastique. Boîte contenant 1 flacon, 1 filtre et 1 dispositif de prélèvement.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 24/09/2024
RIASTAP 1 g, poudre pour solution injectable/ perfusion
Fibrinogène humain
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que RIASTAP 1 g, poudre pour solution injectable/ perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser RIASTAP 1 g, poudre pour solution injectable/ perfusion ?
3. Comment utiliser RIASTAP 1 g, poudre pour solution injectable/ perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver RIASTAP 1 g, poudre pour solution injectable/ perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE RIASTAP 1 g, poudre pour solution injectable/ perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : B02BB01
Qu’est-ce que RIASTAP ?
RIASTAP contient du fibrinogène humain qui est une protéine importante de la coagulation sanguine. Le déficit en fibrinogène signifie que le sang ne coagule pas aussi rapidement qu'il le devrait, ce qui a pour conséquence une plus grande tendance au saignement. Le remplacement du fibrinogène humain par RIASTAP rétablira les mécanismes de coagulation.
Dans quel cas est utilisé RIASTAP ?
RIASTAP est utilisé pour le traitement de l’hémorragie chez les patients présentant un déficit congénital en fibrinogène (hypo- ou afibrinogénémie) avec une tendance au saignement.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER RIASTAP 1 g, poudre pour solution injectable/ perfusion ?
N’utilisez jamais RIASTAP 1 g, poudre pour solution injectable/ perfusion :
· si vous êtes allergique au fibrinogène humain ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Veuillez informer votre médecin si vous êtes allergique à un médicament ou à un aliment.
Avertissements et précautions
· Si vous avez présenté des réactions allergiques à RIASTAP dans le passé. Des antihistaminiques et des corticostéroïdes peuvent être administrés en prophylaxie si votre médecin vous les prescrit.
· Lorsque des réactions allergiques ou de type anaphylactiques surviennent (une réaction allergique grave peut entraîner de sévères difficultés à respirer ou des vertiges). L’administration de RIASTAP devra être interrompue immédiatement.
· en raison du risque accru de coagulation dans les vaisseaux sanguins (thrombose), en particulier :
o en cas de dose élevée ou d’administration répétée
o si vous avez des antécédents d’attaque cardiaque (maladie cardiaque coronaire ou d'infarctus du myocarde)
o si vous présentez une maladie du foie
o si vous venez de subir une opération chirurgicale (patients en postopératoire)
o si vous allez bientôt subir une opération chirurgicale (patients en préopératoire)
o chez les nouveau-nés
o si vous avez des prédispositions à former des caillots de sang plus que la normale (patients à risque thromboembolique ou à risque de coagulation intravasculaire disséminée).
Votre médecin prendra soigneusement en compte le bénéfice du traitement avec RIASTAP par rapport au risque de ces complications.
Sécurité virale
Les mesures habituelles de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent :
· la sélection clinique des donneurs de plasma et de sang pour exclure le risque de transmission d’infection,
· la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de plasma.
Les fabricants de ces produits ont aussi inclus dans le procédé de fabrication des étapes efficaces d’inactivation et d’élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents ou d’autres types d’agents infectieux.
Les mesures mises en place sont considérées efficaces vis-à-vis des virus enveloppés comme le virus de l’immunodéficience humaine (VIH, les virus du SIDA), le virus de l’hépatite B et celui de l’hépatite C (inflammation du foie) ainsi que contre les virus non enveloppés comme le virus de l’hépatite A (inflammation du foie) et le parvovirus B19.
Votre médecin peut vous recommander de vous vacciner contre les hépatites A et B si vous recevez régulièrement/de façon répétée des médicaments dérivés du plasma humain.
Il est fortement recommandé que chaque fois que RIASTAP est administré, votre médecin enregistre la date d’administration, le numéro du lot et le volume injecté.
Autres médicaments et RIASTAP 1 g, poudre pour solution injectable/ perfusion
· Veuillez indiquer à votre médecin ou à votre pharmacien si vous prenez ou avez pris récemment un autre médicament, même s'il s'agit d'un médicament obtenu sans ordonnance.
· RIASTAP ne doit pas être mélangé avec d’autres médicaments, à l’exception de ceux mentionnés dans la rubrique « Les informations suivantes sont destinées exclusivement aux professionnels de santé : ».
· Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou vous planifiez de l’être, demandez conseil à votre médecin ou à votre pharmacien avant de prendre ce médicament
· Au cours de la grossesse et de l’allaitement, RIASTAP ne doit être utilisé qu’en cas de nécessité absolue.
Conduite de véhicules et utilisation de machines
RIASTAP n’a pas d’effet ou qu’un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
RIASTAP 1 g, poudre pour solution injectable/ perfusion contient du sodium
RIASTAP contient jusqu’à 164 mg (7,1 mmol) de sodium par flacon, soit 11.5 mg (0,5 mmol) de sodium par kg de poids corporel chez un patient recevant la posologie initiale de 70 mg/kg de poids corporel. Veuillez le prendre en considération si vous suivez un régime hyposodé.
3. COMMENT UTILISER RIASTAP 1 g, poudre pour solution injectable/ perfusion ?
Le traitement doit être initié et surveillé par un médecin expérimenté dans ce type de maladie.
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Posologie
La dose de fibrinogène humain dont vous avez besoin et la durée du traitement dépendront de :
· la sévérité de la maladie
· la localisation et l’importance de l’hémorragie
· l’état clinique du patient
Mode d’administration
Si vous avez toute autre question sur l'utilisation de ce médicament, demandez conseil à votre médecin ou à votre pharmacien (voir rubrique « Les informations suivantes sont destinées exclusivement aux professionnels de santé ").
Si vous avez utilisé plus de RIASTAP 1 g, poudre pour solution injectable/ perfusion que vous n’auriez dû
Votre médecin surveillera régulièrement votre coagulation sanguine pendant le traitement. En cas de surdosage, le risque pour développer des complications thromboemboliques est augmenté.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Veuillez informer immédiatement votre médecin :
· si vous présentez un des effets indésirables
· si vous remarquez des effets indésirables non mentionnés dans cette notice.
L’effet indésirable suivant a été observé très fréquemment (pouvant affecter plus d’1 patient sur 10 :
· augmentation de la température corporelle
L'effet indésirable suivant a été observé peu fréquemment (pouvant affecter jusqu'à 1 personne sur 100) :
· Une réaction allergique soudaine (tels que rougeur de la peau, des éruptions cutanées sur tout le corps, chute de la pression artérielle, difficulté à respirer).
L'effet indésirable suivant a été observé fréquemment (pouvant affecter jusqu'à 1 personne sur 10, mais l'incidence a été plus élevée chez les patients ne recevant pas de fibrinogène) :
· Risque d'augmentation de la formation de caillots sanguins (voir la rubrique 2 « Quelles sont les informations à connaitre avant d’utiliser RIASTAP 1 g, poudre pour solution injectable/ perfusion ? »).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER RIASTAP 1 g, poudre pour solution injectable/ perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois. À conserver à une température ne dépassant pas 25°C.
Ne pas congeler.
Conserver le flacon dans l'emballage extérieur à l'abri de la lumière.
La solution reconstituée doit, de préférence, être utilisée immédiatement.
Cependant, si le produit reconstitué n’est pas administré immédiatement, la durée de conservation ne doit pas dépasser 8 heures à température ambiante (maximum + 25°C).
La solution reconstituée ne doit pas être conservée au réfrigérateur.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient RIASTAP 1 g, poudre pour solution injectable/ perfusion
· La substance active est :
Fibrinogène humain (1 g/flacon ; après reconstitution avec 50 ml d’eau pour préparations injectables, approximativement 20 mg/ml).
Pour plus d’informations, voir la rubrique « Les informations suivantes sont destinées exclusivement aux professionnels de santé ».
· Les autres composants sont :
Albumine humaine, Chlorure de sodium, Chlorhydrate de L-arginine, citrate de sodium déshydraté, Hydroxyde de sodium (pour l’ajustement du pH).
Voir le dernier paragraphe de la rubrique 2 « Les informations importantes concernant certains composants de RIASTAP 1 g ».
RIASTAP se présente sous forme d’une poudre blanche.
Après reconstitution avec de l’eau pour préparations injectables, le produit doit être claire à légèrement opalescent, des bulles peuvent être observées à la lumière mais la solution ne doit pas contenir de particules.
Présentation
Boîte de 1 g (Figure 1)
1. Un flacon contenant1 g de fibrinogène humain.
2. Filtre : Filtre seringue Pall®
3. Dispositif de prélèvement : Dispositif de prélèvement Mini-Spike®
 Figure 1
Figure 1
Titulaire de l’autorisation de mise sur le marché
EMIL-VON-BEHRING STRASSE 76
35041 MARBURG
ALLEMAGNE
Exploitant de l’autorisation de mise sur le marché
CARRE SUFFREN
31-35 RUE DE LA FEDERATION
75015 PARIS
FRANCE
EMIL-VON-BEHRING STRASSE 76
35041 MARBURG
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé:
Posologie
La concentration (fonctionnelle) en fibrinogène doit être déterminée afin de calculer la posologie individuelle. La posologie et la fréquence de l'administration doivent être déterminées sur une base individuelle par la mesure régulière de la concentration plasmatique en fibrinogène et le contrôle continu de l'état clinique du patient et d’autres traitements de substitution utilisés.
Les valeurs normales sont comprises entre 1,5 à 4,5 g/l. Le seuil critique de fibrinogène plasmatique au-dessous duquel les hémorragies peuvent se produire est de 0,5 à 1,0 g/l. En cas d'intervention chirurgicale majeure, la surveillance précise du traitement de substitution par des analyses de coagulation est essentielle.
Dose initiale :
Si la concentration en fibrinogène du patient n'est pas connue, la dose recommandée est de 70 mg par kilogramme de poids corporel administrée par voie intraveineuse.
Dose suivante :
La concentration cible (1g/l) pour des événements mineurs (par exemple épistaxis, saignement intramusculaire ou ménorragie) devrait être maintenue pendant au moins trois jours.
La concentration cible (1,5 g/l) pour des événements majeurs (par exemple le trauma crânien ou l'hémorragie intracrânienne) devrait être maintenue pendant sept jours.
|
Dose de fibrinogène = |
[Concentration cible (g/l) – Concentration mesurée (g/l)](g/l)] |
|
(mg/kg de poids corporel) |
0,017 (g/l par mg/kg de poids corporel) |
Posologie pour les nouveau-nés, les enfants en bas âge et les enfants :
Les données disponibles obtenues lors des études cliniques portant sur les posologies de RIASTAP chez l’enfant sont limitées. Compte tenu de ces études et de la longue expérience clinique avec les spécialités contenant du fibrinogène, la posologie recommandée dans le traitement chez l’enfant est la même que celle chez l’adulte.
Mode et voie d’administration
Instructions générales
· La reconstitution et le prélèvement doivent être effectués en conditions aseptiques.
· Les produits reconstitués doivent être inspectés visuellement avant administration pour mettre en évidence la présence éventuelle de particules ou un changement de coloration.
· La solution est incolore à jaunâtre, claire à légèrement opalescente et de pH neutre. N’utilisez pas de solutions troubles ou contenant des dépôts.
Reconstitution
· Amener le flacon de solvant et celui de poudre non ouverts à la température de la pièce ou à la température corporelle (ne pas dépasser +37°C)
· Riastap doit être reconstitué avec de l’eau pour préparations injectables (50 ml, non fourni).
· Se laver les mains ou mettre des gants avant de reconstituer le produit
· Retirer la capsule protectrice du flacon de Riastap pour découvrir la partie centrale du bouchon de perfusion.
· Nettoyer la surface du bouchon de perfusion avec une solution antiseptique et laisser sécher.
· Transférer le solvant dans le flacon à l’aide d’un dispositif de transfert approprié. S’assurer de l'imprégnation complète de la poudre.
· Remuer le flacon avec un léger mouvement de rotation jusqu'à dissolution complète de la poudre et obtention de la solution prête à administrer. Eviter de secouer le flacon car cela pourrait générer l’apparition de mousse. Généralement, la poudre se dissout en 5 minutes environ. Cela ne devrait pas prendre plus de 15 minutes pour se dissoudre complétement.
· Ouvrir le blister en plastique contenant le dispositif de prélèvement (dispositif de prélèvement Mini-Spike®) fourni avec Riastap (Figure 2).
 Figure 2
Figure 2
· Prendre le dispositif de prélèvement fourni et l’insérer dans le bouchon du flacon contenant le produit reconstitué (Figure 3).
 Figure 3
Figure 3
· Après avoir inséré le dispositif de prélèvement, retirer le capuchon. Après avoir retiré le capuchon, ne pas toucher la surface exposée.
· Ouvrir le blister du filtre (Filtre seringue Pall®) fourni avec Riastap (Figure 4).
 Figure 4
Figure 4
· Visser la seringue sur le filtre (Figure 5).
 Figure 5
Figure 5
· Visser la seringue avec le filtre sur le dispositif de prélèvement (Figure 6).
 Figure 6
Figure 6
· Aspirer le produit reconstitué dans la seringue (Figure 7).
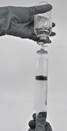 Figure 7
Figure 7
· Une fois terminé, retirer le filtre, le dispositif de prélèvement et le flacon vide de la seringue, les jeter selon la réglementation en vigueur et procéder à l’administration comme d’habitude.
· Le produit reconstitué doit être administré immédiatement par une ligne séparée d’injection / de perfusion.
· Eviter que du sang ne pénètre dans les seringues remplies de produit.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Administration
L’utilisation d’un kit de perfusion standard est recommandée pour l’administration de la solution reconstituée à température ambiante. La solution reconstituée doit être injecté ou perfusé lentement à un débit confortable pour le patient. Le débit d'injection ou de perfusion ne devrait pas dépasser approximativement 5 ml par minute.