Dernière mise à jour le 01/06/2026
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable
Indications thérapeutiques
Classe pharmacothérapeutique : Agents antinéoplasiques, analogues de la pyrimidine, Code ATC : L01BC07
Qu’est-ce que AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est un agent anticancéreux qui appartient à un groupe de médicaments appelés « antimétabolites ». AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable contient la substance active « azacitidine ».
Dans quels cas AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est-il utilisé ?
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est utilisé chez les adultes qui ne peuvent pas recevoir une greffe de cellules souches afin de traiter :
· Les syndromes myélodysplasiques (SMD) de risque élevé.
· La leucémie myélomonocytaire chronique (LMMC).
· La leucémie aiguë myéloblastique (LAM).
Ces maladies touchent la moelle osseuse et peuvent altérer la production de cellules sanguines normales.
Comment agit AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
AZACITIDINE HIKMA agit en empêchant les cellules cancéreuses de se développer. L’azacitidine pénètre dans le matériel génétique présent dans les cellules (acide ribonucléique [ARN] et acide désoxyribonucléique [ADN]). Elle agit en modifiant la façon dont les cellules activent et désactivent les gènes et en interférant avec la synthèse d’ARN et d’ADN. Ces actions corrigent les problèmes de croissance et de maturation des jeunes cellules sanguines dans la moelle osseuse qui sont responsables des syndromes myélodysplasiques et tuent les cellules cancéreuses dans les leucémies. Pour toutes questions sur la façon dont AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable agit ou la raison pour laquelle ce médicament vous a été prescrit, adressez-vous à votre médecin ou infirmier/ère.
Présentations
> 1 flacon(s) en verre de 100 mg
Code CIP : 34009 302 567 7 0
Déclaration de commercialisation : 31/05/2024
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : HIKMA FARMACEUTICA (Portugal) SA
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription hospitalière
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 111 801 3
ANSM - Mis à jour le : 11/03/2024
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 100 mg d’azacitidine.
Après reconstitution, chaque mL de suspension contient 25 mg d’azacitidine.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour suspension injectable.
Poudre blanche lyophilisée.
4.1. Indications thérapeutiques
AZACITIDINE HIKMA est indiqué dans le traitement des patients adultes non éligibles pour une greffe de cellules souches hématopoïétiques (GCSH) et présentant :
· un syndrome myélodysplasique (SMD) de risque intermédiaire-2 ou élevé selon l’index pronostique international (International Prognostic Scoring System, IPSS),
· une leucémie myélomonocytaire chronique (LMMC) avec 10 à 29 % de blastes médullaires sans syndrome myéloprolifératif,
· une leucémie aiguë myéloblastique (LAM) avec 20 à 30 % de blastes et dysplasie de lignées multiples, selon la classification de l’Organisation Mondiale de la Santé (OMS),
· une LAM avec > 30 % de blastes médullaires selon la classification de l’OMS.
4.2. Posologie et mode d'administration
Le traitement par AZACITIDINE HIKMA doit être instauré et poursuivi sous la surveillance d’un médecin ayant l’expérience de l’utilisation des agents chimiothérapeutiques. Les patients doivent recevoir une prémédication par des antiémétiques contre les nausées et les vomissements.
Posologie
La dose initiale recommandée pour le premier cycle de traitement, chez tous les patients, indépendamment des valeurs hématologiques de base, est de 75 mg/m2 de surface corporelle, par injection sous-cutanée, quotidiennement pendant 7 jours, suivis d’une période de repos de 21 jours (cycle de traitement de 28 jours).
Il est recommandé d’administrer au patient un minimum de 6 cycles de traitement. Le traitement doit être poursuivi tant qu’il apporte des bénéfices au patient ou jusqu’à progression de la maladie.
Le rapport réponse/toxicité hématologique et la toxicité rénale doivent être surveillés chez le patient (voir rubrique 4.4) ; il pourra être nécessaire de différer le début du cycle suivant ou de réduire la dose comme indiqué ci-dessous.
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ne doit pas être utilisé de façon interchangeable avec l'azacitidine orale. En raison des différences en matière d'exposition, la posologie et le schéma thérapeutique de l'azacitidine orale sont différents de celles de l'azacitidine injectable. Il est recommandé aux professionnels de santé de vérifier le nom du médicament, la dose et la voie d'administration.
Analyses de laboratoire
Un bilan hépatique et une mesure de la créatinine sérique et du bicarbonate sérique doivent être effectués avant de commencer le traitement et avant chaque cycle de traitement.
Une numération sanguine complète doit être réalisée avant de commencer le traitement et, si nécessaire, pour contrôler la réponse et la toxicité, mais dans tous les cas, au minimum avant chaque cycle de traitement.
Ajustement posologique lié à la toxicité hématologique
La toxicité hématologique se définit comme la numération sanguine la plus basse atteinte (nadir) au cours d’un cycle donné si les plaquettes ≤ 50,0 x 109/L et/ou que la numération des polynucléaires neutrophiles (PNN) ≤ 1 x 109/L.
La récupération correspond à une augmentation du nombre de cellules de la/des lignée(s) cellulaire(s) affectée(s) par la toxicité hématologique à hauteur d’au moins le nadir plus la moitié de la différence absolue entre le nadir et la numération de base (soit : numération sanguine après récupération ≥ nadir + (0,5 x [| numération de base – nadir |]).
Chez les patients dont les valeurs hématologiques de base ne sont pas diminuées (c-à-d. numération leucocytaire ≥ 3,0 x 109/L et PNN ≥ 1,5 x 109/L, et plaquettes ≥ 75,0 x 109/L) avant initiation du traitement.
Si une toxicité hématologique est observée suite au traitement par AZACITIDINE HIKMA, le cycle de traitement suivant doit être différé jusqu’à récupération de la numération plaquettaire et des PNN. Si la récupération est obtenue dans un délai de 14 jours, aucun ajustement posologique n’est nécessaire. En revanche, si la récupération ne se produit pas dans ce délai de 14 jours, la dose doit être réduite comme indiqué dans le tableau suivant. Après ces modifications posologiques, la durée du cycle sera ramenée à 28 jours.
|
Taux de PNN par cycle |
Dose lors du cycle suivant, si la récupération* n’est pas obtenue dans les 14 jours (%) |
|
|
PNN (x 109/L) |
Plaquettes (x 109/L) |
|
|
≤ 1,0 |
≤ 50,0 |
50 % |
|
> 1,0 |
> 50,0 |
100 % |
*Récupération = numérations ≥ nadir + (0,5 x [numération de base – nadir])
Chez les patients dont les valeurs hématologiques de base sont diminuées (c-à-d. numération leucocytaire < 3,0 x 109/L ou PNN < 1,5 x 109/L ou plaquettes < 75,0 x 109/L) avant initiation du traitement
Si, à la suite du traitement par AZACITIDINE HIKMA, la réduction de la numération leucocytaire ou des PNN ou des plaquettes par rapport aux numérations antérieures au traitement est ≤ 50 %, ou supérieure à 50 % mais qu’elle s’accompagne d’une amélioration d’une lignée cellulaire, le cycle suivant ne doit pas être différé et aucun ajustement posologique n’est requis.
Si la réduction de la numération leucocytaire, ou des PNN, ou des plaquettes, est supérieure à 50 % par rapport aux numérations antérieures au traitement mais ne s’accompagne d’aucune amélioration de la différenciation cellulaire, le cycle de traitement suivant par AZACITIDINE HIKMA doit être différé jusqu’à récupération de la numération plaquettaire et des PNN. Toutefois, si la récupération est obtenue dans un délai de 14 jours, aucun ajustement posologique n’est nécessaire. En revanche, si la récupération ne se produit pas dans ce délai de 14 jours, la cellularité de la moelle osseuse doit être déterminée. Si la cellularité de la moelle osseuse est > 50 %, aucun ajustement posologique n’est requis. Si la cellularité de la moelle osseuse est ≤ 50 %, le traitement doit être différé et la dose réduite comme indiqué dans le tableau suivant :
|
Cellularité de la moelle osseuse |
Dose lors du cycle suivant si la récupération n’est pas obtenue dans les 14 jours (%) |
|
|
Récupération* ≤ 21 jours |
Récupération* > 21 jours |
|
|
15-50 % |
100 % |
50 % |
|
< 15 % |
100 % |
33 % |
*Récupération = numérations ≥ nadir + (0,5 x [numération de base – nadir])
Après ces modifications posologiques, la durée du cycle suivant sera ramenée à 28 jours.
Populations particulières
Sujets âgés
Aucun ajustement posologique spécifique n’est recommandé chez les patients âgés. La probabilité d’une insuffisance rénale étant plus importante chez les patients âgés, il pourra être utile de contrôler la fonction rénale.
Patients présentant une insuffisance rénale
Chez les patients présentant une insuffisance rénale, l’azacitidine peut être administrée sans ajustement posologique initial (voir rubrique 5.2). En cas de diminution inexpliquée du taux de bicarbonate sérique en dessous de 20 mmol/L, la dose doit être réduite de 50 % lors du cycle suivant. En cas d’augmentation inexpliquée de la créatinine sérique ou de l’urée sanguine à hauteur de ≥ 2 fois la valeur de base et la limite supérieure de la normale (LSN), le cycle suivant doit être différé jusqu’à ce que les valeurs reviennent à la normale ou à leur niveau de base et la dose doit être réduite de 50 % lors du cycle de traitement suivant (voir rubrique 4.4).
Patients présentant une insuffisance hépatique
Aucune étude formelle n’a été menée chez les patients atteints d’insuffisance hépatique (voir rubrique 4.4). En cas d’insuffisance hépatique sévère, les patients doivent être surveillés attentivement afin de détecter les événements indésirables. Aucune modification spécifique de la dose initiale n’est recommandée chez les patients atteints d’insuffisance hépatique avant le début du traitement ; les ajustements posologiques ultérieurs devront se faire sur la base des valeurs hématologiques. AZACITIDINE HIKMA est contre-indiqué chez les patients atteints de tumeurs hépatiques malignes avec un stade avancé (voir rubriques 4.3 et 4.4)
Population pédiatrique
La sécurité et l’efficacité de AZACITIDINE HIKMA chez les enfants âgés de 0 à 17 ans n’ont pas encore été établies. Les données actuellement disponibles sont décrites aux rubriques 4.8, 5.1 et 5.2 mais aucune recommandation sur la posologie ne peut être donnée.
Mode d’administration
Une fois reconstitué, AZACITIDINE HIKMA doit être injecté par voie sous-cutanée dans le haut du bras, la cuisse ou l’abdomen. Les sites d’injection doivent être alternés. Chaque nouvelle injection doit être pratiquée à au moins 2,5 cm de distance du site précédent et en aucun cas sur une zone sensible, présentant une ecchymose, une rougeur ou une induration.
Après reconstitution, la suspension ne doit pas être filtrée. Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Tumeur hépatique maligne à un stade avancé (voir rubrique 4.4).
Allaitement (voir rubrique 4.6).
4.4. Mises en garde spéciales et précautions d'emploi
Toxicité hématologique
Le traitement par l’azacitidine est associé à des cas d’anémie, de neutropénie et de thrombocytopénie, en particulier au cours des 2 premiers cycles (voir rubrique 4.8). Une numération sanguine complète doit être réalisée si nécessaire pour contrôler la réponse et la toxicité, mais dans tous les cas, au moins avant chaque cycle de traitement. Après administration de la dose recommandée pour le premier cycle, la dose utilisée lors des cycles suivants devra être réduite ou son administration différée en fonction du nadir des numérations et de la réponse hématologique (voir rubrique 4.2). Il devra être conseillé aux patients de signaler rapidement tout épisode fébrile. Il est également conseillé aux patients et à leurs médecins d’être attentifs aux signes et symptômes d’hémorragie.
Insuffisance hépatique
Aucune étude formelle n’a été menée chez les patients atteints d’insuffisance hépatique. Chez les patients présentant une charge tumorale élevée due à une atteinte métastatique, des cas de coma hépatique progressif et de décès sous traitement par l’azacitidine ont été signalés, en particulier lorsque le taux de base d’albumine sérique de ces patients était < 30 g/L. L’azacitidine est contre-indiquée chez les patients atteints de tumeurs hépatiques malignes à un stade avancé (voir rubrique 4.3).
Insuffisance rénale
Des anomalies rénales allant de l’augmentation du taux de créatinine sérique à l’insuffisance rénale et au décès ont été signalées chez des patients traités par l’azacitidine en voie intraveineuse en association avec d’autres agents chimiothérapeutiques. Par ailleurs, une acidose tubulaire rénale définie par la chute du bicarbonate sérique à < 20 mmol/L associée à une urine alcaline et à une hypokaliémie (potassium sérique < 3 mmol/L), est survenue chez 5 sujets atteints de leucémie myéloïde chronique (LMC) traités par l’azacitidine et l’étoposide. En cas de diminution inexpliquée du bicarbonate sérique (< 20 mmol/L) ou d’augmentation de la créatinine sérique ou de l’urée sanguine, la dose doit être réduite ou son administration différée (voir rubrique 4.2).
Les patients doivent être informés qu’ils doivent signaler immédiatement à leur médecin une oligurie et une anurie.
Bien qu’il n’ait pas été observé de différences cliniquement significatives dans la fréquence des effets indésirables entre les patients ayant une fonction rénale normale et ceux présentant une insuffisance rénale, les patients doivent être surveillés attentivement afin de détecter toute toxicité car l’azacitidine et/ou ses métabolites sont excrétés principalement par les reins (voir rubrique 4.2).
Analyses de laboratoire
Un bilan hépatique et une mesure de la créatinine et du bicarbonate sériques doivent être effectués avant de commencer le traitement et avant chaque cycle de traitement. Une numération sanguine complète doit être réalisée avant de commencer le traitement et, si nécessaire, pour contrôler la réponse et la toxicité, mais dans tous les cas, au minimum avant chaque cycle de traitement, voir également rubrique 4.8.
Affections cardiaques et pulmonaires
La sécurité et l’efficacité de l’azacitidine n’ont pas été établies chez les patients présentant des antécédents d’insuffisance cardiaque congestive sévère, d’affection cardiaque cliniquement instable ou d’affection pulmonaire, ces patients ayant été exclus des études pivots d’enregistrement (AZA PH GL 2003 CL 001 et AZA-AML-001). Les données récentes d’une étude clinique chez des patients ayant des antécédents connus de maladie cardiovasculaire ou pulmonaire ont montré une augmentation significative de l’incidence des événements cardiaques avec l’azacitidine (voir rubrique 4.8). La prudence est donc recommandée en cas de prescription d’azacitidine chez ces patients. Un bilan cardio-pulmonaire doit être envisagé avant et pendant le traitement.
Fasciite nécrosante
Des cas de fasciite nécrosante, dont certains d’issue fatale, ont été rapportés chez des patients traités par AZACITIDINE HIKMA. Le traitement par AZACITIDINE HIKMA doit être arrêté chez les patients qui développent une fasciite nécrosante et un traitement approprié doit être instauré immédiatement.
Syndrome de lyse tumorale
Les patients présentant des risques de syndrome de lyse tumorale sont ceux qui ont une charge tumorale élevée avant le traitement. Ces patients doivent être étroitement surveillés et les précautions appropriées doivent être prises.
Syndrome de différenciation
Des cas de syndrome de différenciation (également connu sous le nom de syndrome de l'acide rétinoïque) ont été signalés chez des patients recevant de l’azacitidine injectable. Le syndrome de différenciation peut être mortel et les symptômes et les observations cliniques incluent une détresse respiratoire, des infiltrats pulmonaires, de la fièvre, une éruption cutanée, un œdème pulmonaire, un œdème périphérique, une prise de poids rapide, des épanchements pleuraux, des épanchements péricardiques, une hypotension et un dysfonctionnement rénal (voir rubrique 4.8). Un traitement par des corticostéroïdes IV à forte dose et une surveillance hémodynamique doivent être envisagés dès l'apparition des premiers symptômes ou signes évocateurs d'un syndrome de différenciation.
L'arrêt temporaire du traitement par azacitidine injectable doit être envisagé jusqu'à disparition des symptômes et, en cas de reprise du traitement, la prudence est recommandée.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
D’après les données in vitro, le métabolisme de l’azacitidine ne semble pas être médié par les isoenzymes du cytochrome P450 (CYP), les UDP-glucuronosyl-transférases (UGT), les sulfotransférases (SULT) et les glutathion transférases (GST) ; la survenue d’interactions liées à ces enzymes de métabolisation in vivo est donc jugée improbable.
La survenue d’effets inhibiteurs ou inducteurs cliniquement significatifs de l’azacitidine sur les enzymes du cytochrome P450 est improbable (voir rubrique 5.2).
Aucune étude clinique formelle des interactions médicamenteuses de l’azacitidine n’a été réalisée.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer/Contraception chez les hommes et les femmes
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et pendant au moins 6 mois après l’arrêt du traitement. Les hommes doivent être informés qu’ils ne doivent pas concevoir pendant le traitement et qu’ils doivent utiliser une contraception efficace pendant le traitement et pendant au moins 3 mois après l’arrêt du traitement.
Grossesse
Il n’existe pas de données suffisamment pertinentes sur l’utilisation de l'azacitidine chez la femme enceinte. Des études effectuées chez la souris ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Le risque potentiel en clinique n’est pas connu chez l’Homme. Compte tenu des résultats des études chez l’animal et de son mécanisme d’action, l’azacitidine ne doit pas être utilisée pendant la grossesse, en particulier pendant le premier trimestre, à moins d’une nécessité absolue. Les effets bénéfiques du traitement doivent être évalués au cas par cas au regard des risques éventuels encourus par le fœtus.
Allaitement
L’excrétion de l’azacitidine ou ses métabolites dans le lait maternel est inconnue. Compte tenu des effets indésirables graves possibles chez l’enfant allaité, l’allaitement est contre-indiqué pendant le traitement par l’azacitidine.
Fertilité
Il n’existe pas de données concernant les effets de l'azacitidine sur la fertilité humaine. Chez l’animal, des effets indésirables sur la fertilité des mâles ont été décrits avec l’azacitidine (voir rubrique 5.3).. Avant de commencer le traitement, il est conseillé aux patients de sexe masculin de se renseigner sur les procédures de conservation du sperme.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
L’azacitidine a une influence mineure ou modérée sur l’aptitude à conduire des véhicules et à utiliser des machines. Une fatigue a été rapportée pendant le traitement par l’azacitidine. La prudence est donc recommandée en cas de conduite ou d’utilisation de machines.
Résumé du profil de sécurité
Population de patients adultes présentant un SMD, une LMMC ou une LAM (avec 20 à 30 % de blastes médullaires)
Des effets indésirables jugés potentiellement ou probablement en rapport avec l’administration de l’azacitidine sont survenus chez 97 % des patients.
Les effets indésirables graves relevés le plus fréquemment lors de l’étude pivot (AZA PH GL 2003 CL 001) ont été notamment des neutropénies fébriles (8,0 %) et des anémies (2,3 %) qui ont également été signalés lors des études complémentaires (CALGB 9221 et CALGB 8921). Les autres effets indésirables graves observés dans ces trois études ont été notamment des infections telles que septicémies sur neutropénie (0,8 %) et des pneumonies (2,5 %) (certaines d’issue fatale), des thrombocytopénies (3,5 %), des réactions d’hypersensibilité (0,25 %) et des événements hémorragiques (par exemple hémorragie cérébrale [0,5 %], hémorragie gastro-intestinale [0,8 %] et hémorragie intracrânienne [0,5 %]).
Les effets indésirables signalés le plus fréquemment lors du traitement par l’azacitidine ont été les réactions hématologiques (71,4 %), notamment la thrombocytopénie, la neutropénie et la leucopénie (généralement de grade 3 à 4), les événements gastro-intestinaux (60,6 %), notamment les nausées et les vomissements (généralement de grade 1 à 2), et les réactions au site d’injection (77,1 % ; généralement de grade 1 à 2).
Population de patients adultes âgés de 65 ans et plus présentant une LAM avec > 30 % de blastes médullaires
Les effets indésirables graves les plus fréquents (≥ 10 %) observés dans le bras de traitement par l’azacitidine de l’étude AZA-AML-001 ont été notamment des neutropénies fébriles (25,0 %), des pneumonies (20,3 %) et des pyrexies (10,6 %). Les autres effets indésirables graves signalés moins fréquemment dans le bras de traitement par l’azacitidine ont été notamment : septicémie (5,1 %), anémie (4,2 %), septicémie sur neutropénie (3,0 %), infection urinaire (3,0 %), thrombocytopénie (2,5 %), neutropénie (2,1 %), cellulite (2,1 %), vertiges (2,1 %) et dyspnée (2,1 %).
Les effets indésirables signalés le plus fréquemment (≥ 30 %) lors du traitement par l’azacitidine ont été des événements gastro-intestinaux, notamment des constipations (41,9 %), des nausées (39,8 %) et des diarrhées (36,9 %) , (généralement de grade 1 ou 2), des troubles généraux et anomalies au site d’administration, notamment de la fièvre (37,7 % ; généralement de grade 1 ou 2) et des événements hématologiques, notamment des neutropénies fébriles (32,2 %) et des neutropénies (30,1 %), (généralement de grade 3 ou 4).
Liste tabulée des effets indésirables
Le tableau 1 ci-dessous liste les effets indésirables associés au traitement par l’azacitidine qui ont été observés dans les principales études cliniques menées dans les SMD et la LAM et rapportés dans le cadre de la pharmacovigilance.
Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité. Les effets indésirables sont présentés dans le tableau ci-dessous en fonction de la fréquence la plus élevée observée dans l’une des principales études cliniques.
Tableau 1 : Effets indésirables observés chez des patients présentant un SMD ou une LAM traités par l’azacitidine (rapportés dans le cadre des études cliniques et de la pharmacovigilance)
|
Classe de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Fréquence indéterminée |
|
Infections et infestations |
pneumonie* (y compris bactérienne, virale et fongique), rhinopharyngite |
septicémie* (y compris bactérienne, virale et fongique), septicémie sur neutropénie*, infection respiratoire (y compris des voies respiratoires supérieures et bronchite), infection urinaire, cellulite, diverticulite, mycose buccale, sinusite, pharyngite, rhinite, herpès simplex, infection cutanée |
fasciite nécrosante* |
||
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
|
|
|
|
syndrome de différenciation*, a |
|
Affections hématologiques et du système lymphatique |
neutropénie fébrile*, neutropénie, leucopénie, thrombocytopénie, anémie |
pancytopénie*, aplasie médullaire |
|||
|
Affections du système immunitaire |
réactions d’hypersensibilité |
||||
|
Troubles du métabolisme et de la nutrition |
anorexie, diminution de l’appétit, hypokaliémie |
déshydratation |
syndrome de lyse tumorale |
||
|
Affections psychiatriques |
insomnie |
état confusionnel, anxiété |
|||
|
Affections du système nerveux |
vertiges, céphalées |
hémorragie intracrânienne*, syncope, somnolence, léthargie |
|||
|
Affections oculaires |
hémorragie oculaire, hémorragie conjonctivale |
||||
|
Affections cardiaques |
épanchement péricardique |
péricardite |
|||
|
Affections vasculaires |
hypotension*, hypertension, hypotension orthostatique, hématome |
||||
|
Affections respiratoires, thoraciques et médiastinales |
dyspnée, épistaxis |
épanchement pleural, dyspnée d’effort, douleur pharyngo-laryngée |
pneumo- pathie intersti- tielle |
||
|
Affections gastro- intestinales |
diarrhée, vomissements, constipation, nausées, douleurs abdominales (y compris inconfort aux niveaux supérieur et inférieur de l’abdomen) |
hémorragie gastro-intestinale* (y compris hémorragie buccale), hémorragie hémorroïdaire, stomatite, hémorragie gingivale, dyspepsie |
|||
|
Affections hépatobiliaires |
insuffisance hépatique*, coma hépatique progressif |
||||
|
Affections de la peau et du tissu sous-cutané |
pétéchies, prurit (y compris prurit généralisé), éruption cutanée, ecchymoses |
purpura, alopécie, urticaire, érythème, éruption maculaire |
dermatose neutrophilique fébrile aiguë, pyoderma gangrenosum |
Vascularite cutanée |
|
|
Affections musculo- squelettiques et systémiques |
arthralgie, douleurs musculo-squelettiques (y compris dorsalgies, douleurs osseuses et douleurs aux extrémités) |
spasmes musculaires, myalgie |
|||
|
Affections du rein et des voies urinaires |
insuffisance rénale*, hématurie, augmentation du taux de créatinine sérique |
acidose tubulaire rénale |
|||
|
Troubles généraux et anomalies au site d’administration |
fièvre*, fatigue, asthénie, douleurs thoraciques, érythème au site d’injection, douleur au site d’injection, réaction (non précisée) au site d’injection |
ecchymose, hématome, induration, éruption cutanée, prurit, inflammation, décoloration, nodule et hémorragie (au site d’injection), malaise, frissons, hémorragie au site du cathéter |
nécrose (au site d’injection) |
||
|
Investigations |
perte de poids |
* = de rares cas fatals ont été rapportés.
a = voir rubrique 4.4
Description de certains effets indésirables
Effets indésirables hématologiques
Les effets indésirables hématologiques signalés le plus fréquemment (≥ 10 %) en association avec le traitement par l’azacitidine sont : anémie, thrombocytopénie, neutropénie, neutropénie fébrile et leucopénie, et sont généralement de grade 3 ou 4. Le risque de survenue de ces événements est plus important pendant les 2 premiers cycles, après quoi ils deviennent moins fréquents chez le patient dont la fonction hématologique se rétablit. Dans la plupart des cas, les effets indésirables hématologiques ont été pris en charge par le biais d’une surveillance régulière des numérations sanguines complètes et, si nécessaire, en différant l’administration de l’azacitidine lors du cycle suivant, à l’aide d’une prophylaxie antibiotique et/ou d'un traitement de support par facteur de croissance (G-CSF, par exemple) pour la neutropénie et de transfusions pour l’anémie ou la thrombocytopénie.
Infections
L’insuffisance médullaire peut entraîner une neutropénie et un risque accru d’infection. Des effets indésirables graves, tels que des septicémies, y compris des septicémies sur neutropénie et des pneumonies, dont certaines d’issue fatale, ont été signalés chez des patients recevant de l’azacitidine. Les infections peuvent être prises en charge en utilisant des agents anti-infectieux associés à un traitement de support par facteur de croissance (G-CSF, par exemple) pour la neutropénie.
Hémorragies
Des hémorragies peuvent se produire chez les patients sous azacitidine. Des effets indésirables graves tels que des hémorragies gastro-intestinales et des hémorragies intracrâniennes ont été rapportés. Les patients doivent être surveillés afin de détecter les signes et symptômes d’hémorragie, en particulier en cas de thrombocytopénie préexistante ou liée au traitement.
Hypersensibilité
De graves réactions d’hypersensibilité ont été décrites chez des patients sous azacitidine. En cas de réaction de type anaphylactique, le traitement par l’azacitidine doit être immédiatement interrompu et un traitement symptomatique adapté doit être instauré.
Effets indésirables cutanés et du tissu sous-cutané
Les effets indésirables cutanés et sous-cutanés sont liés majoritairement au site d’injection. Aucun de ces effets indésirables n’a nécessité l’interruption du traitement par l’azacitidine ou la réduction de la dose d’azacitidine lors des études pivots. Les effets indésirables sont survenus majoritairement au cours des 2 premiers cycles de traitement et ont eu tendance à diminuer lors des cycles suivants. Les effets indésirables sous-cutanés tels que éruption/inflammation/prurit au site d’injection, éruption cutanée, érythème et lésion cutanée peuvent nécessiter un traitement concomitant par des antihistaminiques, des corticostéroïdes et des anti-inflammatoires non stéroïdiens (AINS) notamment. Ces réactions cutanées doivent être différenciées des infections des tissus mous survenant parfois au site d’injection. Des infections des tissus mous, incluant une cellulite et une fasciite nécrosante d’issue fatale dans de rares cas, ont été rapportées avec l’azacitidine dans le cadre de la pharmacovigilance. Pour la prise en charge clinique des effets indésirables infectieux, voir la rubrique 4.8 « Infections ».
Effets indésirables gastro-intestinaux
Les effets indésirables gastro-intestinaux signalés le plus fréquemment en association avec le traitement par l’azacitidine ont été notamment la constipation, la diarrhée, les nausées et les vomissements. Ces effets indésirables ont été pris en charge à l’aide d’un traitement symptomatique par des antiémétiques pour les nausées et les vomissements, des anti-diarrhéiques pour la diarrhée et des laxatifs et/ou émollients fécaux pour la constipation.
Effets indésirables rénaux
Des anomalies rénales, allant de l’augmentation du taux de créatinine sérique et d’une hématurie à l’acidose tubulaire rénale, à l’insuffisance rénale et au décès, ont été signalées chez des patients traités par l’azacitidine (voir rubrique 4.4).
Effets indésirables hépatiques
Chez les patients présentant une charge tumorale élevée due à une atteinte métastatique, des cas d’insuffisance hépatique, de coma hépatique progressif et de décès ont été rapportés pendant le traitement par l’azacitidine (voir rubrique 4.4).
Événements cardiaques
Les données d’une étude clinique permettant l’inclusion de patients ayant des antécédents connus de maladie cardiovasculaire ou pulmonaire ont montré une augmentation des événements cardiaques chez les patients présentant une LAM nouvellement diagnostiquée traités par l’azacitidine (voir rubrique 4.4).
Population âgée
Les données de sécurité concernant l’utilisation de l’azacitidine chez les patients ≥ 85 ans sont limitées (avec 14 patients [5,9 %] ≥ 85 ans traités au cours de l’étude AZA-AML-001).
Population pédiatrique
Dans l’étude AZA-JMML-001, 28 patients pédiatriques (âgés de 1 mois à moins de 18 ans) ont été traités par l’azacitidine pour un SMD (n = 10) ou une leucémie myélomonocytaire juvénile (LMMJ) (n = 18) (voir rubrique 5.1).
Les 28 patients ont tous présenté au moins 1 événement indésirable et 17 (60,7 %) ont présenté au moins un événement indésirable lié au traitement. Les événements indésirables les plus fréquemment rapportés pour la totalité de la population pédiatrique étaient la fièvre, des événements hématologiques comme l’anémie, la thrombopénie et la neutropénie fébrile, et des événements gastrointestinaux tels que la constipation et les vomissements.
Trois (3) sujets ont souffert d’un événement lié au traitement, ayant conduit à un arrêt du traitement (fièvre, progression de la maladie et douleurs abdominales).
Dans l’étude AZA-AML-004, 7 patients pédiatriques (âgés de 2 à 12 ans) ont été traités par l’azacitidine pour une LAM en rechute moléculaire après une première rémission complète [RC1] (voir rubrique 5.1).
Les 7 patients ont tous présenté au moins 1 événement indésirable lié au traitement. Les événements indésirables les plus fréquemment rapportés étaient la neutropénie, les nausées, la leucopénie, la thrombopénie, la diarrhée et l’élévation de l’alanine aminotransférase (ALAT). Deux patients ont présenté un événement lié au traitement ayant entraîné l’interruption de ce dernier (neutropénie et neutropénie fébrile).
Aucun nouveau signal de sécurité n’a été identifié parmi le nombre limité de patients pédiatriques traités par l’azacitidine au cours de l’étude clinique. Le profil de sécurité global est cohérent avec celui de la population adulte.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Un cas de surdosage de l’azacitidine a été signalé lors des études cliniques. Un patient a présenté une diarrhée, des nausées et des vomissements après avoir reçu une dose intraveineuse unique de 290 mg/m2 environ, soit près de 4 fois la dose initiale recommandée.
En cas de surdosage, l’état du patient devra être surveillé par des numérations sanguines appropriées et un traitement d’appoint devra être mis en œuvre au besoin. Aucun antidote spécifique à l’azacitidine en cas de surdosage n’est connu.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Agents antinéoplasiques, analogues de la pyrimidine, Code ATC : L01BC07
Mécanisme d’action
L’azacitidine pourrait exercer ses effets antinéoplasiques par des mécanismes multiples comprenant une cytotoxicité directe à l’encontre des cellules hématopoïétiques anormales de la moelle osseuse et une hypométhylation de l’ADN. Les effets cytotoxiques de l’azacitidine pourraient résulter de mécanismes multiples, comprenant l’inhibition de la synthèse de l’ADN, de l’ARN et de protéines de synthèse, son incorporation dans l’ARN et l’ADN, et l’activation des voies de dégradation de l’ADN. Les cellules non prolifératives sont relativement insensibles à l’azacitidine. L’incorporation de l’azacitidine dans l’ADN entraîne l’inactivation des ADN méthyltransférases, ce qui engendre une hypométhylation de l’ADN. L’hypométhylation de l’ADN des gènes qui sont impliqués dans la régulation du cycle cellulaire normal, la différentiation et les voies de l’apoptose, et qui présentent une méthylation aberrante peut entraîner une réexpression des gènes suppresseurs de tumeurs et une restauration de leurs fonctions. L’importance relative de l’hypométhylation de l’ADN par rapport à la cytotoxicité ou aux autres activités de l’azacitidine en termes de résultats cliniques observés n’a pas été établie.
Efficacité et sécurité cliniques
Population de patients adultes (SMD, LMMC et LAM [avec 20 à 30 % de blastes médullaires])
L’efficacité et la sécurité de l’azacitidine ont été étudiées dans une étude comparative, internationale, multicentrique, contrôlée, ouverte, randomisée, sur groupes parallèles, de phase 3 (AZA PH GL 2003 CL 001) chez des patients adultes atteints de : SMD à risque intermédiaire-2 ou élevé selon l’index pronostique international (International Prognostic Scoring System, IPSS), anémie réfractaire avec excès de blastes (AREB), anémie réfractaire avec excès de blastes en transformation (AREB-T) et leucémie myélomonocytaire chronique (LMMC) modifiée selon le système de classification franco-américano-britannique (FAB). L’AREB-T (21 à 30 % de blastes) est désormais classée parmi les LAM d’après le système de classification actuel de l’OMS. L’azacitidine ajoutée au traitement symptomatique optimal (Best Supportive Care, BSC) (n = 179) a été comparée aux traitements classiques (Conventional Care Regimens, CCR). Les CCR étaient constitués du BSC seul (n = 105), de cytarabine à faible dose plus le BSC (n = 49) ou d’une chimiothérapie d’induction standard plus le BSC (n = 25). Le choix d’un des trois CCR a été fait par le médecin pour chaque patient avant la randomisation. Les patients ont reçu le CCR préalablement sélectionné s’ils n’étaient pas randomisés dans le groupe de traitement par l’azacitidine. Parmi les critères d’inclusion, les patients devaient présenter un indice de performance ECOG (Eastern Cooperative Oncology Group) de 0 à 2. Les patients atteints de SMD secondaire ont été exclus de l’étude. Le critère d’évaluation principal de l’étude était la durée de survie globale. L’azacitidine a été administrée par injection sous-cutanée à la dose de 75 mg/m2 quotidiennement pendant 7 jours, suivis d’une période de repos de 21 jours (cycle de traitement de 28 jours) pendant une durée médiane de 9 cycles (intervalle = 1 à 39) et une durée moyenne de 10,2 cycles. Au sein de la population en intention de traiter (ITT), l’âge médian était de 69 ans (intervalle : 38 à 88 ans).
Dans la population en ITT, constituée de 358 patients (179 sous azacitidine et 179 sous CCR), le traitement par l’azacitidine a été associé à une durée de survie médiane de 24,46 mois contre 15,02 mois chez les patients sous CCR, soit une différence de 9,4 mois, avec une valeur de p = 0,0001 avec le test du log-rank stratifié. Le rapport de risque (RR) correspondant aux effets de ce traitement a été de 0,58 (IC à 95 % : 0,43 ; 0,77). Le taux de survie à deux ans a été de 50,8 % chez les patients sous azacitidine contre 26,2 % chez les patients sous CCR (p < 0,0001).
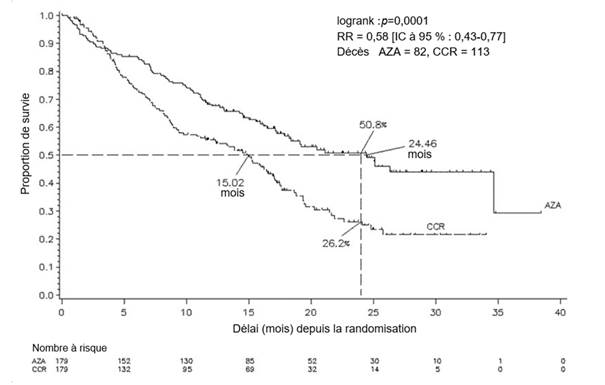
Abréviations : AZA = azacitidine ; CCR = traitements classiques (conventional care regimens) ; IC = intervalle de confiance ; RR = Rapport de risque
Les bénéfices de l’azacitidine en termes de survie ont été cohérents indépendamment du traitement CCR choisi (BSC seul, cytarabine à faible dose plus BSC ou chimiothérapie d’induction standard plus BSC) dans le groupe témoin.
Lorsque les différents sous-groupes cytogénétiques IPSS ont été analysés, des résultats similaires ont été obtenus en termes de survie globale médiane dans tous les groupes (profils cytogénétiques de bon pronostic, de pronostic intermédiaire ou de mauvais pronostic y compris les monosomies 7).
Les analyses des sous-groupes par classes d’âge ont fait apparaître une augmentation de la survie globale médiane dans tous les groupes (< 65 ans, ≥ 65 ans et ≥ 75 ans).
Le traitement par l’azacitidine a été associé à un délai médian avant décès ou transformation en LAM de 13,0 mois contre 7,6 mois chez les patients sous CCR, soit une amélioration de 5,4 mois avec une valeur de p = 0,0025 avec le test du log-rank stratifié.
Le traitement par l’azacitidine a également été associé à une réduction des cytopénies et des symptômes associés. Le traitement par l’azacitidine a entraîné une réduction du besoin en transfusions de plaquettes et de globules rouges. Sur l’ensemble des patients du groupe sous azacitidine qui étaient initialement dépendants des transfusions de globules rouges, 45,0 % sont devenus indépendants des transfusions de globules rouges pendant la période de traitement, contre 11,4 % dans les groupes sous CCR poolés (soit une différence statistiquement significative (p < 0,0001) de 33,6 % (IC à 95 % : 22,4 ; 44,6). Parmi les patients initialement dépendants des transfusions de globules rouges devenus indépendants, la durée médiane de cette indépendance a été de 13 mois dans le groupe sous azacitidine.
La réponse au traitement a été évaluée par l’investigateur ou par le Comité de revue indépendant (CRI). La réponse globale (rémission complète [RC] + rémission partielle [RP]) déterminée par l’investigateur a été de 29 % dans le groupe sous azacitidine et de 12 % dans les groupes sous CCR poolés (p = 0,0001). La réponse globale (RC + RP) déterminée par le CRI dans l’étude AZA PH GL 2003 CL 001 a été de 7 % (12/179) dans le groupe sous azacitidine contre 1 % (2/179) dans les groupes sous CCR poolés (p = 0,0113). Les différences entre les évaluations du CRI et de l’investigateur s’expliquent par l’utilisation des critères du groupe de travail international (International Working Group, IWG), lesquels requièrent une amélioration des numérations sanguines périphériques et le maintien de cette amélioration pendant au minimum 56 jours. Un bénéfice en termes de survie a également été démontré chez les patients n’ayant pas obtenu de réponse complète/partielle suite au traitement par l’azacitidine. Une amélioration hématologique (majeure ou mineure) déterminée par le CRI a été obtenue chez 49 % des patients sous azacitidine contre 29 % des patients dans les groupes sous CCR poolés (p < 0,0001).
Sur l’ensemble des patients présentant initialement une ou plusieurs anomalies cytogénétiques, le pourcentage de patients ayant bénéficié d’une réponse cytogénétique majeure a été similaire dans le groupe sous azacitidine et dans les groupes sous CCR poolés. Le taux de réponses cytogénétiques mineures a été supérieur de façon statistiquement significative (p = 0,0015) dans le groupe sous azacitidine (34 %) par rapport aux groupes sous CCR poolés (10 %).
Population de patients adultes âgés de 65 ans et plus présentant une LAM avec > 30 % de blastes médullaires
Les résultats présentés ci-dessous représentent la population en intention de traiter évaluée dans l’étude AZA-AML-001 (voir la rubrique 4.1 pour l’indication autorisée).
L’efficacité et la sécurité de l’azacitidine ont été étudiées dans une étude de phase 3 internationale, multicentrique, contrôlée, ouverte, sur groupes parallèles, chez des patients âgés de 65 ans et plus présentant une LAM de novo ou secondaire récemment diagnostiquée avec > 30 % de blastes médullaires selon la classification de l’OMS, qui n’étaient pas éligibles à une GCSH. L’azacitidine plus BSC (n = 241) a été comparé aux CCR. Les CCR consistaient en BSC seul (n = 45), cytarabine à faible dose plus BSC (n = 158) ou chimiothérapie intensive standard avec cytarabine et anthracycline plus BSC (n = 44). Le choix d’un des trois CCR a été fait par le médecin pour chaque patient avant la randomisation. Les patients ont reçu le CCR préalablement sélectionné s’ils n’étaient pas randomisés dans le bras de traitement par l’azacitidine. Parmi les critères d’inclusion, les patients devaient avoir un indice de performance ECOG de 0 à 2 et présenter des anomalies cytogénétiques de risque intermédiaire ou défavorable. L’objectif principal de l’étude était d’évaluer la durée de survie globale.
L’azacitidine a été administrée par voie SC à la dose de 75 mg/m2/jour pendant 7 jours, suivis d’une période de repos de 21 jours (cycle de traitement de 28 jours), pendant un nombre médian de 6 cycles (intervalle : 1 à 28) ; les patients du groupe BSC seul ont été traités pendant un nombre médian de 3 cycles (intervalle : 1 à 20), les patients du groupe cytarabine à faible dose pendant un nombre médian de 4 cycles (intervalle : 1 à 25) et les patients du groupe chimiothérapie intensive standard pendant un nombre médian de 2 cycles (intervalle : 1 à 3, cycle d’induction plus 1 ou 2 cycle(s) de consolidation).
Les paramètres individuels initiaux étaient comparables entre les groupes l’azacitidine et CCR. L'âge médian des patients était de 75,0 ans (intervalle : 64 à 91 ans), 75,2 % étaient caucasiens et 59,0 % étaient des hommes. Lors de l’inclusion, 60,7 % des patients présentaient une LAM sans spécification particulière, 32,4 % une LAM avec anomalies associées aux myélodysplasies, 4,1 % une néoplasie myéloïde secondaire à un traitement et 2,9 % une LAM avec anomalies cytogénétiques récurrentes selon la classification de l’OMS.
Dans l'analyse en ITT portant sur 488 patients (241 patients traités par l’azacitidine et 247 patients recevant un CCR), le traitement par l’azacitidine a été associé à une durée de survie médiane de 10,4 mois versus 6,5 mois chez les patients recevant un CCR, soit une différence de 3,8 mois, avec une valeur de p = 0,1009 selon un test du log-rank stratifié (bilatéral). Le rapport de risque (RR) pour l’effet du traitement a été de 0,85 (IC à 95 % = 0,69 ; 1,03). Les taux de survie à un an ont été de 46,5 % chez les patients recevant l’azacitidine versus 34,3 % chez les patients recevant un CCR.
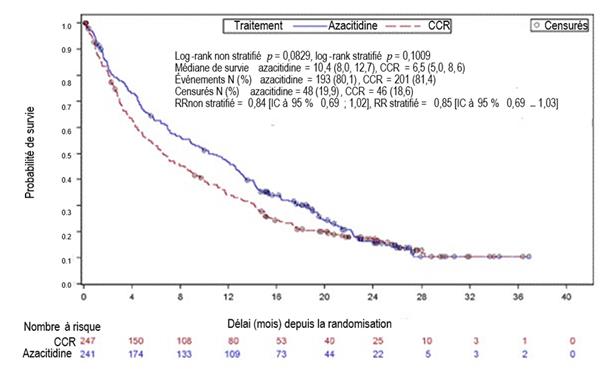
Le modèle à risques proportionnels de Cox ajusté pour les facteurs pronostiques initiaux prédéfinis a montré un RR de 0,80 pour l’azacitidine versus CCR (IC à 95 % = 0,66 ; 0,99, p = 0,0355).
De plus, bien que l’étude n’ait pas eu la puissance nécessaire pour démontrer une différence statistiquement significative dans la comparaison du groupe traité par l’azacitidine aux groupes recevant les CCR présélectionnés, la survie chez les patients traités par l’azacitidine a été plus longue comparé aux options de traitement CCR, BSC seul et cytarabine à faible dose plus BSC, et comparable à celle observée dans le bras recevant la chimiothérapie intensive standard plus BSC.
Dans tous les sous-groupes prédéfinis (âge [< 75 ans et ≥ 75 ans], sexe, groupe ethnique, indice de performance ECOG [0 ou 1 et 2], risque cytogénétique lors de l’inclusion [intermédiaire et défavorable], région géographique, classification OMS de la LAM [incluant LAM avec anomalies associées aux myélodysplasies], numération leucocytaire lors de l’inclusion [≤ 5 x 109/L et > 5 x 109/L], taux de blastes médullaires lors de l'inclusion [≤ 50 % et > 50 %] et antécédents de SMD), il a été observé une tendance à un bénéfice de SG en faveur de l’azacitidine.
Le RR pour la SG a atteint la significativité statistique dans quelques sous-groupes prédéfinis, dont les patients ayant un risque cytogénétique défavorable, les patients présentant une LAM avec anomalies associées aux myélodysplasies, les patients âgés de moins de 75 ans, les femmes et les patients blancs.
Les réponses hématologique et cytogénétique ont été évaluées par les investigateurs et par le CRI avec des résultats comparables. Le taux de réponse globale (rémission complète [RC] + rémission complète avec récupération incomplète de la numération sanguine [RCi]) déterminé par le CRI a été de 27,8 % dans le bras recevant l’azacitidine et de 25,1 % dans les bras sous CCR poolés (p = 0,5384). Chez les patients ayant obtenu une RC ou une RCi, la durée de rémission médiane a été de 10,4 mois (IC à 95 % = 7,2 ; 15,2) chez les patients recevant l’azacitidine et de 12,3 mois (IC à 95 % = 9,0 ; 17,0) chez les patients recevant un CCR. Un bénéfice en termes de survie a également été démontré pour l’azacitidine par rapport aux CCR chez les patients qui n’avaient pas obtenu de réponse complète.
Le traitement par l’azacitidine a amélioré les numérations sanguines périphériques et a entraîné une diminution du besoin de transfusions de globules rouges (GR) et de plaquettes. Un patient était considéré comme dépendant des transfusions de globules rouges ou de plaquettes lors de l’inclusion s’il avait reçu une ou plusieurs transfusions de GR ou de plaquettes au cours des 56 jours (8 semaines) précédant ou suivant la randomisation respectivement. Un patient était considéré comme indépendant des transfusions de GR ou de plaquettes pendant la période de traitement s’il n’avait pas reçu de transfusions de GR ou de plaquettes pendant 56 jours consécutifs au cours de la période de notification.
Chez les patients du bras l’azacitidine qui étaient initialement dépendants des transfusions de globules rouges, 38,5 % (IC à 95 % = 31,1 ; 46,2) sont devenus indépendants des transfusions de GR pendant la période de traitement contre 27,6 % (IC à 95 % = 20,9 ; 35,1) des patients des groupes CCR poolés. Chez les patients qui étaient initialement dépendants des transfusions de globules rouges et qui sont devenus indépendants des transfusions pendant la période de traitement, la durée médiane d’indépendance aux transfusions de GR a été de 13,9 mois dans le bras l’azacitidine et n’a pas été atteinte dans le bras CCR.
Chez les patients du bras l’azacitidine qui étaient dépendants des transfusions de plaquettes lors de l’inclusion, 40,6 % (IC à 95 % = 30,9 ; 50,8) sont devenus indépendants des transfusions pendant la période de traitement contre 29,3 % (IC à 95 %= 19,7 ; 40,4) des patients des groupes CCR poolés. Chez les patients qui étaient initialement dépendants des transfusions de plaquettes et qui sont devenus indépendants des transfusions pendant la période de traitement, la durée médiane d’indépendance aux transfusions de plaquettes a été de 10,8 mois dans le bras l’azacitidine et de 19,2 mois dans le bras CCR.
La qualité de vie liée à la santé (QdVLS) a été évaluée à l’aide du questionnaire de base sur la qualité de vie de l’European Organization for Research and Treatment of Cancer (EORTC QLQ-C30). Les données de QdVLS ont pu être analysées pour un sous-groupe de la population totale de l’étude. Malgré des limites dans l’analyse, les données disponibles semblent indiquer que les patients ne présentent pas une détérioration significative de la qualité de vie pendant le traitement par l’azacitidine.
Population pédiatrique
L’étude AZA-JMML-001 était une étude de phase II, internationale, multicentrique, en ouvert, visant à évaluer la pharmacocinétique, la pharmacodynamique, la sécurité et l’activité de l’azacitidine avant une GCSH chez des patients pédiatriques atteints d’un SMD ou d’une LMMJ avancé(e) nouvellement diagnostiqué(e). L’objectif principal de l’étude clinique était d’évaluer l’effet de l’azacitidine sur le taux de réponse au Cycle 3, Jour 28.
Les patients (SMD, n = 10 ; LMMJ, n = 18, 3 mois à 15 ans ; 71 % de garçons) ont été traités par 75 mg/m² de l’azacitidine par voie intraveineuse, quotidiennement les Jours 1 à 7 d’un cycle de 28 jours pendant un minimum de 3 cycles et un maximum de 6 cycles.
L’inclusion dans le bras SMD de l’étude a été arrêtée après 10 patients SMD en raison du manque d’efficacité : aucune réponse confirmée n’a été rapportée pour ces 10 patients.
Dans le bras LMMJ de l’étude, 18 patients (13 avec la mutation somatique PTPN11, 3 avec NRAS et 1 avec KRAS, et 1 diagnostic clinique de neurofibromatose de type 1 [NF-1]) ont été inclus. Seize patients ont effectué 3 cycles de traitement et 5 ont terminé les 6 cycles. Un total de 11 patients LMMJ a présenté une réponse clinique au Cycle 3, Jour 28, et parmi ces 11 sujets, 9 (50 %) avaient une réponse clinique confirmée (3 sujets avec une RCc et 6 sujets avec une RPc).
Parmi la cohorte de patients LMMJ traités par l’azacitidine, 7 patients (43,8 %) ont montré une réponse plaquettaire soutenue (numération ≥ 100 x 109/L) et 7 patients (43,8 %) ont nécessité une transfusion à la GCSH. La GCSH a pu être effectuée chez 17 patients sur les 18.
En raison de sa conception (petit nombre de patients et différents facteurs de confusion), cette étude ne permet pas de conclure si l’azacitidine avant la GCSH améliore la survie chez les patients LMMJ.
L’étude AZA-AML-004 était une étude de phase II, multicentrique et en ouvert, visant à évaluer la sécurité, la pharmacodynamique et l’efficacité de l’azacitidine par rapport à l’absence de tout traitement anticancéreux chez l’enfant et le jeune adulte présentant une LAM en rechute moléculaire après une RC1.
Sept patients (âge médian : 6,7 ans [intervalle : 2 à 12 ans] ; 71,4 % de garçons) ont été traités par 100 mg/m² de l’azacitidine par voie intraveineuse quotidiennement les Jours 1 à 7 de chaque cycle de 28 jours pendant un maximum de 3 cycles.
Cinq patients ont passé une évaluation de la maladie résiduelle minimale (MRM) le Jour 84. 4 d’entre eux ont atteint une stabilisation moléculaire (n = 3) ou une amélioration moléculaire (n = 1) et 1 patient a présenté une rechute clinique. Six des 7 patients (90 % [IC à 95 % = 0,4 ; 1,0]) traités par l’azacitidine avaient reçu une greffe de cellules souches hématopoïétiques (GCSH).
En raison de la faible taille de l’échantillon, l’efficacité de l’azacitidine chez les patients pédiatriques présentant une LAM ne peut pas être établie.
Voir la rubrique 4.8 pour des informations sur la sécurité.
5.2. Propriétés pharmacocinétiques
Absorption
Après administration sous-cutanée d’une dose unique de 75 mg/m2, l’azacitidine a été rapidement absorbée, avec un pic de concentration plasmatique de 750 ± 403 ng/mL atteint 0,5 h après l’administration (premier prélèvement effectué). La biodisponibilité absolue de l’azacitidine administrée par voie sous-cutanée par rapport à l’azacitidine administrée par voie intraveineuse (doses uniques de 75 mg/m2) a été d’approximativement 89 % d’après l’aire sous la courbe (ASC).
L’aire sous la courbe et la concentration plasmatique maximale (Cmax) de l’azacitidine administrée par voie sous-cutanée ont été approximativement proportionnelles dans l’intervalle de doses de 25 à 100 mg/m2.
Distribution
Suite à l’administration intraveineuse, le volume de distribution moyen a été de 76 ± 26 L et la clairance systémique de 147 ± 47 L/h.
Biotransformation
D’après les données in vitro, le métabolisme de l’azacitidine ne semble pas être médié par les isoenzymes du cytochrome P450 (CYP), les UDP-glucuronosyl-transférases (UGT), les sulfotransférases (SULT) et les glutathion transférases (GST).
L’azacitidine subit une hydrolyse spontanée et une désamination par la cytidine désaminase. Au niveau des fractions hépatiques humaines S9, la formation des métabolites a été indépendante du NADPH, ce qui implique que les isoenzymes du cytochrome P450 ne contribuent pas au métabolisme de l’azacitidine. Une étude in vitro de l’azacitidine sur des cultures d’hépatocytes humains indique qu’à des concentrations de 1,0 µM à 100 µM (c.-à-d. jusqu’à 30 fois environ la concentration atteinte en pratique clinique), l’azacitidine n’a pas d’effet inducteur sur les CYP 1A2, 2C19, 3A4 ou 3A5. Lors d’études visant à évaluer l’inhibition d’une série d’isoenzymes du cytochrome P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 et 3A4), l’azacitidine à des concentrations allant jusqu’à 100 µM n’a pas eu d’effet inhibiteur. Il est donc improbable que l’azacitidine entraîne une induction ou une inhibition des isoenzymes CYP aux concentrations plasmatiques atteintes en pratique clinique.
Élimination
L’azacitidine est rapidement éliminée du plasma, avec une demi-vie d’élimination (t½) moyenne, après administration sous-cutanée, de 41 ± 8 minutes. Aucune accumulation n’est observée après l’administration sous-cutanée de 75 mg/m2 d’azacitidine une fois par jour pendant 7 jours.
L’élimination de l’azacitidine et/ou de ses métabolites se fait principalement par excrétion urinaire. Après administration intraveineuse et sous-cutanée de 14C-azacitidine, respectivement 85 % et 50 % de la radioactivité administrée ont été retrouvés dans les urines contre < 1 % dans les selles.
Populations particulières
Les effets de l’insuffisance hépatique (voir rubrique 4.2), du sexe, de l’âge ou des origines ethniques sur les propriétés pharmacocinétiques de l’azacitidine n’ont pas été formellement étudiés.
Population pédiatrique
Dans l’étude AZA-JMML-001, l’analyse pharmacocinétique a été réalisée le Jour 7 du Cycle 1 auprès de 10 patients pédiatriques SMD et 18 patients LMMJ (voir rubrique 5.1). L’âge médian (intervalle) des patients SMD était de 13,3 ans (1,9-15) et de 2,1 ans (0,2-6,9) pour les patients LMMJ.
Après administration intraveineuse d’une dose de 75 mg/m², l’azacitidine atteint rapidement la Cmax en 0,083 heure, tant dans la population SMD que LMMJ. Les moyennes géométriques de la Cmax étaient de 1 797,5 et de 1 066,3 ng/mL, et les moyennes géométriques de l’ASC0-∞ étaient de 606,9 et 240,2 ng.h/mL, pour les patients SMD et LMMJ, respectivement. Les moyennes géométriques du volume de distribution des sujets SMD et LMMJ étaient de 103,9 et 61,1 L, respectivement. Il est apparu que l’exposition plasmatique totale du l’azacitidine était supérieure chez les sujets SMD ; toutefois, une variabilité inter-patient modérée à élevée a été observée tant pour l’ASC que pour la Cmax.
Les moyennes géométriques de t1/2 étaient de 0,4 et de 0,3 heure, et les moyennes géométriques de la clairance étaient de 166,4 et de 148,3 L/h pour SMD et LMMJ, respectivement.
Les données pharmacocinétiques de l’étude AZA-JMML-001 ont été rassemblées et comparées aux données pharmacocinétiques de 6 sujets adultes SMD ayant reçu 75 mg/m² de l’azacitidine par voie intraveineuse de l’étude AZA-2002-BA-002. La Cmax et l’ASC0-t moyenne étaient similaires entre les patients adultes et les patients pédiatriques après administration intraveineuse (2 750 ng/mL comparé à 2 841 ng/mL et 1 025 ng.h/mL comparé à 882,1 ng.h/mL, respectivement).
Dans l’étude AZA-AML-004, une analyse pharmacocinétique a été réalisée auprès de 6 des 7 patients pédiatriques qui présentaient au moins une concentration pharmacocinétique mesurable après l’administration d’une dose (voir rubrique 5.1). L’âge médian (intervalle) des patients présentant une LAM était de 6,7 ans (2-12).
Après administration de plusieurs doses de 100 mg/m², les moyennes géométriques de la Cmax et de l’ASC0-tau le Jour 7 du Cycle 1 étaient de 1 557 ng/mL et de 899,6 ng.h/mL respectivement, avec l’observation d’une forte variabilité interindividuelle (%CV de 201,6 % et de 87,8 % respectivement). Après l’administration intraveineuse, l’azacitidine a rapidement atteint la Cmax (délai médian de 0,090 heure) avant de diminuer (moyenne géométrique de t1/2 de 0,380 heure). Les moyennes géométriques de la clairance et du volume de distribution étaient de 127,2 L/h et de 70,2 L respectivement.
L’exposition pharmacocinétique (à l’azacitidine) observée chez les enfants présentant une LAM en rechute moléculaire après une RC1 était comparable non seulement à l’exposition relevée à partir de données groupées portant sur 10 enfants présentant un SMD et sur 18 enfants présentant une LMMJ, mais également à l’exposition à l’azacitidine documentée chez des adultes présentant un SMD.
Insuffisance rénale
L’insuffisance rénale n’a pas d’effet majeur sur les paramètres d’exposition pharmacocinétique de l’azacitidine après administration sous-cutanée de doses uniques et répétées. Après administration sous-cutanée d’une dose unique de 75 mg/m2, les valeurs d’exposition moyennes (ASC et Cmax) chez les patients présentant une insuffisance rénale légère, modérée et sévère étaient augmentées de respectivement 11 à 21 %, 15 à 27 % et 41 à 66 % par rapport aux sujets ayant une fonction rénale normale. Cependant, l’exposition était dans le même intervalle général d’expositions que celles qui sont observées chez les sujets ayant une fonction rénale normale. Chez les patients présentant une insuffisance rénale, l’azacitidine peut être administrée sans ajustement posologique initial, sous réserve que ces patients soient surveillés afin de détecter toute toxicité car l’azacitidine et/ou ses métabolites sont excrétés principalement par les reins.
Caractéristiques pharmacogénomiques
Les effets des polymorphismes connus de la cytidine désaminase sur le métabolisme de l’azacitidine n’ont pas été formellement étudiés.
5.3. Données de sécurité préclinique
L’azacitidine induit des mutations génétiques et des aberrations chromosomiques dans les systèmes cellulaires de bactéries et de mammifères in vitro. La carcinogénicité de l’azacitidine a été évaluée chez la souris et le rat. L’azacitidine a induit des tumeurs dans le système hématopoïétique des souris femelles, lorsqu’elle a été administrée par voie intrapéritonéale 3 fois par semaine pendant 52 semaines. Une augmentation de l’incidence des tumeurs du système lymphoréticulaire, des poumons, des glandes mammaires et de la peau a été observée chez les souris traitées par l’azacitidine administrée par voie intrapéritonéale pendant 50 semaines. Une étude du potentiel tumorigène chez le rat a fait apparaître une augmentation de l’incidence des tumeurs testiculaires.
Les études d’embryotoxicité précoce chez la souris ont révélé une fréquence de 44 % de décès embryonnaires intra-utérins (résorption accrue) suite à une injection unique d’azacitidine par voie intrapéritonéale en cours d’organogenèse. Des anomalies du développement cérébral ont été détectées chez la souris sous azacitidine pendant ou avant la soudure de la voûte palatine. Chez le rat, l’azacitidine n’a provoqué aucun effet indésirable lorsqu’elle était administrée avant l’implantation mais elle s’est révélée clairement embryotoxique lorsqu’elle était administrée pendant l’organogenèse. Les anomalies fœtales survenues en cours d’organogenèse chez le rat ont été notamment : anomalies du SNC (exencéphalie/encéphalocèle), anomalies des membres (micromélie, pied bot, syndactylie, oligodactylie) et autres (microphtalmie, micrognathie, laparoschisis, œdème et anomalies costales).
L’administration d’azacitidine chez la souris mâle avant l’accouplement avec une femelle non traitée a entraîné une réduction de la fertilité et des pertes dans la descendance lors du développement embryonnaire et post-natal ultérieur. Le traitement des rats mâles a engendré une réduction de la masse testiculaire et des épididymes, une diminution de la numération des spermatozoïdes, une réduction des taux de grossesse, une augmentation du nombre d’embryons anormaux et des pertes embryonnaires accrues chez les femelles fécondées (voir rubrique 4.6).
Mannitol (E421).
Ce médicament ne doit pas être mélangé avec d’autres médicaments à l’exception de ceux mentionnés dans la rubrique 6.6.
Flacon de poudre non ouvert :
4 ans.
Après reconstitution :
Lorsque AZACITIDINE HIKMA est reconstitué en utilisant de l’eau pour préparations injectables qui n’a pas été réfrigérée, la stabilité physicochimique en cours d’utilisation du médicament reconstitué a été démontrée 45 minutes à 25 °C et de 8 heures entre 2 °C et 8 °C.
La durée de conservation du médicament reconstitué peut être prolongée en utilisant de l’eau pour préparations injectables réfrigérée (2 °C à 8 °C) pour la reconstitution. Lorsque AZACITIDINE HIKMA est reconstitué en utilisant de l’eau pour préparations injectables réfrigérée (2 °C à 8 °C), la stabilité physicochimique en cours d’utilisation du médicament reconstitué a été démontrée 22 heures entre 2 °C et 8 °C.
D’un point de vue microbiologique, le produit reconstitué doit être utilisé immédiatement. En cas d’utilisation non immédiate, la durée et les conditions de conservation du médicament reconstitué avant administration relèvent de la responsabilité de l’utilisateur et ne doivent en aucun cas dépasser 8 heures entre 2 °C et 8 °C en cas de reconstitution avec de l’eau pour préparations injectables non réfrigérée ou 22 heures en cas de reconstitution avec de l’eau pour préparations injectables réfrigérée (2 °C à 8 °C).
6.4. Précautions particulières de conservation
Flacons non ouverts
Ce médicament ne nécessite pas de précautions particulières de conservation.
Suspension reconstituée
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon en verre incolore de type I fermé hermétiquement par un bouchon en caoutchouc et une capsule en aluminium type flip-off contenant 100 mg d’azacitidine.
Présentation : 1 flacon.
6.6. Précautions particulières d’élimination et de manipulation
Recommandations pour une manipulation en toute sécurité
AZACITIDINE HIKMA est un médicament cytotoxique et, comme pour toute autre substance potentiellement toxique, la manipulation et la préparation de la suspension d’azacitidine doivent être réalisées avec précaution. Les procédures appropriées de manipulation et d’élimination applicables aux médicaments anticancéreux doivent être respectées.
Si l’azacitidine reconstituée entre en contact avec la peau, rincer immédiatement et abondamment avec de l’eau et du savon. Si elle entre en contact avec les muqueuses, rincer abondamment avec de l’eau.
Procédure de reconstitution
AZACITIDINE HIKMA doit être reconstitué avec de l’eau pour préparations injectables. La durée de conservation du médicament reconstitué peut être prolongée en utilisant de l’eau pour préparations injectables réfrigérée (2 °C à 8 °C) pour la reconstitution. Des informations sur la conservation du médicament reconstitué figurent ci-dessous.
1. Réunir les éléments suivants : flacon(s) d’azacitidine ; flacon(s) d’eau pour préparations injectables ; gants chirurgicaux non stériles ; lingettes désinfectantes ; seringue(s) pour injection de 5 mL avec aiguille(s).
2. Aspirer 4 mL d’eau pour préparations injectables dans la seringue, en veillant à expulser toute bulle d’air présente dans la seringue.
3. Introduire l’aiguille de la seringue contenant les 4 mL d’eau pour préparations injectables dans le bouchon en caoutchouc du flacon d’azacitidine et injecter l’eau pour préparations injectables dans le flacon.
4. Retirer la seringue et l’aiguille, agiter vigoureusement le flacon jusqu’à obtenir une suspension trouble uniforme. Après reconstitution, chaque mL de suspension contient 25 mg d’azacitidine (100 mg/4 mL). Le produit reconstitué se présente sous la forme d’une suspension trouble homogène dépourvue d’agglomérats. Jeter la suspension si elle contient de grosses particules ou des agglomérats. Ne pas filtrer la suspension après reconstitution car cela pourrait éliminer la substance active. Tenir compte du fait que certains adaptateurs, dispositifs sans aiguille de type spikes et systèmes fermés sont équipés de filtres ; ces dispositifs ou systèmes ne doivent donc pas être utilisés pour l’administration du médicament reconstitué.
5. Nettoyer le dessus du bouchon et introduire une nouvelle seringue avec aiguille dans le flacon. Retourner le flacon et s’assurer que l’extrémité de l’aiguille se situe en dessous de la surface du liquide. Tirer le piston afin d’aspirer le volume de médicament correspondant à la dose appropriée, en veillant à expulser toute bulle d’air présente dans la seringue. Retirer la seringue avec aiguille du flacon et jeter l’aiguille.
6. Fixer solidement une aiguille pour injection sous-cutanée neuve (calibre 25 Gauge recommandé) sur la seringue. Afin de réduire l’incidence des réactions locales au site d’injection, l’aiguille ne doit pas être purgée avant l’injection.
7. Lorsque plus d’un flacon est nécessaire, réitérer les étapes ci-dessus pour achever la préparation de la suspension. Si la dose requiert plus d’un flacon, elle doit être répartie de façon égale, par exemple, pour une dose de 150 mg = 6 mL, 2 seringues de 3 mL chacune. Une petite quantité de suspension peut rester dans le flacon et l’aiguille et il peut ne pas être possible d’aspirer la totalité de la suspension du flacon.
8. Le contenu de la seringue doit être remis en suspension immédiatement avant l’administration. La seringue contenant la suspension reconstituée doit être laissée à température ambiante pendant 30 minutes au maximum avant l’administration jusqu’à ce qu’elle atteigne une température d’environ 20 °C à 25 °C. Si ce délai de 30 minutes est dépassé, la suspension doit être éliminée de façon appropriée et une nouvelle dose doit être préparée. Pour remettre le produit en suspension, faire rouler la seringue vigoureusement entre les paumes de la main jusqu’à obtenir une suspension trouble uniforme. Jeter la suspension si elle contient de grosses particules ou des agglomérats.
Conservation du médicament reconstitué
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
Calcul d’une dose spécifique
La dose totale basée sur la surface corporelle peut être calculée ainsi :
Dose totale (mg) = dose (mg/m2) x surface corporelle (m²)
Le tableau suivant est proposé uniquement à titre d’exemple pour montrer comment calculer une dose d’azacitidine spécifique pour une surface corporelle moyenne de 1,8 m2.
|
Dose en mg/m2 (% de la dose initiale recommandée) |
Dose totale basée sur une surface corporelle de 1,8 m2 |
Nombre de flacons nécessaires |
Volume total de suspension reconstituée requis |
|
75 mg/m2 (100 %) |
135 mg |
2 flacons |
5,4 mL |
|
37,5 mg/m2 (50 %) |
67,5 mg |
1 flacon |
2,7 mL |
|
25 mg/m2 (33 %) |
45 mg |
1 flacon |
1,8 mL |
Mode d’administration
Une fois reconstitué, AZACITIDINE HIKMA doit être injecté par voie sous-cutanée (introduire l’aiguille avec un angle de 45 à 90°) à l’aide d’une aiguille de calibre 25 Gauge dans le haut du bras, la cuisse ou l’abdomen.
Les doses supérieures à 4 mL doivent être injectées dans deux sites différents.
Les sites d’injection doivent être alternés. Chaque nouvelle injection doit être pratiquée à au moins 2,5 cm de distance du site précédent et en aucun cas sur une zone sensible, présentant une ecchymose, une rougeur ou une induration.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
HIKMA FARMACÊUTICA (PORTUGAL), S.A.
ESTRADA DO RIO DA MÓ, 8, 8A E 8B
FERVENÇA
2705-906 TERRUGEM SNT
PORTUGAL
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 567 7 0 : Poudre en flacon (verre). Boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation : {JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 11/03/2024
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable
Azacitidine
· Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu’est-ce que AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d’utiliser AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
3. Comment utiliser AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
6. Contenu de l’emballage et autres informations
1. QU’EST-CE QUE AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Agents antinéoplasiques, analogues de la pyrimidine, Code ATC : L01BC07
Qu’est-ce que AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est un agent anticancéreux qui appartient à un groupe de médicaments appelés « antimétabolites ». AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable contient la substance active « azacitidine ».
Dans quels cas AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est-il utilisé ?
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est utilisé chez les adultes qui ne peuvent pas recevoir une greffe de cellules souches afin de traiter :
· Les syndromes myélodysplasiques (SMD) de risque élevé.
· La leucémie myélomonocytaire chronique (LMMC).
· La leucémie aiguë myéloblastique (LAM).
Ces maladies touchent la moelle osseuse et peuvent altérer la production de cellules sanguines normales.
Comment agit AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
AZACITIDINE HIKMA agit en empêchant les cellules cancéreuses de se développer. L’azacitidine pénètre dans le matériel génétique présent dans les cellules (acide ribonucléique [ARN] et acide désoxyribonucléique [ADN]). Elle agit en modifiant la façon dont les cellules activent et désactivent les gènes et en interférant avec la synthèse d’ARN et d’ADN. Ces actions corrigent les problèmes de croissance et de maturation des jeunes cellules sanguines dans la moelle osseuse qui sont responsables des syndromes myélodysplasiques et tuent les cellules cancéreuses dans les leucémies. Pour toutes questions sur la façon dont AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable agit ou la raison pour laquelle ce médicament vous a été prescrit, adressez-vous à votre médecin ou infirmier/ère.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
N’utilisez jamais AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable
· si vous êtes allergique à l’azacitidine ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6 ;
· si vous souffrez d’un cancer du foie à un stade avancé ;
· si vous allaitez.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou infirmier/ère avant de prendre AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable :
· si vous avez un nombre réduit de plaquettes sanguines, de globules rouges ou de globules blancs ;
· si vous avez une maladie des reins ;
· si vous avez une maladie du foie ;
· si vous avez eu dans le passé une affection cardiaque ou une crise cardiaque ou si vous avez des antécédents de maladie pulmonaire.
AZACITIDINE HIKMA 25 mg/mL, poudre pour solution injectable peut provoquer une réaction immunitaire grave appelée « syndrome de différentiation » (voir rubrique 4).
Analyses de sang
Vous ferez des analyses sanguines avant de commencer le traitement par AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable et au début de chaque période de traitement (appelée « cycle »). Elles visent à vérifier que vos cellules sanguines sont en nombre suffisant et que votre foie et vos reins fonctionnent correctement.
Enfants et adolescents
L’utilisation de AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable chez les enfants et adolescents de moins de 18 ans n’est pas recommandée.
Autres médicaments et AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament. AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable peut interférer avec l’action de certains autres médicaments. Inversement, certains autres médicaments peuvent interférer avec l’action de AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable.
Grossesse, allaitement et fertilité
Grossesse
Vous ne devez pas utiliser AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable pendant la grossesse car il pourrait être nocif pour votre enfant.
Si vous êtes une femme en âge de procréer (susceptible d'être enceinte), vous devez utiliser une méthode de contraception efficace pendant votre traitement par AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable et pendant 6 mois après l'arrêt du traitement par AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable.
Prévenez immédiatement votre médecin si vous devenez enceinte pendant le traitement par AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Allaitement
Vous ne devez pas allaiter pendant le traitement par AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable. On ne sait pas si ce médicament passe dans le lait maternel.
Fertilité
Les hommes ne doivent pas concevoir d’enfant pendant le traitement par AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable. Les hommes doivent utiliser une méthode de contraception efficace pendant le traitement par AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable et pendant 3 mois après l’arrêt du traitement par AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable.
Si vous souhaitez conserver un échantillon de votre sperme avant de recevoir ce traitement, parlez-en à votre médecin.
Conduite de véhicules et utilisation de machines
Ne conduisez pas de véhicules et n’utilisez pas d’outils ou de machines si vous présentez des effets indésirables tels que la fatigue.
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable contient
Sans objet.
3. COMMENT utiliser AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
Avant l’administration de AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable, votre médecin vous donnera un autre médicament pour vous éviter les nausées et les vomissements au début de chaque cycle de traitement.
· La dose recommandée est de 75 mg par m² de surface corporelle. Votre médecin déterminera votre dose de médicament en fonction de votre état général, de votre taille et de votre poids. Votre médecin surveillera l’évolution de votre état et pourra modifier la dose si nécessaire.
· AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est administré chaque jour pendant une semaine, suivie d’une période de repos de 3 semaines. Ce « cycle de traitement » sera répété toutes les 4 semaines. En général, au moins 6 cycles de traitement vous seront administrés.
Ce médicament vous sera administré en injection sous-cutanée (sous la peau) par un médecin ou un(e) infirmier/ère. Il pourra être injecté sous la peau de votre cuisse, de votre ventre ou dans le haut de votre bras.
Si vous avez utilisé plus d’AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable que vous n’auriez dû
Sans objet.
Si vous oubliez d’utiliser AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable
Sans objet.
Si vous arrêtez d’utiliser AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Prévenez immédiatement votre médecin si vous remarquez l’un des effets indésirables suivants :
· Somnolence, tremblements, ictère (« jaunisse »), ballonnements abdominaux et tendance aux ecchymoses (« bleus »). Ils peuvent être des symptômes d’insuffisance hépatique pouvant mettre en jeu la vie du patient.
· Gonflement des jambes et des pieds, douleurs dans le dos, diminution du volume des urines, soif excessive, pouls rapide, étourdissements et nausées, vomissements ou diminution de l’appétit, et sensations de confusion, agitation ou fatigue. Ils peuvent être des symptômes d’insuffisance rénale pouvant mettre en jeu la vie du patient.
· Fièvre. Cela pourrait être dû à une infection résultant du nombre réduit de globules blancs, qui peut mettre en jeu la vie du patient.
· Douleur dans la poitrine ou essoufflement, pouvant s’accompagner de fièvre. Cela peut être dû à une infection des poumons appelée « pneumonie » qui peut mettre en jeu la vie du patient.
· Saignements. Par exemple, un saignement dans le crâne ou une présence de sang dans les selles due à un saignement dans l’estomac ou les intestins. Cela peut être des symptômes d’un taux faible de plaquettes dans le sang.
· Difficultés à respirer, gonflement des lèvres, démangeaisons ou éruption cutanée. Cela peut être dû à une réaction allergique (hypersensibilité).
Les autres effets indésirables sont :
Effets indésirables très fréquents (pouvant affecter plus de 1 patient sur 10) :
· Nombre réduit de globules rouges (anémie). Vous pouvez vous sentir fatigué et pâle.
· Nombre réduit de globules blancs, qui peut s’accompagner de fièvre. Vous présentez également un risque accru d’infections.
· Nombre réduit de plaquettes sanguines (thrombocytopénie). Vous êtes plus facilement sujet aux saignements et ecchymoses.
· Constipation, diarrhée, nausées, vomissements.
· Pneumonie.
· Douleur dans la poitrine, essoufflement.
· Fatigue.
· Réaction au site d’injection, avec notamment une rougeur, une douleur ou une réaction cutanée.
· Perte d’appétit.
· Douleurs articulaires.
· Ecchymose (bleu).
· Éruption cutanée.
· Points rouges ou violets sous la peau.
· Maux de ventre (douleur abdominale).
· Démangeaisons.
· Fièvre.
· Douleurs dans le nez et maux de gorge.
· Étourdissements.
· Maux de tête.
· Troubles du sommeil (insomnie).
· Saignements de nez (épistaxis).
· Douleurs musculaires.
· Faiblesse (asthénie).
· Perte de poids.
· Faible taux de potassium dans le sang.
Effets indésirables fréquents (pouvant affecter jusqu’à 1 patient sur 10) :
· Saignement à l’intérieur de la tête.
· Infection du sang provoquée par une bactérie (septicémie). Cela peut être dû au nombre réduit de globules blancs dans votre sang.
· Moelle osseuse défaillante. Cela peut entraîner une diminution du nombre de globules rouges et blancs et de plaquettes.
· Type d’anémie avec une diminution du nombre de globules rouges, de globules blancs et de plaquettes.
· Infection urinaire.
· Infection virale responsable de boutons de fièvre (herpès).
· Saignement des gencives, saignement dans l’estomac ou les intestins, saignement au niveau du rectum dû aux hémorroïdes (hémorragie hémorroïdaire), saignement dans les yeux, saignement sous la peau ou dans la peau (hématome).
· Présence de sang dans les urines.
· Aphtes dans la bouche ou sur la langue.
· Altérations de la peau au niveau du site d’injection. Il peut s’agir d’un gonflement, d’une masse dure, d’un bleu, d’un saignement dans la peau (hématome), d’une éruption cutanée, de démangeaisons ou d’un changement de couleur de la peau.
· Rougeur de la peau.
· Infection de la peau (cellulite).
· Infection du nez et de la gorge, ou maux de gorge.
· Douleur ou écoulement dans le nez ou les sinus (sinusite).
· Pression artérielle élevée ou basse (hypertension ou hypotension).
· Essoufflement pendant l’effort.
· Douleur dans la gorge et le larynx.
· Indigestion.
· Léthargie.
· Sensation de malaise général.
· Anxiété.
· Confusion.
· Perte de cheveux.
· Insuffisance rénale.
· Déshydratation.
· Dépôts blanchâtres sur la langue, l’intérieur des joues et parfois le palais, les gencives et les amygdales (mycose buccale).
· Évanouissement.
· Chute de la pression artérielle provoquant des vertiges lors du passage en position debout ou assise (hypotension orthostatique).
· Envie de dormir (somnolence).
· Saignement causé par le cathéter.
· Maladie touchant l’intestin pouvant provoquer une fièvre, des vomissements et des douleurs abdominales (diverticulite).
· Présence de liquide autour des poumons (épanchement pleural).
· Tremblements (frissons).
· Spasmes musculaires.
· Éruption de plaques en relief sur la peau accompagnées de démangeaisons (urticaire).
· Accumulation de liquide autour du cœur (épanchement péricardique).
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 patient sur 100) :
· Réaction allergique (hypersensibilité).
· Tremblements.
· Insuffisance hépatique.
· Grandes plaques violacées surélevées douloureuses sur la peau accompagnées de fièvre.
· Ulcérations cutanées douloureuses (pyoderma gangrenosum).
· Inflammation de la membrane enveloppant le cœur (péricardite).
Effets indésirables rares (pouvant affecter jusqu’à 1 patient sur 1 000) :
· Toux sèche.
· Gonflement indolore de l’extrémité des doigts (hippocratisme digital).
· Syndrome de lyse tumorale : des complications métaboliques peuvent survenir pendant le traitement du cancer et même parfois sans traitement. Ces complications sont causées par les substances libérées par les cellules cancéreuses détruites. Elles peuvent inclure : des modifications du bilan sanguin ; des taux élevés de potassium, de phosphore, d’acide urique et de faibles taux de calcium entraînant des modifications de la fonction rénale et du rythme cardiaque, des convulsions et dans certains cas le décès.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
· Infection des couches profondes de la peau qui s’étend rapidement en provoquant une destruction de la peau et des tissus, susceptible d’engager le pronostic vital (fasciite nécrosante).
· Réaction immunitaire grave (syndrome de différenciation) qui peut provoquer de la fièvre, de la toux, des difficultés respiratoires, des éruptions cutanées, une diminution de la quantité d'urine, une pression sanguine basse (hypotension), un gonflement des bras ou des jambes et une prise de poids rapide.
· Inflammation des vaisseaux sanguins dans la peau pouvant entraîner une éruption cutanée (vascularite cutanée)
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette du flacon et la boîte. La date de péremption fait référence au dernier jour de ce mois.
Votre médecin, votre pharmacien ou votre infirmier/ère se chargera de conserver AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable. Ils s’occuperont également de préparer et d’éliminer de façon appropriée le reste de AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable inutilisé.
Flacons non ouverts : ce médicament ne nécessite pas de précautions particulières de conservation.
Lorsque AZACITIDINE HIKMA est reconstitué en utilisant de l’eau pour préparations injectables qui n’a pas été réfrigérée, la stabilité physicochimique en cours d’utilisation du médicament reconstitué a été démontrée 45 minutes à 25 °C et de 8 heures entre 2 °C et 8 °C.
La durée de conservation du médicament reconstitué peut être prolongée en utilisant de l’eau pour préparations injectables réfrigérée (2 °C à 8 °C) pour la reconstitution. Lorsque AZACITIDINE HIKMA est reconstitué en utilisant de l’eau pour préparations injectables réfrigérée (2 °C à 8 °C), la stabilité physicochimique en cours d’utilisation du médicament reconstitué a été démontrée 22 heures entre 2 °C et 8 °C.
Avant l’administration, laisser la suspension atteindre la température ambiante (20 °C-25 °C) pendant 30 minutes.
Si la suspension contient de grosses particules, elle ne doit pas être utilisée.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable
· La substance active est l’azacitidine. Un flacon contient 100 mg d’azacitidine. Après reconstitution avec 4 mL d’eau pour préparations injectables, la suspension reconstituée contient 25 mg/mL d’azacitidine.
· L’autre composant est le mannitol (E421).
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est une poudre pour suspension injectable de couleur blanche conditionnée dans un flacon en verre contenant 100 mg d’azacitidine.
Chaque boîte contient un flacon de AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable.
Titulaire de l’autorisation de mise sur le marché
HIKMA FARMACÊUTICA (PORTUGAL), S.A.
ESTRADA DO RIO DA MÓ, 8, 8A E 8B
FERVENÇA
2705-906 TERRUGEM SNT
PORTUGAL
Exploitant de l’autorisation de mise sur le marché
HIKMA France
105 RUE MARCEL DASSAULT
92100 BOULOGNE-BILLANCOURT
FRANCE
THYMOORGAN PHARMAZIE GMBH
SCHIFFGRABEN 23
38690 GOSLAR-VIENENBURG
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen et au Royaume-Uni (Irlande du Nord) sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Recommandations pour une manipulation en toute sécurité
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable est un médicament cytotoxique et, comme pour toute autre substance potentiellement toxique, la manipulation et la préparation de la suspension d’azacitidine doivent être réalisées avec précaution. Les procédures appropriées de manipulation et d’élimination applicables aux médicaments anticancéreux doivent être respectées.
Si l’azacitidine reconstituée entre en contact avec la peau, rincer immédiatement et abondamment avec de l’eau et du savon. Si elle entre en contact avec les muqueuses, rincer abondamment avec de l’eau.
Incompatibilités
Ce médicament ne doit pas être mélangé avec d’autres médicaments à l’exception de ceux mentionnés ci-dessous (voir « Procédure de reconstitution »).
Procédure de reconstitution
AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable doit être reconstitué avec de l’eau pour préparations injectables. La durée de conservation du médicament reconstitué peut être prolongée en utilisant de l’eau pour préparations injectables réfrigérée (entre 2 °C et 8 °C) pour la reconstitution. Des informations sur la conservation du médicament reconstitué figurent ci-dessous.
1. Réunir les éléments suivants :
Flacon(s) d’azacitidine ; flacon(s) d’eau pour préparations injectables ; gants chirurgicaux non stériles ; lingettes désinfectantes ; seringue(s) pour injection de 5 mL avec aiguille(s).
2. Aspirer 4 mL d’eau pour préparations injectables dans la seringue, en veillant à expulser toute bulle d’air présente dans la seringue.
3. Introduire l’aiguille de la seringue contenant les 4 mL d’eau pour préparations injectables dans le bouchon en caoutchouc du flacon d’azacitidine et injecter l’eau pour préparations injectables dans le flacon.
4. Retirer la seringue et l’aiguille, agiter vigoureusement le flacon jusqu’à obtenir une suspension trouble uniforme. Après reconstitution, chaque mL de suspension contient 25 mg d’azacitidine (100 mg/4 mL). Le produit reconstitué se présente sous la forme d’une suspension trouble homogène dépourvue d’agglomérats. Jeter la suspension si elle contient de grosses particules ou des agglomérats. Ne pas filtrer la suspension après reconstitution car cela pourrait éliminer la substance active. Tenir compte du fait que certains adaptateurs, dispositifs sans aiguille de type spikes et systèmes fermés sont équipés de filtres ; ces dispositifs ou systèmes ne doivent donc pas être utilisés pour l’administration du médicament reconstitué.
5. Nettoyer le dessus du bouchon et introduire une nouvelle seringue avec aiguille dans le flacon. Retourner le flacon et s’assurer que l’extrémité de l’aiguille se situe en dessous de la surface du liquide. Tirer le piston afin d’aspirer le volume de médicament correspondant à la dose appropriée, en veillant à expulser toute bulle d’air présente dans la seringue. Retirer la seringue avec aiguille du flacon et jeter l’aiguille.
6. Fixer solidement une aiguille pour injection sous-cutanée neuve (calibre 25 Gauge recommandé) sur la seringue. Afin de réduire l’incidence des réactions locales au site d’injection, l’aiguille ne doit pas être purgée avant l’injection.
7. Lorsque plus d’un flacon est nécessaire, réitérer les étapes ci-dessus pour achever la préparation de la suspension. Si la dose requiert plus d’un flacon, elle doit être répartie de façon égale, par exemple, pour une dose de 150 mg = 6 mL, 2 seringues de 3 mL chacune. Une petite quantité de suspension peut rester dans le flacon et l’aiguille et il peut ne pas être possible d’aspirer la totalité de la suspension du flacon.
8. Le contenu de la seringue doit être remis en suspension immédiatement avant l’administration. Au moment de l’injection, la température de la suspension doit atteindre environ 20 °C à 25 °C. Pour remettre le produit en suspension, faire rouler la seringue vigoureusement entre les paumes de la main jusqu’à obtenir une suspension trouble uniforme. Jeter la suspension si elle contient de grosses particules ou des agglomérats.
Conservation du médicament reconstitué
Pour une utilisation immédiate
La suspension d’AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable peut être préparée immédiatement avant utilisation et la suspension reconstituée doit être administrée dans les 45 minutes. Si ce délai de 45 minutes est dépassé, la suspension reconstituée doit être éliminée de façon appropriée et une nouvelle dose doit être préparée.
Pour une utilisation ultérieure
En cas de reconstitution avec de l’eau pour préparations injectables qui n’a pas été réfrigérée, la suspension reconstituée doit être placée immédiatement au réfrigérateur (entre 2 °C et 8 °C) après reconstitution et y être conservée pendant 8 heures maximum. Si ce délai de 8 heures dans le réfrigérateur est dépassé, la suspension doit être éliminée de façon appropriée et une nouvelle dose doit être préparée.
En cas de reconstitution avec de l’eau pour préparations injectables réfrigérée (entre 2 °C et 8 °C), la suspension reconstituée doit être placée immédiatement au réfrigérateur (entre 2 °C et 8 °C) après reconstitution et y être conservée pendant 22 heures maximum. Si ce délai de 22 heures dans le réfrigérateur est dépassé, la suspension doit être éliminée de façon appropriée et une nouvelle dose doit être préparée.
La seringue contenant la suspension reconstituée doit être laissée à température ambiante pendant 30 minutes avant l’administration jusqu’à ce qu’elle atteigne une température d’environ 20°C à 25 °C. Si ce délai de 30 minutes est dépassé, la suspension doit être éliminée de façon appropriée et une nouvelle dose doit être préparée.
Calcul d’une dose spécifique
La dose totale basée sur la surface corporelle peut être calculée ainsi :
Dose totale (mg) = dose (mg/m2) x surface corporelle (m²)
Le tableau suivant est proposé uniquement à titre d’exemple pour montrer comment calculer une dose d’azacitidine spécifique pour une surface corporelle moyenne de 1,8 m2.
|
Dose en mg/m2 (% de la dose initiale recommandée) |
Dose totale basée sur une surface corporelle de 1,8 m2 |
Nombre de flacons nécessaires |
Volume total de suspension reconstituée requis |
|
75 mg/m2 (100 %) |
135 mg |
2 flacons |
5,4 mL |
|
37,5 mg/m2 (50 %) |
67,5 mg |
1 flacon |
2,7 mL |
|
25 mg/m2 (33 %) |
45 mg |
1 flacon |
1,8 mL |
Mode d’administration
Ne pas filtrer la suspension après reconstitution.
Une fois reconstitué, AZACITIDINE HIKMA 25 mg/mL, poudre pour suspension injectable doit être injecté par voie sous-cutanée (introduire l’aiguille avec un angle de 45 à 90°) à l’aide d’une aiguille de calibre 25 Gauge dans le haut du bras, la cuisse ou l’abdomen.
Les doses supérieures à 4 mL doivent être injectées dans deux sites différents.
Les sites d’injection doivent être alternés. Chaque nouvelle injection doit être pratiquée à au moins 2,5 cm de distance du site précédent et en aucun cas sur une zone sensible, présentant une ecchymose, une rougeur ou une induration.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.